

Abstract
Brain tumors represent the most common solid tumor of childhood, with gliomas comprising the largest fraction of these cancers. Several features distinguish them from their adult counterparts, including their natural history, causative genetic mutations, and brain locations. These unique properties suggest that the cellular and molecular etiologies that underlie their development and maintenance might be different from those that govern adult gliomagenesis and growth. In this review, we discuss the genetic basis for pediatric low-grade and high-grade glioma in the context of developmental neurobiology, and highlight the differences between histologically-similar tumors arising in children and adults.
Keywords: pilocytic astrocytoma, diffuse astrocytoma, glioblastoma, DIPG, pediatric glioma
Unique biological and clinical features highlight childhood glioma as a developmental disease
The commonest solid tumors in childhood are brain tumors. Among adults, the situation is very different; the majority of cancers are carcinomas, and primary brain tumors account for only 2% of the total cancer burden. In addition, the types of primary brain tumor arising in adults and children differ significantly. Meningiomas and the high-grade glioblastoma dominate in adults, while low-grade gliomas (LGGs) and embryonal tumors, such as the medulloblastoma, are most numerous in children (Ostrom et al. 2014). A further clear distinction between brain tumors in adults and children is where they arise. Approximately 75% of adult tumors form in the supratentorial compartment, while the same proportion of pediatric tumors develops in the posterior fossa. Common sites for pediatric brain tumors are the cerebellum, brainstem, and diencephalon, but these are all rare sites for adult tumors (Jones and Baker 2014; Sturm et al. 2014). Finally, the biological behavior of histopathologically-related tumors in children and adults can vary considerably, as exemplified by the diffuse astrocytoma (DA), which is classified as a grade II tumor according to the World Health Organization (WHO) classification system (Louis et al. 2007b). DAs are infiltrative LGGs composed of atypical cells with an inconsistent astrocytic phenotype. DAs presenting in childhood look microscopically like their adult counterparts, yet exhibit distinct biological behavior. Very few pediatric DAs undergo pathological progression to a diffuse astrocytic tumor of higher grade, an anaplastic astrocytoma (WHO grade III) or glioblastoma (WHO grade IV) (Broniscer et al. 2007). In contrast, up to 75% of adult DAs in a separate patient series transformed into high-grade astrocytic tumors.
Gliomas are the most frequently encountered pediatric brain tumor. From histopathological and therapeutic perspectives, they are also heterogeneous, encompassing low-grade tumors that are generally curable, such as cerebellar pilocytic astrocytoma (PA), and fatal high-grade tumors, like diffuse intrinsic pontine glioma (DIPG). Most common among LGGs is the PA (WHO grade I), accounting for 85% of these tumors. PAs arise mainly in the cerebellum and in the hypothalamic/chiasmatic region. Most of the remaining LGGs are DAs and pleomorphic xanthoastrocytomas (PXAs); a diffuse oligodendroglioma or oligoastrocytoma is rare in children. The diffuse LGGs share an infiltrative nature with pediatric high-grade gliomas (HGGs), anaplastic astrocytoma and glioblastoma. The diffuse gliomas occur principally in the cerebral hemispheres, thalamus, and brainstem. In the brainstem, diffuse gliomas nearly always occur within the pons (DIPG) (Louis et al. 2007a), in contrast to circumscribed WHO grade I tumors, which occur most frequently in the midbrain and cervicomedullary region. Clinicopathological distinctions between pediatric and adult brain tumors reflect fundamental differences in oncogenesis – particularly the range and nature of genetic alterations, cellular environment, and cell of origin.
Postnatal gliogenesis and unique progenitor cell pools
The cellular composition and growth environment are profoundly different in the pediatric brain relative to its adult counterpart. There is significant experimental evidence that neural stem cells, glial progenitor cells, oligodendrocyte progenitor cells, and possibly differentiated astrocytes can serve as cells of origin for glioma (Zong et al. 2015). The pools of neural stem and progenitor cells and their rates of proliferation vary widely between the developing versus mature brain (Rowitch and Kriegstein 2010). Gliogenesis occurs in several waves, which have been most extensively studied in rodents (Figure 1). A shift in differentiation patterns of radial glia from neurogenesis to gliogenesis occurs in the ventricular zone at embryonic day 16–18 in the developing mouse brain, giving rise to an initial burst of intermediate glial progenitors. However, the majority of gliogenesis occurs in the first 3 weeks postnatally (reviewed in (Molofsky and Deneen 2015; Takebayashi and Ikenaka 2015)). Oligodendrocyte progenitor cells (OPCs) arise from the medial ganglionic eminence and anterior entopeduncular area and migrate in streams to populate the telencephalon including the developing cerebral cortex around E12, followed by a second wave of OPCs produced from the lateral and caudal ganglionic eminences around E15. A third wave of OPCs appears in the cortex around birth, and these cells rapidly expand in number to undergo terminal differentiation into myelinating oligodendrocytes within the maturing brain. A significant proportion of the oligodendrocytes generated from the earliest embryonic stages are eliminated and replaced by those generated postnatally (Kessaris et al. 2006). Gliogenesis has been much less intensively studied in the brainstem compared to cortical development. In the rat, there is a greater density of the presumptive oligodendrocyte progenitors expressing NG2, PDGFRα and Olig2 in the early postnatal brainstem compared with the cerebrum (Masui et al. 2010). In mice, the postnatal glial progenitors of the ventral pons are a sonic hedgehog-responsive population of Sox2-expressing cells (Monje et al. 2011).
Figure 1. Developmental origins of glia in the brain.
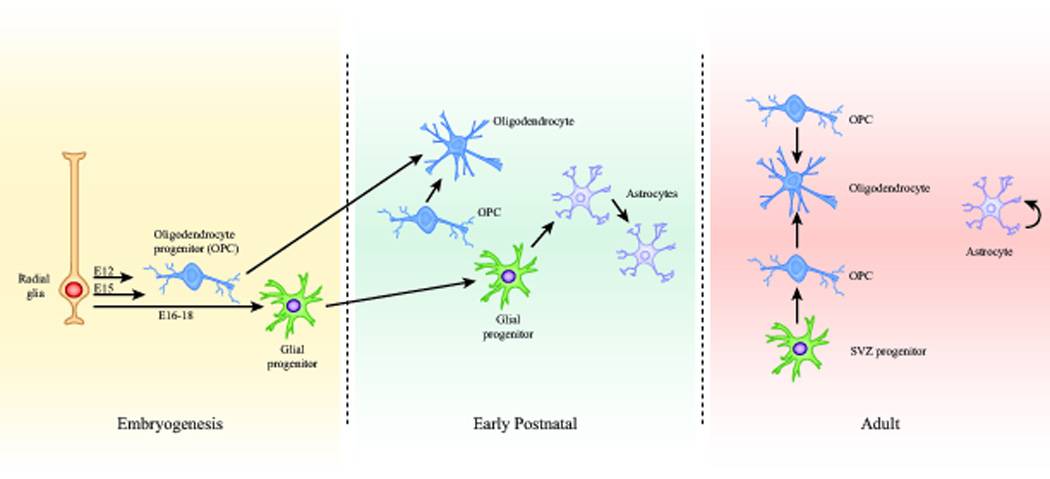
(A) Ventricular zone stem cells or radial glia (RG) function as the embryonic sources of glial progenitors. Two separate waves of oligodendrocyte progenitor cells (OPCs) arise from specific ventricular zone regions at E12 and E15, and migrate in streams to populate the telencephalon. Radial glia shift from neurogenesis to gliogenesis at E16 to generate intermediate glial progenitors. The majority of gliogenesis occurs during the first 3 weeks postnatally. The predominant source of oligodendrocytes in the postnatal brain comes from a third wave of OPCs that appears in the cortex around birth, expands, and differentiates into myelinating oligodendrocytes. Intermediate glial progenitors migrate into the cortex and differentiate into astrocytes. These differentiated astrocytes also undergo symmetric division during the first three weeks of age. In the adult, differentiated astrocytes can generate additional astrocytes, especially in response to injury. New oligodendrocytes are formed in the adult brain from resident OPCs in the cortex as well as OPCs arising from intermediate progenitors of the subventricular zone.
The first wave of astrogliogenesis is initiated with the burst of glial intermediate progenitors arising from radial glia at E16–18. However, astrocyte populations expand 6- to 8-fold in the rodent brain during the first 3 weeks postnatally, as they arise from progenitors in the postnatal subventricular zone (SVZ) that migrate and undergo asymmetric division to generate differentiated astrocytes. Strikingly, this pool of differentiated astrocytes which populates the cortex then proceeds to undergo symmetric division, such that ~50% of cortical astrocytes are generated from resident differentiated astrocytes (Ge et al. 2012). This rapid expansion of astrocytes is heavily dependent on local environmental cues, and stops abruptly by the end of three weeks of life in the rodent brain (Bandeira et al. 2009).
The ordered developmental processes underlying brain maturation proceed in an analogous fashion in rodents and humans, although they are distributed across a very different time scale. In humans, gliogenesis peaks during late gestation and continues postnatally through the first year of life (Roessmann and Gambetti 1986). Myelination is an ongoing process, initiated in late gestation and progressing in an ordered pattern from inferior to superior and posterior to anterior brain regions. Although there is substantial myelination of major tracts during early childhood, this ongoing process continues through young adulthood, (Brody et al. 1987; Kinney et al. 1988; Lebel et al. 2012; Semple et al. 2013; Tasker 2006). In the developing human brainstem, Nestin-expressing precursor cells are found in the ventral pons, which is the presumptive site of origin of DIPGs. This precursor cell population was found at highest densities in neonates and diminished by age 2, and then increased again around age 6, corresponding roughly to the median age of DIPG onset from ages 6–8. (Monje et al. 2011).
Several sources of proliferative cells persist in later stages of development and throughout adulthood, and are likely cells of origin of adult brain tumors. NSC and progenitor cells continue to divide within the adult SVZ. Similarly, OPCs persist and slowly divide in the adult brain (Dawson et al. 2003; Geha et al. 2010), where neuronal activity can heavily influence their proliferation, differentiation and myelination (Gibson et al. 2014). Finally, differentiated astrocytes also have the capacity to proliferate in the adult brain, especially in response to injury (Bardehle et al. 2013).
Taken together, the absolute numbers of proliferating cells in the developing brain vastly outnumber those in the adult brain. This extent of normal developmental proliferation provides an opportunity for the clonal expansion of somatic mutations, increasing the likelihood that multiple mutations may be acquired within a single cell that function together to initiate and promote tumorigenesis. Furthermore, the physiological requirements for these cell populations in the developing brain, which are programmed to expand, migrate and differentiate to generate all of the structures and integrated functions of a developing brain, are significantly different from the stem/progenitor cells in the adult brain that are more directed towards homeostatic maintenance, response to damage, and plasticity. Moreover, epigenetic programming plays critical roles in regulating differentiation states (Burton and Torres-Padilla 2014; Hu et al. 2014), and there are unambiguous connections between chromatin regulation and pediatric brain tumor pathogenesis.
The innate differences in the role of neural stem/progenitor cells at different developmental stages likely also confer intrinsically different susceptibilities to transformation. This may explain in large part why the genomic landscape of childhood gliomas is substantially less complex than that observed in adult gliomas (Jones and Baker 2014). Notably, childhood and adult brain tumors are distinguished by unique mutations with precise neuroanatomical and age-dependent associations, highlighting changing selective pressures driving gliomagenesis in different developmental contexts.
Pediatric low-grade gliomas
Pediatric low-grade gliomas comprise two different histological grades with distinct genetic alterations, brain locations, and clinical manifestations. As defined by the current World Health Organization classification, both grade I (pilocytic astrocytoma) and grade II (diffuse astrocytoma) are considered low-grade glial neoplasms (Louis et al. 2007b; Louis et al. 2014).
Pilocytic astrocytoma
Pilocytic astrocytomas (PAs) are typically infratentorial tumors in children, where they usually arise in the cerebellum, optic pathway, and brainstem (Ostrom et al. 2014; Pfister and Witt 2009). These low-grade tumors may develop sporadically (without a known inherited basis) or form in the context of the Neurofibromatosis type 1 (NF1) tumor predisposition syndrome. Those arising in children with NF1 have unique features from their sporadic counterparts (Guillamo et al. 2003; Listernick et al. 1995; Listernick et al. 1997). In this regard, sporadic PAs most commonly develop in the cerebellum, whereas those in children with NF1 predominate in the optic pathways (optic nerve, chiasm, tracks, and radiations). Because of these differences in brain location, the presenting signs and symptoms range from visual impairment and early onset puberty (optic pathway PAs) to headaches and hydrocephalus (cerebellum, brainstem PAs). Moreover, PAs tend to arise at younger ages in children with NF1.
Histologically, PAs have a low cell density and may contain Rosenthal fibers and eosinophilic granular bodies. Classic examples demonstrate biphasic architecture, with areas of disaggregated cells or small cysts alternating with more compact regions. Consistent with their classification as low-grade glial neoplasms, mitotic figures are rare. Immunohistochemical studies reveal immunoreactivity for proteins associated with glial lineage cells, including glial fibrillary acidic protein (GFAP). PAs are highly vascular, contributing to their robust enhancement following contrast agent administration on magnetic resonance imaging (MRI). While involvement of the subarachnoid space is common, these tumors rarely transform and remain grade I tumors throughout the lifetime of the individual.
Molecular analyses have shown that PAs arising in children with NF1 harbor bi-allelic inactivation of the NF1 tumor suppressor gene (Gutmann et al. 2000; Kluwe et al. 2001; Wimmer et al. 2002). As such, patients with NF1 are born with one mutated copy of the NF1 gene (germline NF1 gene mutation) and develop tumors following somatic (acquired) loss of the remaining NF1 allele in the appropriate cell type (neuroglial progenitor cell). Whole genome sequencing of NF1-PA tumors demonstrated the presence of non-neoplastic cells (microglia) harboring only the germline NF1 gene mutation and tumor cells with alterations affecting both NF1 alleles (germline NF1 gene mutation and somatically-acquired NF1 gene mutation) (Gutmann et al. 2013). Since the majority of NF1-PAs are not biopsied for diagnostic purposes, the full spectrum of genetic alterations in these tumors is not known. However, rare cases of NF1-PAs have been reported containing other potential cooperating genetic changes, including the common BRAF rearrangement (see below) and hemizygous loss of the PTEN tumor suppressor gene (Rodriguez et al. 2012).
In contrast, NF1 gene mutations are rare in sporadic PAs, where genomic rearrangement in many tumors results in the generation of a fusion transcript in which the kinase domain of the BRAF gene is fused to a gene of unknown function (KIAA1549) (Forshew et al. 2009; Jones et al. 2008; Pfister et al. 2008; Yu et al. 2009). These fusion BRAF alterations create a BRAF molecule lacking the regulatory amino terminal domain, leading to deregulated (increased) BRAF activation of the downstream MEK signaling cascade. While KIAA1549 is the most commonly reported fusion partner, other genomic regions can undergo rearrangement to generate alternate fusion BRAF molecules (e.g., FAM131B, RNF130) (Cin et al. 2011; Jones et al. 2013; Zhang et al. 2013). It should be noted that fusion BRAF mutations predominate in PAs arising in the cerebellum (Ida et al. 2012).
Further genomic studies revealed the presence of other potential driver mutations in sporadic PA, including mutations in the KRAS, FGFR1, PTPN11, and NTRK2 genes. KRAS and PTPN11 mutations are rare, occurring in ~2% of tumors sequenced (Jones et al. 2013; Zhang et al. 2013). Similarly, fusion rearrangements involving the TRKB receptor tyrosine kinase gene (NTRK2) are uncommon. However, ~5% of sporadic PAs harbored mutations in the fibroblast growth factor receptor (FGFR1; residues K655, K656, N546, T568), which potentially leads to constitutive activation of this receptor tyrosine kinase protein. Importantly, all FGFR1-mutant tumors reported to date arose outside of the cerebellum, typically in midline structures (e.g., brainstem, third ventricle), suggesting regional heterogeneity in sporadic PA driver events.
Taken together, the genetic changes reported for sporadic and NF1-associated PA occur in genes whose protein products function to regulate cell growth through the RAS downstream signaling pathway (Figure 2). In this respect, the NF1 protein, neurofibromin, functions to inactivate RAS by accelerating its conversion from its active GTP-bound to an inactive GDP-bound state (Brossier and Gutmann 2015), while activating mutations in the PTPN11 gene increase the conversion of GDP-bound to GTP-bound RAS. RAS transmits its growth promoting signal through downstream effectors, including the RAF/MEK/MAPK and PI3K/AKT pathways (Liu et al. 2011b). Activated MEK or AKT increases progression through the cell cycle, leading to increased cell growth. While these downstream effectors can function independently of each other, both converge on the mammalian target of rapamycin (mTOR) complex to result to increased mTOR-driven proliferation (Jiang et al. 2015; Kaul et al. 2012). In this manner, mTOR hyperactivation, as a consequence of increased RAS, AKT, or RAF/MEK function, is a major driver of glioma growth, and therefore represents a logical target for future therapeutic trials.
Figure 2. Low-grade glioma growth control pathways.
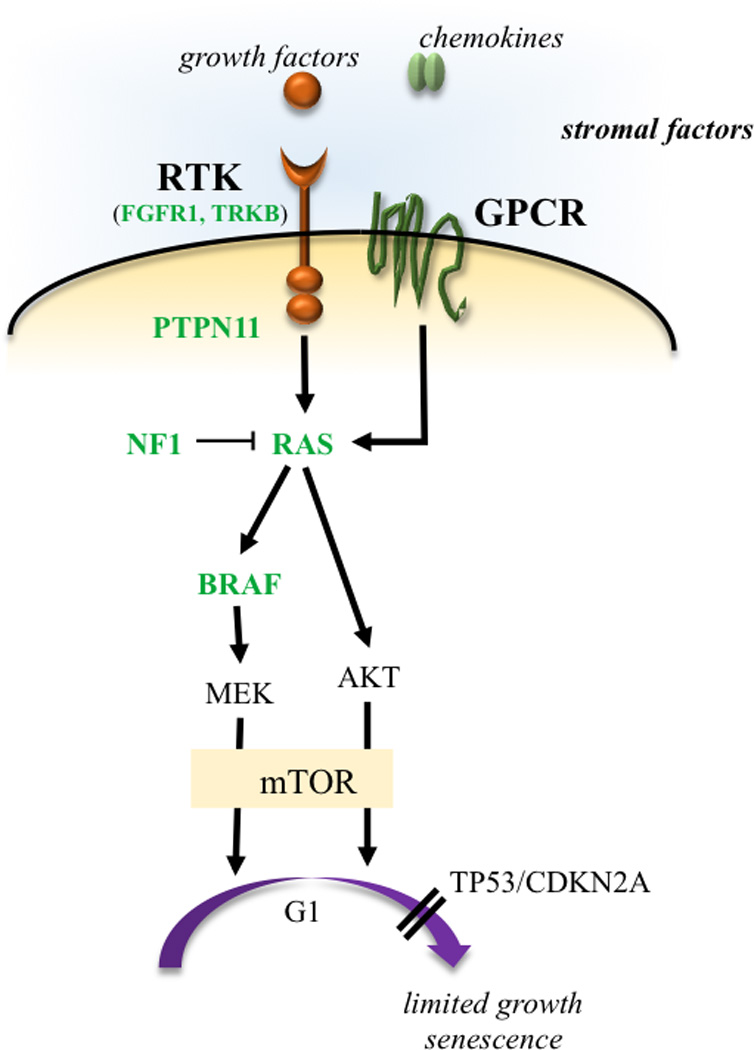
LGGs arise from mutations in genes whose protein products regulate RAS pathway signaling. In this manner, mutations in genes encoding receptor tyrosine kinases (RTK), which transduce extracellular signals, as well as the downstream effectors PTPN11, RAS, BRAF/RAF, and lead to hyperactivation of mitogenic signaling downstream through the MAP kinase and PI3-kinase pathways. In addition, mutational inactivation of the neurofibromatosis type 1 (NF1) tumor suppressor gene results in increased RAS activity. MEK and AKT can shorten G1 transition through mammalian target of rapamycin (mTOR)-dependent and -independent signaling. The presence of stromal growth factors and chemokines in the tumor microenvironment transduce signals through tumor cell receptors and further enhance glioma growth in a paracrine fashion. Reported mutations are denoted in green.
Pediatric grade II glioma – diffuse astrocytomas
DAs are rarer than PAs, constituting fewer than 10% of pediatric LGGs. DAs can arise throughout the entire neuraxis, but are particularly prevalent in the cerebral cortex and thalamus. The infiltrative nature of DAs fundamentally distinguishes this tumor from the relatively circumscribed PA, with important therapeutic implications: DAs are hardly ever resectable, leaving adjuvant therapy as the major therapeutic modality for disease stabilization. In addition, DAs can be difficult to distinguish histopathologically from PAs. As such, their invasive nature is not always clearly demonstrated on histopathological examination, and some PAs can infiltrate at the tumor margins. Cytological features also can overlap, since both glial neoplasms have low proliferative indices, and DAs can contain microcysts. The indolent clinical course of the pediatric-type DA, which rarely undergoes anaplastic progression, is in sharp contrast to the situation for adult-type DA, which frequently transforms into a high-grade astrocytic tumor.
The genetic alterations that characterize pediatric DAs are distinct from those observed in PAs and adult-type DAs, and contribute to the clinicopathological differences described above. A mutation in one of three genes characterizes approximately two thirds of pediatric DAs: MYB, BRAF, and FGFR1 (Zhang et al. 2013). MYB is involved in fusions with a variety of partner genes (e.g., PCDHGA1 and QKI), while, in some tumors, MYB is amplified (Tatevossian et al. 2010) or the MYB-related gene MYBL1 is rearranged (Ramkissoon et al. 2013). About 25% of pediatric DAs harbor a BRAFV600E mutation, whereas alterations of the FGFR1 gene characterize a lower proportion of these tumors. FGFR1 contains a duplication of the tyrosine kinase domain in some tumors, and a FGFR1-TACC1 fusion occurs in others (Zhang et al. 2013). In striking contrast, diffuse LGGs in adults harbor an entirely different set of mutations (Brat et al. 2015): an IDH1 mutation occurs in most adult LGGs and is accompanied by an ATRX or TP53 mutation in many astrocytomas. An IDH2 mutation occurs in place of an IDH1 mutation in rare adult-type diffuse gliomas, while in diffuse oligodendrogliomas, IDH1/2 mutation is accompanied by co-deletion of chromosomes 1p and 19q and often a CIC gene mutation. Because of the rarity of pediatric diffuse LGGs and the small number of tumors included in genomic analyses so far, it is possible that further novel genetic alterations will be discovered and the frequency of known alterations will change with time.
Dissecting low-grade glioma biology
Understanding what determines the unusual biology of low-grade glioma is challenging when studying human pathological specimens, as these analyses do not provide insights into the mechanisms underlying tumor formation or progression and cannot easily distinguish between causative molecular changes and bystander, non-pathogenic alterations. For this reason, investigators have leveraged both primary human tumor specimens and novel genetically-engineered mouse models to address these questions.
The use of primary human low-grade glioma cell lines has been hampered by the relative paucity of these resources and their limited characterization as authentic representations of their native human counterparts. Moreover, studies using glioma stem cells from PA tumors have revealed premature senescence and loss of the responsible driver mutation (e.g. fusion BRAF) with prolonged in vitro adaptation (Jacob et al. 2011; Raabe et al. 2011). Similarly, others have engineered normal fetal human astrocyte lines or neural stem cells with specific genetic mutations to define the contributions of these alterations to signaling pathway activation and growth regulation in vitro. Efforts in numerous laboratories are focused on refining the methods used to isolate or engineer lines de novo for future translational research.
In the absence of tractable human cell models for pediatric low-grade glioma, researchers have attempted to develop small-animal models to better understand the principles and determinants that underlie tumor pathogenesis. These types of pursuits would not be possible using cell lines, since these lines lack the appropriate cellular and molecular context provided by the intact animal during the process of brain development. To date, small animal modeling has been focused on NF1-, RAS- and BRAF-driven low-grade tumors, although work is progressing on other causative genetic mutations.
In this regard, the expression of an oncogenic KRAS allele in GFAP+ neuroglial progenitor cells results in the formation of an infiltrative intermediate-grade glioma arising from the subventricular zone (SVZ) (Abel et al. 2009). The resulting tumors in these mice are multi-focal and might more accurately model a condition termed gliomatosis cerebri (malignant glioma), rather than PA or DA. Interestingly, other investigators using different transgenesis approaches (postnatal induction) failed to generate gliomas following the expression of an oncogenic KRAS allele (Holland et al. 2000; Munoz et al. 2013; Splinter et al. 1990; Uhrbom et al. 2002). In addition, transgenic mice harboring an oncogenic HRAS mutation in neuroglial progenitor cells developed high-grade gliomas (Ding et al. 2001; Shannon et al. 2005). These malignant brain tumors were often multi-focal and dependent on the RAS gene dosage. Further work using specific cell types originating from particular brain locations may help to resolve these apparent conflicting results.
Early work focused on BRAF gene alterations has demonstrated that oncogenic BRAFV600E expression in neuroglial progenitor cells alone is not sufficient to generate gliomas (Dasgupta et al. 2005), but does result in high-grade tumors when coupled with Cdkn2a loss (Uhrbom et al. 2002; Yu et al. 2009). However, the introduction of the kinase domain of an oncogenic BRAFV600 molecule into nestin+ neuroglial progenitor cells within the cerebral hemispheres is sufficient to produce low-grade gliomas with features of pilocytic astrocytoma (Gronych et al. 2011). While a truncated and mutated BRAF gene is not observed in human PAs and supratentorial locations are not typical for PAs, this model represents one of the most relevant representations of this human tumor type to date. Using another approach, the human KIAA1549:BRAF fusion transcript was introduced into cerebellar neural stem cells, and these transduced cells were transplanted into the cerebella of syngeneic mice to generate low-grade glioma-like lesions (Kaul et al. 2012). Current investigations in several laboratories aim to generate conditional mouse strains or explant model systems for preclinical drug studies.
Finally, Nf1 genetically-engineered mouse strains have been leveraged to develop models for NF1-associated optic glioma. Mice harboring a germline inactivating mutation in the Nf1 gene coupled with somatic Nf1 gene inactivation in neuroglial progenitors develop low-grade gliomas of the optic nerve and chiasm by 3 months of age (Bajenaru et al. 2003; Hegedus et al. 2008; Zhu et al. 2005). These tumors have low proliferative indices, microglial infiltration, and robust GFAP and Olig2 expression, and are detectable on small-animal MRI (Bajenaru et al. 2005). Moreover, since these tumors arise in the optic pathway, the resulting conditional Nf1 mutant mice develop optic nerve dysfunction, retinal ganglion cell (RGC) loss, retinal nerve fiber layer thinning, and reduced visual acuity (Hegedus et al. 2009; Kaul et al. 2015). These mice, along with the KIAA1549:BRAF neural stem cell explant model, provide new insights into disease pathogenesis, including the contributions of the brain location and cell of origin, microenvironmental cell types and molecules, and potential sexual dimorphic effects (Diggs-Andrews et al. 2014; Gutmann 2014).
Seminal work in GEM strains in which multiple tumor suppressor genes are simultaneously deleted resulted in the generation of a number of models of adult high-grade glioma, and suggested that high-grade glioma (glioblastoma) formation can be driven by the introduction of mutations in cells from multiple differentiation states and diverse brain regions (Alcantara Llaguno et al. 2009; Bachoo et al. 2002; Chow et al. 2011; Friedmann-Morvinski et al. 2012; Galvao et al. 2014; Lei et al. 2011; Liu et al. 2011a; Wang et al. 2013; Zong et al. 2015). In one model of glioblastoma, combined deletion of Nf1 and Tp53 was induced in neural stem cells, however tumors developed from oligodendrocyte progenitor cells that were descendants of the mutated stem cells (Liu et al. 2011a). Thus tumor cell of origin may be difficult to pinpoint precisely, however inducing mutations in a range of cell types can result in models of adult glioblastoma. In striking contrast, there appears to be exquisite selectivity in both the specific cell type and the region of the central nervous system when single low-grade glioma-associated mutations are involved. In this regard, ectopic expression of the KIAA1549:BRAF fusion protein increases the proliferation of neural stem cells (NSCs), but not astrocytes, in vitro and in vivo (Kaul et al. 2012; Kaul et al. 2013). Moreover, this sporadic PA-associated mutation has brain region-specific effects, such that cerebellar and brainstem NSCs exhibit increased proliferation, whereas those from the cortex or lateral ventricle subventricular zone (lv-SVZ) do not. Similar regional differences have been reported in response to Nf1 gene inactivation in vitro and in vivo (Lee da et al. 2012; Lee da et al. 2010). Neurofibromin loss in NSCs in the third ventricle and brainstem increases proliferation and glial cell differentiation, while Nf1 gene inactivation in cortical or lv-SVZ NSCs does not. Collectively, these findings suggest that the cell of origin and the brain location are critical determinants partly responsible for low-grade glioma temporal and spatial patterning.
The contribution of the tumor microenvironment to tumorigenesis and growth is also nicely illustrated by Nf1 GEM strains (Figure 3). Whereas Nf1 loss in neuroglial progenitor cells alone is not sufficient for optic glioma formation (Bajenaru et al. 2002), neuroglial progenitor Nf1 gene inactivation in mice heterozygous for a germline Nf1 gene mutation results in tumor development (Bajenaru et al. 2003). The requirement for Nf1+/− cells has also been reported for benign nerve sheath tumors (neurofibromas) arising in the context of NF1 (Staser et al. 2012; Yang et al. 2008). While mast cells and macrophages are critical for NF1-associated neurofibroma formation in mice, microglia are the responsible stromal cell type in optic glioma pathogenesis. In this respect, 30–50% of the cells in both NF1-associated and sporadic PA tumors are microglia (Simmons et al. 2011). Formal demonstration for a requirement for microglia in Nf1 mouse optic glioma development and maintenance derives from both genetic (Cx3cr1 mutant and CD11b–TK mice) and pharmacological (minocycline, JNK inhibition) silencing experiments in which impaired microglial function is sufficient to delay optic glioma formation and inhibit established tumor proliferation (Daginakatte et al. 2008; Daginakatte and Gutmann 2007; Pong et al. 2013; Simmons et al. 2011). Ongoing studies in one of our laboratories (D.H.G.), employing advanced RNA-sequencing methods to discover those essential microglia-produced growth factors and cytokines (gliomagens) have led to the identification of a limited number of gliomagens whose silencing has a profound effect on Nf1 murine optic glioma maintenance. Coupled with a growing body of evidence in high-grade glioma mouse models (Feng et al. 2015; Hu et al. 2015; Pyonteck et al. 2013; Zhai et al. 2011), the identification and therapeutic targeting of microglia could be considered as a potential adjuvant approach to reducing low-grade glioma growth in children.
Figure 3. The glioma ecosystem.
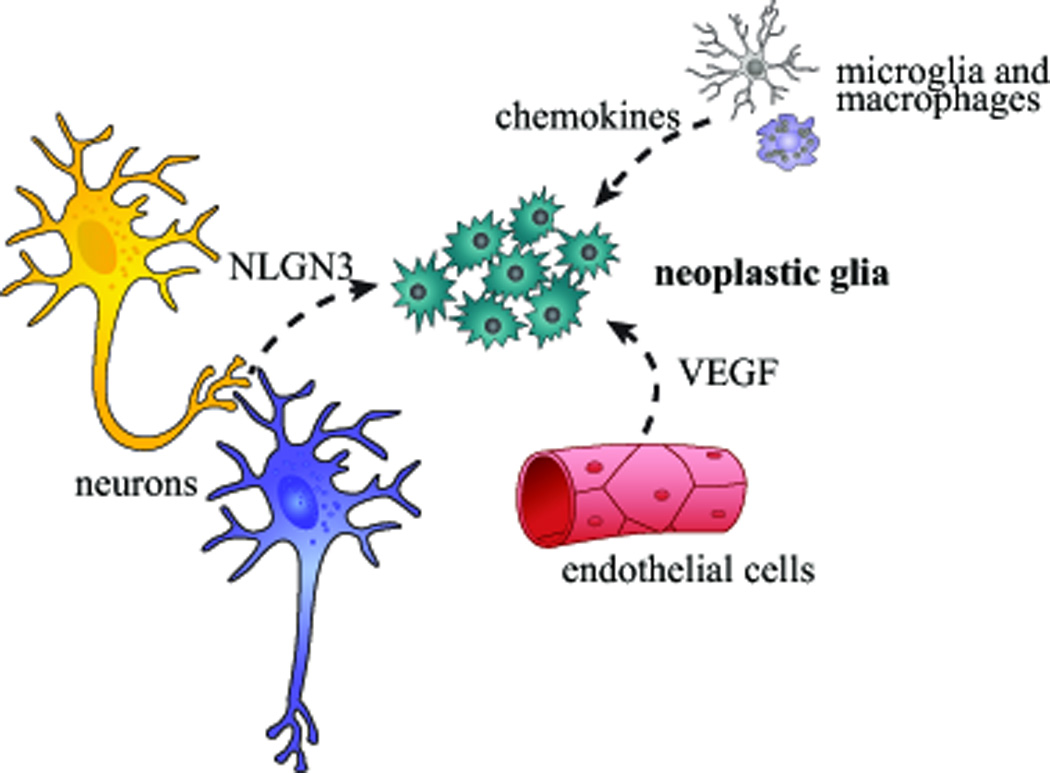
Gliomas form and are maintained by a circuit of cellular and acellular elements, including neurons, endothelial cells, and monocytes (microglia and macrophages), which each produce growth factors (neuroligin-3 [NLGN3], vascular endothelial-derived growth factor [VEGF]) and chemokines that stimulate neoplastic glia.
Children with NF1-associated PAs are prone to impaired visual acuity as a consequence of optic glioma-induced retinal ganglion cell (RGC) dysfunction (Daginakatte et al. 2008; Daginakatte and Gutmann 2007). While both boys and girls with NF1 develop optic pathway gliomas (OPGs) at equivalent frequencies, girls with NF1-associated optic nerve gliomas are 5–10 times more likely to experience visual decline as a result of their tumor and therefore require chemotherapy (Diggs-Andrews et al. 2014). This apparent sexual dimorphism is also observed in Nf1 mutant mice, such that only female Nf1+/−GFAP CKO mice exhibit visual acuity loss as a result of greater RGC death, despite nearly identical optic glioma volumes in male and female mice. Current studies are focused on defining the predictive and therapeutic implications of this potential sexually-dimorphic effect.
The impact of genomics on dictating NF1 low-grade glioma formation derives from both human and mouse studies. Using mice heterozygous for a germline mutation in the Nf1 and p53 genes (NPCis mice), Reilly and colleagues have demonstrated striking strain-dependent effects on gliomagenesis (Reilly et al. 2000; Reilly et al. 2004). In these experiments, NPCis mice maintained on a C57BL/6 background develop gliomas, whereas those on a 129sv background form brain tumors at a significantly lower rate. Further dissection of the responsible genomic loci has revealed numerous distinct genes responsible for spinal and brain astrocytomas that operate in a sex-dependent manner (Amlin-Van Schaick et al. 2012; Walrath et al. 2009). Analogously, single nucleotide polymorphisms in the adenylate cyclase 8 (ADCY8) gene correlate with glioma risk in a sex-specific manner in children with NF1 (Warrington et al. 2015). In this latter report, these sexually-dimorphic effects reflect differences in cyclic AMP regulation in males and female mice. In support of genomic effects, epidemiologic studies in children with NF1 as well as the general population have demonstrated racial differences in glioma risk. For example, Caucasian children are 1.5–2.1 fold more likely to develop a glioma than their non-Caucasian counterparts, whereas non-Caucasian children with NF1 are ~7 times less likely to develop a brain tumor (Abadin et al. 2015). The identification of modifier genes that predict risk for glioma formation would significantly impact on the management of children with cancer predisposition syndromes, like NF1, by providing prognostic tools to guide our surveillance.
High-grade diffuse gliomas of childhood: Similar histology but unique molecular pathogenesis
High-grade diffuse gliomas (HGGs, grades III and IV) are distinguished from the low-grade diffuse gliomas by defined histological criteria detailed in the WHO classification of nervous system tumors, including brisk mitotic activity and nuclear pleiomorphism in grades III and IV glial neoplasms, and with the addition of microvascular proliferation and necrosis in the grade IV tumors, also known as glioblastoma (Louis et al. 2007b). In addition to morphological similarities, gene expression signatures also show strong similarity between childhood and adult HGG, with an unsupervised analysis of pediatric HGG identifying three major expression subgroups that correlate with the Proneural, Proliferative and Mesenchymal expression subgroups identified in adults (Paugh et al. 2011; Paugh et al. 2010; Puget et al. 2012). HGGs comprise 15–20% of all CNS tumors in children and have a 2-year survival rate of only 10–30%. The standard of care involves surgery and irradiation, and the strongest predictor of survival remains the extent of surgical resection. In adults, the addition of temozolomide treatment results in an increased survival time, however, no chemotherapeutic agents, including temozolomide, have shown any impact on survival times in childhood HGG (Cohen et al. 2011a; Cohen et al. 2011b; Warren 2012). For all ages, although HGGs respond initially to radiation, these highly infiltrative tumors inevitably recur.
Although pediatric high-grade gliomas carry a larger mutation burden than the low-grade tumors, they still have approximately 3- to 4-fold fewer coding alterations than adult glioblastoma (Jones and Baker 2014). High-grade gliomas in infants have a much better prognosis, and have an extremely low mutation burden, similar to low-grade glioma, suggesting that fewer mutations are required to generate a high-grade glioma if it arises earlier in development (Qaddoumi et al. 2009; Wu et al. 2014).
Clear connections with neural development are strongly demonstrated by the markedly different location spectrum of high-grade gliomas across different age groups, and the intriguing specificity of recurrent mutations in particular subgroups (Figure 4). Pediatric high-grade gliomas commonly arise in the cerebral hemispheres, which is the site of origin for the vast majority of glioblastomas in adults. However, approximately half of all high-grade gliomas in children are diffuse intrinsic pontine glioma (DIPG), which arise from the ventral pons in the brainstem and occur almost exclusively in children. High-grade gliomas in other midline structures, most commonly the thalamus, but also the cerebellum and spinal cord, are also much more common in children than in adults. DIPGs carry the worse prognosis of all pediatric HGGs, with a 2-year survival of less than 10%. While this dismal outcome is likely due, at least in part, to the fact that DIPGs cannot be surgically resected because of their vital location, a wealth of genome-wide sequencing data has also shown clear biological differences between DIPGs and pediatric HGGs at other locations (Jones and Baker 2014; Sturm et al. 2014).
Figure 4. Spatiotemporal association of mutations in pediatric high-grade glioma.
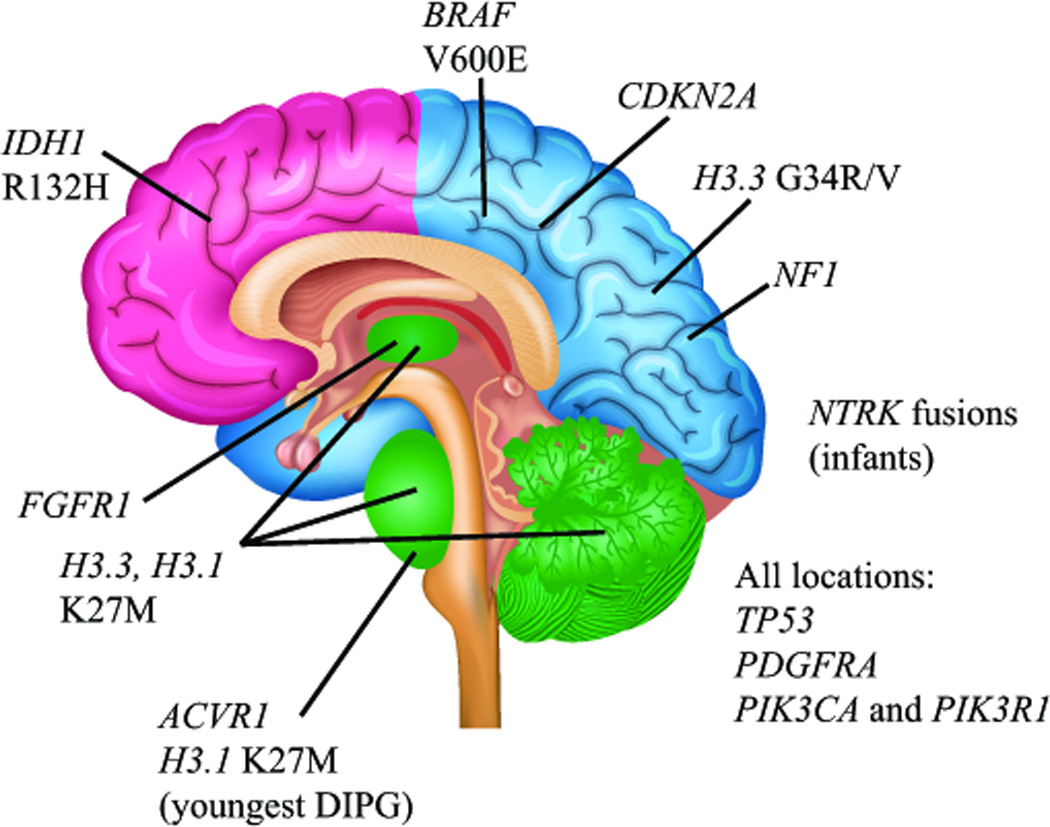
Clear associations between specific mutations and age- and location-dependent subgroups of pediatric HGG indicate a strong connection between brain development and gliomagenesis in children. Histone H3 K27M mutations are found predominantly in DIPGs, and other midline HGGs including thalamic and cerebellar (green), with the majority of mutations occurring in the H3.3 isoform. The youngest DIPG patients show a predominance of ACVR1 mutations and K27M mutations in the H3.1 isoform. Histone H3.3 G34R/V mutations are predominantly found in cortical HGGs of late adolescence, most commonly those arising in the parietal, occipital, or temporal lobes (blue). In contrast, frontal lobe HGGs of late adolescence and young adults are more commonly associated with IDH1 R132H mutation (pink). BRAFV600E mutations and CDKN2A homozygous deletions are found in non-brainstem HGGs and not DIPGs, and NF1 mutations are more frequent in cortical HGGs than DIPG. NTRK fusion genes have been reported in non-brainstem HGGs as well as DIPGs, but appear to occur at higher frequency in HGGs arising outside the brainstem in children less than 3 years of age. FGFR1 mutations are found predominantly in thalamic HGGs. Mutations in TP53, PDGFRA, PIK3CA and PIK3R1 are found in HGGs from all locations.
The identification of a recurrent hotspot mutation in histone H3 encoding a substitution of lysine 27 with methionine (K27M) in nearly 80% of DIPGs provided conclusive evidence that the molecular pathogenesis of DIPGs is distinct from cortical HGGs in children or adults (Wu et al. 2012). Among 16 genes encoding 3 different isoforms of histone H3, the mutations are found in mutually exclusive patterns with 75% of H3 mutations targeting H3F3A, one of two genes encoding the H3.3 isoform, and 25% of mutations targeting HIST1H3B, or rarely HIST1H3C, two of 10 genes encoding the H3.1 isoform (Buczkowicz et al. 2014; Fontebasso et al. 2014; Taylor et al. 2014; Wu et al. 2014). H3 K27M mutations were also identified in non-brainstem HGGs arising in midline structures such as thalamic tumors as well as less common cerebellar or spinal cord HGGs. Strikingly, an alternate mutation was identified in H3F3A in cortical HGGs mostly arising in adolescents and young adults; a glycine 34 substitution with either arginine or valine (G34R/V) (Schwartzentruber et al. 2012; Wu et al. 2012). A number of other tumor types contain recurrent mutations in enzymes that catalyze or remove post-translational modifications of histone H3 (Simo-Riudalbas and Esteller 2014), however, many of these enzymes may also regulate other proteins, therefore the precise connection between cancer and histone H3 regulation was not entirely clear. Direct mutation of histone H3 in pediatric HGG, including DIPG, was the first identification of histone mutation in human cancer, making an unambiguous connection between histone regulation and oncogenesis. Further, H3.3 and H3.1 mutations are mutually exclusive. The recurrent heterozygous mutations in only one of 16 genes comprising a highly redundant gene family clearly indicate that these mutations function in dominant fashion. In this regard, the H3 K27M mutant protein comprises only 5–15% of the total histone H3 in a tumor cell; however, the expression of this mutant causes a dramatic and dominant loss of lysine 27 trimethylation (H3K27me3) on the wild-type H3 co-expressed from the histone H3 gene family in these tumors (Bender et al. 2013; Chan et al. 2013; Lewis et al. 2013; Venneti et al. 2013). This post-translational modification is associated with transcriptionally-silent chromatin, and in tumors or cell lines containing K27M mutation, a low level of residual H3K27me3 is detected, where its distribution in the genome marks a different subset of genes than in tumors without the K27M mutation (Bender et al. 2013; Chan et al. 2013). There is no clear dominant mechanism of action associated with G34R/V mutations, although elevated MYCN expression is noted in some of these tumors, perhaps due to altered localization of H3K36me3 chromatin marks (Bjerke et al. 2013).
Tumors carrying histone H3 K27M or G34R/V mutations were each associated with characteristic global DNA methylation signatures. These differential methylation signatures as well as their associated gene expression signatures in tumors with K27M relative to those harboring the G34R/V mutation highlighted genes associated with hindbrain versus cortical development respectively, consistent with the developmental origins of the tumors (Sturm et al. 2012). Similar gene expression signatures between DIPG and thalamic tumors support a likely common developmental origin among these tumors of midline structures (Bjerke et al. 2013; Sturm et al. 2012).
The age distribution of cortical glioblastomas with H3F3A G34R/V mutation, from late adolescence into early adulthood, overlaps with adult secondary glioblastomas with IDH1 R132H mutation, which occur over a broader age range from late adolescence into adulthood with a mean age of 33–45 (Ohgaki and Kleihues 2013). H3F3A G34R/V and IDH1 R132H mutations are mutually exclusive, and there are a number of biological and molecular features that distinguish tumors with these mutations. Glioblastomas with IDH1 mutation most frequently arise in the frontal lobe, while those with H3F3A G34R/V predominantly arise in the other lobes of the cerebral cortex, implying different developmental origins (Sturm et al. 2012). IDH1 mutations are the molecular hallmark of secondary glioblastomas, which arise from the malignant transformation of a lower grade astrocytoma, while H3F3A G34R/V mutant glioblastomas, like the vast majority of pediatric glioblastomas, arise as primary or de novo malignancies without evidence of a precursor lower-grade lesion (Broniscer et al. 2007; Ohgaki and Kleihues 2013).
On a molecular level, the IDH1 R132H mutation results in catalysis of alpha-ketoglutarate to R-2-hydroxyglutarate (R-2HG) (Dang et al. 2009). One effect of this elevated R-2HG level is inhibition of TET2, resulting in increased DNA methylation (Xu et al. 2011). IDH1 mutant tumors are associated with a global DNA hypermethylation signature known as the glioma-CpG island methylator phenotype (G-CIMP), in contrast to the relatively global hypomethylation signatures associated with histone H3 mutant tumors (Sturm et al. 2012). Therefore, these histologically-similar glioblastomas found in overlapping age groups likely arise within discrete developmental contexts with differing disease pathogenesis mechanisms involving selective pressures for distinct effects on the epigenome.
Activating somatic mutations in ACVR1, a type I receptor in the bone morphogenetic protein (BMP) signaling pathway, are found in 25–30% of DIPG patients, and are typically found in patients less than 5 years of age. ACVR1 mutations almost always co-occur with HIST1H3B K27M, encoding mutant histone H3.1, which is also associated with younger age. Although H3.1 K27M mutations are also found in thalamic HGGs, ACVR1 mutation has only been identified in DIPG (Buczkowicz et al. 2014; Fontebasso et al. 2014; Taylor et al. 2014; Wu et al. 2014). Several of the identical missense substitutions found as somatic mutations in DIPG occur as heterozygous germline alterations in patients who inherit the disease fibrodysplasia ossificans progressiva (FOP), which is a disorder of abnormal cell fate in which different cell types inappropriately differentiate into osteoblasts and form ectopic bony lesions at sites of inflammation (Shore et al. 2006). BMP signaling has a diverse range of outcomes that are highly dependent on developmental context. In the developing brain, overexpression of BMP4 also influenced cell fate, inducing astrocyte differentiation at the expense of oligodendrocytic differentiation (Gomes et al. 2003). Although activation of BMP signaling through ACVR1 mutation may contribute to DIPG formation, BMP treatment induces differentiation and inhibits tumorigenesis in medulloblastoma (Zhao et al. 2008). Therefore, in two different pediatric brain tumors arising in infratentorial locations, activation of BMP signaling may have opposing consequences.
Pediatric HGGs also contain mutations in pathways that are common to many tumor types. In almost all adult glioblastomas, three major cancer signaling pathways are concomitantly deregulated; the TP53, RB and receptor tyrosine kinase (RTK)/RAS/PI3K pathways (Parsons et al. 2008; TCGA 2008). These signal transduction networks are of central importance in diverse tumors types, and also play key roles in pediatric high-grade glioma, but the specific effectors within each pathway vary with context. The TP53 checkpoint is compromised through TP53 mutations in more than half of pediatric HGG, whereas mutually-exclusive mutations in PPM1D, a TP53 target gene involved in DNA damage response, are found in an additional 10–20% of DIPGs and midline HGGs (Buczkowicz et al. 2014; Fontebasso et al. 2014; Schwartzentruber et al. 2012; Taylor et al. 2014; Wu et al. 2014). The CyclinD/CDK4 or CDK6 complex phosphorylates RB to regulate G1 cell cycle progression. The importance of this cell cycle regulator is underscored by the observation that approximately one quarter of non-brainstem HGGs harbor homozygous CDKN2A deletion, which encodes the CDK4/6 inhibitor (INK4A) as well as the p19ARF protein, which controls p53 function through MDM2 (Bax et al. 2010; Paugh et al. 2010; Qu et al. 2010). In DIPG, the RB pathway is subverted by an alternative genetic mechanism, focal amplifications of the genes encoding the Cyclin D family, CDK4 and/or CDK6 (Paugh et al. 2011; Zarghooni et al. 2010).
Alterations in the RTK-RAS-PI3K pathway are also very common. The most frequent RTK alterations are focal amplifications and/or activating mutations of PDGFRA, which are found in approximately 30% of tumors (Paugh et al. 2011; Paugh et al. 2010; Paugh et al. 2013; Puget et al. 2012; Qu et al. 2010; Zarghooni et al. 2010). Downstream mutations in PIK3CA or PIK3R1, encoding the catalytic and regulatory subunits of PI3-kinase (PI3K), respectively, occur in 10–25% of pediatric HGGs regardless of location. Sporadic NF1 gene mutations are similarly seen in 5–25% of pediatric HGGs, with higher frequencies in cortical HGGs (Buczkowicz et al. 2014; Fontebasso et al. 2014; Schwartzentruber et al. 2012; Taylor et al. 2014; Wu et al. 2014). However, it is not clear whether complete loss of NF1 gene expression and function is observed in these tumors characterized by NF1 gene mutation. Other components of PI3K signaling are mutated in more selective patterns. FGFR1 activating mutations are found predominantly in thalamic HGGs, but not in other locations (Fontebasso et al. 2014). Approximately 10% of cortical pediatric HGGs contain BRAFV600E mutations, but these mutations have not been reported in DIPG (Huillard et al. 2012). Interestingly, the BRAF gene duplications found at extremely high frequency in pilocytic astrocytomas are not found in high-grade gliomas. Structural variants generating activating fusions of the NTRK family of neurotrophin receptors are also found in ~5% of brainstem and non-brainstem pediatric HGGs, with an increased incidence among HGGs arising in children younger than three years of age. These HGGs from very young children have much quieter genomes than those arising in older children, and more closely resemble the bland genetic landscape of pediatric low-grade diffuse glioma (Wu et al. 2014). It is intriguing that NTRK fusion genes in the context of few other genetic alterations are found in both low-grade and high-grade gliomas, but with different prognoses (Frattini et al. 2013; Jones et al. 2013; Wu et al. 2014; Zhang et al. 2013). In these tumors with minimal genetic alterations, it is more readily apparent that different cells of origin and/or microenvironments must contribute strongly to tumor biology.
Dissecting pediatric high-grade glioma biology
A number of different approaches have been used to generate genetically engineered mouse models to illuminate the biology of high-grade glioma. Using combinations of loss of function mutations of Tp53, Rb, Pten or Nf1, there is strong evidence that high-grade gliomas can arise from neural stem and/or progenitor cells in the subventricular zone, as well as outside of proliferative niches, most likely from either astrocyte or oligodendrocyte progenitor cells (Alcantara Llaguno et al. 2009; Chow et al. 2011; Liu et al. 2011a; Ozawa et al. 2014; Sugiarto et al. 2011). Given the central roles of these tumor suppressor pathways in HGGs across all age groups and locations, these models address some unifying features of all HGGs including potential cells of origin, gene expression signatures shared across age groups, synergism and cross-talk between signaling pathways in gliomagenesis, and patterns of glioma infiltration. However, the PI3K pathway is typically activated by mutations other than PTEN in childhood HGGs, and many of the mouse models introduced the mutations in adult mice, therefore these models likely have greatest relevance to adult glioblastoma.
The clear association of particular mutations with age and specific brain regions in pediatric HGG indicates that these mutations provide selective advantages only in particular developmental contexts. Modeling the oncogenic effects of these mutations may therefore require a more in-depth consideration of relevant developmental setting than is needed for cancer mutations that occur commonly in many different tumor types, such as Tp53. Several different model systems have been developed to address this issue.
A small number of mouse models of brainstem glioma have been generated through introduction of exogenous PDGF-B. PDGF-B overexpression in NG2-expressing cells of the brainstem delivered by retrovirus into early postnatal wild-type mice resulted in malignant glioma in two-thirds of animals (Masui et al. 2010). Similarly, retroviral delivery of PDGF-B into Ink4A–ARF-null nestin-positive cells of the brainstem induced malignant gliomas with complete penetrance. These mutations would be expected to deregulate the same pathways as disrupted by PDGFRA and Tp53 mutations, which are common in human DIPG (Becher et al. 2010). Retroviral expression of H3.3 K27M in p53-deficient neural progenitor cells induced proliferation of the surrounding cells in the neonatal mouse brainstem, without glioma formation or a cell-autonomous increase of proliferation in the cells expressing the mutant H3.3 (Lewis et al. 2013). Intriguingly, human DIPG cells also induced a paracrine effect, with orthotopic transplantation of human DIPG cells into mouse pons resulting in the development of pontine tumors of mouse origin (Caretti et al. 2014). Although these model systems were not ideal to evaluate mutant H3.3-driven glioma formation, they revealed an intimate interaction between H3.3 mutant cells and the surrounding microenvironment that may be relevant to tumorigenesis. Intriguing paracrine effects of the tumor microenvironment on HGG growth were recently shown in an elegant experiment in which optogenetic stimulation of neuronal activity increased proliferation of surrounding glioma xenograft cells in vivo. This effect could be recapitulated using conditioned medium from stimulated cortical slices to promote in vitro proliferation of a number of patient-derived HGG cell cultures including DIPG, pediatric cortical HGG and adult HGG. Surprisingly, a secreted form of the synaptic protein neuroligin-3 (NLGN3) was identified as the factor released by stimulated neurons that was necessary and sufficient to drive enhanced HGG proliferation (Venkatesh et al. 2015). These findings imply that neuronal activity, which varies significantly with developmental stages, and between normal and neuropathological states, may play a significant role in gliomagenesis.
Comparing the consequences of H3.3 K27M in multiple differentiation states revealed interesting selective phenotypes. Expression of H3.3 K27M accelerated proliferation of neural progenitor cells derived from human embryonic stem cells, but not of undifferentiated embryonic stem cells or differentiated astrocytes. Combined expression of H3.3 K27M with a mutant PDGFRα and dominant negative Tp53 increased cell migration, inhibited differentiation of neural progenitor cells into astrocytes, and demonstrated infiltrative growth in the pons when implanted into immunodeficient mice. The combined expression of all three genes was required for oncogenesis. Overall, the gene expression changes and genomic distribution of H3K27me3 were consistent with resetting neural precursor cells to a more primitive stem cell state (Funato et al. 2014). Thus, the functional consequences of the expression of this oncogene appear to be highly dependent on differentiation state of the cell. Given the general association of BMP signaling with differentiation and cell fate decisions, it seems highly likely that ACVR1 mutations, which are limited to the youngest of DIPG patients, contributes to gliomagenesis by influencing cell differentiation within the developing pons. It is perhaps not unexpected that mutations that are strongly influenced by developmental context may indeed function to directly impact developmental processes.
Opportunities for precision oncology
As we begin to develop precision medicine approaches for the treatment of pediatric glioma, it is important to consider the factors that influence gliomagenesis and continued tumor growth in children. These include cell-intrinsic, cell-extrinsic (stromal), and genomic influences. A detailed dissection of these factors is likely to provide new insights relevant to both disease risk assessment and therapeutic drug design.
In pediatric glioma, cell-intrinsic factors that distinguish these childhood brain tumors from their adult counterparts include not only the specific genetic mutations that drive gliomagenesis and maintain tumor growth, but also the putative cell of origin. Multiple studies in murine models of adult high-grade glioma have revealed that the cell of origin may be less important, since these malignancies have been generated by introducing mutations in neurons, oligodendrocyte precursor cells, astrocytes, and neural stem cells with the same driver genetic alterations (Alcantara Llaguno et al. 2009; Bachoo et al. 2002; Chow et al. 2011; Friedmann-Morvinski et al. 2012; Galvao et al. 2014; Lei et al. 2011; Liu et al. 2011a; Wang et al. 2013; Zong et al. 2015). In contrast, the timing and cell of origin may be more constrained in pediatric low-grade glioma, where driver mutations operate in specific cell types and during a narrower temporal window. Similar to normal brain development, pediatric gliomagenesis is likely to be influenced by developmental context and the effect of specific genetic mutations on particular cell types. In this manner, the signature PA KIAA1549:BRAF fusion gene has little effect on astrocyte growth, but increases the proliferation of neural stem cells from the cerebellum as opposed to the cortex (Kaul et al. 2012).
Cell-extrinsic factors are emerging as important drivers of both adult and pediatric glioma growth (Charles et al. 2011) (Figure 3). With the recognition that monocytes comprise a significant percent of the cells in these tumors (Graeber et al. 2002), numerous studies have revealed the importance of targeting microglia and their paracrine growth-promoting factors (gliomagens) as alternative therapeutic strategies. While the majority of these studies have focused on high-grade gliomas that model adult glioblastoma (Feng et al. 2015; Hu et al. 2015; Pyonteck et al. 2013; Zhai et al. 2011; Zhang et al. 2009), there is emerging evidence that silencing microglia function may disrupt critical cellular relationships with the tumor ecosystem to profoundly impair glioma growth (Solga A & Gutmann DH, unpublished observations). Importantly, microglia are highly dynamic immune system-like cells which respond to changes in their local environment (Solga et al. 2015). The discovery of future stroma-directed therapies will need to specifically focus on glioma-associated monocytes isolated directly from the tumor. In this regard, not only are microglia reprogrammed by the tumor ecosystem, but other cell types, including cancer stem cells, may be altered in the context of a glioma. Recent studies have revealed that Nf1-deficient mouse low-grade glioma stem cells are molecularly and functionally distinct from their Nf1-deficient non-neoplastic neural stem cell counterparts (Chen et al. 2015). This observation creates a unique opportunity to develop treatments that target the cancer stem cell, rather than non-neoplastic neural stem cells in the developing brains of children.
Finally, the influence of background genomics has not been fully investigated, but represents an essential element that dictate how mutations impact on preneoplastic cells and how non-neoplastic stromal cells respond to an evolving glioma. Elegant studies in murine Nf1 models have uncovered several genomic modifier loci that influence the penetrance or location of glioma (Reilly 2009). While less is known about these genomic modifiers in humans, early studies in children with NF1 have revealed several potential single nucleotide polymorphisms which influence optic glioma formation (Warrington et al. 2015). The mechanisms underlying these effects are currently unknown, but could reflect the impact of specific NF1 gene mutations on neurofibromin expression and function in the context of NF1-associated tumors (Anastasaki et al. 2015; Hawes et al. 2007) or sporadic gliomas harboring other genetic changes. Similarly, the intriguing protective effect of asthma and allergic skin conditions on glioma incidence (Chen et al. 2011; Linos et al. 2007) suggests that genomic modifiers of immune system cell (e.g., monocyte) function might differentially affect stromal cells important for glioma formation and continued growth.
Together with new insights that derive from comprehensive genetic analyses of human pediatric gliomas, the availability of accurate small-animal models of glioma afford tractable experimental platforms to critically evaluate each contributing factor in isolation, define its responsible mechanism, and translate these findings into improved risk stratification models and treatment strategies for children with low-grade and high-grade glioma.
Main Points.
Pediatric gliomas comprise a heterogeneous collection of glial neoplasms driven by different molecular events. Herein, we discuss the intersection between developmental neurobiology and pediatric neuro-oncology focusing on childhood glial neoplasms.
Acknowledgments
This work was partially funded by grants from the National Brain Tumor Society (D.H.G.) and the National Cancer Institute (1-R01-CA195692-01 to DHG, R01-CA188516 to SJB and P01-CA096832 to SJB and DWE).
References
- Abadin SS, Zoellner NL, Schaeffer M, Porcelli B, Gutmann DH, Johnson KJ. Racial/Ethnic Differences in Pediatric Brain Tumor Diagnoses in Patients with Neurofibromatosis Type 1. J Pediatr. 2015 doi: 10.1016/j.jpeds.2015.04.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel TW, Clark C, Bierie B, Chytil A, Aakre M, Gorska A, Moses HL. GFAP-Cre-mediated activation of oncogenic K-ras results in expansion of the subventricular zone and infiltrating glioma. Mol Cancer Res. 2009;7(5):645–653. doi: 10.1158/1541-7786.MCR-08-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, Alvarez-Buylla A, Parada LF. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell. 2009;15(1):45–56. doi: 10.1016/j.ccr.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amlin-Van Schaick JC, Kim S, DiFabio C, Lee MH, Broman KW, Reilly KM. Arlm1 is a male-specific modifier of astrocytoma resistance on mouse Chr 12. Neuro Oncol. 2012;14(2):160–174. doi: 10.1093/neuonc/nor206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasaki C, Woo AS, Messiaen LM, Gutmann DH. Elucidating the impact of neurofibromatosis-1 germline mutations on neurofibromin function and dopamine-based learning. Hum Mol Genet. 2015;24(12):3518–3528. doi: 10.1093/hmg/ddv103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, Tang Y, DeFrances J, Stover E, Weissleder R, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1(3):269–277. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- Bajenaru ML, Garbow JR, Perry A, Hernandez MR, Gutmann DH. Natural history of neurofibromatosis 1-associated optic nerve glioma in mice. Ann Neurol. 2005;57(1):119–127. doi: 10.1002/ana.20337. [DOI] [PubMed] [Google Scholar]
- Bajenaru ML, Hernandez MR, Perry A, Zhu Y, Parada LF, Garbow JR, Gutmann DH. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 2003;63(24):8573–8577. [PubMed] [Google Scholar]
- Bajenaru ML, Zhu Y, Hedrick NM, Donahoe J, Parada LF, Gutmann DH. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol. 2002;22(14):5100–5113. doi: 10.1128/MCB.22.14.5100-5113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandeira F, Lent R, Herculano-Houzel S. Changing numbers of neuronal and non-neuronal cells underlie postnatal brain growth in the rat. Proc Natl Acad Sci U S A. 2009;106(33):14108–14113. doi: 10.1073/pnas.0804650106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardehle S, Kruger M, Buggenthin F, Schwausch J, Ninkovic J, Clevers H, Snippert HJ, Theis FJ, Meyer-Luehmann M, Bechmann I, et al. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat Neurosci. 2013;16(5):580–586. doi: 10.1038/nn.3371. [DOI] [PubMed] [Google Scholar]
- Bax DA, Mackay A, Little SE, Carvalho D, Viana-Pereira M, Tamber N, Grigoriadis AE, Ashworth A, Reis RM, Ellison DW, et al. A distinct spectrum of copy number aberrations in pediatric high-grade gliomas. Clin Cancer Res. 2010;16(13):3368–3377. doi: 10.1158/1078-0432.CCR-10-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher OJ, Hambardzumyan D, Walker TR, Helmy K, Nazarian J, Albrecht S, Hiner RL, Gall S, Huse JT, Jabado N, et al. Preclinical evaluation of radiation and perifosine in a genetically and histologically accurate model of brainstem glioma. Cancer Res. 2010;70(6):2548–2557. doi: 10.1158/0008-5472.CAN-09-2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, et al. Reduced H3K27me3 and DNA Hypomethylation Are Major Drivers of Gene Expression in K27M Mutant Pediatric High-Grade Gliomas. Cancer Cell. 2013;24(5):660–672. doi: 10.1016/j.ccr.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Bjerke L, Mackay A, Nandhabalan M, Burford A, Jury A, Popov S, Bax DA, Carvalho D, Taylor KR, Vinci M, et al. Histone H3.3 Mutations Drive Pediatric Glioblastoma through Upregulation of MYCN. Cancer Discov. 2013 doi: 10.1158/2159-8290.CD-12-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, Morozova O, et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med. 2015;372(26):2481–2498. doi: 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody BA, Kinney HC, Kloman AS, Gilles FH. Sequence of central nervous system myelination in human infancy. I. An autopsy study of myelination. J Neuropathol Exp Neurol. 1987;46(3):283–301. doi: 10.1097/00005072-198705000-00005. [DOI] [PubMed] [Google Scholar]
- Broniscer A, Baker SJ, West AN, Fraser MM, Proko E, Kocak M, Dalton J, Zambetti GP, Ellison DW, Kun LE, et al. Clinical and molecular characteristics of malignant transformation of low-grade glioma in children. J Clin Oncol. 2007;25(6):682–689. doi: 10.1200/JCO.2006.06.8213. [DOI] [PubMed] [Google Scholar]
- Brossier NM, Gutmann DH. Improving outcomes for neurofibromatosis 1-associated brain tumors. Expert Rev Anticancer Ther. 2015;15(4):415–423. doi: 10.1586/14737140.2015.1009043. [DOI] [PubMed] [Google Scholar]
- Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Bouffet E, Bartels U, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet. 2014;46(5):451–456. doi: 10.1038/ng.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton A, Torres-Padilla ME. Chromatin dynamics in the regulation of cell fate allocation during early embryogenesis. Nat Rev Mol Cell Biol. 2014;15(11):723–734. doi: 10.1038/nrm3885. [DOI] [PubMed] [Google Scholar]
- Caretti V, Sewing AC, Lagerweij T, Schellen P, Bugiani M, Jansen MH, van Vuurden DG, Navis AC, Horsman I, Vandertop WP, et al. Human pontine glioma cells can induce murine tumors. Acta Neuropathol. 2014;127(6):897–909. doi: 10.1007/s00401-014-1272-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013;27(9):985–990. doi: 10.1101/gad.217778.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2011;59(8):1169–1180. doi: 10.1002/glia.21136. [DOI] [PubMed] [Google Scholar]
- Chen C, Xu T, Chen J, Zhou J, Yan Y, Lu Y, Wu S. Allergy and risk of glioma: a meta-analysis. Eur J Neurol. 2011;18(3):387–395. doi: 10.1111/j.1468-1331.2010.03187.x. [DOI] [PubMed] [Google Scholar]
- Chen YH, McGowan LD, Cimino PJ, Dahiya S, Leonard JR, Lee da Y, Gutmann DH. Mouse low-grade gliomas contain cancer stem cells with unique molecular and functional properties. Cell Rep. 2015;10(11):1899–1912. doi: 10.1016/j.celrep.2015.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow LM, Endersby R, Zhu X, Rankin S, Qu C, Zhang J, Broniscer A, Ellison DW, Baker SJ. Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell. 2011;19(3):305–316. doi: 10.1016/j.ccr.2011.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cin H, Meyer C, Herr R, Janzarik WG, Lambert S, Jones DT, Jacob K, Benner A, Witt H, Remke M, et al. Oncogenic FAM131B–BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol. 2011;121(6):763–774. doi: 10.1007/s00401-011-0817-z. [DOI] [PubMed] [Google Scholar]
- Cohen KJ, Heideman RL, Zhou T, Holmes EJ, Lavey RS, Bouffet E, Pollack IF. Temozolomide in the treatment of children with newly diagnosed diffuse intrinsic pontine gliomas: a report from the Children's Oncology Group. Neuro Oncol. 2011a;13(4):410–416. doi: 10.1093/neuonc/noq205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen KJ, Pollack IF, Zhou T, Buxton A, Holmes EJ, Burger PC, Brat DJ, Rosenblum MK, Hamilton RL, Lavey RS, et al. Temozolomide in the treatment of high-grade gliomas in children: a report from the Children's Oncology Group. Neuro Oncol. 2011b;13(3):317–323. doi: 10.1093/neuonc/noq191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daginakatte GC, Gianino SM, Zhao NW, Parsadanian AS, Gutmann DH. Increased c-Jun-NH2-kinase signaling in neurofibromatosis-1 heterozygous microglia drives microglia activation and promotes optic glioma proliferation. Cancer Res. 2008;68(24):10358–10366. doi: 10.1158/0008-5472.CAN-08-2506. [DOI] [PubMed] [Google Scholar]
- Daginakatte GC, Gutmann DH. Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum Mol Genet. 2007;16(9):1098–1112. doi: 10.1093/hmg/ddm059. [DOI] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta B, Li W, Perry A, Gutmann DH. Glioma formation in neurofibromatosis 1 reflects preferential activation of K-RAS in astrocytes. Cancer Res. 2005;65(1):236–245. [PubMed] [Google Scholar]
- Dawson MR, Polito A, Levine JM, Reynolds R. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci. 2003;24(2):476–488. doi: 10.1016/s1044-7431(03)00210-0. [DOI] [PubMed] [Google Scholar]
- Diggs-Andrews KA, Brown JA, Gianino SM, Rubin JB, Wozniak DF, Gutmann DH. Sex Is a major determinant of neuronal dysfunction in neurofibromatosis type 1. Ann Neurol. 2014;75(2):309–316. doi: 10.1002/ana.24093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Roncari L, Shannon P, Wu X, Lau N, Karaskova J, Gutmann DH, Squire JA, Nagy A, Guha A. Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res. 2001;61(9):3826–3836. [PubMed] [Google Scholar]
- Feng X, Szulzewsky F, Yerevanian A, Chen Z, Heinzmann D, Rasmussen RD, Alvarez-Garcia V, Kim Y, Wang B, Tamagno I, et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget. 2015;6(17):15077–15094. doi: 10.18632/oncotarget.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, Bechet D, Faury D, De Jay N, Ramkissoon LA, et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet. 2014;46(5):462–466. doi: 10.1038/ng.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forshew T, Tatevossian RG, Lawson AR, Ma J, Neale G, Ogunkolade BW, Jones TA, Aarum J, Dalton J, Bailey S, et al. Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol. 2009;218(2):172–181. doi: 10.1002/path.2558. [DOI] [PubMed] [Google Scholar]
- Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, Keir ST, Ji AX, Zoppoli P, Niola F, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. 2013;45(10):1141–1149. doi: 10.1038/ng.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O, Ellisman MH, Verma IM. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012;338(6110):1080–1084. doi: 10.1126/science.1226929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato K, Major T, Lewis PW, Allis CD, Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science. 2014;346(6216):1529–1533. doi: 10.1126/science.1253799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvao RP, Kasina A, McNeill RS, Harbin JE, Foreman O, Verhaak RG, Nishiyama A, Miller CR, Zong H. Transformation of quiescent adult oligodendrocyte precursor cells into malignant glioma through a multistep reactivation process. Proc Natl Acad Sci U S A. 2014;111(40):E4214–E4223. doi: 10.1073/pnas.1414389111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge WP, Miyawaki A, Gage FH, Jan YN, Jan LY. Local generation of glia is a major astrocyte source in postnatal cortex. Nature. 2012;484(7394):376–380. doi: 10.1038/nature10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geha S, Pallud J, Junier MP, Devaux B, Leonard N, Chassoux F, Chneiweiss H, Daumas-Duport C, Varlet P. NG2+/Olig2+ cells are the major cycle-related cell population of the adult human normal brain. Brain Pathol. 2010;20(2):399–411. doi: 10.1111/j.1750-3639.2009.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G, Zuchero JB, et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science. 2014;344(6183):1252304. doi: 10.1126/science.1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes WA, Mehler MF, Kessler JA. Transgenic overexpression of BMP4 increases astroglial and decreases oligodendroglial lineage commitment. Dev Biol. 2003;255(1):164–177. doi: 10.1016/s0012-1606(02)00037-4. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Scheithauer BW, Kreutzberg GW. Microglia in brain tumors. Glia. 2002;40(2):252–259. doi: 10.1002/glia.10147. [DOI] [PubMed] [Google Scholar]
- Gronych J, Korshunov A, Bageritz J, Milde T, Jugold M, Hambardzumyan D, Remke M, Hartmann C, Witt H, Jones DT, et al. An activated mutant BRAF kinase domain is sufficient to induce pilocytic astrocytoma in mice. J Clin Invest. 2011;121(4):1344–1348. doi: 10.1172/JCI44656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillamo JS, Creange A, Kalifa C, Grill J, Rodriguez D, Doz F, Barbarot S, Zerah M, Sanson M, Bastuji-Garin S, et al. Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): a retrospective study of 104 patients. Brain. 2003;126(Pt 1):152–160. doi: 10.1093/brain/awg016. [DOI] [PubMed] [Google Scholar]
- Gutmann DH. Eliminating barriers to personalized medicine: learning from neurofibromatosis type 1. Neurology. 2014;83(5):463–471. doi: 10.1212/WNL.0000000000000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutmann DH, Donahoe J, Brown T, James CD, Perry A. Loss of neurofibromatosis 1 (NF1) gene expression in NF1-associated pilocytic astrocytomas. Neuropathol Appl Neurobiol. 2000;26(4):361–367. doi: 10.1046/j.1365-2990.2000.00258.x. [DOI] [PubMed] [Google Scholar]
- Gutmann DH, McLellan MD, Hussain I, Wallis JW, Fulton LL, Fulton RS, Magrini V, Demeter R, Wylie T, Kandoth C, et al. Somatic neurofibromatosis type 1 (NF1) inactivation characterizes NF1-associated pilocytic astrocytoma. Genome Res. 2013;23(3):431–439. doi: 10.1101/gr.142604.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawes JJ, Tuskan RG, Reilly KM. Nf1 expression is dependent on strain background: implications for tumor suppressor haploinsufficiency studies. Neurogenetics. 2007;8(2):121–130. doi: 10.1007/s10048-006-0078-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus B, Banerjee D, Yeh TH, Rothermich S, Perry A, Rubin JB, Garbow JR, Gutmann DH. Preclinical cancer therapy in a mouse model of neurofibromatosis-1 optic glioma. Cancer Res. 2008;68(5):1520–1528. doi: 10.1158/0008-5472.CAN-07-5916. [DOI] [PubMed] [Google Scholar]
- Hegedus B, Hughes FW, Garbow JR, Gianino S, Banerjee D, Kim K, Ellisman MH, Brantley MA, Jr, Gutmann DH. Optic nerve dysfunction in a mouse model of neurofibromatosis-1 optic glioma. J Neuropathol Exp Neurol. 2009;68(5):542–551. doi: 10.1097/NEN.0b013e3181a3240b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25(1):55–57. doi: 10.1038/75596. [DOI] [PubMed] [Google Scholar]
- Hu F, a Dzaye OD, Hahn A, Yu Y, Scavetta RJ, Dittmar G, Kaczmarek AK, Dunning KR, Ricciardelli C, Rinnenthal JL, et al. Glioma-derived versican promotes tumor expansion via glioma-associated microglial/macrophages Toll-like receptor 2 signaling. Neuro Oncol. 2015;17(2):200–210. doi: 10.1093/neuonc/nou324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu N, Strobl-Mazzulla PH, Bronner ME. Epigenetic regulation in neural crest development. Dev Biol. 2014;396(2):159–168. doi: 10.1016/j.ydbio.2014.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huillard E, Hashizume R, Phillips JJ, Griveau A, Ihrie RA, Aoki Y, Nicolaides T, Perry A, Waldman T, McMahon M, et al. Cooperative interactions of BRAFV600E kinase and CDKN2A locus deficiency in pediatric malignant astrocytoma as a basis for rational therapy. Proc Natl Acad Sci U S A. 2012;109(22):8710–8715. doi: 10.1073/pnas.1117255109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ida CM, Lambert SR, Rodriguez FJ, Voss JS, Mc Cann BE, Seys AR, Halling KC, Collins VP, Giannini C. BRAF alterations are frequent in cerebellar low-grade astrocytomas with diffuse growth pattern. J Neuropathol Exp Neurol. 2012;71(7):631–639. doi: 10.1097/NEN.0b013e31825c448a. [DOI] [PubMed] [Google Scholar]
- Jacob K, Quang-Khuong DA, Jones DT, Witt H, Lambert S, Albrecht S, Witt O, Vezina C, Shirinian M, Faury D, et al. Genetic aberrations leading to MAPK pathway activation mediate oncogene-induced senescence in sporadic pilocytic astrocytomas. Clin Cancer Res. 2011;17(14):4650–4660. doi: 10.1158/1078-0432.CCR-11-0127. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Scheinberg JA, Senevirathne R, Cutter CN. The efficacy of short and repeated high-pressure processing treatments on the reduction of non-O157:H7 Shiga-toxin producing Escherichia coli in ground beef patties. Meat Sci. 2015;102:22–26. doi: 10.1016/j.meatsci.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Jones C, Baker SJ. Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat Rev Cancer. 2014;14(10) doi: 10.1038/nrc3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Hutter B, Jager N, Korshunov A, Kool M, Warnatz HJ, Zichner T, Lambert SR, Ryzhova M, Quang DA, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45(8):927–932. doi: 10.1038/ng.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, Collins VP. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68(21):8673–8677. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul A, Chen YH, Emnett RJ, Dahiya S, Gutmann DH. Pediatric glioma-associated KIAA1549:BRAF expression regulates neuroglial cell growth in a cell type-specific and mTOR-dependent manner. Genes Dev. 2012;26(23):2561–2566. doi: 10.1101/gad.200907.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul A, Chen YH, Emnett RJ, Gianino SM, Gutmann DH. Conditional KIAA1549:BRAF mice reveal brain region- and cell type-specific effects. Genesis. 2013;51(10):708–716. doi: 10.1002/dvg.22415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul A, Toonen JA, Cimino PJ, Gianino SM, Gutmann DH. Akt- or MEK-mediated mTOR inhibition suppresses Nf1 optic glioma growth. Neuro Oncol. 2015;17(6):843–853. doi: 10.1093/neuonc/nou329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, Richardson WD. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci. 2006;9(2):173–179. doi: 10.1038/nn1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney HC, Brody BA, Kloman AS, Gilles FH. Sequence of central nervous system myelination in human infancy. II. Patterns of myelination in autopsied infants. J Neuropathol Exp Neurol. 1988;47(3):217–234. doi: 10.1097/00005072-198805000-00003. [DOI] [PubMed] [Google Scholar]
- Kluwe L, Hagel C, Tatagiba M, Thomas S, Stavrou D, Ostertag H, von Deimling A, Mautner VF. Loss of NF1 alleles distinguish sporadic from NF1-associated pilocytic astrocytomas. J Neuropathol Exp Neurol. 2001;60(9):917–920. doi: 10.1093/jnen/60.9.917. [DOI] [PubMed] [Google Scholar]
- Lebel C, Gee M, Camicioli R, Wieler M, Martin W, Beaulieu C. Diffusion tensor imaging of white matter tract evolution over the lifespan. Neuroimage. 2012;60(1):340–352. doi: 10.1016/j.neuroimage.2011.11.094. [DOI] [PubMed] [Google Scholar]
- Lee da Y, Gianino SM, Gutmann DH. Innate neural stem cell heterogeneity determines the patterning of glioma formation in children. Cancer Cell. 2012;22(1):131–138. doi: 10.1016/j.ccr.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee da Y, Yeh TH, Emnett RJ, White CR, Gutmann DH. Neurofibromatosis-1 regulates neuroglial progenitor proliferation and glial differentiation in a brain region-specific manner. Genes Dev. 2010;24(20):2317–2329. doi: 10.1101/gad.1957110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei L, Sonabend AM, Guarnieri P, Soderquist C, Ludwig T, Rosenfeld S, Bruce JN, Canoll P. Glioblastoma models reveal the connection between adult glial progenitors and the proneural phenotype. PLoS One. 2011;6(5):e20041. doi: 10.1371/journal.pone.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340(6134):857–861. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linos E, Raine T, Alonso A, Michaud D. Atopy and risk of brain tumors: a meta-analysis. J Natl Cancer Inst. 2007;99(20):1544–1550. doi: 10.1093/jnci/djm170. [DOI] [PubMed] [Google Scholar]
- Listernick R, Darling C, Greenwald M, Strauss L, Charrow J. Optic pathway tumors in children: the effect of neurofibromatosis type 1 on clinical manifestations and natural history. J Pediatr. 1995;127(5):718–722. doi: 10.1016/s0022-3476(95)70159-1. [DOI] [PubMed] [Google Scholar]
- Listernick R, Louis DN, Packer RJ, Gutmann DH. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143–149. doi: 10.1002/ana.410410204. [DOI] [PubMed] [Google Scholar]
- Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, Foreman O, Bronson RT, Nishiyama A, Luo L, et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011a;146(2):209–221. doi: 10.1016/j.cell.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu KW, Feng H, Bachoo R, Kazlauskas A, Smith EM, Symes K, Hamilton RL, Nagane M, Nishikawa R, Hu B, et al. SHP-2/PTPN11 mediates gliomagenesis driven by PDGFRA and INK4A/ARF aberrations in mice and humans. J Clin Invest. 2011b;121(3):905–917. doi: 10.1172/JCI43690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO Classification of Tumours of the Central Nervous System. 2007a doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007b;114(2):97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis DN, Perry A, Burger P, Ellison DW, Reifenberger G, von Deimling A, Aldape K, Brat D, Collins VP, Eberhart C, et al. International Society Of Neuropathology--Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol. 2014;24(5):429–435. doi: 10.1111/bpa.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui K, Suzuki SO, Torisu R, Goldman JE, Canoll P, Iwaki T. Glial progenitors in the brainstem give rise to malignant gliomas by platelet-derived growth factor stimulation. Glia. 2010;58(9):1050–1065. doi: 10.1002/glia.20986. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Deneen B. Astrocyte development: A Guide for the Perplexed. Glia. 2015;63(8):1320–1329. doi: 10.1002/glia.22836. [DOI] [PubMed] [Google Scholar]
- Monje M, Mitra SS, Freret ME, Raveh TB, Kim J, Masek M, Attema JL, Li G, Haddix T, Edwards MS, et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci U S A. 2011;108(11):4453–4458. doi: 10.1073/pnas.1101657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz DM, Tung T, Agnihotri S, Singh S, Guha A, Zadeh G, Hawkins C. Loss of p53 cooperates with K-ras activation to induce glioma formation in a region-independent manner. Glia. 2013;61(11):1862–1872. doi: 10.1002/glia.22563. [DOI] [PubMed] [Google Scholar]
- Ohgaki H, Kleihues P. The definition of primary and secondary glioblastoma. Clin Cancer Res. 2013;19(4):764–772. doi: 10.1158/1078-0432.CCR-12-3002. [DOI] [PubMed] [Google Scholar]
- Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, Wolinsky Y, Kruchko C, Barnholtz-Sloan J. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2014;16(Suppl 4):iv1–iv63. doi: 10.1093/neuonc/nou223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K, Charles N, Michor F, Holland EC. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014;26(2):288–300. doi: 10.1016/j.ccr.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paugh BS, Broniscer A, Qu C, Miller CP, Zhang J, Tatevossian RG, Olson JM, Geyer JR, Chi SN, da Silva NS, et al. Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J Clin Oncol. 2011;29(30):3999–4006. doi: 10.1200/JCO.2011.35.5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, Bax DA, Coyle B, Barrow J, Hargrave D, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol. 2010;28(18):3061–3068. doi: 10.1200/JCO.2009.26.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paugh BS, Zhu X, Qu C, Endersby R, Diaz AK, Zhang J, Bax DA, Carvalho D, Reis RM, Onar-Thomas A, et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res. 2013;73(20):6219–6229. doi: 10.1158/0008-5472.CAN-13-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N, Toedt G, Wittmann A, Kratz C, Olbrich H, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest. 2008;118(5):1739–1749. doi: 10.1172/JCI33656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister S, Witt O. Pediatric gliomas. Recent Results Cancer Res. 2009;171:67–81. doi: 10.1007/978-3-540-31206-2_4. [DOI] [PubMed] [Google Scholar]
- Pong WW, Higer SB, Gianino SM, Emnett RJ, Gutmann DH. Reduced microglial CX3CR1 expression delays neurofibromatosis-1 glioma formation. Ann Neurol. 2013;73(2):303–308. doi: 10.1002/ana.23813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puget S, Philippe C, Bax DA, Job B, Varlet P, Junier MP, Andreiuolo F, Carvalho D, Reis R, Guerrini-Rousseau L, et al. Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS One. 2012;7(2):e30313. doi: 10.1371/journal.pone.0030313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qaddoumi I, Sultan I, Gajjar A. Outcome and prognostic features in pediatric gliomas: a review of 6212 cases from the Surveillance, Epidemiology, and End Results database. Cancer. 2009;115(24):5761–5770. doi: 10.1002/cncr.24663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu HQ, Jacob K, Fatet S, Ge B, Barnett D, Delattre O, Faury D, Montpetit A, Solomon L, Hauser P, et al. Genome-wide profiling using single-nucleotide polymorphism arrays identifies novel chromosomal imbalances in pediatric glioblastomas. Neuro Oncol. 2010;12(2):153–163. doi: 10.1093/neuonc/nop001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raabe EH, Lim KS, Kim JM, Meeker A, Mao XG, Nikkhah G, Maciaczyk J, Kahlert U, Jain D, Bar E, et al. BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res. 2011;17(11):3590–3599. doi: 10.1158/1078-0432.CCR-10-3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramkissoon LA, Horowitz PM, Craig JM, Ramkissoon SH, Rich BE, Schumacher SE, McKenna A, Lawrence MS, Bergthold G, Brastianos PK, et al. Genomic analysis of diffuse pediatric low-grade gliomas identifies recurrent oncogenic truncating rearrangements in the transcription factor MYBL1. Proc Natl Acad Sci U S A. 2013;110(20):8188–8193. doi: 10.1073/pnas.1300252110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly KM. Brain tumor susceptibility: the role of genetic factors and uses of mouse models to unravel risk. Brain Pathol. 2009;19(1):121–131. doi: 10.1111/j.1750-3639.2008.00236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000;26(1):109–113. doi: 10.1038/79075. [DOI] [PubMed] [Google Scholar]
- Reilly KM, Tuskan RG, Christy E, Loisel DA, Ledger J, Bronson RT, Smith CD, Tsang S, Munroe DJ, Jacks T. Susceptibility to astrocytoma in mice mutant for Nf1 and Trp53 is linked to chromosome 11 and subject to epigenetic effects. Proc Natl Acad Sci U S A. 2004;101(35):13008–13013. doi: 10.1073/pnas.0401236101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez FJ, Ligon AH, Horkayne-Szakaly I, Rushing EJ, Ligon KL, Vena N, Garcia DI, Cameron JD, Eberhart CG. BRAF duplications and MAPK pathway activation are frequent in gliomas of the optic nerve proper. J Neuropathol Exp Neurol. 2012;71(9):789–794. doi: 10.1097/NEN.0b013e3182656ef8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessmann U, Gambetti P. Astrocytes in the developing human brain. An immunohistochemical study. Acta Neuropathol. 1986;70(3–4):308–313. doi: 10.1007/BF00686089. [DOI] [PubMed] [Google Scholar]
- Rowitch DH, Kriegstein AR. Developmental genetics of vertebrate glial-cell specification. Nature. 2010;468(7321):214–222. doi: 10.1038/nature09611. [DOI] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol. 2013;106–107:1–16. doi: 10.1016/j.pneurobio.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Sabha N, Lau N, Kamnasaran D, Gutmann DH, Guha A. Pathological and molecular progression of astrocytomas in a GFAP:12 V-Ha-Ras mouse astrocytoma model. Am J Pathol. 2005;167(3):859–867. doi: 10.1016/S0002-9440(10)62057-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38(5):525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- Simmons GW, Pong WW, Emnett RJ, White CR, Gianino SM, Rodriguez FJ, Gutmann DH. Neurofibromatosis-1 heterozygosity increases microglia in a spatially and temporally restricted pattern relevant to mouse optic glioma formation and growth. J Neuropathol Exp Neurol. 2011;70(1):51–62. doi: 10.1097/NEN.0b013e3182032d37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simo-Riudalbas L, Esteller M. Cancer genomics identifies disrupted epigenetic genes. Hum Genet. 2014;133(6):713–725. doi: 10.1007/s00439-013-1373-5. [DOI] [PubMed] [Google Scholar]
- Solga AC, Pong WW, Walker J, Wylie T, Magrini V, Apicelli AJ, Griffith M, Griffith OL, Kohsaka S, Wu GF, et al. RNA-sequencing reveals oligodendrocyte and neuronal transcripts in microglia relevant to central nervous system disease. Glia. 2015;63(4):531–548. doi: 10.1002/glia.22754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splinter WM, Stewart JA, Muir JG. Large volumes of apple juice preoperatively do not affect gastric pH and volume in children. Can J Anaesth. 1990;37(1):36–39. doi: 10.1007/BF03007481. [DOI] [PubMed] [Google Scholar]
- Staser K, Yang FC, Clapp DW. Pathogenesis of plexiform neurofibroma: tumor-stromal/hematopoietic interactions in tumor progression. Annu Rev Pathol. 2012;7:469–495. doi: 10.1146/annurev-pathol-011811-132441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm D, Bender S, Jones DT, Lichter P, Grill J, Becher O, Hawkins C, Majewski J, Jones C, Costello JF, et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer. 2014;14(2):92–107. doi: 10.1038/nrc3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22(4):425–437. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- Sugiarto S, Persson AI, Munoz EG, Waldhuber M, Lamagna C, Andor N, Hanecker P, Ayers-Ringler J, Phillips J, Siu J, et al. Asymmetry-defective oligodendrocyte progenitors are glioma precursors. Cancer Cell. 2011;20(3):328–340. doi: 10.1016/j.ccr.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebayashi H, Ikenaka K. Oligodendrocyte generation during mouse development. Glia. 2015;63(8):1350–1356. doi: 10.1002/glia.22863. [DOI] [PubMed] [Google Scholar]
- Tasker RC. Changes in white matter late after severe traumatic brain injury in childhood. Dev Neurosci. 2006;28(4–5):302–308. doi: 10.1159/000094156. [DOI] [PubMed] [Google Scholar]
- Tatevossian RG, Tang B, Dalton J, Forshew T, Lawson AR, Ma J, Neale G, Shurtleff SA, Bailey S, Gajjar A, et al. MYB upregulation and genetic aberrations in a subset of pediatric low-grade gliomas. Acta Neuropathol. 2010;120(6):731–7343. doi: 10.1007/s00401-010-0763-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KR, Mackay A, Truffaux N, Butterfield YS, Morozova O, Philippe C, Castel D, Grasso CS, Vinci M, Carvalho D, et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet. 2014;46(5):457–461. doi: 10.1038/ng.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TCGA. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhrbom L, Dai C, Celestino JC, Rosenblum MK, Fuller GN, Holland EC. Ink4a–Arf loss cooperates with KRas activation in astrocytes and neural progenitors to generate glioblastomas of various morphologies depending on activated Akt. Cancer Res. 2002;62(19):5551–5558. [PubMed] [Google Scholar]
- Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S, Gibson EM, Mount CW, Polepalli J, Mitra SS, et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell. 2015;161(4):803–816. doi: 10.1016/j.cell.2015.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti S, Garimella MT, Sullivan LM, Martinez D, Huse JT, Heguy A, Santi M, Thompson CB, Judkins AR. Evaluation of Histone 3 Lysine 27 Trimethylation (H3K27me3) and Enhancer of Zest 2 (EZH2) in Pediatric Glial and Glioneuronal Tumors Shows Decreased H3K27me3 in H3F3A K27M Mutant Glioblastomas. Brain Pathol. 2013 doi: 10.1111/bpa.12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walrath JC, Fox K, Truffer E, Gregory Alvord W, Quinones OA, Reilly KM. Chr 19(A/J) modifies tumor resistance in a sex- and parent-of-origin-specific manner. Mamm Genome. 2009;20(4):214–223. doi: 10.1007/s00335-009-9179-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Bushman J, Wang X, Mayer-Proschel M, Johnson M, Noble M. Oligodendrocyte/type-2 astrocyte progenitor cells and glial-restricted precursor cells generate different tumor phenotypes in response to the identical oncogenes. J Neurosci. 2013;33(42):16805–16817. doi: 10.1523/JNEUROSCI.0546-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren KE. Diffuse intrinsic pontine glioma: poised for progress. Front Oncol. 2012;2:205. doi: 10.3389/fonc.2012.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrington NM, Sun T, Luo J, McKinstry RC, Parkin PC, Ganzhorn S, Spoljaric D, Albers AC, Merkelson A, Stewart DR, et al. The cyclic AMP pathway is a sex-specific modifier of glioma risk in type I neurofibromatosis patients. Cancer Res. 2015;75(1):16–21. doi: 10.1158/0008-5472.CAN-14-1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer K, Eckart M, Meyer-Puttlitz B, Fonatsch C, Pietsch T. Mutational and expression analysis of the NF1 gene argues against a role as tumor suppressor in sporadic pilocytic astrocytomas. J Neuropathol Exp Neurol. 2002;61(10):896–902. doi: 10.1093/jnen/61.10.896. [DOI] [PubMed] [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44(3):251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46(5):444–450. doi: 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang FC, Ingram DA, Chen S, Zhu Y, Yuan J, Li X, Yang X, Knowles S, Horn W, Li Y, et al. Nf1-dependent tumors require a microenvironment containing Nf1+/-- and c-kit-dependent bone marrow. Cell. 2008;135(3):437–448. doi: 10.1016/j.cell.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Deshmukh H, Gutmann RJ, Emnett RJ, Rodriguez FJ, Watson MA, Nagarajan R, Gutmann DH. Alterations of BRAF and HIPK2 loci predominate in sporadic pilocytic astrocytoma. Neurology. 2009;73(19):1526–1531. doi: 10.1212/WNL.0b013e3181c0664a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarghooni M, Bartels U, Lee E, Buczkowicz P, Morrison A, Huang A, Bouffet E, Hawkins C. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J Clin Oncol. 2010;28(8):1337–1344. doi: 10.1200/JCO.2009.25.5463. [DOI] [PubMed] [Google Scholar]
- Zhai H, Heppner FL, Tsirka SE. Microglia/macrophages promote glioma progression. Glia. 2011;59(3):472–485. doi: 10.1002/glia.21117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, Orisme W, Punchihewa C, Parker M, Qaddoumi I, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. 2013;45(6):602–612. doi: 10.1038/ng.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Alizadeh D, Van Handel M, Kortylewski M, Yu H, Badie B. Stat3 inhibition activates tumor macrophages and abrogates glioma growth in mice. Glia. 2009;57(13):1458–1467. doi: 10.1002/glia.20863. [DOI] [PubMed] [Google Scholar]
- Zhao H, Ayrault O, Zindy F, Kim JH, Roussel MF. Post-transcriptional down-regulation of Atoh1/Math1 by bone morphogenic proteins suppresses medulloblastoma development. Genes Dev. 2008;22(6):722–727. doi: 10.1101/gad.1636408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Harada T, Liu L, Lush ME, Guignard F, Harada C, Burns DK, Bajenaru ML, Gutmann DH, Parada LF. Inactivation of NF1 in CNS causes increased glial progenitor proliferation and optic glioma formation. Development. 2005;132(24):5577–5588. doi: 10.1242/dev.02162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong H, Parada LF, Baker SJ. Cell of origin for malignant gliomas and its implication in therapeutic development. Cold Spring Harb Perspect Biol. 2015;7(5) doi: 10.1101/cshperspect.a020610. [DOI] [PMC free article] [PubMed] [Google Scholar]