

Abstract
Neurodegenerative diseases are largely defined by protein aggregates in affected tissues. Aggregates contain some shared components as well as proteins thought to be specific for each disease. Aggregation has not previously been reported in the normal, aging heart or the hypertensive heart. Detergent-insoluble protein aggregates were isolated from mouse heart and characterized on 2D gels. Their levels increased markedly and significantly with aging and following sustained angiotensin-II-induced hypertension. Of the aggregate components identified by high-resolution proteomics, half changed in abundance with age (392/787) or with sustained hypertension (459/824), while 30% (273/901) changed concordantly in both, each P<0.05. One fifth of these proteins were previously associated with age-progressive neurodegenerative and/or cardiovascular diseases — e.g. ApoE, ApoJ, ApoA-IV, clusterin, complement C3, and others involved in stress-response and protein-homeostasis pathways. Since fibrosis is a characteristic of both aged and hypertensive hearts, we posited that aging of fibroblasts may contribute to the aggregates observed in cardiac tissue. Indeed, as cardiac myofibroblasts “senesced” (approached their replicative limit) in vitro, they accrued aggregates with many of the same constituent proteins observed in vivo during natural aging or sustained hypertension. In summary, we have shown for the first time that compact (detergent-insoluble) protein aggregates accumulate during natural aging, chronic hypertension and in vitro myofibroblast senescence, sharing many common proteins. Thus, aggregates that arise from disparate causes (aging, hypertension, replicative senescence) may have common underlying mechanisms of accrual.
Keywords: Aging (cardiac), Hypertension, Fibroblasts (cardiac), Protein aggregation, Protein homeostasis
Introduction
Deaths from atherosclerotic cardiovascular disease (CVD) comprise 31% of all mortality world-wide. Age and hypertension are the major risk factors for atherosclerotic CVD, and both are associated with increased stiffness of the heart. This rigidity, resulting in diastolic dysfunction, is largely attributed to myofibroblast growth and collagen deposition between cardiomyocytes. Knowledge of the mechanisms that contribute to cardiovascular aging would have profound clinical implication for CVD prevention, early detection, and development of therapies.
Most proteins adopt, either spontaneously or with the help of other proteins, specific folded structures with limited degrees of freedom. Chemically altered or misfolded structures, when they occur, are vulnerable to aggregation with other unstructured proteins (1). Although protein damage and misfolding are inevitable, multiple “proteostasis” systems are devoted to the repair or clearance of damaged proteins. The heart, in particular, is subject to continuous mechanical and metabolic stress; as a result, the cardiac proteome may be especially reliant on multi-level quality control to ensure proper folding and integrity of proteins (2). In diverse neuro-degenerative disorders, insoluble protein aggregates accrue in neuronal cytoplasm or nuclei (3–5) which are believed to comprise misfolded proteins that were not cleared by either chaperone-mediated refolding, the ubiquitin-proteasome pathway, or autophagy (6). Genetic disruption of these pathways can also lead to heritable cardiomyopathies that feature aggregate foci (7;8) While protein aggregation has been studied extensively in neurodegenerative diseases (9;10), aggregates that form during normal cardiac aging or sporadic CVD have not previously been characterized.
Aging is accompanied by a state of chronic inflammation termed “inflammaging” (11), which is also characteristic of many age-associated diseases such as atherosclerosis, neurodegenerative diseases and hypertension (12–14). Both cardiac aging and sustained hypertension feature elevated levels of reactive oxygen species (ROS), which have been implicated in CVD, chronic inflammation, and reduced biosynthesis and availability of nitric oxide (15).
The ubiquitin-proteasome system is the first line of defense for clearing misfolded and aggregated proteins from many tissues, including heart muscle (2;16). Despite extensive research on protein aggregation as a mediator of age-dependent functional decline, consideration of its possible role in atherosclerotic CVD has emerged only quite recently (17).
In the present study, we isolated and quantified compact aggregates from the hearts of young-adult and aged mice, and identified their protein constituents. To ask whether the hypertensive state itself disrupts proteostasis and thus mimics aging, we compared protein aggregates from hearts of young mice that were either hypertensive or normotensive. We also examined protein aggregation in early- and late-passage cardiac myofibroblasts, to assess whether their proteostasis is impaired during in vitro senescence and thus may contribute to cardiac senescence in vivo. Protein components of these aggregates were identified and quantified by high-resolution mass spectrometry, revealing a remarkable overlap in constituent proteins among aggregates formed during normal aging and angiotensin-II-induced hypertension, and (to a lesser extent) in myofibroblast replicative senescence.
Methods
We analyzed protein aggregates from young, aged, hypertensive hearts, and fibroblasts isolated from mice hearts. See online-only Data Supplement for detailed material and methods.
Results
Cardiac protein aggregates increase in abundance and complexity with age
Proteins were recovered and solubilized from cardiac-tissue aggregates, and resolved on 2-D gels (isoelectric focusing followed by SDS/acrylamide-gel electrophoresis). Results of representative experiments are shown in Fig. 1, panels A and B. Heart tissue from aged mice (30 months, panel B) showed a dramatic increase in the number and abundance of aggregated protein components, relative to those from young-adult mice (12 months, panel A). Combining data from three independent comparisons (panel C), hearts of older mice contained 4.5-fold more aggregated proteins than hearts from young-adult mice (P = 0.01 by 2-tailed t-test; each N=3).
Fig. 1.
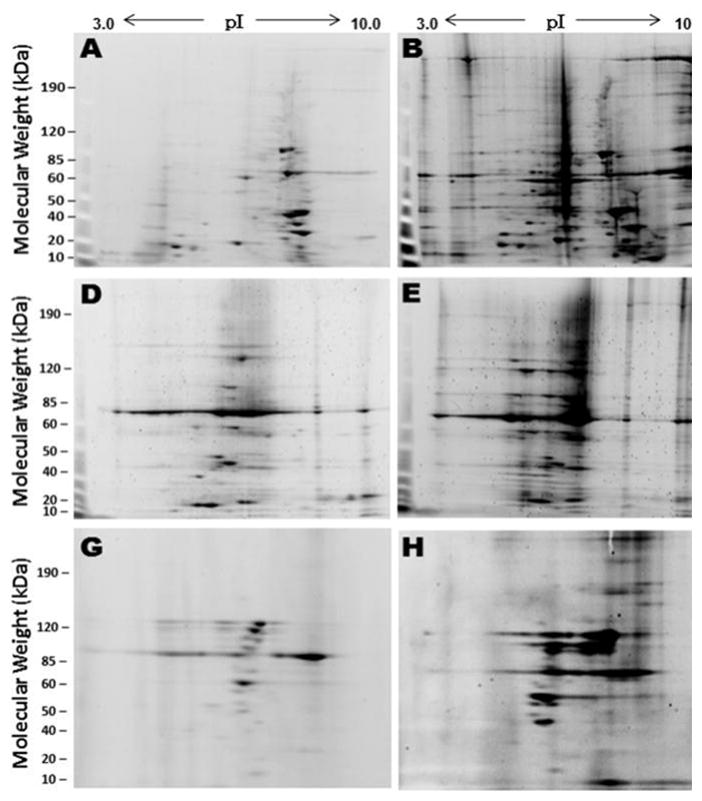
Aggregated proteins increase in cardiac cells with natural aging, hypertension, and in vitro senescence. Aggregates were isolated by differential centrifugation and sarcosyl-insolubility. Panels A, B, D, E, G and H show typical separations of proteins from hearts of young mice (A, 3.5 months old) or aged mice (B, 30 months), with data summarized in panel C; saline-infused normotensive young mice (D), angiotensin-II infused hypertensive young mice (E), data summarized in F. Aggregate proteins from mouse cardiac myofibroblasts at 3 MPD (early-passage, G) and 15 MPD (”senescent”, H), with combined data summarized in panel I. Histograms (panels C, F and I) present data as mean ± SD, for ImageJ quantitation of aggregate-protein signal from 3 different mice or in-vitro cultures.
Hypertension increases the quantity and diversity of aggregated cardiac proteins
Chronic hypertension mimics and accelerates many of the clinical indices of cardiac aging. Since Ang II infusion induces hypertension and initiates cardiac remodeling (cardiomyocyte hypertrophy and extensive fibrosis) (18), and hypertensive hearts show functional declines similar to those observed in naturally aged hearts (e.g. loss of compliance), we asked whether mouse hearts exposed to sustained hypertension might also show protein aggregates resembling those of aged hearts. Indeed, cardiac tissues of Ang II-infused mice showed a marked elevation in aggregated proteins relative to control (saline-treated) mice of the same age (Fig. 1, panels D and E). This increase averaged 3.2-fold for three independent comparisons (P<0.01 by 2-tailed t-test; see Fig. 1F). The patterns of aggregated proteins observed in 2D gels also became more complex and diverse after Ang II infusion relative to control hearts, but were not as complex as those of cardiac tissue from 30-month-old mice. Although it is not possible to correlate specific protein spots between these 2D profiles, the results suggest that young hypertensive hearts may contain a subset of the aggregate components that appear with normal aging.
Protein aggregation accompanies “replicative aging” of cardiac myofibroblasts
Fibroblasts play important role in maintaining cardiac structure and function, and predominate during cardiac remodeling that occurs with aging and pressure-overload situations such as sustained hypertension (21). We isolated fibroblasts from hearts of young mice (2.5 months), and propagated them in vitro for 15 mean population doublings (MPD), by which time their inter-division time had increased 2.5-fold, indicating replicative senescence. We isolated protein aggregates from myofibroblasts at 3 and 15 MPD, and separated their detergent-insoluble fractions from less compact and/or less stable protein complexes. Protein intensity and complexity increased substantially as cells approached their replicative limit. Representative examples of aggregated proteins from cardiac myofibroblasts are shown in Fig. 1, panels G and H. While the 2D protein patterns for cardiac myofibroblasts are rather different from those of heart tissue (Fig. 1, panels A,B,D and E), both the pattern complexity and abundance (intensity) of aggregated proteins increased with myofibroblast senescence, paralleling the changes observed in aged and hypertensive hearts. The mean increase (for two replicate cultures per group, expanded independently) in aggregate protein content with in vitro senescence was >2-fold (see Fig. 1; P = 0.01 by 2-tailed t-test).
Identification of aggregated proteins from hearts of normal young, aged, or hypertensive mice
Sarcosyl-insoluble protein aggregates isolated from mouse hearts (two per group) and cultured cardiac myofibroblasts (two cultures per group) were dissolved by heating in Laemmli buffer, containing a strong ionic detergent (SDS) and a strong reducing agent (β-mercaptoethanol). Constituent proteins of aggregates were then electrophoresed on denaturing polyacrylamide gels, digested with trypsin in excised gel slices, and peptides identified by high-resolution tandem mass spectrometry coupled to nanoflow liquid chromatography. Proteins with Mascot scores >90 (indicating >95% likelihood of correct identification) were considered to be present, and relative quantities of those proteins were estimated from spectral counts (peptide “hits”) summed for all peptides of each protein. This provides a crude measure of relative protein abundance, uncorrected for protein size (which affects hit frequency but can be neglected when comparing samples for abundance of the same protein).
As mice aged from 3.5 to 30 months, 155 proteins became more abundant while 237 proteins became less abundant in aggregates isolated from cardiac tissue (considering only changes that were significant at P<0.05, Fig. 2), far more than would be expected by chance in a total of 787 proteins identified. Hypertension led to similar shifts in the composition of cardiac aggregates, with 223 proteins increasing in abundance and 236 declining, relative to normotensive mice (out of 824 identified proteins). Among proteins that increased their abundance in aged hearts, 44% also increased in hearts from mice with sustained hypertension (considering only changes significant at P<0.05, Fig. 2). Moreover, 56% of proteins that diminished in aged hearts were also significantly reduced in hypertensive hearts. Conversely, 34% of proteins that became more abundant with hypertension also increased in aged hearts, while 61% of proteins that declined in hypertensive hearts also fell in aged hearts. Thus, there was substantial overlap between the shifts in the protein composition of aggregates accompanying aging and hypertension.
Fig. 2.
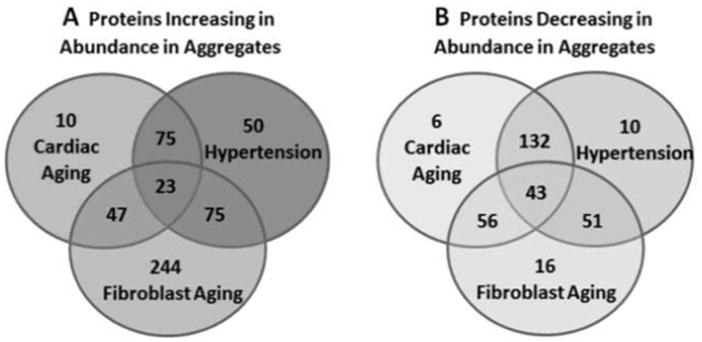
Venn diagrams indicate the overlap of identified proteins that increased (A) or decreased (B) significantly in abundance within cardiac aggregates, as mice aged naturally, or were made hypertensive by angiotensin-II infusion, or as cardiac myofibroblasts underwent replicative aging in vitro.
All proteins included in these totals differed with at least nominal significance (chi-squared P<0.05) for the indicated comparisons.
Although proteomics indicates more aggregate proteins decreasing than increasing with age (and nearly equal numbers with hypertension), there is no disagreement with the net declines consistently seen in 2D gel analyses (Fig. 1). Aggregates were initially scarce in hearts of young, untreated mice (Fig. 1, A and D), so the marked increases that accompany aging and angiotensin-II treatment were far more conspicuous than the declines.
Selection of proteins for validation
Six of the proteins identified in the aggregate proteomes of heart tissue (HSP90, fibronectin, cytochrome C, 14-3-3, ApoE and clusterin) were assessed by western blotting of fresh aggregates isolated from young, aged and hypertensive mouse hearts. As shown in Figure 3, the levels of these proteins indicated by immunodetection altered with age and hypertension, fully corroborating the proteomics data (indicated by “hit counts” superimposed over the histogram bars).
Fig. 3.

Representative western blots for HSP90, fibronectin, vimentin, 14-3-3, ApoE and Clusterin from young, aged and hypertensive mice. Multiple isoforms (e.g. for HSP90, 14-3-3, ApoE) are not distinguished. ImageJ quantitations of each protein are indicated by bars and the spectral counts identified by mass spectrometry are indicated by numbers above each bar. For each gel lane with maximum intensity is used as 100 percent. The significance is indicated by *, **= ≥0.01 and *= ≥0.05. Molecular weight of each protein HSP90 =90kD, Fibronectin -220kD, Cytochrome C - 15kD, 14-3-3 = 29kD, Apo E - 44kD and clusterin - 36kD.
Identification of aggregated proteins from cardiac myofibroblasts aging in vitro
A rather different picture emerged from the analysis of proteins in aggregates isolated from cultured cardiac myofibroblasts: 389 proteins increased in abundance with replicative senescence, while 166 declined. Overlaps among the aggregated proteins that displayed differential abundance in each comparison are shown as Venn diagrams (Fig. 2). The proteins that declined in senescent myofibroblasts were also likely to have decreased in aged hearts (58–60%), whereas proteins that rose in abundance coincided less frequently (15–18%).
Overlapping profiles of aggregate proteins that change in abundance with aging, hypertension, or in vitro senescence
Many of the proteins from sarcosyl-insoluble aggregates, identified by proteomics with high confidence (False Discovery Rate q<0.05) also shifted in abundance during aging and/or sustained hypertension (considering only changes significant at P<0.05, Fig. 2). The overlap of these sets was highly enriched for proteins previously shown to play roles in CVD or Alzheimer’s disease; these are listed in Table S1 along with their known functions and spectral counts (Supplementary Data). Abundance ratios for all proteins found in sarcosyl-insoluble aggregates, contrasting young-adult vs. aged, or normotensive vs. hypertensive hearts, or cultured cardiac myofibroblasts at 3 or 15 population doublings are shown in Table S2. Of 1393 total proteins identified with confidence (q<0.05) in any sample, 836 were enriched in at least one comparison (2267 significant changes, summed for three comparisons), whereas <70 per contrast (<210 for all three) would be expected to differ by chance at P<0.05. A substantial fraction of the proteins that accrued in aggregates from aged and hypertensive hearts had been previously implicated in atherosclerotic CVD (12.1%) and/or neurodegenerative diseases (6.2%) or other age-dependent diseases (2.1%), for a total of 20.4% (see Table S1 for references).
Proteins that were highly abundant in the aged heart, but rare or undetected in hypertension and senescent myofibroblasts, include apolipoprotein E, clusterin/ApoJ, complement C3, histones H1t, H2A and H2B, periostin, vitronectin and von Willebrand factor A protein. Among these, ApoE, clusterin and C3 have been implicated in both CVD and Alzheimer’s disease (ref. 38 and citations in Table S1).
Of particular interest are the 98 proteins that increased in heart aggregates with both natural aging and sustained hypertension; these include Apolipoprotein A-IV, atrial natriuretic factor, cardiac phospholamban, filamin’s A–C, lamin-A/C, myosin-binding protein C, myotilin, obscurin, 9 proteasome subunits, serotransferrin, synaptopodin-2-like, tropomyosin α-4 chain, vinculin, vitronectin, and xin/actin-binding protein 1.
Proteins that declined dramatically with natural aging and sustained hypertension include 14-3-3 zeta and epsilon (3 other isoforms also declined somewhat), α-actinin 3, annexin A6, cadherin 2, EH-domain protein 2, γ-enolase, HSP75, myosin 1, neurofilament L chain, sarcalumenin, sarcoplasmic/endoplasmic recticulum calcium ATPase 1 (SRCA1), Ser/Thr phosphatase 2A, SET & MYND-domain-containing protein 1, and ubiquitin-like modifier-activating enzyme 1. Two highly abundant proteins, myosin 1 and SRCA1, and five less-abundant ones (α-actinin-3, annexin A6, EH-domain protein 2, γ-enolase, HSP75,), were identified only in aggregates from young hearts. Many proteins that declined in aggregate abundance with age and/or hypertension are involved in stress responses and protein homeostasis, which are thought to be protective; these include SRCA1, α-actinin-3, γ enolase, glycogen phosphorylase, and heat shock proteins.
Gene ontology (GO, functional annotation) analysis has been used to identify pathways and processes implicated by transcriptomics and other “omics” analyses, by looking for enrichment of differentially represented molecular species in each category. We applied DAVID (v.6.7, david.abcc.ncifcrf.gov) analysis to proteins that were either over-represented or (separately) under-represented in aged hearts relative to young-adult hearts. The most significantly age-enriched categories (GO terms or pathways) are listed in Table S3, along with the corresponding data for GO/pathway enrichment in hypertensive heart and in vitro-aged myofibroblasts. Protein categories that were significantly enriched in all three contrasts, due to increased protein abundance in aggregates (Table S3A) and/or decreased abundance (Table S3C), include contractile fiber, proteasome complex, non-membrane bounded organelle, Alzheimer disease, actin binding, methylation, phosphoprotein, UBL conjugation, mitochondrion, transit peptide, acetylation, cytoskeleton, and ATP-binding. Categories significantly enriched with hypertension but not with cardiac aging are also listed in Table S3, B and D.
Discussion
Aging, hypertension, and proteostasis
Protein misfolding is a phenomenon observed across all taxa, from bacteria to humans. Nearly a third of newly synthesized proteins misfold, and fail to refold with the help of chaperones (22). Under normal conditions, irreversibly misfolded proteins are cleared by proteasomes; however, aging alters rates of synthesis, modification, folding, processing and degradation of proteins (3). Deficiencies in protein quality control can lead to accumulation of aggregates, which are believed to underlie the development of diverse neurodegenerative diseases (23;24;). In these disease states, proteins coalesce into insoluble aggregates that accumulate with age and/or disease progression. Examples include beta-amyloid and tau aggregates (Alzheimer’s disease); α-synuclein, parkin, DJ-1 and PINK-1 (Parkinson’s disease); huntingtin, ataxins and other proteins with long polyglutamine tracts (Huntington’s disease); mutant forms of actinin-4 (renal failure); and superoxide dismutase (ALS). Cytotoxicity of these protein aggregates has been attributed to various factors, such as membrane permeabilization (25), oxidative and endoplasmic-reticulum stress (26;27), and mitochondrial dysfunction (28).
Proteasome activity and quality-control have also been reported to be impaired in the aged heart (16;29;30). Since aging is a prominent risk factor for both atherosclerotic cardiovascular disease and neurodegenerative diseases, we asked whether age-dependent protein aggregation might occur in the heart. Indeed, we observed that aggregates do form in mouse heart and accumulate with age (in vivo and in vitro), and likewise with sustained hypertension. We identified many proteins in cardiac aggregates that are components of the ubiquitin-proteasome system and/or autophagy (see Tables S1 and S2), clearance systems for misfolded proteins that undergo functional declines in cardiac aging and disease (32;32). Analysis of the protein composition of aggregates revealed nearly 400 constituent proteins that differ markedly in abundance between young and old mouse hearts, despite the paucity of age-dependent differences in the total cardiac proteome (30). Proteins that increased with age were on average 5.1-fold (range of 2- to 50-fold) more abundant in hearts from aged mice, and those that decreased in abundance with age declined by nearly 3-fold (range, 16% to 25-fold). Changes in protein abundance with hypertension were almost as impressive, averaging 4.6-fold for those that increased, and 2-fold for those that decreased. It is noteworthy that roughly half of the proteins that increased (or decreased) in aged hearts were also enriched (or depleted) in hypertensive hearts. These observations strongly suggest that aging and hypertension feature similar disruptions of protein homeostasis. Some of the interesting differential hits and their roles in diverse physiological processes are compiled in Tables S1–S3.
Previous studies have sought changes with age in the total proteome of cardiac tissues from mice and rats. Significant shifts in abundance were remarkably rare, comprising <0.01% to 3% of the total proteins identified, and these changes were relatively modest in degree, ranging from 1.2- to 2.5-fold (30;33). No significant changes were seen in protein turnover, among >800 proteins monitored in hearts of young and aged rodents (34). In contrast, we found far larger and more frequent changes with age, among proteins isolated from “compact” (detergent-insoluble) protein aggregates. The striking contrast in outcomes for whole-proteome vs. “aggregome” discovery implies that aging primarily alters the susceptibility of proteins to aggregate, rather than their synthesis, degradation, or steady-state levels. Further, our observation of significant overlap in protein aggregates between hypertensive and aging hearts suggests that hypertension may in some respects be considered to accelerate or mimic cardiac aging. Physiologically, many functional characteristics of the hypertensive heart mirror those seen with cardiac aging, such as fibrosis and impaired diastolic relaxation (35;36).
Since fibrosis results from proliferation and collagen synthesis by fibroblasts, we speculated that protein aggregates in senescent fibroblasts might bear some similarity to those in hypertensive and aged hearts. Although there were many differences, a surprisingly large proportion of aggregated proteins that decreased with myofibroblast aging (150 out of 166, or 90%) also declined in heart aggregates with aging and/or hypertension. Since most of these proteins appear likely to play protective roles, we interpret this as evidence that replicative senescence of cardiac myofibroblasts has one critical feature in common with the heart’s responses to aging and hypertension: the deterioration of proteostasis-maintenance machinery.
Implications of specific proteins found to accumulate in aggregates
Proteomic analysis of aggregated proteins indicated marked alterations with age and/or hypertension in many proteins previously associated with cardiovascular pathology: these comprise the bulk of proteins listed in Table S1. Some of these, and numerous additional proteins (see Tables S1 – S3), have been implicated in neurodegenerative diseases such as Alzheimer’s; these include specific 14-3-3 proteins and apolipoprotein E, clusterin, complement C3, H1 histones, HSP90 α & β, catalase, laminin γ-1, NADH-ubiquinone oxidoreductase, PAI-1, peripherin, proteasome α subunits, transthyretin, vimentin, and vitamin D-binding protein.
Specific apolipoprotein species have been implicated in multiple age-related diseases including Alzheimer disease, certain cancers, diabetes, and renal disease (37–40). Apolipoprotein A-I (ApoA1) predicts CVD risk, while ApoA-IV has antioxidant and anti-atherogenic properties. ApoE isoforms are valuable biomarkers of susceptibility to atherosclerotic cardiovascular disease (41;42); the ApoEε4 allele markedly increases (and the ε2 allele decreases) risk of both CVD and Alzheimer Disease (43). In the current study, apolipoproteins A-I and A-IV were enriched in aggregates of hypertensive hearts, whereas ApoE was exclusively abundant in aged heart (164 hits, vs. 0 for young and HT). Clusterin (apolipoprotein J), an extracellular chaperone thought to oppose aggregation of plasma proteins, was similarly quite abundant in aggregates from old and hypertensive heart, perhaps indicating its induction by nacent aggregates, but was absent from young cardiac aggregates.
Protective/proteostatic heat-shock proteins HSP70, HSP90α and HSP90β were significantly depleted in aggregates from aged hearts, while HSP75, a mitochondrial HSP that protects against cardiac hypertrophy and fibrosis (44;45), was absent from both aged and hypertensive heart aggregates (Table S1). Several large heat shock proteins, especially HSP70 and HSP60, are elevated in the offspring of centenarians relative to age-matched controls (46) supporting protective functions. In contrast, the small heat shock proteins HSP-β6, -β7 and -β8 are more abundant in aged-heart aggregates (see Tables S1, S2). They co-localize with amyloid plaques in neurodegen-erative diseases [47–48], suggesting entrapment in aggregates and/or roles in reducing aggregate toxicity.
Previous reports have indicated that expression in heart declines with rat age for actin, tropomyosin and troponin — proteins responsible for cardiac contractility [32]. We found more tropomyosin in the aggregates of aged and hypertensive hearts, suggesting that increased sequestration of tropomyosin within aggregates may contribute to the decreased contractility of aged and hypertensive hearts. Periostin is also more abundant in heart with both aging and hypertension, and in cardiac myofibroblasts with replicative senescence. Periostin is a major TGFβ target involved in tissue remodeling and angiogenesis following injury (49;50); indeed, cardiac healing is impaired in periostin-knockout mice after acute myocardial infarction (51). Genomewide association studies found periostin alleles to be strongly associated with early-onset atherosclerosis (52). Sequestration of periostin in aggregates may contribute to age- and hypertension-impaired healing of cardiac injury.
Of 11 proteasome subunits identified in cardiac aggregates, 9 were enriched (and 2 depleted) with aging, and 10 were enriched with hypertension (Tables S1 and S2). Proteasome activity declines with age and under conditions of high protein aggregation, and appears to involve entrapment of proteasomes within aggregates [1]. Aggregation of proteasomes would impair protein degradation, consistent with the reported decline in proteasome activity with cardiac aging (32). Complement C3, a marker of inflammation, was elevated 14-fold in aggregates from aged heart. C3 is activated in age-related macular degeneration (53–55), and is associated with increased amyloid-plaque deposition (56).
Lessons from GO meta-analysis
GO analysis of the aggregate proteome can provide insight into specific pathways, processes, and cell structures that contribute to aggregation. By their nature, however, they ignore “random” events as well as processes that depend on unannotated features of proteins, such as intrinsic disorder, low thermal stability, hydrophobicity, or propensity for disruptive post-synthetic modifications. Meta-analysis of aggregate proteins that increased with age (Table S3A) highlighted categories for extracellular matrix, contractile fiber, nucleosome, protein:DNA complex, and proteasome complex (enriched 19- to 27-fold; each P<2E–9). Intriguingly, Alzheimer disease-related proteins were enriched 7.4-fold (P<2E–8). Among categories of proteins decreasing with age (Table S3B), mitochondrion, acetylation, generation of precursor metabolites, transit peptides, and cell respiration were enriched 5.5- to 37-fold (each P<E–15). Proteins associated with Parkinson Disease (11-fold, P≈2E–12) and Huntington Disease (8-fold, P≈3E–10) were not far behind, suggesting that proteostasis-protection pathways commonly defective in those diseases are also impaired with cardiac aging.
Hypertension as a caricature of normal cardiovascular aging
GO categories (Table S3), like individual proteins (Tables S1 and S2), underscore the many commonalities in aggregate composition between cardiac aging and hypertension, and (to a lesser extent) in vitro aging of cardiac myofibroblasts (Fig. 2). The overlap in protein-aggregate composition between hypertensive and aged hearts is especially remarkable, suggesting that hypertension, a known risk factor for cardiovascular disease, is linked to age-associated CVD risk through protein aggregation. The observation that angiotensin-induced hypertension recapitulated many of the same aggregate characteristics seen in normal aging, but at a young age, implies that hypertension may be the proximal culprit, contributing many causal factors exacerbated by aging. Although we cannot formally exclude the alternative possibility that hypertension itself accelerates cardiac aging, this would be a tenuous hypothesis on purely mechanistic grounds. Further, some age-dependent changes in protein aggregation were also seen during myofibroblast senescence. With both aging and sustained hypertension, fibroblasts proliferate and contribute to fibrosis; this could account for part of the observed overlap in protein-aggregate composition between cardiac aging and hypertension. Other differences, not explained by myofibroblasts, could be attributed to changes in other cell types, e.g. cardiomyocytes and/or endothelial cells.
Aggregation links cardiovascular aging to age-progressive neuropathies
We were particularly intrigued by similarities in protein-aggregation levels and constituents in aged or hypertensive hearts, to those reported in neurodegenerative diseases (Table S1). It was recently speculated that cardiac aging may be considered as “Alzheimer’s disease of the heart” [17], and our observations may be the strongest evidence to date for this proposition. A further, unforeseen finding is that hypertensive hearts have much in common with two other aggregation-defined neurodegenerative diseases, Parkinson’s and Huntington’s (Table S3B). This implies that cardiac aggregation accompanying aging or hypertension reflects a molecular pathology remarkably similar to progressive neurological diseases. The current studies provide, for the first time, a plausible mechanism to explain this puzzling convergence between such disparate but highly age-associated diseases: Aggregates form as a concomitant of aging, but produce distinct pathologies in each susceptible tissue.
Clinical perspective
Protein damage and misfolding are common features of aging-related neurologic disorders like Alzheimer’s disease and Huntington chorea. These disorders in “proteostasis” may be the basis of aging in general, and may affect the heart as well. Hypertensive and aged hearts show similar dysfunction of the heart related most likely to the development of fibrosis. The present study revealed that indeed there are significant changes in proteostasis in the aged heart as well as in the hypertensive heart. Further, there was a significant overlap in upregulated and downregulated proteins in aged heart and the hypertensive heart. Senescent myofibroblasts in culture also showed a significant overlap in disorders of proteostasis in the aged heart and the hypertensive heart. Lastly, the aged and hypertensive heart shared some features of the aged brain in neurologic disorders. Thus hypertension appears to be caricature of normal cardiovascular aging, and both conditions may be considered “Alzheimer’s” of the heart.
Supplementary Material
Novelty and Significance.
What Is New?
There are significant changes in proteostasis in the aged heart and in the hypertensive heart. Both involve the formation of detergent-insoluble aggregates, not previously reported.
There was a significant overlap of aggregated proteins in aged heart and the hypertensive heart.
Senescent myofibroblasts in culture showed a lesser but significant overlap in aggregate proteins with those of aged and hypertensive hearts.
What Is Relevant?
Based on this proteomic study, hypertension appears to accelerate or mimic some aspects of normal physiologic aging.
Aggregates formed in the aged heart and the hypertensive heart share some features with the aged brain, in particular features that are markedly increased in neurologic disorders.
Summary
Both aging and chronic hypertension lead to the accumulation of similar aggregates in the heart, many of which overlap the aggregates that occur in the cortex of Alzheimer’s patients.
Acknowledgments
Funding sources
This research was supported by NIH grants R01GM106024, R33CA173264, UL1TR000039, P30GM103450 and P20GM103429 (to JLM); VA Merit Review grants (to RJSR and JLM); a VA Senior Research Career Scientist Award (to RJSR); and a subaward to SA from an NIA Claude Pepper Center grant P30-AG028718 (J. Wei, P.I.).
We acknowledge support from the UAMS Proteomics Core Facility, within the UAMS Department of Biochemistry and Molecular Biology. We thank Dr. Steven W. Barger (UAMS) for antibodies to apolipoprotein E, fibronectin and 14-3-3 proteins. We also thank Ramani Alla for superb technical assistance.
Footnotes
Disclosures
The authors have no conflicts of interest with regard to this study. The views presented herein do not necessarily reflect those of the Department of Veteran Affairs
Reference List
- 1.Ayyadevara S, Balasubramaniam M, Gao Y, Yu LR, Alla R, Shmookler RR. Proteins in aggregates functionally impact multiple neurodegenerative disease models by forming proteasome-blocking complexes. Aging Cell. 2014;14:35–48. doi: 10.1111/acel.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glembotski CC. Clarifying the cardiac proteasome paradox: protein quality control. Circ Res. 2012;111:509–512. doi: 10.1161/CIRCRESAHA.112.275917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morawe T, Hiebel C, Kern A, Behl C. Protein homeostasis, aging and Alzheimer’s disease. Mol Neurobiol. 2012;46:41–54. doi: 10.1007/s12035-012-8246-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nussbaum-Krammer CI, Morimoto RI. Caenorhabditis elegans as a model system for studying non-cell-autonomous mechanisms in protein-misfolding diseases. Dis Model Mech. 2014;7:31–39. doi: 10.1242/dmm.013011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soto C, Estrada LD. Protein misfolding and neurodegeneration. Arch Neurol. 2008;65:184–189. doi: 10.1001/archneurol.2007.56. [DOI] [PubMed] [Google Scholar]
- 6.Marques C, Guo W, Pereira P, Taylor A, Patterson C, Evans PC, Shang F. The triage of damaged proteins: degradation by the ubiquitin-proteasome pathway or repair by molecular chaperones. FASEB J. 2006;20:741–743. doi: 10.1096/fj.05-5080fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boelens WC. Cell biological roles of alphaB-crystallin. Prog Biophys Mol Biol. 2014;115:3–10. doi: 10.1016/j.pbiomolbio.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 8.Tajsharghi H, Oldfors A. Myosinopathies: pathology and mechanisms. Acta Neuropathol. 2013;125:3–18. doi: 10.1007/s00401-012-1024-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hindle JV. Ageing, neurodegeneration and Parkinson’s disease. Age Ageing. 2010;39:156–161. doi: 10.1093/ageing/afp223. [DOI] [PubMed] [Google Scholar]
- 10.Meyerhof W, Richter D. Neurodegeneration--from multiple sclerosis to Alzheimer’s disease. FEBS J. 2013;280:4337. doi: 10.1111/febs.12430. [DOI] [PubMed] [Google Scholar]
- 11.Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128:92–105. doi: 10.1016/j.mad.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 12.Li Z, Jansen M, Pierre SR, Figueiredo-Pereira ME. Neurodegeneration: linking ubiquitin/proteasome pathway impairment with inflammation. Int J Biochem Cell Biol. 2003;35:547–552. doi: 10.1016/s1357-2725(02)00384-9. [DOI] [PubMed] [Google Scholar]
- 13.Sesso HD, Buring JE, Rifai N, Blake GJ, Gaziano JM, Ridker PM. C-reactive protein and the risk of developing hypertension. JAMA. 2003;290:2945–2951. doi: 10.1001/jama.290.22.2945. [DOI] [PubMed] [Google Scholar]
- 14.Spagnoli LG, Bonanno E, Sangiorgi G, Mauriello A. Role of inflammation in atherosclerosis. J Nucl Med. 2007;48:1800–1815. doi: 10.2967/jnumed.107.038661. [DOI] [PubMed] [Google Scholar]
- 15.Cachofeiro V, Goicochea M, de Vinuesa SG, Oubina P, Lahera V, Luno J. Oxidative stress and inflammation, a link between chronic kidney disease and cardiovascular disease. Kidney Int Suppl. 2008:S4–S9. doi: 10.1038/ki.2008.516. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Robbins J. Heart failure and protein quality control. Circ Res. 2006;99:1315–1328. doi: 10.1161/01.RES.0000252342.61447.a2. [DOI] [PubMed] [Google Scholar]
- 17.Willis MS, Patterson C. Proteotoxicity and cardiac dysfunction--Alzheimer’s disease of the heart? N Engl J Med. 2013;368:455–464. doi: 10.1056/NEJMra1106180. [DOI] [PubMed] [Google Scholar]
- 18.Hu C, Dandapat A, Sun L, Marwali MR, Inoue N, Sugawara F, Inoue K, Kawase Y, Jishage K, Suzuki H, Hermonat PL, Sawamura T, Mehta JL. Modulation of angiotensin II-mediated hypertension and cardiac remodeling by lectin-like oxidized low-density lipoprotein receptor-1 deletion. Hypertension. 2008;52:556–562. doi: 10.1161/HYPERTENSIONAHA.108.115287. [DOI] [PubMed] [Google Scholar]
- 19.Chen J, Li D, Zhang X, Mehta JL. Tumor necrosis factor-alpha-induced apoptosis of human coronary artery endothelial cells: modulation by the peroxisome proliferator-activated receptor-gamma ligand pioglitazone. J Cardiovasc Pharmacol Ther. 2004;9:35–41. doi: 10.1177/107424840400900i106. [DOI] [PubMed] [Google Scholar]
- 20.Byrum SD, Larson SK, Avaritt NL, Moreland LE, Mackintosh SG, Cheung WL, Tackett AJ. Quantitative Proteomics Identifies Activation of Hallmark Pathways of Cancer in Patient Melanoma. J Proteomics Bioinform. 2013;6:43–50. doi: 10.4172/jpb.1000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–278. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 22.Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- 23.Cuervo AM, Wong ES, Martinez-Vicente M. Protein degradation, aggregation, and misfolding. Mov Disord. 2010;25(Suppl 1):S49–S54. doi: 10.1002/mds.22718. [DOI] [PubMed] [Google Scholar]
- 24.Dimcheff DE, Portis JL, Caughey B. Prion proteins meet protein quality control. Trends Cell Biol. 2003;13:337–340. doi: 10.1016/s0962-8924(03)00125-9. [DOI] [PubMed] [Google Scholar]
- 25.Lashuel HA. Membrane permeabilization: a common mechanism in protein-misfolding diseases. Sci Aging Knowledge Environ. 2005;2005(38):e28. doi: 10.1126/sageke.2005.38.pe28. [DOI] [PubMed] [Google Scholar]
- 26.Jiang P, Gan M, Ebrahim AS, Lin WL, Melrose HL, Yen SH. ER stress response plays an important role in aggregation of alpha-synuclein. Mol Neurodegener. 2010;5:56. doi: 10.1186/1750-1326-5-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol. 2010;11:777–788. doi: 10.1038/nrm2993. [DOI] [PubMed] [Google Scholar]
- 28.Xie W, Wan OW, Chung KK. New insights into the role of mitochondrial dysfunction and protein aggregation in Parkinson’s disease. Biochim Biophys Acta. 2010;1802:935–941. doi: 10.1016/j.bbadis.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 29.Su H, Wang X. The ubiquitin-proteasome system in cardiac proteinopathy: a quality control perspective. Cardiovasc Res. 2010;85:253–262. doi: 10.1093/cvr/cvp287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walther DM, Mann M. Accurate quantification of more than 4000 mouse tissue proteins reveals minimal proteome changes during aging. Mol Cell Proteomics. 2011;10:M110. doi: 10.1074/mcp.M110.004523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bulteau AL, Szweda LI, Friguet B. Age-dependent declines in proteasome activity in the heart. Arch Biochem Biophys. 2002;397:298–304. doi: 10.1006/abbi.2001.2663. [DOI] [PubMed] [Google Scholar]
- 32.De Meyer GR, De Keulenaer GW, Martinet W. Role of autophagy in heart failure associated with aging. Heart Fail Rev. 2010;15:423–430. doi: 10.1007/s10741-010-9166-6. [DOI] [PubMed] [Google Scholar]
- 33.Grant JE, Bradshaw AD, Schwacke JH, Baicu CF, Zile MR, Schey KL. Quantification of protein expression changes in the aging left ventricle of Rattus norvegicus. J Proteome Res. 2009;8:4252–4263. doi: 10.1021/pr900297f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dai DF, Karunadharma PP, Chiao YA, Basisty N, Crispin D, Hsieh EJ, Chen T, Gu H, Djukovic D, Raftery D, Beyer RP, MacCoss MJ, Rabinovitch PS. Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell. 2014;13:529–539. doi: 10.1111/acel.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan V, Fenning A, Levick SP, Loch D, Chunduri P, Iyer A, Teo YL, Hoey A, Wilson K, Burstow D, Brown L. Cardiovascular changes during maturation and ageing in male and female spontaneously hypertensive rats. J Cardiovasc Pharmacol. 2011;57:469–478. doi: 10.1097/FJC.0b013e3182102c3b. [DOI] [PubMed] [Google Scholar]
- 36.Cheitlin MD. Cardiovascular physiology-changes with aging. Am J Geriatr Cardiol. 2003;12:9–13. doi: 10.1111/j.1076-7460.2003.01751.x. [DOI] [PubMed] [Google Scholar]
- 37.Bonomini F, Filippini F, Hayek T, Aviram M, Keidar S, Rodella LF, Coleman R, Rezzani R. Apolipoprotein E and its role in aging and survival. Exp Gerontol. 2010;45:149–157. doi: 10.1016/j.exger.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 38.Keeney JT, Swomley AM, Forster S, Harris JL, Sultana R, Butterfield DA. Apolipoprotein A-I: insights from redox proteomics for its role in neurodegeneration. Proteomics Clin Appl. 2013;7:109–122. doi: 10.1002/prca.201200087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kulminski AM, Culminskaya I, Arbeev KG, Ukraintseva SV, Arbeeva L, Yashin AI. Trade-off in the effect of the APOE gene on the ages at onset of cardiocascular disease and cancer across ages, gender, and human generations. Rejuvenation Res. 2013;16:28–34. doi: 10.1089/rej.2012.1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soiland H, Soreide K, Janssen EA, Korner H, Baak JP, Soreide JA. Emerging concepts of apolipoprotein D with possible implications for breast cancer. Cell Oncol. 2007;29:195–209. doi: 10.1155/2007/487235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Erqou S, Thompson A, Di AE, Saleheen D, Kaptoge S, Marcovina S, Danesh J. Apolipoprotein(a) isoforms and the risk of vascular disease: systematic review of 40 studies involving 58,000 participants. J Am Coll Cardiol. 2010;55:2160–2167. doi: 10.1016/j.jacc.2009.10.080. [DOI] [PubMed] [Google Scholar]
- 42.Pedro-Botet J, Senti M, Auguet T, Nogues X, Rubies-Prat J, Aubo C, Vidal-Barraquer F. Apolipoprotein(a) genetic polymorphism and serum lipoprotein(a) concentration in patients with peripheral vascular disease. Atherosclerosis. 1993;104:87–94. doi: 10.1016/0021-9150(93)90179-x. [DOI] [PubMed] [Google Scholar]
- 43.Hofman A, Ott A, Breteler MM, Bots ML, Slooter AJ, van HF, van Duijn CN, Van BC, Grobbee DE. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer’s disease in the Rotterdam Study. Lancet. 1997;349:151–154. doi: 10.1016/S0140-6736(96)09328-2. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y, Jiang DS, Yan L, Cheng KJ, Bian ZY, Lin GS. HSP75 protects against cardiac hypertrophy and fibrosis. J Cell Biochem. 2011;112:1787–1794. doi: 10.1002/jcb.23091. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Jiang DS, Yan L, Cheng KJ, Bian ZY, Lin GS. HSP75 protects against cardiac hypertrophy and fibrosis. J Cell Biochem. 2011;112:1787–1794. doi: 10.1002/jcb.23091. [DOI] [PubMed] [Google Scholar]
- 46.Terry DF, McCormick M, Andersen S, Pennington J, Schoenhofen E, Palaima E, Bausero M, Ogawa K, Perls TT, Asea A. Cardiovascular disease delay in centenarian offspring: role of heat shock proteins. Ann N Y Acad Sci. 2004;1019:502–505. doi: 10.1196/annals.l297.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilhelmus MM, Boelens WC, Otte-Holler I, Kamps B, de Waal RM, Verbeek MM. Small heat shock proteins inhibit amyloid-beta protein aggregation and cerebrovascular amyloid-beta protein toxicity. Brain Res. 2006;1089:67–78. doi: 10.1016/j.brainres.2006.03.058. [DOI] [PubMed] [Google Scholar]
- 48.Wilhelmus MM, Otte-Holler I, Wesseling P, de Waal RM, Boelens WC, Verbeek MM. Specific association of small heat shock proteins with the pathological hallmarks of Alzheimer’s disease brains. Neuropathol Appl Neurobiol. 2006;32:119–130. doi: 10.1111/j.1365-2990.2006.00689.x. [DOI] [PubMed] [Google Scholar]
- 49.Hakuno D, Kimura N, Yoshioka M, Mukai M, Kimura T, Okada Y, Yozu R, Shukunami C, Hiraki Y, Kudo A, Ogawa S, Fukuda K. Periostin advances atherosclerotic and rheumatic cardiac valve degeneration by inducing angiogenesis and MMP production in humans and rodents. J Clin Invest. 2010;120:2292–2306. doi: 10.1172/JCI40973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lindner V, Wang Q, Conley BA, Friesel RE, Vary CP. Vascular injury induces expression of periostin: implications for vascular cell differentiation and migration. Arterioscler Thromb Vasc Biol. 2005;25:77–83. doi: 10.1161/01.ATV.0000149141.81230.c6. [DOI] [PubMed] [Google Scholar]
- 51.Shimazaki M, Nakamura K, Kii I, Kashima T, Amizuka N, Li M, Saito M, Fukuda K, Nishiyama T, Kitajima S, Saga Y, Fukayama M, Sata M, Kudo A. Periostin is essential for cardiac healing after acute myocardial infarction. J Exp Med. 2008;205:295–303. doi: 10.1084/jem.20071297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hixson JE, Shimmin LC, Montasser ME, Kim DK, Zhong Y, Ibarguen H, Follis J, Malcom G, Strong J, Howard T, Langefeld C, Liu Y, Rotter JI, Johnson C, Herrington D. Common variants in the periostin gene influence development of atherosclerosis in young persons. Arterioscler Thromb Vasc Biol. 2011;31:1661–1667. doi: 10.1161/ATVBAHA.111.224352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Despriet DD, van Duijn CM, Oostra BA, Uitterlinden AG, Hofman A, Wright AF, ten Brink JB, Bakker A, de Jong PT, Vingerling JR, Bergen AA, Klaver CC. Complement component C3 and risk of age-related macular degeneration. Ophthalmology. 2009;116:474–480. doi: 10.1016/j.ophtha.2008.09.055. [DOI] [PubMed] [Google Scholar]
- 54.Hoh KJ, Lenassi E, Malik TH, Pickering MC, Jeffery G. Complement component C3 plays a critical role in protecting the aging retina in a murine model of age-related macular degeneration. Am J Pathol. 2013;183:480–492. doi: 10.1016/j.ajpath.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 55.Seddon JM, Yu Y, Miller EC, Reynolds R, Tan PL, Gowrisankar S, Goldstein JI, Triebwasser M, Anderson HE, Zerbib J, Kavanagh D, Souied E, Katsanis N, Daly MJ, Atkinson JP, Raychaudhuri S. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat Genet. 2013;45:1366–1370. doi: 10.1038/ng.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res. 2001;73:887–896. doi: 10.1006/exer.2001.1094. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.