

Abstract
There is currently no effective treatment for multiorgan failure following shock other than alleviation supportive care. A better understanding of the pathogenesis of these sequelae to shock is required. The intestine plays a central role in multiorgan failure. It was previously suggested that bacteria and their toxins are responsible for the organ failure seen in circulatory shock, but clinical trials in septic patients have not confirmed this hypothesis. Instead, we review here evidence that the digestive enzymes, synthesized in the pancreas and discharged into the small intestine as requirement for normal digestion, may play a role in multi-organ failure. These powerful enzymes are non-specific, highly concentrated and fully activated in the lumen of the intestine. During normal digestion they are compartmentalized in the lumen of the intestine by the mucosal epithelial barrier. However, if this barrier becomes permeable, e.g. in an ischemic state, the digestive enzymes escape into the wall of the intestine. They digest tissues in the mucosa and generate small molecular weight cytotoxic fragments such as unbound free fatty acids. Digestive enzymes may also escape into the systemic circulation and activate other degrading proteases. These proteases have the ability to clip the ectodomain of surface receptors and compromise their function; for example cleaving the insulin receptor causing insulin resistance. The combination of digestive enzymes and cytotoxic fragments leaking into the central circulation causes cell and organ dysfunction, and ultimately may lead to complete organ failure and death. We summarize current evidence suggesting that enteral blockade of digestive enzymes inside the lumen of the intestine may serve to reduce acute cell and organ damage and improve survival in experimental shock.
Keywords: Intestinal epithelium, sepsis, pancreatic digestive enzyme, intestinal mucosa, permeability, mucin, unbound free fatty acids
Introduction
Organ failure after shock often involves damage to remote organs that are not necessarily part of the initial injury. An example is intestinal ischemia that is followed by failure of the lung (1,2), heart, brain and other organs (3,4). The relationship of the intestine to remote organ failure is illustrated by enterectomy, which has been found to protect against the irreversible progression of multiorgan failure and death in acute shock (5). However, the molecular pathways by which the intestine causes remote organ injury are not clearly understood.
The fundamental function of the intestine is digestion of food to provide nutritive requirements to the body. During digestion the lumen of the intestine is filled with digestive enzymes, degraded food components, and ambient microorganisms and viruses. Shock research has focused for decades on the role of bacterial microorganisms (i.e. the microbiome), but no treatment to prevent the irreversible progression to organ failure in shock has been demonstrated. Instead, in the following discussion we will focus on the other components in the gut, mainly (a) digestive enzymes and (b) products they produce during food degradation; both may play a role in organ injury mediated by the intestine (6,7). The evidence available to date is derived from preclinical studies unless stated otherwise.
Digestion requires pancreatic enzymes that are discharged into the proximal small intestine where they mix with food entering from the stomach. Pancreatic digestive enzymes in the small intestine are relatively non-specific, fully activated and present in high concentrations. These enzymes are able to hydrolyse daily hundreds of grams of complex biological compounds (8,9). This is the dominating degrading process in the body and the key requirement to derive nutrients from the environment. Any discussion about the intestine in shock needs to be concerned with digestive enzymes and starts with a fundamental question:
“How is it possible that food (which may consist of intestine) is degraded in the small intestine, and yet the intestine itself is not – or is only minimally – digested.”
In other words, how are the intestine and other organs protected against autodigestion?
One of the main protection mechanisms against autodigestion of the intestine is provided by the mucosal epithelial barrier. This barrier prevents leakage of contents from the intestine, including digestive enzymes, from entering into the wall of the intestine. Consequently, breakdown of mucosal barrier integrity may allow digestive enzymes to escape past the mucosal barrier and into the wall of the intestine where they can begin the autodigestion process.
Different forms of shock including hemorrhage, trauma and sepsis are accompanied by markers for an inflammatory cascade (10) whose fundamental purpose is to repair damaged tissue after an injury. We will focus on initial mechanisms that may cause tissue injury and thereby evoke a repair mechanism, i.e. an inflammatory cascade. The approach serves as basis for new strategies to minimize tissue injury starting from early stages of shock.
Digestive Enzymes
We obtained initial insight into a potential role for digestive enzymes in cell and microvascular injury from studies using organ homogenates. These studies in the rat indicate that homogenates derived from the pancreas and the small intestine, as compared to other organs, are a major source of cytotoxic and inflammatory mediators (11). The intestine generates these mediators if the lumenal contents are present but not when the lumen is flushed and cleared of material. The addition of selected pancreatic enzymes to a flushed intestine restores the generation of inflammatory mediators, similar to the native environment of the intestine. This evidence clearly points to the digestive enzymes in the small intestine as instrumental in the generation of inflammatory mediators. Further analysis showed that among the major families of pancreatic digestive enzymes (proteases, lipases, amylases, and nucleases) proteases and lipases can generate mediators that are able to cause acute cell injury and organ dysfunction (12,13). In the following, we will limit the discussion to digestive proteases; other degrading enzymes are currently less explored.
Digestive proteases originate by biosynthesis in the acinar cells of the pancreas and are released in the form of proenzymes. Digestive proteases are transported via the pancreatic ducts into the duodenum and activated by enterokinases (14). In the lumen of the small intestine digestive proteases facilitate degradation of proteins and peptides from food into amino acids that can be taken up by the mucosal epithelial transporters (15). In the small intestine they form a powerful degrading system due to their high concentrations and relatively non-specific ability to hydrolyze proteins from different food sources, which suggests that the barrier is controlled by several factors to protect the gut against its contents. Thus, there is a fundamental need for an intrinsic protection mechanism in the small intestine against the action of digestive enzymes and autodigestion.
Mucosal Barrier
The mucosal barrier is well recognized for its ability to prevent undigested food or bacteria from passing across the epithelial cells into the mucosal space of the intestinal villi (16,17). In the context of the current discussion, the intestinal mucosal barrier serves to compartmentalize the digestive enzymes inside the lumen of the intestine. The mucosal barrier consists of a mucus layer, composed chiefly of mucins, covering the epithelium on the villi of the intestine. This layer, in conjunction with the epithelium, forms a barrier to the entry of digestive enzymes (18–20) and larger molecular weight food fragments while allowing the uptake of relatively low molecular weight nutrients (e.g. ions, amino acids, monosaccharides). There are multiple isoforms of mucin that protect the intestine via different mechanisms. In the rat, Mucin 2 is secreted by the goblet cells to cover the epithelium. During intestinal peristalsis this mucin is carried with food and detaches from the cells, helping facilitate movement along the length of the intestine. In contrast, Mucin 13 is bound to the epithelial membrane, protecting membrane receptors against extracellular cleavage by digestive proteases. This protection mechanism against ectodomain receptor cleavage is essential for normal nutrient absorption by the gut epithelium because if these proteins were nonfunctional, normal digestion would be hindered.
Breakdown of the Mucosal Barrier: A Hallmark for Shock
There is evidence that epithelial cells at the tips of the intestinal villi are subject to cell damage even in the absence of an apparent challenge that may be associated with shock (i.e., villous ischemia); in fact apoptosis is consistently observed in these cells even in apparently normal animals (21,22).
Experimental breakdown of the mucus barrier (e.g. with mucolytic N-acetylcysteine treatment) allows digestive enzymes to enter the intestinal wall, a process that is followed by severe damage to the intestinal mucosa (20) and may lead to death even in the absence of any other systemic challenge (19).
Damage to the mucosal barrier is consistently observed in shock. Irrespective of the particular form of injury (e.g. splanchnic artery occlusion, hemorrhagic, endotoxic shock, peritonitis), the resulting shock state is accompanied by a breakdown of the mucosal barrier. Even relatively short periods of intestinal ischemia (~15 min) are associated with degradation of the epithelial villi, including the underlying connective tissue and capillaries. Longer periods of ischemia lead to a more complete degradation of the villi, and even complete destruction of the entire villus structure (23), leaving the intestinal wall fully exposed to digestive enzymes and other luminal contents (e.g. partially digested food, bacteria, viruses). As the lamina propria degrades, not only are the epithelial cells detaching and apoptotic, but also the remaining cells are subject to a proteolytic degradation of their membrane receptors. We demonstrated this for the inter-epithelial adhesion molecules (e.g. occludin, E-cadherin) (24). The loss of the ectodomain of these adhesion molecules reduces the ability of epithelial cells to remain attached and maintain a tight barrier. New biosynthesis of these adhesion molecules in the absence of degrading digestive enzymes in the extracellular space is required to restore the epithelial barrier.
Entry of Digestive Enzymes into the Systemic Circulation
If the mucosal barrier is compromised, digestive proteases are transported from the intestinal lumen into the wall of the intestine (Figure 1). They may be further carried into venous blood vessels and intestinal lymphatics, and even across the full thickness of the small intestine directly into the peritoneal cavity (25–27). The escape of pancreatic proteases from the small intestine is accompanied by an increase in digestive protease activity in plasma and tissues, such as the liver, lung and heart (25,27), suggesting that endogenous inhibitors of digestive proteases may become fully bound and their ability to block proteases has become saturated (28–30). In experimental shock, proteases circulate in plasma and exhibit elevated activities as detected by cleavage of fluorescently quenched substrates (27,31), irrespective of whether caused by a directly ischemic state (e.g. hemorrhagic shock, splanchnic artery occlusion), by exposure to endotoxin or digested food in the intestine (e.g. endotoxic shock, cecal ligation shock) or by generation of inflammatory mediators in burns (e.g. complements (32)). In addition, there may be release of digestive enzymes directly from the pancreas, the magnitude of which in specific models of shock remains to be determined.
Figure 1.
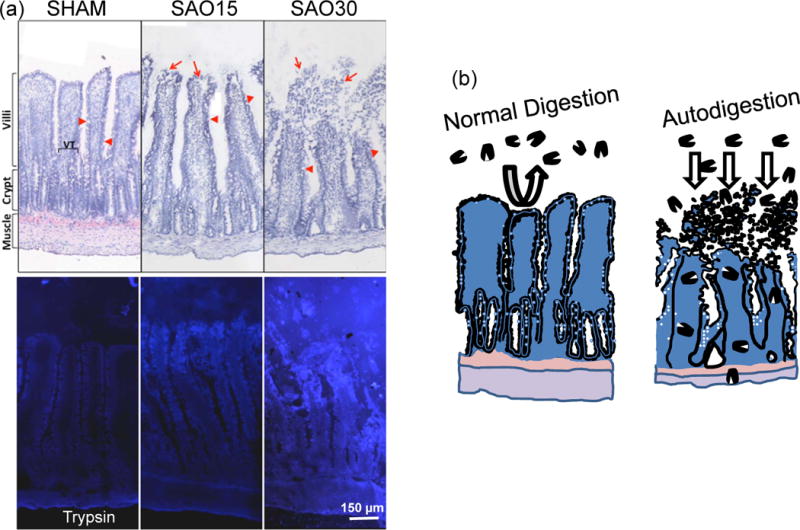
(a) Example for escape of a digestive enzyme (trypsin) into the wall of the small intestine in the case of splenic artery occlusion (SAO). Top panels show intestinal villi morphology (frozen section labeled with Evans blue) before (Sham) and 15 and 30 min after SAO (SAO15, SAO30, respectively). Bottom panels show corresponding pancreatic trypsin activity (by in-situ zymography according to Methods described in (47)). Bright blue fluorescence indicated cleavage of trypsin specific substrate and is visible inside the villi and across the full thickness of the intestinal wall to the level of the muscle and outer serosa. (Modified from Reference (47)).
(b) Normal Digestion: Schematic illustration of normal containment of digestive enzymes in the lumen of the intestine by the mucosal barrier (mucin layer in conjunction with the epithelium). Autodigestion: entry of digestive enzymes into the wall of the villi and small intestine and destruction of mucosal barrier.
Generation of inflammatory and cytotoxic fragments by digestive enzymes
Entry of digestive enzymes into the intestinal wall leads to generation of lipid fragments with cytotoxic activity (13). This is observed in the intestinal wall during ischemia (but not without ischemia) and under conditions of elevated permeability of the mucosal barrier with entry of digestive proteases into the intestinal wall. Cytotoxic mediators are also generated by trypsin, chymotrypsin and elastase and are undetectable if digestive enzymes in the lumen of the intestine are inhibited. While digestive enzymes may generate many water soluble protein fragments, such fragments may stimulate cells but collectively have low cytotoxicity (13). Instead, the major cytotoxic mediators are lipid in nature, especially unbound free fatty acids, which may cause severe destruction of membranes even at low concentrations. Free fatty acid binding proteins, e.g. albumin, bind unbound free fatty acid and may prevent their cytotoxic actions unless the albumin is also degraded by proteases (6). Mesenteric lymph draining the intestinal wall following ischemia is toxic due to the presence of free fatty acids (20,33) and may be involved in lung damage in shock due to the fact that mesenteric lymph enters the subclavian vein via the thoracic duct, which empties directly into the venous return and the lungs (20).
Another source of free fatty acids may be in food itself within the lumen of the intestine after exposure to pancreatic lipases and proteases, as suggested by in-vitro studies (13). The evidence is consistent with the protection provided to the intestine if the lumen is emptied and food absent prior to ischemia (4,26).
Matrix Metalloproteinases – Trigger for elevated permeability
Another family of proteases that is prominently involved in the inflammatory cascade and tissue repair is matrix metalloproteinases (MMPs) (34,35). They are present in tissues in a pro-form and can be activated within minutes, and therefore may play a role in the early stages of shock. ProMMPs are activated under ischemic conditions or by other proteases, including the pancreatic proteases in the intestine (e.g. trypsin) (36). MMP activity is encountered in the extracellular matrix, on endothelial and epithelial cells and mast cells and derived from activated neutrophils in the circulation.
MMPs can increase endothelial and epithelial permeability by proteolytic cleavage of the ectodomain of junctional proteins and opening of intercellular junctions (25,37,38), increasing mucosal permeability early in shock. MMPs also have the ability to digest the basement membrane of endothelium (39), thereby allowing characteristic tissue lesion formation due to escape of plasma and blood cells into the surrounding tissue (27). MMPs can also process lymphokines and cytokines that contribute to the inflammatory cascade (40), which illustrates their dual functions in tissue injury and tissue repair.
MMP activity and inhibition have been studied in models of organ ischemia and in shock and trauma (41–43), observed in acute lung (44) and heart injury (45) and in vascular refractoriness to different contractile agents (46). MMP inhibition in human shock conditions and as a therapy that may serve to minimize breakdown of the mucosal barrier remains to be examined.
Protease Activity and Receptor Cleavage
One consequence of enhanced proteolytic activity in the circulation and the extracellular space is that proteins and specifically receptors on the surface of cells may be degraded (18,24,47) (Figure 2). Receptor extracellular domains (“ectodomains”) may be clipped, leading to a loss of cell function. There appear to be multiple receptors subject to ectodomain cleavage (e.g. the TLR4 in the bowel), in addition to the inter-endothelial or inter-epithelial adhesion molecules (e.g. VE- cadherin, E-cadherin) (26,47).
Figure 2.
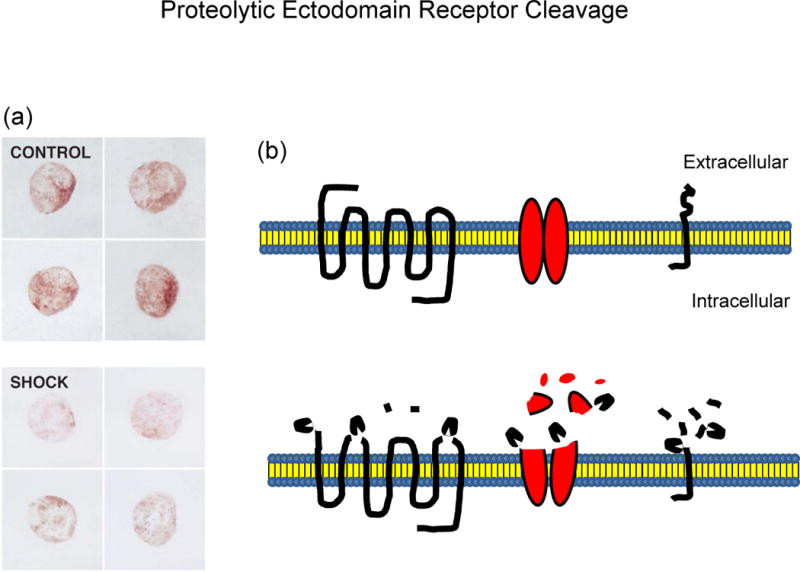
(a) Example of proteolytic receptor destruction as detected by immune-label density measurement. Receptor density on typical circulating leukocytes after labeling with a primary Ab against the extracellular domain of the insulin receptor before (CONTROL) and after hemorrhagic shock (SHOCK), according to Methods described in (27). Note the reduction in receptor density, a phenomenon associated with reduced insulin signaling and preventable by enteral blockade of the digestive proteases. Adopted from Reference (27)
(b) Schematic of extracellular receptor cleavage hypothesis by destruction of the extracellular domain of 7-transmembrane spanning receptor by cleavage of extracellular connecting loops, by extracellular destruction of membrane ion transporters, and by extracellular cleavage of single transmembrane receptors (e.g. insulin receptor).
One interesting case in this regard is the insulin receptor. Critically ill patients exhibit a decrease in insulin response, i.e. acute insulin resistance (48,49). The ectodomain of this receptor is readily cleaved by proteases, such as MMPs or serine proteases, yielding extracellular (“soluble”) receptor fragments (50). This action renders the receptor unable to signal after insulin binding and therefore contributes to an insulin-resistant state. Indeed, analysis of the molecular mechanisms for acute insulin resistance after hemorrhagic shock indicates proteolytic cleavage of the insulin receptor on endothelium and other cells as possibly cause for acute insulin resistance (51). In addition, there may also be a possible proteolytic degradation of the insulin molecule itself; this remains to be investigated.
Other models of acute inflammation provide evidence that receptor degradation by ectodomain cleavage may be a common mechanism for decreased intracellular signaling (50). Since many membrane receptors have potential cleavage sites in their extracellular domains, the phenomenon may play a major role in the multiple organ dysfunction characteristic of shock. Proteolytic destruction of membrane receptor ectodomains may also compromise pharmacological interventions that are receptor dependent, and therefore the receptor cleavage mechanism may underlie hemodynamic instability in shock where patients are less responsive to treatment.
It has yet to be demonstrated in animal models or human subjects, but an increase in soluble receptor fragments or reduced signaling of key surface receptors on endothelial cells may be responsible for inadequate cellular function in response to a receptor-mediated signal (52–54). A diminished response to stimulation of an extracellular receptor points to the possibility that either the receptor signaling is not properly in place or the receptor itself is missing. Cellar membrane receptors have specific structures including loops and chains that are necessary for binding to ligands. If these binding sites are disrupted, their agonists may not bind properly. However, there is difficulty in conducting experiments that differentiate between the extracellular and intracellular domain of receptors due to specific antibody availability and it remains a challenge to detect low levels of peptides in shock plasma that may have been cleaved from receptors. The development of a downregulated state of the immune system in sepsis (55) is consistent with such receptor cleavage by proteases. Many membrane ligand binding- or adhesion-receptors have potential cleavage sites in their ectodomain for proteases like trypsin or MMPs. The development of intestinal mucosa apoptotic markers and lesions in septic patients is also consistent with an uncontrolled proteolytic activity (56,57).
Inhibition of Digestive Protease Activity
Recent evidence from our lab and others on rats and pigs indicate that enteral inhibition of digestive enzymes attenuates a wide range of organ complications in shock. Protection against intestinal damage is observed in hemorrhagic shock, shock after splanchnic artery occlusion, endotoxin shock, and peritonitis (by peritoneal injection of cecal material) (3,4,23,27,58–62). Irrespective of the particular (serine) protease inhibitor used, the mortality after shock is significantly reduced in these shock models (27). Animal mobility and responsiveness in the recovery phase after shock is also improved (60). Histologically, damage to the intestinal villi is significantly reduced, as well as injury to remote organs such as the lung, liver, and heart (27). If digestive proteases are inhibited inside the lumen of the small intestine no signs of insulin resistance (discussed above) or insulin receptor cleavage are detected after hemorrhagic shock (51). This evidence in rats and pigs is in line with the basic hypothesis advanced in this review for autodigestion in shock. The evidence is also in line with protection provided to a septic patient treated on consent basis for the first time by enteral blockade of digestive enzymes (63). Addition of a free radical scavenger to protease inhibitors given enterally does not provide enhanced protection against tissue damage in intestinal ischemia (64), indicating that the protease inhibition per se is the major contributor to the protection.
An important issue is that inhibitors of the digestive proteases have to be applied directly into the lumen of the small intestine (“enterally’); intravenous application is ineffective (4,58). The requirement for the enteral route of administration is due to the high concentrations of the digestive enzymes in the lumen of the intestine (at an order of magnitude of 100 μM and higher). Such high concentrations need to be matched if competitive inhibitors are used and would be with side effects if used intravenously. Furthermore, intravenous inhibitors will not readily reach the lumen of the intestine if the microcirculation in the intestinal wall is compromised.
Another important requirement for effective enteral blockade of digestive enzymes is that enzymatic activity needs to be inhibited over the entire length of the small intestine. If digestive proteases are not inhibited in even short segments of the small intestine, such segments may be subject to significant intestinal damage and generate multi-organ failure (27). Regions of higher enzyme concentration may be not homogenously distributed across the intestine’s length, causing a bias in regions that are more susceptible to damage.
The enteral protease inhibitor treatment serves to minimize destruction of the intestine, but it should be noted that enteral digestive enzyme inhibition is not a treatment to repair damaged intestine. Repair of the damaged intestine requires a program of proliferation and differentiation of mucosal stem cells, epithelial growth factors and inflammatory repair mechanisms; inhibition of digestive enzymes merely stops continued elevation of mucosal permeability and autodigestion of the intestinal wall.
Conclusions, Clinical Implications and Future Work
The current evidence is consistent with the hypothesis that an important complication following elevation of the mucosal barrier permeability is escape of pancreatic digestive enzymes from the lumen of the intestine into the intestinal wall, peritoneum, lymph and circulation (Figure 1). Inside the wall of the intestine there is inadequate endogenous blockade of the high concentrations of digestive enzymes. The consequence is autodigestion of the intestinal wall. Digestive enzymes as well as cytotoxic products they generate escape into the systemic circulation, activate other degrading proteases, such as MMPs, proteolytically degrade membrane proteins and consequently cause loss of various cell functions (Figure 2). Degrading proteases can be derived not only from the pancreas and the lumen of the intestine, but also from circulating cells, mast cells, endothelial and epithelial cells, the extracellular matrix and bacteria in the intestine. Their role in opening of the mucosal barrier and resulting escape of digestive enzymes from the lumen of the intestine remains to be determined. Besides enteral blockade of the digestive enzymes, this new insight may open additional opportunities to minimize escape of digestive enzymes from the lumen of the intestine. MMP inhibition may serve to attenuate elevation of the mucosal barrier permeability, reduce the cytotoxic actions of free fatty acids by minimizing lipase activity and/or attachment to free fatty acid binding proteins (e.g. albumin).
Enteral blockade of digestive enzymes in shock patients may be feasible by way of a nasal gastric tube (63). However, the degree to which enteral blockade of digestive enzymes may serve to improve clinical outcomes in shock patients remain to be determined and depends in part on the magnitude of intestinal damage at the time an intervention is possible. If severe prolonged autodigestion and organ damage has already developed, enteral blockade of the digestive enzymes may not be sufficient to prevent organ failure. The earlier the blockade is initiated, the lower the level of subsequent organ damage. Ideal in this respect are elective surgery scenarios, in which pretreatment with digestive enzyme or MMP inhibitors is an option to minimize damage due to an ischemic intestine and autodigestion.
In shock research, many interventions that exhibit significant protection in preclinical studies ended up in human clinical trials demonstrating little or no efficacy. To help understand this discrepancy, it may be relevant to note that preclinical studies carried out in otherwise healthy animals may not simulate the degree of intestinal damage seen in critically ill patients, especially such comorbidities as prolonged surgery, previous infections or bowel diseases. The degree to which the intestine of patients is damaged and allows escape of digestive enzymes requires new measurement techniques.
Several lines of independent investigations on shock and acute organ failure point to proteolytic injury of cells and tissues as one of the early injury mechanisms. This provides an opportunity to understand and possibly prevent early injury as compared to interventions against the downstream inflammatory cascade, which in fact is often part of the tissue repair mechanisms.
Acknowledgments
We thank Drs. Alex Penn, Marisol Chang, and Frank A. DeLano for discussions and suggestions regarding the autodigestion hypothesis. Supported by NIH Grant GM 85072, Career Development Award (CDA2) 1IK2BX001277-01A1 from the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development.
Footnotes
Conflict of Interest: GWSS owns equity in InflammaGen, a company by Leading Bioscience Inc, which develops therapy for shock patients. For the remaining authors none were declared.
References
- 1.Magnotti LJ, Upperman JS, Xu DZ, Lu Q, Deitch EA. Gut-derived mesenteric lymph but not portal blood increases endothelial cell permeability and promotes lung injury after hemorrhagic shock. Ann Surg. 1998;228:518–527. doi: 10.1097/00000658-199810000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waisman D, Brod V, Wolff R, Sabo E, Chernin M, Weintraub Z, Rotschild A, Bitterman H. Effects of hyperoxia on local and remote microcirculatory inflammatory response after splanchnic ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2003;285:H643–652. doi: 10.1152/ajpheart.00900.2002. [DOI] [PubMed] [Google Scholar]
- 3.Mitsuoka H, Kistler EB, Schmid-Schönbein GW. Protease inhibition in the intestinal lumen: attenuation of systemic inflammation and early indicators of multiple organ failure in shock. Shock. 2002;17:205–209. doi: 10.1097/00024382-200203000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Mitsuoka H, Kistler EB, Schmid-Schönbein GW. Generation of in vivo activating factors in the ischemic intestine by pancreatic enzymes. Proc Natl Acad Sci U S A. 2000;97:1772–1777. doi: 10.1073/pnas.97.4.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang TW. Improvement of survival from hemorrhagic shock by enterectomy in rats: finding to implicate the role of the gut for irreversibility of hemorrhagic shock. J Trauma. 1997;42:223–230. doi: 10.1097/00005373-199702000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Penn AH, Schmid-Schönbein GW. The intestine as source of cytotoxic mediators in shock: free fatty acids and degradation of lipid-binding proteins. Am J Physiol Heart Circ Physiol. 2008;294:H1779–1792. doi: 10.1152/ajpheart.00902.2007. [DOI] [PubMed] [Google Scholar]
- 7.Fishman JE, Sheth SU, Levy G, Alli V, Lu Q, Xu D, Qin Y, Qin X, Deitch EA. Intraluminal nonbacterial intestinal components control gut and lung injury after trauma hemorrhagic shock. Ann Surg. 2014;260:1112–1120. doi: 10.1097/SLA.0000000000000631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rothman S, Liebow C, Isenman L. Conservation of digestive enzymes. Physiol Rev. 2002;82:1–18. doi: 10.1152/physrev.00022.2001. [DOI] [PubMed] [Google Scholar]
- 9.Whitcomb DC, Lowe ME. Human pancreatic digestive enzymes. Dig Dis Sci. 2007;52:1–17. doi: 10.1007/s10620-006-9589-z. [DOI] [PubMed] [Google Scholar]
- 10.Girard TD, Ware LB, Bernard GR, Pandharipande PP, Thompson JL, Shintani AK, Jackson JC, Dittus RS, Ely EW. Associations of markers of inflammation and coagulation with delirium during critical illness. Intensive Care Med. 2012;38:1965–1973. doi: 10.1007/s00134-012-2678-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kistler EB, Lefer AM, Hugli TE, Schmid-Schönbein GW. Plasma activation during splanchnic arterial occlusion shock. Shock. 2000;14:30–34. doi: 10.1097/00024382-200014010-00006. [DOI] [PubMed] [Google Scholar]
- 12.Kramp WJ, Waldo S, Schmid-Schönbein GW, Hoyt D, Coimbra R, Hugli TE. Characterization of two classes of pancreatic shock factors: functional differences exhibited by hydrophilic and hydrophobic shock factors. Shock. 2003;20:356–362. doi: 10.1097/01.shk.0000082442.66379.90. [DOI] [PubMed] [Google Scholar]
- 13.Penn AH, Hugli TE, Schmid-Schönbein GW. Pancreatic enzymes generate cytotoxic mediators in the intestine. Shock. 2007;27:296–304. doi: 10.1097/01.shk.0000235139.20775.7f. [DOI] [PubMed] [Google Scholar]
- 14.Mann NS, Mann SK. Enterokinase. Proc Soc Exp Biol Med. 1994;206:114–118. doi: 10.3181/00379727-206-43728. [DOI] [PubMed] [Google Scholar]
- 15.Pickel VM, Nirenberg MJ, Chan J, Mosckovitz R, Udenfriend S, Tate SS. Ultrastructural localization of a neutral and basic amino acid transporter in rat kidney and intestine. Proc Natl Acad Sci U S A. 1993;90:7779–7783. doi: 10.1073/pnas.90.16.7779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 17.Fishman JE, Levy G, Alli V, Zheng X, Mole DJ, Deitch EA. The intestinal mucus layer is a critical component of the gut barrier that is damaged during acute pancreatitis. Shock. 2014;42:264–270. doi: 10.1097/SHK.0000000000000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang M, Kistler EB, Schmid-Schönbein GW. Disruption of the mucosal barrier during gut ischemia allows entry of digestive enzymes into the intestinal wall. Shock. 2012;37:297–305. doi: 10.1097/SHK.0b013e318240b59b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kistler EB, Alsaigh T, Chang M, Schmid-Schönbein GW. Impaired small-bowel barrier integrity in the presence of lumenal pancreatic digestive enzymes leads to circulatory shock. Shock. 2012;38:262–267. doi: 10.1097/SHK.0b013e31825b1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharpe SM, Qin X, Lu Q, Feketeova E, Palange DC, Dong W, Sheth SU, Lee MA, Reino D, Xu DZ, et al. Loss of the intestinal mucus layer in the normal rat causes gut injury but not toxic mesenteric lymph nor lung injury. Shock. 2010;34:475–481. doi: 10.1097/SHK.0b013e3181dc3ff5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westcarr S, Farshori P, Wyche J, Anderson WA. Apoptosis and differentiation in the crypt-villus unit of the rat small intestine. J Submicrosc Cytol Pathol. 1999;31:15–30. [PubMed] [Google Scholar]
- 23.Fitzal F, DeLano FA, Young C, Rosario HS, Schmid-Schönbein GW. Pancreatic protease inhibition during shock attenuates cell activation and peripheral inflammation. J Vasc Res. 2002;39:320–329. doi: 10.1159/000065544. [DOI] [PubMed] [Google Scholar]
- 24.Altshuler AE, Lamadrid I, Li D, Ma SR, Kurre L, Schmid-Schönbein GW, Penn AH. Transmural intestinal wall permeability in severe ischemia after enteral protease inhibition. PLoS One. 2014;9:e96655. doi: 10.1371/journal.pone.0096655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Altshuler AE, Penn AH, Yang JA, Kim GR, Schmid-Schönbein GW. Protease activity increases in plasma, peritoneal fluid, and vital organs after hemorrhagic shock in rats. PLoS One. 2012;7:e32672. doi: 10.1371/journal.pone.0032672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altshuler AE, Richter MD, Modestino AE, Penn AH, Heller MJ, Schmid-Schönbein GW. Removal of luminal content protects the small intestine during hemorrhagic shock but is not sufficient to prevent lung injury. Physiol Rep. 2013;1:e00109. doi: 10.1002/phy2.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeLano FA, Hoyt DB, Schmid-Schönbein GW. Pancreatic digestive enzyme blockade in the intestine increases survival after experimental shock. Sci Transl Med. 2013;5:169ra111. doi: 10.1126/scitranslmed.3005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuroiwa K, Nakatsuyama S, Katayama K, Nagasawa T. Determination of alpha 2-macroglobulin-trypsin complex with a new synthetic substrate. Clin Chem. 1989;35:2169–2172. [PubMed] [Google Scholar]
- 29.Borgstrom A, Lasson A. Trypsin-alpha 1-protease inhibitor complexes in serum and clinical course of acute pancreatitis. Scand J Gastroenterol. 1984;19:1119–1122. [PubMed] [Google Scholar]
- 30.Aubry M, Bieth J. A kinetic study of the inhibition of human and bovine trypsins and chymotrypsins by the inter-alpha-inhibitor from human plasma. Biochim Biophys Acta. 1976;438:221–230. doi: 10.1016/0005-2744(76)90238-2. [DOI] [PubMed] [Google Scholar]
- 31.Gobbetti T, Cenac N, Motta JP, Rolland C, Martin L, Andrade-Gordon P, Steinhoff M, Barocelli E, Vergnolle N. Serine protease inhibition reduces post-ischemic granulocyte recruitment in mouse intestine. Am J Pathol. 2012;180:141–152. doi: 10.1016/j.ajpath.2011.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gelfand JA, Donelan M, Burke JF. Preferential activation and depletion of the alternative complement pathway by burn injury. Ann Surg. 1983;198:58–62. doi: 10.1097/00000658-198307000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qin X, Dong W, Sharpe SM, Sheth SU, Palange DC, Rider T, Jandacek R, Tso P, Deitch EA. Role of lipase-generated free fatty acids in converting mesenteric lymph from a noncytotoxic to a cytotoxic fluid. Am J Physiol Gastrointest Liver Physiol. 2012;303:G969–978. doi: 10.1152/ajpgi.00290.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yabluchanskiy A, Ma Y, Iyer RP, Hall ME, Lindsey ML. Matrix metalloproteinase-9: Many shades of function in cardiovascular disease. Physiology (Bethesda) 2013;28:391–403. doi: 10.1152/physiol.00029.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vandooren J, Van den Steen PE, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): the next decade. Crit Rev Biochem Mol Biol. 2013;48:222–272. doi: 10.3109/10409238.2013.770819. [DOI] [PubMed] [Google Scholar]
- 36.Rosario HS, Waldo SW, Becker SA, Schmid-Schönbein GW. Pancreatic trypsin increases matrix metalloproteinase-9 accumulation and activation during acute intestinal ischemia-reperfusion in the rat. Am J Pathol. 2004;164:1707–1716. doi: 10.1016/S0002-9440(10)63729-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Munjal C, Tyagi N, Lominadze D, Tyagi SC. Matrix metalloproteinase-9 in homocysteine-induced intestinal microvascular endothelial paracellular and transcellular permeability. J Cell Biochem. 2012;113:1159–1169. doi: 10.1002/jcb.23451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu DY, Yu WH, Yeh WL, Tang CH, Leung YM, Wong KL, Chen YF, Lai CH, Fu WM. Hypoxia-induced matrix metalloproteinase-13 expression in astrocytes enhances permeability of brain endothelial cells. J Cell Physiol. 2009;220:163–173. doi: 10.1002/jcp.21746. [DOI] [PubMed] [Google Scholar]
- 39.Kargozaran H, Yuan SY, Breslin JW, Watson KD, Gaudreault N, Breen A, Wu MH. A role for endothelial-derived matrix metalloproteinase-2 in breast cancer cell transmigration across the endothelial-basement membrane barrier. Clin Exp Metastasis. 2007;24:495–502. doi: 10.1007/s10585-007-9086-6. [DOI] [PubMed] [Google Scholar]
- 40.Van Lint P, Libert C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J Leukoc Biol. 2007;82:1375–1381. doi: 10.1189/jlb.0607338. [DOI] [PubMed] [Google Scholar]
- 41.Santibanez-Gallerani AS, Barber AE, Williams SJ, Davis S, Zhao Y, Shires GT. Matrix metalloproteinase inhibition protects hepatic integrity in hemorrhagic shock. J Gastrointest Surg. 2000;4:536–541. doi: 10.1016/s1091-255x(00)80098-0. [DOI] [PubMed] [Google Scholar]
- 42.Hu J, Van den Steen PE, Dillen C, Opdenakker G. Targeting neutrophil collagenase/matrix metalloproteinase-8 and gelatinase B/matrix metalloproteinase-9 with a peptidomimetic inhibitor protects against endotoxin shock. Biochem Pharmacol. 2005;70:535–544. doi: 10.1016/j.bcp.2005.04.047. [DOI] [PubMed] [Google Scholar]
- 43.Alexander JS, Elrod JW. Extracellular matrix, junctional integrity and matrix metalloproteinase interactions in endothelial permeability regulation. J Anat. 2002;200:561–574. doi: 10.1046/j.1469-7580.2002.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsu AT, Barrett CD, DeBusk GM, Ellson CD, Gautam S, Talmor DS, Gallagher DC, Yaffe MB. Kinetics and Role of Plasma Matrix Metalloproteinase-9 Expression in Acute Lung Injury and the Acute Respiratory Distress Syndrome. Shock. 2015;44:128–136. doi: 10.1097/SHK.0000000000000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lalu MM, Gao CQ, Schulz R. Matrix metalloproteinase inhibitors attenuate endotoxemia induced cardiac dysfunction: a potential role for MMP-9. Mol Cell Biochem. 2003;251:61–66. [PubMed] [Google Scholar]
- 46.de Souza P, Schulz R, da Silva-Santos JE. Matrix metalloproteinase inhibitors prevent sepsis-induced refractoriness to vasoconstrictors in the cecal ligation and puncture model in rats. Eur J Pharmacol. 2015;765:164–170. doi: 10.1016/j.ejphar.2015.08.030. [DOI] [PubMed] [Google Scholar]
- 47.Chang M, Alsaigh T, Kistler EB, Schmid-Schönbein GW. Breakdown of mucin as barrier to digestive enzymes in the ischemic rat small intestine. PLoS One. 2012;7:e40087. doi: 10.1371/journal.pone.0040087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu F, Yang X, Lu Z, Kuang F. Evaluation of glucose metabolic disorder: insulin resistance and insulin receptors in critically ill children. Chin Med J (Engl) 1996;109:807–809. [PubMed] [Google Scholar]
- 49.Marik PE, Raghavan M. Stress-hyperglycemia, insulin and immunomodulation in sepsis. Intensive Care Med. 2004;30:748–756. doi: 10.1007/s00134-004-2167-y. [DOI] [PubMed] [Google Scholar]
- 50.Schmid-Schönbein GW. An emerging role of degrading proteinases in hypertension and the metabolic syndrome: autodigestion and receptor cleavage. Curr Hypertens Rep. 2012;14:88–96. doi: 10.1007/s11906-011-0240-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DeLano FA, Schmid-Schönbein GW. Pancreatic digestive enzyme blockade in the small intestine prevents insulin resistance in hemorrhagic shock. Shock. 2014;41:55–61. doi: 10.1097/SHK.0000000000000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cartier A, Cote M, Bergeron J, Almeras N, Tremblay A, Lemieux I, Despres JP. Plasma soluble tumour necrosis factor-alpha receptor 2 is elevated in obesity: specific contribution of visceral adiposity. Clin Endocrinol (Oxf) 2010;72:349–357. doi: 10.1111/j.1365-2265.2009.03671.x. [DOI] [PubMed] [Google Scholar]
- 53.Ostrowski SR, Sorensen AM, Windelov NA, Perner A, Welling KL, Wanscher M, Larsen CF, Johansson PI. High levels of soluble VEGF receptor 1 early after trauma are associated with shock, sympathoadrenal activation, glycocalyx degradation and inflammation in severely injured patients: a prospective study. Scand J Trauma Resusc Emerg Med. 2012;20:27. doi: 10.1186/1757-7241-20-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsao PN, Chan FT, Wei SC, Hsieh WS, Chou HC, Su YN, Chen CY, Hsu WM, Hsieh FJ, Hsu SM. Soluble vascular endothelial growth factor receptor-1 protects mice in sepsis. Crit Care Med. 2007;35:1955–1960. doi: 10.1097/01.CCM.0000275273.56547.B8. [DOI] [PubMed] [Google Scholar]
- 55.Perl M, Chung CS, Garber M, Huang X, Ayala A. Contribution of anti-inflammatory/immune suppressive processes to the pathology of sepsis. Front Biosci. 2006;11:272–299. doi: 10.2741/1797. [DOI] [PubMed] [Google Scholar]
- 56.Coutinho HB, Robalinho TI, Coutinho VB, Amorim AM, Furtado AF, Ferraz A, Ferraz E, Walker F, King G, Sewell HF, et al. Intra-abdominal sepsis: an immunocytochemical study of the small intestine mucosa. J Clin Pathol. 1997;50:294–298. doi: 10.1136/jcp.50.4.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haglund U, Hulten L, Ahren C, Lundgren O. Mucosal lesions in the human small intestine in shock. Gut. 1975;16:979–984. doi: 10.1136/gut.16.12.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deitch ED, Shi HP, Lu Q, Feketeova E, Xu DZ. Serine proteases are involved in the pathogenesis of trauma-hemorrhagic shock-induced gut and lung injury. Shock. 2003;19:542–456. doi: 10.1097/01.shk.0000048899.46342.f6. [DOI] [PubMed] [Google Scholar]
- 59.Doucet JJ, Hoyt DB, Coimbra R, Schmid-Schönbein GW, Junger WG, Paul LW, Loomis WH, Hugli TE. Inhibition of enteral enzymes by enteroclysis with nafamostat mesilate reduces neutrophil activation and transfusion requirements after hemorrhagic shock. J Trauma. 2004;56:501–510. doi: 10.1097/01.ta.0000114536.98447.f7. discussion 510-501. [DOI] [PubMed] [Google Scholar]
- 60.Kim HD, Malinoski DJ, Borazjani B, Patel MS, Chen J, Slone J, Nguyen XM, Steward E, Schmid-Schönbein GW, Hoyt DB. Inhibition of intraluminal pancreatic enzymes with nafamostat mesilate improves clinical outcomes after hemorrhagic shock in Swine. J Trauma. 2010;68:1078–1083. doi: 10.1097/TA.0b013e3181da78b1. [DOI] [PubMed] [Google Scholar]
- 61.Fitzal F, Delano FA, Young C, Rosario HS, Junger WG, Schmid-Schönbein GW. Pancreatic enzymes sustain systemic inflammation after an initial endotoxin challenge. Surgery. 2003;134:446–456. doi: 10.1067/s0039-6060(03)00168-5. [DOI] [PubMed] [Google Scholar]
- 62.Fitzal F, DeLano FA, Young C, Schmid-Schönbein GW. Improvement in early symptoms of shock by delayed intestinal protease inhibition. Arch Surg. 2004;139:1008–1016. doi: 10.1001/archsurg.139.9.1008. [DOI] [PubMed] [Google Scholar]
- 63.Lee YT, Wei J, Chuang YC, Chang CY, Chen IC, Weng CF, Schmid-Schönbein GW. Successful treatment with continuous enteral protease inhibitor in a patient with severe septic shock. Transplant Proc. 2012;44:817–819. doi: 10.1016/j.transproceed.2012.03.032. [DOI] [PubMed] [Google Scholar]
- 64.Mitsuoka H, Schmid-Schönbein GW. Mechanisms for blockade of in vivo activator production in the ischemic intestine and multi-organ failure. Shock. 2000;14:522–527. doi: 10.1097/00024382-200014050-00005. [DOI] [PubMed] [Google Scholar]