

Abstract
The reconstitution of lost bone is a subject that is germane to many orthopaedic conditions including fractures and non-unions, infection, inflammatory arthritis, osteoporosis, osteonecrosis, metabolic bone disease, tumors, and periprosthetic particle-associated osteolysis. In this regard, the processes of acute and chronic inflammation play an integral role. Acute inflammation is initiated by endogenous or exogenous adverse stimuli, and can become chronic in nature if not resolved by normal homeostatic mechanisms. Dysregulated inflammation leads to increased bone resorption and suppressed bone formation. Crosstalk amongst inflammatory cells (polymorphonuclear leukocytes and cells of the monocyte-macrophage-osteoclast lineage) and cells related to bone healing (cells of the mesenchymal stem cell-osteoblast lineage and vascular lineage) is essential to the formation, repair and remodeling of bone. In this review, the authors provide a comprehensive summary of the literature related to inflammation and bone repair. Special emphasis is placed on the underlying cellular and molecular mechanisms, and potential interventions that can favorably modulate the outcome of clinical conditions that involve bone repair.
Keywords: Inflammation, Bone Repair, Fracture Healing, Tissue Engineering
1. Introduction and scope of the problem
Bone loss and subsequent repair are important issues in orthopaedics and related specialties. A clear understanding of the principles underlying bone loss and repair is essential for the treatment of traumatic injuries (fractures and non-unions), patients with bone infection, osteonecrosis, arthritis, osteoporosis, spinal fusion, wear particle associated osteolysis, metabolic bone disease, tumors and other diseases affecting bone. The subject of bone loss and repair has great clinical and economic importance. Approximately 100,000 fractures develop a non-union each year in the USA [1]. The average cost for treatment of an established non-union is approximately US$11,333 [2]. Fragility fractures secondary to senile osteoporosis are a major source of pain and disability, and affect 50% of women and 25% of men over age 50 [3]. Medical care for these fractures will cost over US$25 billion by 2025. The number of surgical cases that use auto- or allograft bone to repair bone defects or obtain a robust fusion totals approximately 1.5 million cases in the USA, with an additional 2.2 million cases worldwide per year [4]. In 2011 alone, there were about 465,070 spinal fusion procedures performed in the USA, the majority of which use bone grafts or byproducts [5]. These are but a few examples of the social and financial burden that bone loss and repair places on our society, and the urgent need for a deeper understanding of the etiology, biological mechanisms, and methods for prevention of fracture non-unions and healing of bone.
Although bone loss and repair were once simply thought to involve only osteoblasts and osteoclasts, currently there has been great emphasis on more complex interactions among cells of the mesenchymal stem cell-osteoblast lineage, and the monocyte-macrophage-osteoclast lineage. Indeed it is now generally appreciated that crosstalk amongst inflammatory cells and cells related to bone healing is essential to the formation repair and remodeling of bone [6]. This fact is not surprising, given that acute inflammation has been recognized as the first stage of fracture healing [7].
The processes of bone healing are biologically intertwined with those of acute inflammation and the innate immune system. When humans or lower organisms experience a perturbing stimulus that may potentially jeopardize their existence or function, the innate immune system is activated in order to re-establish the normal homeostatic state [8–11]. Local and circulating cells of the monocyte/macrophage lineage function as tissue sentinels that become activated and respond immediately to serious adverse stimuli via a pre-programmed non-antigen specific series of events. Monocyte/macrophages sense and regulate subsequent biological events to mitigate the adverse stimulus and re-establish pre-morbid local anatomy and physiology. If this does not occur, permanent tissue alterations may result due to ongoing active inflammation, fibrosis, or chronic inflammation, in which active inflammation, fibrosis and attempts at repair all occur simultaneously [12].
Bone is a complex organ with numerous functions including hematopoiesis, regulation and storage of key minerals, the protection of vital life-sustaining organs, facilitation of locomotion etc. When bone is subjected to injurious, pro-inflammatory stimuli (trauma, infection and so forth), the same biological processes regulated by the innate immune system ensue, as with other tissues and organ systems, to effect local repair and bone healing. These events necessitate ongoing communication between cells of the monocyte-macrophage-osteoclast lineage, which directly confronts the offending stimulus (such as with infection), but then initiates repair through the process of macrophage transformation (polarization) into a pro-healing phenotype, and through the liberation of cytokines, chemokines and other factors that promote angiogenesis and the homing of cells of the mesenchymal stem cell-osteoblast lineage [6, 8, 10, 13–15]. In addition, mesenchymal-derived cells modulate inflammatory cells to promote resolution of pro-inflammatory activities, and reconstitution of normal tissue.
This review will summarize the fundamental principles of bone healing and repair after exposure to adverse physical and biological trauma, elucidate the mechanisms by which this occurs, emphasize the important interactions and cross-talk among cells of the monocyte-macrophage-osteoclast and mesenchymal stem cell-osteoblast lineages, and provide discussion on new opportunities for enhancing bone repair by modulating the processes of inflammation.
2. Bone Healing and Repair
2.1. Types of Fracture Healing
Bone is a highly dynamic tissue that undergoes a constant process of remodeling to accommodate changing mechanical stresses, and to repair developing fatigue fractures. In addition to this process of remodeling, bone has a remarkable potential for regeneration. Indeed, under optimal conditions bone can heal completely without fibrous scar formation into a form and function that is indistinguishable from the state prior to the injury. The process of fracture healing is highly complex, and in many respects poorly understood. Several fundamental principles governing bone regeneration have, however, been well established as have several key factors that critically influence the outcome of healing. Indeed, optimizing the conditions for healing is the basis and the goal of all fracture treatment.
One of the best recognized factors that influence outcome and also the type of bone repair is the degree of displacement between the fractured bone ends as well as the mechanical stability of the fracture environment [16–18]. While optimal fracture healing requires proximity of the fracture ends as well as a degree of mechanical stability achieved e.g. with splinting, instability and major displacement at the fracture site interfere with healing. This is presumably caused by repeated mechanical trauma exceeding the durability of the provisional tissues, resulting in repeated cell and tissue damage, chronic inflammation, and ultimately in a non-union. Perfectly rigid fixation with no micromotion can also lead to suboptimal bone regeneration; the reasons for this phenomenon are poorly understood. Thus some amount of motion is required for bone regeneration but what is the optimal amount of motion is still unclear.
Rigidly fixed fractures with good reduction and inter-fragmentary compression typically achieved with plates and screws are characterized by a minimal fracture gap and inter-fragmentary movement. Under these conditions bone can heal directly, via a process known as primary fracture healing. In a case of exact reduction, bone heals via direct contact healing which resembles the process of normal bone remodeling: osteoclast mediated bone resorption advances directly through the fracture line, followed by new bone deposition by osteoblasts thus leading directly to re-establishment of the Haversian system [19, 20]. Gap healing refers to a similar mechanically stable situation but with a slightly larger gap existing between the bone fragments; this void is filled with direct deposition of intramembranous woven bone and the mature bone Haversian system is re-established via the osteoclast-mediated remodeling process [21, 22].
In most fractures, including the ones treated with external splinting, intramedullary nails or external fixator devices, complete rigidity is typically not achieved resulting in more motion at the fracture site and a degree of intermittent displacement between the bone ends [18, 23]. In these cases, the healing progresses via a multi-staged process known as secondary fracture healing. Secondary fracture healing advances via multiple histologically and biochemically distinct stages namely: inflammation, callus formation, and remodeling (see below) and involves bone formation via both direct intramembranous and indirect endochondral ossification routes [7, 18, 23, 24] (Table 1). It has been postulated that the goal of this complex process is a stepwise increase in the mechanical stability of the fracture site that is achieved by progressively replacing fragile provisional tissues with more stable ones, eventually reaching a point that allows vascular ingrowth and mineralization processes to advance [20, 23].
Table 1.
Key events during secondary fracture healing
| 1) Hematoma |
| Activation of blood coagulation cascade |
| Formation of provisional fibrin matrix |
| Release of danger signal molecules |
| Activation of local macrophages |
| 2) Acute inflammation |
| Recruitment of neutrophils and macrophages |
| Clearance of necrotic tissue and provisional matrix |
| Production of pro-inflammatory cytokines, chemokines, growth factors |
| Recruitment and activation of MSCs, osteoprogenitors, fibroblasts |
| 3) Granulation tissue |
| Active proliferation of progenitor cells |
| Deposition of immature fibrotic extracellular matrix |
| Angiogenesis |
| 4) Callus formation |
| Differentiation of progenitor cells into chondrocytes and production of fibrocartilage (central areas) |
| Differentiation of progenitor cells into osteoblasts and production of woven bone (periosteum) |
| Fracture stabilization by fibrocartilage callus |
| Apoptosis of chondrocytes, fibrocartilage calcification |
| Vascular ingrowth, recruitment of osteoprogenitors cell |
| Woven bone deposition on cartilage scaffold |
| 5) Remodeling |
| Formation of chondroclasts and osteoclasts |
| Resorption of cartilage and woven bone |
| Restoration of Haversian system |
Despite the fact that this classification of secondary fracture healing into separate phases is somewhat artificial, it can still be considered as a useful framework to conceptualize the complicated sequence of events and the multiple simultaneous processes occurring during the process of bone regeneration. It is noteworthy, however, that the classic phases of secondary fracture healing are most commonly observed during the healing of cortical bone, for example, in the case of long bone diaphyseal fractures. Fractures at the bone metaphysis characterized by a dense network of trabecular bone heal mainly via intramembranous bone formation leading directly to deposition of new bone trabeculae between the fracture ends and production of new on bone on top of the existing trabeculae in the immediate proximity of the fracture [25–27]; formation of callus and endochondral bone formation are not typically observed. This is likely due to ample vasculature, immediate proximity of the bone marrow, larger number of bone trabecula, and ultimately due to larger availability of inflammatory and osteoprogenitor cells at the trabecular compared to the cortical bone. However, under unfavorable mechanical conditions with increased levels of strain and intermittent displacement between the bone fragments, the healing of trabecular bone also assumes characteristics of secondary fracture healing [28]; this highlights the tight interplay between mechanical and cellular factors in the fracture healing process. Indeed it seems that as a general rule micromotion at the fracture site stimulates callus formation and endochondral bone formation, while rigid fixation is associated with direct intramembranous bone formation.
2.2. Stages of Fracture Healing
The first stages in the cascade of secondary fracture healing are hematoma formation closely followed acute inflammation [7, 18, 29]. Fracture of a bone disrupts the local vasculature within bone tissue itself and at the endosteal and periosteal surfaces, in the bone marrow, and in the surrounding soft tissues. This results in the formation of a hematoma, due to activation of the plasma coagulation cascade and the platelets exposed to the extravascular environment. The fibrin network thus created serves as the first provisional matrix for the influx of inflammatory cells that are attracted by platelet-derived factors, complement fragments, as well as multiple danger signals released from necrotic cells, damaged extracellular matrix and local tissue macrophages. The first inflammatory cells to arrive to the fracture site within the first 24 hours are neutrophils.
It is well recognized that both the initial fracture hematoma [29–31] and the subsequent acute inflammation reaction [32–35] are critical for fracture healing. However, the exact role that the neutrophils play in this process has not been well characterized and it has even been suggested that excessive influx of activated neutrophils to the fracture site might be the reason for poor fracture healing observed in the context polytrauma [36]. By secreting inflammatory and chemotactic mediators, such as IL-6 and CCL2 (also known as monocyte chemotactic protein 1, MCP-1), neutrophils recruit the second wave of inflammatory cell infiltration to the fracture site, namely monocyte/macrophages [37, 38]. In addition to these recruited macrophages, a population of bone specific resident macrophages named osteomacs residing in the peri- and endosteum and participating in the regulation of fracture healing has recently been described [39–41].
Macrophages remove the provisional fibrin matrix and necrotic cells via phagocytosis, while monocyte-derived osteoclasts resorb necrotic bone fragments and the necrotic ends of the fractured bone. In addition to removing the cell and extracellular matrix debris, macrophages secrete a repertoire of inflammatory and chemotactic mediators, such as tumor necrosis factor alpha (TNF-α), IL-1β, IL-6, and CCL2, that initiate the recruitment of fibroblasts, mesenchymal stem cells (MSCs), and osteoprogenitor cells from their local niches [42–44]; these include at least local bone marrow, periosteum, and capillary walls (pericytes). Additional MSCs are required from the circulation; SDF-1 is one of the key chemokines mediating the recruitment of MSCs both from local and systemic sources [43, 45]. Platelet and macrophage derived inflammatory mediators and growth factors guide the proliferation, differentiation and extracellular matrix production of recruited MSCs and osteoprogenitor cells with additional growth factors being released from the remodeling extra cellular matrix. In addition to pro-inflammatory cytokines and chemokines, members of the transforming growth factor beta (TGF-β) family, in particular TGF-β1, -β2, -β3 and bone morphogenetic proteins (BMPs), as well as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF) and fibroblast growth factor-2 (FGF-2), are key mediators in the process [24, 46]. As a result, the initial fracture hematoma and subsequent acute inflammatory reaction are cleared in several days to a week after the fracture and replaced by granulation tissue rich in proliferating mesenchymal cells and developing neovasculature embedded in an unorganized extracellular collagen matrix [7, 18, 23, 47].
Due to the disruption of the local vasculature and the subsequent reactive contraction of the arterioles, the fracture site is hypoxic, in particular close to the fracture gap [18, 48–50]. The low oxygen tension, along with a degree of micromotion and various other microenviromental and macrophage-derived signals, guide the differentiation of MSCs along the chondrogenic pathway especially in the more centrally located areas of the fracture gap [18, 24, 46, 51]. Chondrocytes produce cartilage that ultimately extends throughout the fracture gap connecting the ends of the fractured bone several weeks after the injury. Along with accompanying fibrotic tissues, this cartilage tissue commonly known as soft callus, provides initial mechanical stability for the fracture and serves as the scaffold for endochondral bone formation [7, 18, 23, 24, 46]. At the same time as the soft callus is developing, in the local areas that have better blood supply and more mechanical stability, new bone formation advances via the intramembranous route, as recruited MSCs and periosteal osteoprogenitor cells differentiate into osteoblasts that directly layout woven bone. Typically this intramembranous bone formation begins in the inner layer of periosteum at some distance from the injury site, progressively advancing towards the fracture gap [18, 43, 52]. Ultimately woven bone covers the external surface of the fibrocartilaginous callus providing additional mechanical stability and representing the first stages in mineralization of the fibrocartilage scaffold. In a process that resembles the function of a growth plate, chondrocytes in the soft callus hypertrophy and go onto apoptosis, secreting calcium and mediators that stimulate vascular ingrowth ultimately leaving behind partially calcified cartilage extracellular matrix [24, 46]. Extensive vascular ingrowth to the cartilage scaffold-stabilized fracture gap and correspondingly increased blood flow to the healing fracture site is observed, accompanied by differentiation of osteoprogenitor cells into osteoblasts and by deposition of woven bone on the cartilage scaffold [24, 46]. This stage of fracture healing is commonly known as the formation of hard callus [7, 18]. As this process of cartilage mineralization advances, the mechanical stability of the fracture site increases ultimately reaching sufficient stability to independently carry physiological loads; typically this stage is reached several weeks or months after the primary injury [23]. Finally, both the immature woven bone and underlying cartilage matrix are removed by osteoclasts, initiating the process of remodeling that ultimately re-establishes the typical osteon structure and the Haversian system based on the mechanical stresses applied to the bone [18, 23]. This remodeling process that can take several months or even years to complete, but ultimately the process restores the normal form and integrity of the bone completing the process of fracture healing.
2.3. Failure to Heal and Induction of Bone Regeneration
Despite the remarkable potential for regeneration of bone, fracture healing can fail under adverse conditions resulting in painful non-union or pseudoarthrosis. As outlined above, successful fracture healing requires not only mechanical stability and relative proximity of the fracture ends but also influx of MSCs and osteoprogenitor cells, as well as inflammatory cells that regulate the fracture healing process by secreting a repertoire of inflammatory and chemotactic mediators, as well as growth factors. Thus is not surprising that the condition of the local vasculature and the surrounding soft tissues are critical determinants of the outcome of fracture healing, both due to the availability of essential cell populations, oxygen, and nutrients required for bone formation [18, 23, 50]. Several other local and systemic conditions such as the presence of a infection, smoking, and diabetes mellitus adversely influence bone healing and have been discussed in detail elsewhere [53].
Many of the factors that are crucial for bone regeneration are already utilized in clinics to improve fracture healing or induce healing in a setting where it has failed. For example, the gold standard of treating these situations, autologous bone grafting is highly effective in inducing bone formation by introducing a mixture of osteoprogenitor cells and a series of growth factors embedded in a suitable provisional matrix to the defect site. Growth factors applied to the defect site either individually as recombinant proteins, or as a part of allograft bone or demineralized bone matrix are effective in improving bone formation, and techniques to implant autologous MSCs to the defect site are being developed [18, 54]. However, the potential of regulating inflammation, the functions of the relevant inflammatory cell populations or their secreted inflammatory mediators to enhance fracture healing remain largely unknown, but would seem to hold great promise. Indeed, to highlight the importance of inflammation as key regulator of this process, we suggest that the accepted diamond concept of fracture healing [16] should be modified to encompass inflammatory cells and their secreted mediators (Figure 1).
Figure 1. The main factors influencing fracture healing.
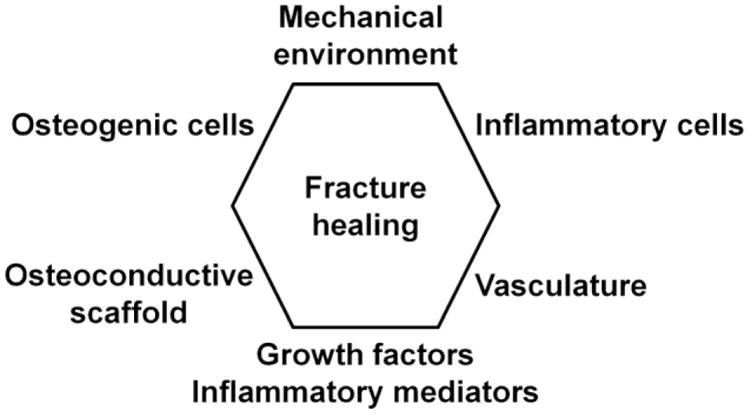
The so-called diamond concept of fracture healing has been modified to encompass the prominent role of inflammatory cells and their secreted mediators. The importance of osteoprogenitor cells, growth factors, osteoconductive scaffold, blood supply, and the mechanical environment in bone regeneration and successful fracture healing has been well documented. Evidence is accumulating that inflammatory cells and their mediators play an equally important role both in the regulation and dysregulation of fracture healing.
3. Interactions Between Inflammatory Cells and Bone Cells
Inflammation is a crucial biological process for eradication of pathogens and maintenance of tissue homeostasis. Successful clearance of inflammatory stimuli is accompanied by anti-inflammatory and reparative cytokines to resolve the inflammatory milieu and re-establish tissue homeostasis [55]. However, if this progression of events goes askew due to the persistence of pro-inflammatory stimuli, the process may progress into a state of chronic inflammation. This unresolved inflammatory response is highlighted by continued secretion of cytokines and other factors, ongoing tissue destruction, and impaired homeostasis. Bone injury elicits an inflammatory response that is beneficial to healing when acute and highly regulated; however, if this response is suppressed, dysregulated, or becomes chronic, inflammation can be detrimental to healing [56–58]. Immune cells, most importantly macrophages, are vital modulators of inflammation [8]. Decades of investigation into the interactions between the immune and skeletal systems have culminated in the coining of the term, osteoimmunology for this emerging field [59]. The close relationship between these two systems is underscored by the fact bone marrow houses both hematopoietic stem cells (HSCs) and MSCs [60]. The former give rise to immunomodulatory macrophages and bone-resorbing osteoclasts and the latter are precursors to bone-forming osteoblasts, which are essential in HSC differentiation [61]. Thus, common cytokines, receptors, signaling molecules, and transcription factors due to shared origin enable a wide range of dynamic cross talk between cells of the monocyte-macrophage-osteoclast and MSC-osteoblast lineages [62].
3.1. Overview of Inflammatory Cells
Since the interactions of MSCs and bone cells with lymphocytes, natural killer cells, and dendritic cells have been investigated at length [63–66], this review will focus on the interactions between macrophages and bone cells.
Macrophages can be described as resident or inflammatory macrophages [40]. Resident macrophages are found in most tissues throughout life whereas recruited inflammatory macrophages, derived from circulating blood monocytes, infiltrate sites of inflammation and are short-lived [40, 67]. Macrophage populations are highly heterogeneous and plastic [68], making them excellent candidates for immunomodulation of tissue remodeling. Macrophages may acquire distinct phenotypes with pro-inflammatory (M1) or anti-inflammatory (M2) functions; this phenomenon is known as macrophage polarization [10, 69]. Classical activation of macrophages with interferon-gamma (IFN-γ) and/or lipopolysaccharide leads to M1 macrophage polarization. M1 macrophages secrete pro-inflammatory cytokines (tumor-necrosis factor-α (TNF-α), IL-1β, etc.) and chemokines (CCL2, macrophage inflammatory protein 1α (MIP-1α), etc.), which results in tissue damage with additional leukocyte infiltration. Alternatively, macrophages exposed to IL-4 or IL-13 are polarized into M2 macrophages, marked by increased arginase-1 and anti-inflammatory cytokine expression including IL-10 and IL-1 receptor antagonist (IL-1Ra). Byproducts of tissue damage and cell debris polarize macrophages into an M2-like anti-inflammatory phenotype [10]. M2 or M2-like macrophages are capable of modulating and terminating the inflammatory response, and are crucial for tissue remodeling and repair. Secretion of VEGF and matrix metalloproteinase (MMP) by M2 macrophages is essential for angiogenesis (new blood vessel formation) and tissue remodeling during the repair process [10]. However, a recent study by Spiller et al reported that the expression of VEGF is dominant in M1 rather than M2 macrophages [70]. These findings suggest that the experimental conditions may be critical to determine VEGF expression in polarized macrophages.
3.2. Overview of Bone Cells
Bone is a highly dynamic organ whose structural integrity is maintained through precise remodeling involving osteoclasts, osteoblasts, and osteocytes. MSCs are pluripotent stromal cells that were initially isolated from bone marrow but can also be found in most tissues including brain, pancreas, muscle, spleen, and lung [71]. Osteoblasts, differentiated from MSCs, secrete the organic bone matrix and induce mineralization [72]. Transcription factors runt-related transcription factor 2 (Runx2) and osterix (Osx) are necessary for osteoblast differentiation [73]. During the final phase of bone remodeling, osteoblasts undergo apoptosis or incorporate themselves into the bone matrix as osteocytes during alkaline phosphatase-mediated calcification [62, 74]. As regulators of mineral metabolism, osteocytes are the primary cells coordinating the activity of osteoblasts and osteoclasts in response to mechanical stimuli [75]. Osteoclasts of hematopoietic origin resorb the bone matrix through acidic decalcification and proteolytic dissolution [76]. Osteoclast differentiation is modulated by both macrophage-colony stimulating factor (M-CSF) and receptor activator of nuclear factor-kappa B (RANK) ligand (RANKL). M-CSF is critical for the proliferation and survival of osteoclast precursor cells while inducing high expression of RANK [61]. When exposed to RANKL, these precursor cells fuse and develop the functions of active osteoclasts [77].
3.3. RANKL/RANK/OPG
The close relationship between the immune and skeletal systems was first underscored by the discovery that RANKL, which enhances T cell growth and dendritic cell function, and osteoclast differentiation factor (ODF) are, in fact, one and the same molecule [78]. RANKL is a TNF superfamily cell-surface cytokine expressed normally by osteoblasts and osteocytes and pathologically by lymphocytes (Figure 2) [60, 79, 80]. RANKL binds to RANK present on osteoclast precursors and dendritic cells [78]. RANK activation promotes osteoclast survival and induces maturation and activation through the intracellular TNF receptor-associated factors (TRAF) 2, 5, and 6, which transduce signals to activate the NF-κB and c-Jun N-terminal kinase (JNK) pathways [59, 60, 81]. Most importantly, RANKL signaling induces the expression of nuclear factor of activated T cells, cytoplasmic 1 (NFATc1), the master regulator of osteoclast differentiation and activation [60, 82]. Osteoprotegerin (OPG), a soluble decoy receptor produced by B cells, dendritic cells, MSCs, and osteoblasts, can block these effects through competitive binding with RANKL [77, 81]. Thus, the RANKL/OPG ratio is an important determinant in osteoclast activity [64]. Interestingly, although osteoblasts are considered the physiologic source of OPG in the skeletal system, RANKL-induced osteoclast progenitors from NFATc1-deficient mice have been shown to express OPG, highlighting the role of NFATc1 in osteoclastogenesis [83].
Figure 2. Crosstalk between inflammatory cells and bone progenitor cells.

Cells of the monocyte-macrophage-osteoclast and MSC-osteoblast lineages modulate each other. Blue lines/arrows indicate inhibition/promotion of differentiation. Red lines/arrows indicate inhibition/promotion of proliferation. Green arrows indicate promotion of migration. Abbreviations: MSC = mesenchymal stem cell, HSC = hematopoietic stem cell, Runx2 = runt-related transcription factor 2, Osx = osterix, OPG = osteoprotegerin, RANKL = receptor activator of nuclear factor κ-B ligand, M-CSF = macrophage colony stimulating factor, IFN-γ = interferon gamma, TNF-α = tumor necrosis factor α, IL = interleukin, TGF = transforming growth factor, VEGF = vascular endothelial growth factor, MCP-1 = monocyte chemoattractant protein-1, RANTES = regulated on activation, normal T expressed and secreted, and BMP = bone morphogenetic protein.
Macrophages can regulate RANKL-induced osteoclastogenesis. RANKL expression is stimulated by osteoclastogenic factors including 1,25-dihydroxyvitamin D3, parathyroid hormone (PTH), prostaglandin E2 (PGE2) and pro-inflammatory cytokines TNF-α, IL-1, and IL-6 (Figure 2) [61, 84]. However, IFN-γ, secreted by activated T cells and macrophages, can inhibit osteoclastogenesis through the subsequent rapid degradation of TRAF6 [79]. Thus, through the secretion of TNF-α, IL-1, IL-6, and IFN-γ, macrophages promote or suppress osteoclast activity [85].
3.4. Inflammatory Cells Modulate Bone Cells
Resident macrophages, termed osteomacs, are present throughout murine and human osteal tissue [86]. Upon bone injury, macrophages release various cytokines, chemokines, and growth factors to recruit additional inflammatory cells, promote neovascularization, direct MSC migration and differentiation, and mediate remodeling [6, 87, 88]. Macrophages stimulate MSC migration through secretion of TNF-α, IL-1, IL-6, CCL2, and Regulated on Activation, Normal T cell Expressed and Secreted (RANTES) (Figure 2) [42, 89]. In vitro, M1 macrophages and their mediators inhibit human MSC viability whereas M2 macrophages and their mediators have the opposite effect [90].
A growing number of reports demonstrate the anabolic effects of macrophages on osteogenesis. Monocytes and macrophages support osteoblast differentiation and proliferation through the release of cytokines including BMP-2, BMP-4, and TGF-β1 (Figure 2) [91–93]. Macrophage-depletion studies have demonstrated macrophages’ central role in normal bone formation and fracture healing. Regarding bone formation, one study found that osteomacs form a canopy structure over osteoblasts at diaphyseal endosteal surfaces of young growing mice, and depletion of these osteomacs in macrophage fas-induced apoptosis (Mafia) transgenic mice completely suppresses this osteoblast surface and thus bone modeling [86]. Another study constitutively depleted macrophages in Mafia mice, which resulted in early skeletal growth retardation and reduced MSCs and their ability to differentiate into osteoblasts [94]. In the same study, macrophage depletion in a tibial fracture model resulted in smaller callus formation, less bone deposition, and more fibrotic tissue [94]. In a tibial injury model, depletion of osteomacs in Mafia mice at the time of injury significantly suppressed woven bone deposition and mineralization and depletion after the primary inflammatory healing phase significantly suppressed new bone formation [39]. M1 macrophages promote mineralization by MSCs and bone healing through the production of oncostatin M [95, 96]. M2-like macrophages, stimulated by β-tricalcium phosphate, also enhance mineralization by murine MSCs [97].
3.4.1. Inflammatory Signals Modulate Bone Cells
The controlled release of pro-inflammatory signals and factors, both spatially and temporally, are essential to optimal bone remodeling [57]. Typically, pro-inflammatory cytokines including TNF-α, IL-1, IL-6, IL-11, and IL-17 promote bone resorption by enhancing osteoclast differentiation and activity, and/or inhibiting osteoblast differentiation, function, collagen synthesis and bone formation [98–100]. On the other hand, anti-inflammatory cytokines IL-10 and IL-13 demonstrate the opposite effect [60, 98].
The paradoxical effects of TNF-α on osteogenesis and bone formation highlight the importance of tightly regulated release of cytokines. Depending on its concentration, cell type, and exposure time, TNF-α can either suppress or promote osteogenesis [15]. Transient TNF-α signaling triggers the release of secondary signaling molecules and the recruitment of MSCs necessary for bone regeneration [57]. Furthermore, TNF-α, synergistically with IL-1β, promotes matrix mineralization by MSCs in vitro and is essential for murine bone regeneration in vivo [32, 33, 101]. Specifically, investigations by Gerstenfeld et al. using TNF-α receptor (p55−/−/p75−/−)-deficient mice demonstrated the central role of TNF-α in both intramembranous and endochondral bone formation, including MSC recruitment, stimulation of chondrocyte apoptosis, and recruitment of osteoclasts [32, 33]. However, high, persistent TNF-α levels systemically are damaging to tissues and can prompt rheumatoid arthritis-like symptoms such as chronic inflammation, reduced bone volume, and diminished bone mechanical strength [102].
The effects of IL-1 on bone healing are similar to those of TNF-α. IL-1 stimulates the production of IL-6 in osteoblasts and participates in fracture healing by promoting the production of the primary cartilaginous callus and angiogenesis at the injured site [42, 81]. However, the absence of IL-1 does not adversely affect fracture healing [103].
Controlled expression of IL-6, which promotes angiogenesis, production of growth factors such as VEGF, and differentiation of osteoblasts and osteoclasts, is critical for normal bone regeneration [6, 15, 34]. IL-6 is essential for the early stages of fracture healing; the absence of IL-6 delays mineralization and remodeling of the fracture callus [34, 57]. On the other hand, aberrantly elevated serum IL-6 levels following bone fracture correlates with decreased lower extremity function in patients [104]. Additionally, osteoblasts obtained from patients with polyarticular juvenile idiopathic arthritis demonstrated reduced osteogenesis in vitro and the only abnormally upregulated serum cytokine was IL-6 [105].
Macrophage-derived cytokines and chemokines can also modulate osteoclast activity. TNF-α and IL-1 synergistically promote osteoclastogenesis directly and indirectly through the stimulation of RANKL expression and downregulation of OPG in osteoblasts and fibroblasts (Figure 2) [106–108]. TNF-α attracts osteoclasts by inducing osteocyte apoptosis [109]. IL-6, though not essential, mediates osteoclastogenesis and is involved in clinical scenarios such as Paget’s disease of bone and rheumatoid arthritis [81, 110].
While inflammatory cells are the primary source of inflammatory signals during the initial phase of bone healing [29], native bone cells such as osteoblasts and chondrocytes are responsible for the release of inflammatory cytokines within 3–7 days of injury [42]. IL-6, for example, is produced by MSCs and osteoblasts, in response to IL-1 and TNF-α [57, 111]. The production of pro-inflammatory cytokines by bone cells may explain why bone healing is not always impaired in the absence of inflammatory cells. While macrophages play a central role in bone repair, in vivo studies report that not all inflammatory cells are necessary for all forms of tissue repair and their absence may even accelerate it. This may depend on the type of inflammatory cell or type of tissue. For example, systemically neutropenic rats via antineutrophil sheep serum injection showed enhanced fracture repair compared with rats injected with normal sheep serum [112]. Recombination activating gene 1 knockout (RAG1−/−) mice, which lack T and B cells, demonstrated accelerated mineralization and remodeling while showing reduced inflammatory TNF-α expression and upregulated anti-inflammatory IL-10 expression in a femoral fracture model [113]. Additionally, PU.1 null mice, which lack macrophages and neutrophils, are able to repair skin wounds similar to wild-type mice [114].
3.5. Bone Cells Modulate Inflammatory Cells
Due in part to their close proximity, bone cells are components of the HSC niche and participate in the regulation of immune cells. MSCs provide stromal support tissue [115]. Osteoblasts regulate HSC maintenance through angiopoietin-1 signaling [116]. Studies have reported that when activated by PTH, an osteoblast subpopulation can increase the number of HSCs in bone marrow through the production of hematopoietic growth factors such as Notch ligand jagged 1 (Figure 2) [117]. Previously, it had been suggested that under stress stimuli, RANKL-activated osteoclasts promote migration of HSCs [118]. However, more recently another study demonstrated that osteoclasts are dispensable for HSC migration using osteoclast-depleted mice via recombinant OPG-fc injections [119]. Further discussion on whether osteoclasts promote HSC mobilization has been provided by Charles and Aliprantis [120].
MSCs have been demonstrated to be highly immunosuppressive. They suppress lymphocyte proliferation and regulate dendritic cell differentiation [121]. MSCs prevent the rejection of transplanted bone marrow, HSCs, and skin allografts [122]. Recent in vitro and in vivo studies reported that MSCs shift macrophage populations from M1 to M2-like phenotype and decrease pro-inflammatory cytokine secretion [115, 121, 123, 124], mediated by PGE2 and TNF-α stimulated gene/protein 6 (TSG-6) [66, 125].
3.6. Summary
The immune and skeletal systems are intimately related due to shared lineages and microenvironments. As more studies demonstrate the anabolic role of macrophages in bone formation and healing, understanding the cross talk between cells of the monocyte-macrophage-osteoclast and MSC-osteoblast lineages is increasingly relevant. While it is known that tightly regulated release of inflammatory cytokines is necessary for optimal bone remodeling, the role of macrophage subtypes and their associated factors in bone healing has not been elucidated. Further understanding of the interactions among inflammatory and bone cells will provide new strategies of harnessing immunomodulation to stimulate bone regeneration.
4. Opportunities for Enhancing Bone Repair by Modulating Inflammation
New therapies to restore homoeostasis in bone diseases have been proposed, including experimental cell-, gene- and biological therapies [126, 127]. All of these treatments are focused either to enhance bone formation by osteoblasts (bone anabolic approaches) or to block bone resorption by osteoclasts (anti-resorptive approaches) [128–130]. While some of these therapies are in clinical use [131], unexpected adverse effects, restricted clinical indications and the need to improve the final formulation has led to restrictions in their use [70, 132, 133].
Bone repair in aged individuals has an increased risk of failure, leading to an increased incidence of fracture non-unions, loosening of implants and chronic disability [126, 127]. This leads to diminished quality of life and increased medical-costs [1–4]. Thus, novel therapies to enhance age-associated impairment in bone healing are sorely needed.
Bone injury site entails both inflammatory and regenerative events, which are critical for the reestablishment of normal bone homeostasis. Therefore, bone healing should be considered an osteo-immunological phenomenon rather than simply an imbalance between bone formation and bone loss. With this consideration, four clinically relevant opportunities to improve impaired bone healing are reviewed that use inflammatory–centered therapies. These clinical conditions include: fragility (osteoporotic) fractures, fracture non-unions, osseointegration of implants and reconstruction of bone defects.
4.1. Fragility (Osteoporotic) Fractures
Fragility fractures are some of the most severe complications associated with primary and secondary osteoporosis [3, 134]. Primary osteoporosis includes hypo-gonadal or postmenopausal osteoporosis, and senile osteoporosis [135]. Secondary osteoporosis is associated with long-term drug treatments (e.g. corticosteroids).
Postmenopausal osteoporosis affects women and is characterized by increased bone loss associated with both the loss of bone-protective role of estrogens and the increased levels of systemic and local pro-inflammatory and pro-resorptive cytokines [136]. Indeed, increased levels of IL-6, IL-1 and TNF-a characterize a subclinical systemic chronic inflammatory state during aging named “inflamm-aging”, which has been linked with postmenopausal bone loss [137–139]. Likewise, increased serum levels of the bone remodeling cytokines RANKL and the ratio RANKL/OPG have been associated with postmenopausal osteoporosis [136, 137]. Interestingly, in postmenopausal osteoporosis, the osteogenic capacity remains unaltered [135]. This imbalance predominantly affects trabecular bone, such as the vertebral bodies [135, 140]. Otherwise, bone resorption normally plays a prominent role in the remodeling of callus during the late stages of fracture healing [40, 141]. Thus, in postmenopausal osteoporosis there would seem to be more benefit for an anti-resorptive approach compared to a bone anabolic one. In contrast, senile osteoporosis affect women and men and it is characterized by a decreased capacity for differentiation of osteoprogenitor cells and decreased bone matrix synthesis by mature osteoblasts due to defects in BMP-2 signaling [142]. The bone resorption capacity remains unaffected by aging [135]. This imbalance leads again to increased bone loss over bone formation and a subsequent decrease in bone mass [143]. Compromised bone formation events lead to dysregulated homeostasis in cortical bone, especially in the femoral neck, leading to increased number of fragility fractures in the aged population [40, 140]. Fragility fractures are associated with disturbed callus formation and an increased risk of delayed fracture healing and non-union [144, 145]. This presents an opportunity to modulate inflammation and impede bone resorption.
4.2. Fracture Non-Unions
Fracture non-union is incomplete fracture consolidation, with absence of progressive radiographic signs of healing over three consecutive months [146]. It constitutes a bone-healing impairment associated with secondary fracture healing, normally characterized by the formation of exuberant callus [146]. From a histologic point of view, non-unions demonstrate persistence of fibrous tissue, woven bone and cartilage at the injury site [146]. As bone healing normally involves interactions between immune system and bone remodeling [2, 147], fracture non-unions present an opportunity for the introduction of novel therapies [146].
Cellular and molecular characterization of non-union tissues has shown that senescent MSCs exhibit a reduced capacity to differentiate into mature, functional osteoblasts [148, 149]. Furthermore, decreased levels of endogenous BMP, a recognized promoter of the differentiation of osteoprogenitors into functional osteoblasts, may lead to decreased responsiveness of MSCs in the fracture non-union site [150]. Indeed, endogenous BMPs and their receptors have been identified in non-union tissues [150]. Interestingly, when MSCs isolated from non-union tissues were stimulated with exogenous BMP, they differentiated into functional osteoblasts [151]. An imbalance between BMPs and their inhibitors such as MMP7 and MMP12; and Dickkopf-1 (Dkk-1), has been suggested as a contributing factor [146]. Thus, the use of exogenous BMP-2 and -7 has demonstrated promising results [146].
Non-unions also entail impairment in the mineralization of the soft connective tissue callus and are sustained by instability at the fracture site. One novel approach is to modulate macrophage phenotypes in the healing tissues, thereby affecting the pro-inflammatory and healing events of bone repair. Several reports have studied the use of autologous bone marrow concentrate of mononuclear cells to facilitate fracture healing [152, 153]. Besides the transference of MSCs and endothelial stem cells (ESCs), this concentrate includes the transference of multi-potent and self-renewal HSCs, capable of differentiating into all hematopoietic cell types, including macrophage precursors [152–154]. Thus, bone marrow concentrate optimizes two of the most desirable clinical outcomes regarding the treatment of non-union fractures: neo-osteogenesis and neo-angiogenesis [155, 156].
4.3. Osseointegration of Endo-osseous Implants
An endo-osseous implant is a foreign biomaterial inserted in a surgically created bone cavity that needs to integrate with the surrounding bone to restore the functionality of the reconstructed segment. From a biological point of view, the implant triggers cell and molecular interactions at the host bone-implant interface, leading to the creation of a dynamic peri-implant bone niche. This particular niche is responsible in part for the implant’s integration and stability within bone and its functionality over time [157]. Thus, during the lifespan of an endo-osseous implant, two coupled biologic phenomena could occur: the enhancement of osseointegration and the breakdown of it.
Enhancing osseointegration
Current insights regarding osseointegration [158] have evolved from viewing this process as one of pure bone healing around the implant, to that of an immune-mediated foreign body reaction (FBR) [159, 160]. The FBR involves a sequence of events including protein adsorption on the surface of the implant, activation of complement and the coagulation system, recruitment of monocyte/macrophages and MSCs, activation and differentiation of these cells into functional M1 and M2 macrophages, osteoclasts, and osteoblasts respectively and the establishment of biological attachments between implant and new bone [160]. Thus, FBR involved in osseointegration represents a foreign body equilibrium (FBE) between osteogenic factors/cells and osteolytic factors/cells around the implant [160, 161].
Breakdown of osseointegration
Once osseointegration has been achieved, the FBE needs to be maintained in the long-term so that the implant preserves its functionality. The continued release of wear debris from total joint replacements (TJRs) and potentially an evolving infection during the lifespan of the implant might perturb this FBE, and induce peri-implant inflammation; this would promote peri-implant osteolysis, aseptic loosening and subsequent implant failure necessitating further surgical intervention [162].
Modulation of inflammation to improve primary osseointegration is generally not needed due to the initial high rates of success [163–165]. However, treatment of aseptic and septic loosening of orthopaedic and dental implants may benefit from considering the role of the immune system [166–168]. For example, blockade of pro-inflammatory mediators such as PGE2 [169], pro-inflammatory cytokines [170], or recruitment of monocyte/macrophage cells by interfering with CCL2/CCR2 axis [171, 172], or inactivation of the pro-inflammatory transcription factor NF-κB [173, 174] has been investigated. Another approach is centered on the modulation of macrophage phenotype from inactivated M0 or pro-inflammatory M1 macrophages toward the pro-remodeling M2 macrophages by using polarizing cytokines [175–177].
4.4. Reconstruction Using Bone Grafts and Tissue Engineering
Large critical sized defects do not heal spontaneously, thereby interfering with normal function. Autologous bone grafts include natural, immune-tolerated bone cells, an osteoconductive scaffold and supportive growth factors [178]. Tissue engineering of bone usually provides a construct composed of a biodegradable scaffold associated with tissue-inductive biomaterials, cells, and/or proteins to promote new bone formation [179]. Although reports of tissue engineering have been centered on the promotion of osteogenic (e.g. Wnt) [180] or vascular (e.g. VEGF) [181] pathways, a growing number of reports involve modulating inflammation to improve tissue [90, 182, 183] and bone repair by biomimetic approaches [70, 94, 96]. In this line, M1 and M2 macrophages have been recently described as promoters of scaffold vascularization in vitro by promoting the sprout formation by endothelial cells and the anastomosis of M1-induced sprouting vessels, respectively [183]. Thus, there are new opportunities to “re-engineer” the current constructs by the functionalization of scaffolds and by direct cell delivery. The functionalization of scaffolds may involve controlled release of biologics to extend their bioactivity, the recruitment and differentiation of autologous stem cells, the local, temporal control of biologics, and to couple scaffold degradation with new bone growth [180, 184–186]. A more recent approach is to reproduce the sequential release of polarizing macrophage factors, that promote the classical activation into M1-macrophage during the first 24 hours (e.g. using IFN-γ) followed by the release of factors that promote the alternative differentiation into M2 macrophage factors (e.g. using IL-4) [70]. This method has been reported as proof-of-concept thus far and although it is an interesting and promising approach, it has not yet been studied in a bone repair model. Another approach involves the local delivery of synthetic peptides that interfere with the signaling of sphyngosine-1 phosphate (S1P), a bioactive lipid with immunomodulatory effects, thereby promoting the polarization of macrophages into an M2 phenotype associated with osteogenic and angiogenic effects [115, 187]. In fact, this method was able to enhance bone regeneration in critical-sized cranial defects using adult male rats [187].
The use of cell therapy in bone reconstruction has been centered on the osteogenic potential of MSCs [126]. Moreover, MSCs exhibit immunomodulatory effects, promoting the alternative activation of macrophages toward the pro-remodeling M2-subset [115, 188]. The use of concentrate of bone marrow mononuclear cells associated with biomaterials to treat pseudoarthroses and optimize bone repair has been reported [152]. Cell based therapies using ex-vivo polarized M2-macrophages to promote tissue repair has shown promise in models of ischemia and multiple sclerosis [189].
Opportunities for modulating inflammation and bone repair are summarized in Figure 3.
Figure 3. Clinical opportunities for enhancing bone healing by an inflammatory-centered approach.

Normal bone healing and the main cells involved in each stage are represented in the upper row. In the middle row, note the persistence of fibrous callus after inflammatory events in fracture healing in the elderly and non-unions. In the lower row, an endo-osseous implant experiment with a foreign body reaction (FBR) at the interface between host bone and the implant, leading to osseointegration. The exposure of wear debris undermines the osseointegrated implant leading to the re-activation of peri-implant inflammation, chronic inflammation and osteolysis, with subsequent loosening of the implant. Tissue engineering and biotherapies (green dashed line) are potential therapeutic interventions to reverse the adverse biological processes by recapitulating early inflammatory events of normal bone healing in fractures in the elderly, non-unions and early stages of periprosthetic osteolysis, respectively.
5. Conclusion
Bone injury elicits an inflammatory response that is beneficial to healing when acute and highly regulated. However, if this response goes askew and inflammation becomes chronic, the process can be detrimental to healing, and in addition, cause additional “bystander” tissue damage [56–58]. The initial acute inflammatory phase of fracture healing is characterized by the influx of neutrophils, macrophages, lymphocytes and other cells, leading to the release of a variety of cytokines, chemokines and growth factors. However, the mechanisms by which these complex signaling cascades trigger bone regeneration still remain unclear. While inflammatory cells are the primary source of the signals during the initial phase of bone healing [29], in the regenerating fracture callus, local derivatives of the mesenchymal stem cell lineage including osteoblasts and chondrocytes are also responsible for the release of inflammatory cytokines within 3–7 days of injury and subsequent phases of healing [42]. Indeed, several in vivo studies have reported that neutrophils, macrophages, and lymphocytes are not always necessary for tissue repair, and their absence may even accelerate healing [29, 113, 114]. Future research to discern the mechanisms underlying inflammatory signaling, as well understanding the cross-talk between inflammatory cells and other cells found at the fracture site are needed. Overall, modulating inflammatory signaling pathways is a promising new strategy for bone regeneration. This could potentially improve the quality of life in cases of bone trauma, infection, osteonecrosis, osteoporosis, ageing associated bone loss, wear particle induced bone defects, and other musculoskeletal injuries.
Highlights.
Fundamental principles of bone healing and repair are summarized.
Crosstalk amongst inflammatory cells and bone cells is important for bone repair.
Opportunities for enhancing bone repair by modulating inflammation are discussed.
Acknowledgments
This work was supported by NIH grants 2R01AR055650 and 1R01AR063717, the Ellenburg Chair in Surgery, and University of Chile CONICYT Becas Chile Award. Funding sources were not directly involved in the manuscript preparation.
Footnotes
Conflicts of interest: None.
Author Contributions
All authors contributed to the drafting and revising of this review. All authors read and approved the final submitted manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Florence Loi, Email: floi@stanford.edu.
Luis A. Córdova, Email: lcordova@stanford.edu.
Jukka Pajarinen, Email: jpajari@stanford.edu.
Tzu-hua Lin, Email: tzuhua@stanford.edu.
Zhenyu Yao, Email: zhenyuy@stanford.edu.
Stuart B. Goodman, Email: goodbone@stanford.edu.
References
- 1.Miranda MA, Moon MS. Treatment strategy for nonunions and malunions. In: Stannard JP, Schmidt AH, Kregor PJ, editors. Surgical Treatment of Orthopaedic Trauma. New York: Thieme; 2007. pp. 77–100. [Google Scholar]
- 2.Hak DJ, Fitzpatrick D, Bishop JA, Marsh JL, Tilp S, Schnettler R, Simpson H, Alt V. Delayed union and nonunions: epidemiology, clinical issues, and financial aspects. Injury. 2014;45(Suppl 2):S3–7. doi: 10.1016/j.injury.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 3.National Osteoporosis Foundation. What is Osteoporosis? [Google Scholar]
- 4.Jahangir AA, Nunley RM, Mehta S, Sharan A, Fellows TWHP. Bone-graft substitutes in orthopaedic surgery. AAOS Now. 2008;2:1–5. [Google Scholar]
- 5.Department of Research & Scientific Affairs AAoOS. Annual Incidence of Common Musculoskeletal Procedures and Treatment. 2014. [Google Scholar]
- 6.Mountziaris PM, Spicer PP, Kasper FK, Mikos AG. Harnessing and modulating inflammation in strategies for bone regeneration. Tissue Eng Part B Rev. 2011;17:393–402. doi: 10.1089/ten.teb.2011.0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marsell R, Einhorn TA. The biology of fracture healing. Injury. 2011;42:551–5. doi: 10.1016/j.injury.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nich C, Takakubo Y, Pajarinen J, Ainola M, Salem A, Sillat T, Rao AJ, Raska M, Tamaki Y, Takagi M, Konttinen YT, Goodman SB, Gallo J. Macrophages-Key cells in the response to wear debris from joint replacements. J Biomed Mater Res A. 2013;101:3033–45. doi: 10.1002/jbm.a.34599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229:176–85. doi: 10.1002/path.4133. [DOI] [PubMed] [Google Scholar]
- 11.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–50. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 12.Ortega-Gomez A, Perretti M, Soehnlein O. Resolution of inflammation: an integrated view. EMBO Mol Med. 2013;5:661–74. doi: 10.1002/emmm.201202382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumagai K, Vasanji A, Drazba JA, Butler RS, Muschler GF. Circulating cells with osteogenic potential are physiologically mobilized into the fracture healing site in the parabiotic mice model. J Orthop Res. 2008;26:165–75. doi: 10.1002/jor.20477. [DOI] [PubMed] [Google Scholar]
- 14.Fong EL, Chan CK, Goodman SB. Stem cell homing in musculoskeletal injury. Biomaterials. 2011;32:395–409. doi: 10.1016/j.biomaterials.2010.08.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Osta B, Benedetti G, Miossec P. Classical and Paradoxical Effects of TNF-a on Bone Homeostasis. Front Immunol. 2014;5:1–9. doi: 10.3389/fimmu.2014.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giannoudis PV, Einhorn TA, Marsh D. Fracture healing: the diamond concept. Injury. 2007;38(Suppl 4):S3–6. doi: 10.1016/s0020-1383(08)70003-2. [DOI] [PubMed] [Google Scholar]
- 17.Jagodzinski M, Krettek C. Effect of mechanical stability on fracture healing--an update. Injury. 2007;38(Suppl 1):S3–10. doi: 10.1016/j.injury.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Claes L, Recknagel S, Ignatius A. Fracture healing under healthy and inflammatory conditions. Nat Rev Rheumatol. 2012;8:133–43. doi: 10.1038/nrrheum.2012.1. [DOI] [PubMed] [Google Scholar]
- 19.McKibbin B. The biology of fracture healing in long bones. J Bone Joint Surg Br. 1978;60-B:150–62. doi: 10.1302/0301-620X.60B2.350882. [DOI] [PubMed] [Google Scholar]
- 20.Perren SM. Physical and biological aspects of fracture healing with special reference to internal fixation. Clin Orthop Relat Res. 1979:175–96. [PubMed] [Google Scholar]
- 21.Shapiro F. Cortical bone repair. The relationship of the lacunar-canalicular system and intercellular gap junctions to the repair process. J Bone Joint Surg Am. 1988;70:1067–81. [PubMed] [Google Scholar]
- 22.Perren SM. Evolution of the internal fixation of long bone fractures. The scientific basis of biological internal fixation: choosing a new balance between stability and biology. J Bone Joint Surg Br. 2002;84:1093–110. doi: 10.1302/0301-620x.84b8.13752. [DOI] [PubMed] [Google Scholar]
- 23.Harwood PJ, Newman JB, Michael ALR. An update on fracture healing and non-union. Orthop Trauma. 2010;24:9–23. [Google Scholar]
- 24.Gerstenfeld LC, Cullinane DM, Barnes GL, Graves DT, Einhorn TA. Fracture healing as a post-natal developmental process: molecular, spatial, and temporal aspects of its regulation. J Cell Biochem. 2003;88:873–84. doi: 10.1002/jcb.10435. [DOI] [PubMed] [Google Scholar]
- 25.Uhthoff HK, Rahn BA. Healing patterns of metaphyseal fractures. Clin Orthop Relat Res. 1981:295–303. [PubMed] [Google Scholar]
- 26.Aspenberg P, Sandberg O. Distal radial fractures heal by direct woven bone formation Per Aspenberg and Olof Sandberg. Acta Orthop. 2013;84:297–300. doi: 10.3109/17453674.2013.792769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen WT, Han da C, Zhang PX, Han N, Kou YH, Yin XF, Jiang BG. A special healing pattern in stable metaphyseal fractures. Acta Orthop. 2015;86:238–42. doi: 10.3109/17453674.2014.1003127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Claes L, Reusch M, Gockelmann M, Ohnmacht M, Wehner T, Amling M, Beil FT, Ignatius A. Metaphyseal fracture healing follows similar biomechanical rules as diaphyseal healing. J Orthop Res. 2011;29:425–32. doi: 10.1002/jor.21227. [DOI] [PubMed] [Google Scholar]
- 29.Kolar P, Schmidt-Bleek K, Schell H, Gaber T, Toben D, Schmidmaier G, Perka C, Buttgereit F, Duda GN. The early fracture hematoma and its potential role in fracture healing. Tissue Eng Part B Rev. 2010;16:427–34. doi: 10.1089/ten.TEB.2009.0687. [DOI] [PubMed] [Google Scholar]
- 30.Grundnes O, Reikeras O. The importance of the hematoma for fracture healing in rats. Acta Orthop Scand. 1993;64:340–2. doi: 10.3109/17453679308993640. [DOI] [PubMed] [Google Scholar]
- 31.Park SH, Silva M, Bahk WJ, McKellop H, Lieberman JR. Effect of repeated irrigation and debridement on fracture healing in an animal model. J Orthop Res. 2002;20:1197–204. doi: 10.1016/S0736-0266(02)00072-4. [DOI] [PubMed] [Google Scholar]
- 32.Gerstenfeld LC, Cho TJ, Kon T, Aizawa T, Cruceta J, Graves BD, Einhorn TA. Impaired intramembranous bone formation during bone repair in the absence of tumor necrosis factor-alpha signaling. Cells Tissues Organs. 2001;169:285–94. doi: 10.1159/000047893. [DOI] [PubMed] [Google Scholar]
- 33.Gerstenfeld LC, Cho TJ, Kon T, Aizawa T, Tsay A, Fitch J, Barnes GL, Graves DT, Einhorn TA. Impaired fracture healing in the absence of TNF-alpha signaling: the role of TNF-alpha in endochondral cartilage resorption. J Bone Miner Res. 2003;18:1584–92. doi: 10.1359/jbmr.2003.18.9.1584. [DOI] [PubMed] [Google Scholar]
- 34.Yang X, Ricciardi BF, Hernandez-Soria A, Shi Y, Pleshko Camacho N, Bostrom MP. Callus mineralization and maturation are delayed during fracture healing in interleukin-6 knockout mice. Bone. 2007;41:928–36. doi: 10.1016/j.bone.2007.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glass GE, Chan JK, Freidin A, Feldmann M, Horwood NJ, Nanchahal J. TNF-alpha promotes fracture repair by augmenting the recruitment and differentiation of muscle-derived stromal cells. Proc Natl Acad Sci U S A. 2011;108:1585–90. doi: 10.1073/pnas.1018501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bastian O, Pillay J, Alblas J, Leenen L, Koenderman L, Blokhuis T. Systemic inflammation and fracture healing. J Leukoc Biol. 2011;89:669–73. doi: 10.1189/jlb.0810446. [DOI] [PubMed] [Google Scholar]
- 37.Xing Z, Lu C, Hu D, Yu YY, Wang X, Colnot C, Nakamura M, Wu Y, Miclau T, Marcucio RS. Multiple roles for CCR2 during fracture healing. Dis Model Mech. 2010;3:451–8. doi: 10.1242/dmm.003186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hurst SM, Wilkinson TS, McLoughlin RM, Jones S, Horiuchi S, Yamamoto N, Rose-John S, Fuller GM, Topley N, Jones SA. Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity. 2001;14:705–14. doi: 10.1016/s1074-7613(01)00151-0. [DOI] [PubMed] [Google Scholar]
- 39.Alexander KA, Chang MK, Maylin ER, Kohler T, Muller R, Wu AC, Van Rooijen N, Sweet MJ, Hume DA, Raggatt LJ, Pettit AR. Osteal macrophages promote in vivo intramembranous bone healing in a mouse tibial injury model. J Bone Miner Res. 2011;26:1517–32. doi: 10.1002/jbmr.354. [DOI] [PubMed] [Google Scholar]
- 40.Raggatt LJ, Wullschleger ME, Alexander KA, Wu AC, Millard SM, Kaur S, Maugham ML, Gregory LS, Steck R, Pettit AR. Fracture healing via periosteal callus formation requires macrophages for both initiation and progression of early endochondral ossification. Am J Pathol. 2014;184:3192–204. doi: 10.1016/j.ajpath.2014.08.017. [DOI] [PubMed] [Google Scholar]
- 41.Pettit AR, Chang MK, Hume DA, Raggatt LJ. Osteal macrophages: a new twist on coupling during bone dynamics. Bone. 2008;43:976–82. doi: 10.1016/j.bone.2008.08.128. [DOI] [PubMed] [Google Scholar]
- 42.Kon T, Cho TJ, Aizawa T, Yamazaki M, Nooh N, Graves D, Gerstenfeld LC, Einhorn TA. Expression of osteoprotegerin, receptor activator of NF-kappaB ligand (osteoprotegerin ligand) and related proinflammatory cytokines during fracture healing. J Bone Miner Res. 2001;16:1004–14. doi: 10.1359/jbmr.2001.16.6.1004. [DOI] [PubMed] [Google Scholar]
- 43.Bielby R, Jones E, McGonagle D. The role of mesenchymal stem cells in maintenance and repair of bone. Injury. 2007;38(Suppl 1):S26–32. doi: 10.1016/j.injury.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 44.Wu AC, Raggatt LJ, Alexander KA, Pettit AR. Unraveling macrophage contributions to bone repair. Bonekey Rep. 2013;2:373. doi: 10.1038/bonekey.2013.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu L, Li G. Circulating mesenchymal stem cells and their clinical implications. J Orthop Translat. 2014;2:1–7. [Google Scholar]
- 46.Tsiridis E, Upadhyay N, Giannoudis P. Molecular aspects of fracture healing: which are the important molecules? Injury. 2007;38(Suppl 1):S11–25. doi: 10.1016/j.injury.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 47.Schindeler A, McDonald MM, Bokko P, Little DG. Bone remodeling during fracture repair: The cellular picture. Semin Cell Dev Biol. 2008;19:459–66. doi: 10.1016/j.semcdb.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 48.Grundnes O, Reikeras O. Blood flow and mechanical properties of healing bone. Femoral osteotomies studied in rats. Acta Orthop Scand. 1992;63:487–91. doi: 10.3109/17453679209154720. [DOI] [PubMed] [Google Scholar]
- 49.Melnyk M, Henke T, Claes L, Augat P. Revascularisation during fracture healing with soft tissue injury. Arch Orthop Trauma Surg. 2008;128:1159–65. doi: 10.1007/s00402-007-0543-0. [DOI] [PubMed] [Google Scholar]
- 50.Keramaris NC, Calori GM, Nikolaou VS, Schemitsch EH, Giannoudis PV. Fracture vascularity and bone healing: a systematic review of the role of VEGF. Injury. 2008;39(Suppl 2):S45–57. doi: 10.1016/S0020-1383(08)70015-9. [DOI] [PubMed] [Google Scholar]
- 51.Thompson Z, Miclau T, Hu D, Helms JA. A model for intramembranous ossification during fracture healing. J Orthop Res. 2002;20:1091–8. doi: 10.1016/S0736-0266(02)00017-7. [DOI] [PubMed] [Google Scholar]
- 52.Malizos KN, Papatheodorou LK. The healing potential of the periosteum molecular aspects. Injury. 2005;36(Suppl 3):S13–9. doi: 10.1016/j.injury.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 53.Gaston MS, Simpson AH. Inhibition of fracture healing. J Bone Joint Surg Br. 2007;89:1553–60. doi: 10.1302/0301-620X.89B12.19671. [DOI] [PubMed] [Google Scholar]
- 54.Komatsu DE, Warden SJ. The control of fracture healing and its therapeutic targeting: improving upon nature. J Cell Biochem. 2010;109:302–11. doi: 10.1002/jcb.22418. [DOI] [PubMed] [Google Scholar]
- 55.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–7. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 56.O’Keefe RJ, Mao J. Bone tissue engineering and regeneration: from discovery to the clinic--an overview. Tissue Eng Part B Rev. 2011;17:389–92. doi: 10.1089/ten.teb.2011.0475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mountziaris PM, Mikos AG. Modulation of the inflammatory response for enhanced bone tissue regeneration. Tissue Eng Part B Rev. 2008;14:179–86. doi: 10.1089/ten.teb.2008.0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Waters RV, Gamradt SC, Asnis P, Vickery BH, Avnur Z, Hill E, Bostrom M. Systemic corticosteroids inhibit bone healing in a rabbit ulnar osteotomy model. Acta Orthop Scand. 2000;71:316–21. doi: 10.1080/000164700317411951. [DOI] [PubMed] [Google Scholar]
- 59.Arron JR, Choi Y. Bone versus immune system. Nature. 2000;408:535–6. doi: 10.1038/35046196. [DOI] [PubMed] [Google Scholar]
- 60.Arboleya L, Castañeda S. Osteoimmunology: The Study of the Relationship Between the Immune System and Bone Tissue. Reumatol Clin. 2013;9:303–315. doi: 10.1016/j.reuma.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 61.Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007;7:292–304. doi: 10.1038/nri2062. [DOI] [PubMed] [Google Scholar]
- 62.Walsh MC, Kim N, Kadono Y, Rho J, Lee SY, Lorenzo J, Choi Y. Osteoimmunology: interplay between the immune system and bone metabolism. Annu Rev Immunol. 2006;24:33–63. doi: 10.1146/annurev.immunol.24.021605.090646. [DOI] [PubMed] [Google Scholar]
- 63.Konnecke I, Serra A, El Khassawna T, Schlundt C, Schell H, Hauser A, Ellinghaus A, Volk HD, Radbruch A, Duda GN, Schmidt-Bleek K. T and B cells participate in bone repair by infiltrating the fracture callus in a two-wave fashion. Bone. 2014;64:155–65. doi: 10.1016/j.bone.2014.03.052. [DOI] [PubMed] [Google Scholar]
- 64.Criscitiello C, Viale G, Gelao L, Esposito A, De Laurentiis M, De Placido S, Santangelo M, Goldhirsch A, Curigliano G. Crosstalk between bone niche and immune system: osteoimmunology signaling as a potential target for cancer treatment. Cancer Treat Rev. 2015;41:61–8. doi: 10.1016/j.ctrv.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 65.Greenblatt MB, Shim JH. Osteoimmunology: a brief introduction. Immune Netw. 2013;13:111–5. doi: 10.4110/in.2013.13.4.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kovach TK, Dighe AS, Lobo PI, Cui Q. Interactions between MSCs and immune cells: implications for bone healing. J Immunol Res. 2015;2015:752510. doi: 10.1155/2015/752510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–74. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brown BN, Ratner BD, Goodman SB, Amar S, Badylak SF. Macrophage polarization: an opportunity for improved outcomes in biomaterials and regenerative medicine. Biomaterials. 2012;33:3792–802. doi: 10.1016/j.biomaterials.2012.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–61. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 70.Spiller KL, Nassiri S, Witherel CE, Anfang RR, Ng J, Nakazawa KR, Yu T, Vunjak-Novakovic G. Sequential delivery of immunomodulatory cytokines to facilitate the M1-to-M2 transition of macrophages and enhance vascularization of bone scaffolds. Biomaterials. 2015;37:194–207. doi: 10.1016/j.biomaterials.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.da Silva Meirelles L, Chagastelles PC, Nardi NB. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J Cell Sci. 2006;119:2204–13. doi: 10.1242/jcs.02932. [DOI] [PubMed] [Google Scholar]
- 72.Harada S, Rodan GA. Control of osteoblast function and regulation of bone mass. Nature. 2003;423:349–55. doi: 10.1038/nature01660. [DOI] [PubMed] [Google Scholar]
- 73.Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 74.Marie PJ, Kassem M. Osteoblasts in osteoporosis: past, emerging, and future anabolic targets. Eur J Endocrinol. 2011;165:1–10. doi: 10.1530/EJE-11-0132. [DOI] [PubMed] [Google Scholar]
- 75.Bellido T. Osteocyte-driven bone remodeling. Calcif Tissue Int. 2014;94:25–34. doi: 10.1007/s00223-013-9774-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–42. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 77.Khosla S. Minireview: the OPG/RANKL/RANK system. Endocrinology. 2001;142:5050–5. doi: 10.1210/endo.142.12.8536. [DOI] [PubMed] [Google Scholar]
- 78.Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95:3597–602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, Takaoka A, Yokochi T, Oda H, Tanaka K, Nakamura K, Taniguchi T. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature. 2000;408:600–5. doi: 10.1038/35046102. [DOI] [PubMed] [Google Scholar]
- 80.Takayanagi H. Inflammatory bone destruction and osteoimmunology. J Periodontal Res. 2005;40:287–93. doi: 10.1111/j.1600-0765.2005.00814.x. [DOI] [PubMed] [Google Scholar]
- 81.Lee SK, Lorenzo J. Cytokines regulating osteoclast formation and function. Curr Opin Rheumatol. 2006;18:411–8. doi: 10.1097/01.bor.0000231911.42666.78. [DOI] [PubMed] [Google Scholar]
- 82.Takayanagi H. Osteoimmunology and the effects of the immune system on bone. Nat Rev Rheumatol. 2009;5:667–76. doi: 10.1038/nrrheum.2009.217. [DOI] [PubMed] [Google Scholar]
- 83.Aliprantis AO, Ueki Y, Sulyanto R, Park A, Sigrist KS, Sharma SM, Ostrowski MC, Olsen BR, Glimcher LH. NFATc1 in mice represses osteoprotegerin during osteoclastogenesis and dissociates systemic osteopenia from inflammation in cherubism. J Clin Invest. 2008;118:3775–89. doi: 10.1172/JCI35711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 85.Takahashi N, Mundy GR, Roodman GD. Recombinant human interferon-gamma inhibits formation of human osteoclast-like cells. J Immunol. 1986;137:3544–9. [PubMed] [Google Scholar]
- 86.Chang MK, Raggatt LJ, Alexander KA, Kuliwaba JS, Fazzalari NL, Schroder K, Maylin ER, Ripoll VM, Hume DA, Pettit AR. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J Immunol. 2008;181:1232–44. doi: 10.4049/jimmunol.181.2.1232. [DOI] [PubMed] [Google Scholar]
- 87.Hoch AI, Binder BY, Genetos DC, Leach JK. Differentiation-dependent secretion of proangiogenic factors by mesenchymal stem cells. PLoS One. 2012;7:e35579. doi: 10.1371/journal.pone.0035579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Porter JR, Ruckh TT, Popat KC. Bone tissue engineering: a review in bone biomimetics and drug delivery strategies. Biotechnol Prog. 2009;25:1539–60. doi: 10.1002/btpr.246. [DOI] [PubMed] [Google Scholar]
- 89.Anton K, Banerjee D, Glod J. Macrophage-associated mesenchymal stem cells assume an activated, migratory, pro-inflammatory phenotype with increased IL-6 and CXCL10 secretion. PLoS One. 2012;7:e35036. doi: 10.1371/journal.pone.0035036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Freytes DO, Kang JW, Marcos-Campos I, Vunjak-Novakovic G. Macrophages modulate the viability and growth of human mesenchymal stem cells. J Cell Biochem. 2013;114:220–9. doi: 10.1002/jcb.24357. [DOI] [PubMed] [Google Scholar]
- 91.Champagne CM, Takebe J, Offenbacher S, Cooper LF. Macrophage cell lines produce osteoinductive signals that include bone morphogenetic protein-2. Bone. 2002;30:26–31. doi: 10.1016/s8756-3282(01)00638-x. [DOI] [PubMed] [Google Scholar]
- 92.Fromigue O, Marie PJ, Lomri A. Bone morphogenetic protein-2 and transforming growth factor-beta2 interact to modulate human bone marrow stromal cell proliferation and differentiation. J Cell Biochem. 1998;68:411–26. [PubMed] [Google Scholar]
- 93.Blom AB, van Lent PL, Holthuysen AE, van der Kraan PM, Roth J, van Rooijen N, van den Berg WB. Synovial lining macrophages mediate osteophyte formation during experimental osteoarthritis. Osteoarthritis Cartilage. 2004;12:627–35. doi: 10.1016/j.joca.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 94.Vi L, Baht GS, Whetstone H, Ng A, Wei Q, Poon R, Mylvaganam S, Grynpas M, Alman BA. Macrophages promote osteoblastic differentiation in-vivo: implications in fracture repair and bone homeostasis. J Bone Miner Res. 2015;30:1090–102. doi: 10.1002/jbmr.2422. [DOI] [PubMed] [Google Scholar]
- 95.Nicolaidou V, Wong MM, Redpath AN, Ersek A, Baban DF, Williams LM, Cope AP, Horwood NJ. Monocytes induce STAT3 activation in human mesenchymal stem cells to promote osteoblast formation. PLoS One. 2012;7:e39871. doi: 10.1371/journal.pone.0039871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Guihard P, Boutet MA, Brounais-Le Royer B, Gamblin AL, Amiaud J, Renaud A, Berreur M, Redini F, Heymann D, Layrolle P, Blanchard F. Oncostatin m, an inflammatory cytokine produced by macrophages, supports intramembranous bone healing in a mouse model of tibia injury. Am J Pathol. 2015;185:765–75. doi: 10.1016/j.ajpath.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 97.Chen Z, Wu C, Gu W, Klein T, Crawford R, Xiao Y. Osteogenic differentiation of bone marrow MSCs by beta-tricalcium phosphate stimulating macrophages via BMP2 signalling pathway. Biomaterials. 2014;35:1507–18. doi: 10.1016/j.biomaterials.2013.11.014. [DOI] [PubMed] [Google Scholar]
- 98.Lorenzo J. Interactions between immune and bone cells: new insights with many remaining questions. J Clin Invest. 2000;106:749–52. doi: 10.1172/JCI11089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gilbert L, He X, Farmer P, Boden S, Kozlowski M, Rubin J, Nanes MS. Inhibition of osteoblast differentiation by tumor necrosis factor-alpha. Endocrinology. 2000;141:3956–64. doi: 10.1210/endo.141.11.7739. [DOI] [PubMed] [Google Scholar]
- 100.Cavagis A, Takamori E, Granjeiro J, Oliveira R, Ferreira C, Peppelenbosch M, Zambuzzi W. TNFalpha contributes for attenuating both Y397FAK and Y416Src phosphorylations in osteoblasts. Oral Dis. 2014;20:780–6. doi: 10.1111/odi.12202. [DOI] [PubMed] [Google Scholar]
- 101.Ding J, Ghali O, Lencel P, Broux O, Chauveau C, Devedjian JC, Hardouin P, Magne D. TNF-alpha and IL-1beta inhibit RUNX2 and collagen expression but increase alkaline phosphatase activity and mineralization in human mesenchymal stem cells. Life Sci. 2009;84:499–504. doi: 10.1016/j.lfs.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 102.Guo R, Yamashita M, Zhang Q, Zhou Q, Chen D, Reynolds DG, Awad HA, Yanoso L, Zhao L, Schwarz EM, Zhang YE, Boyce BF, Xing L. Ubiquitin ligase Smurf1 mediates tumor necrosis factor-induced systemic bone loss by promoting proteasomal degradation of bone morphogenetic signaling proteins. J Biol Chem. 2008;283:23084–92. doi: 10.1074/jbc.M709848200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lange J, Sapozhnikova A, Lu C, Hu D, Li X, Miclau T, 3rd, Marcucio RS. Action of IL-1beta during fracture healing. J Orthop Res. 2010;28:778–84. doi: 10.1002/jor.21061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Miller RR, Cappola AR, Shardell MD, Hawkes WG, Yu-Yahiro JA, Hebel JR, Magaziner J. Persistent changes in interleukin-6 and lower extremity function following hip fracture. J Gerontol A Biol Sci Med Sci. 2006;61:1053–8. doi: 10.1093/gerona/61.10.1053. [DOI] [PubMed] [Google Scholar]
- 105.Caparbo VF, Prada F, Silva CA, Regio PL, Pereira RM. Serum from children with polyarticular juvenile idiopathic arthritis (pJIA) inhibits differentiation, mineralization and may increase apoptosis of human osteoblasts “in vitro”. Clin Rheumatol. 2009;28:71–7. doi: 10.1007/s10067-008-0985-y. [DOI] [PubMed] [Google Scholar]
- 106.Hofbauer LC, Lacey DL, Dunstan CR, Spelsberg TC, Riggs BL, Khosla S. Interleukin-1beta and tumor necrosis factor-alpha, but not interleukin-6, stimulate osteoprotegerin ligand gene expression in human osteoblastic cells. Bone. 1999;25:255–9. doi: 10.1016/s8756-3282(99)00162-3. [DOI] [PubMed] [Google Scholar]
- 107.Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000;106:1481–8. doi: 10.1172/JCI11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jimi E, Nakamura I, Duong LT, Ikebe T, Takahashi N, Rodan GA, Suda T. Interleukin 1 induces multinucleation and bone-resorbing activity of osteoclasts in the absence of osteoblasts/stromal cells. Exp Cell Res. 1999;247:84–93. doi: 10.1006/excr.1998.4320. [DOI] [PubMed] [Google Scholar]
- 109.Tan SD, Kuijpers-Jagtman AM, Semeins CM, Bronckers AL, Maltha JC, Von den Hoff JW, Everts V, Klein-Nulend J. Fluid shear stress inhibits TNFalpha-induced osteocyte apoptosis. J Dent Res. 2006;85:905–9. doi: 10.1177/154405910608501006. [DOI] [PubMed] [Google Scholar]
- 110.Mori G, D’Amelio P, Faccio R, Brunetti G. The Interplay between the bone and the immune system. Clin Dev Immunol. 2013;2013:720504. doi: 10.1155/2013/720504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Girasole G, Jilka RL, Passeri G, Boswell S, Boder G, Williams DC, Manolagas SC. 17 beta-estradiol inhibits interleukin-6 production by bone marrow-derived stromal cells and osteoblasts in vitro: a potential mechanism for the antiosteoporotic effect of estrogens. J Clin Invest. 1992;89:883–91. doi: 10.1172/JCI115668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Grogaard B, Gerdin B, Reikeras O. The polymorphonuclear leukocyte: has it a role in fracture healing? Arch Orthop Trauma Surg. 1990;109:268–71. doi: 10.1007/BF00419942. [DOI] [PubMed] [Google Scholar]
- 113.Toben D, Schroeder I, El Khassawna T, Mehta M, Hoffmann JE, Frisch JT, Schell H, Lienau J, Serra A, Radbruch A, Duda GN. Fracture healing is accelerated in the absence of the adaptive immune system. J Bone Miner Res. 2011;26:113–24. doi: 10.1002/jbmr.185. [DOI] [PubMed] [Google Scholar]
- 114.Martin P, D’Souza D, Martin J, Grose R, Cooper L, Maki R, McKercher SR. Wound healing in the PU. 1 null mouse--tissue repair is not dependent on inflammatory cells. Curr Biol. 2003;13:1122–8. doi: 10.1016/s0960-9822(03)00396-8. [DOI] [PubMed] [Google Scholar]
- 115.Kim J, Hematti P. Mesenchymal stem cell-educated macrophages: a novel type of alternatively activated macrophages. Exp Hematol. 2009;37:1445–53. doi: 10.1016/j.exphem.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, Ito K, Koh GY, Suda T. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118:149–61. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 117.Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR, Milner LA, Kronenberg HM, Scadden DT. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–6. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 118.Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y, Tesio M, Samstein RM, Goichberg P, Spiegel A, Elson A, Lapidot T. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med. 2006;12:657–64. doi: 10.1038/nm1417. [DOI] [PubMed] [Google Scholar]
- 119.Rao M, Supakorndej T, Schmidt AP, Link DC. Osteoclasts are dispensable for hematopoietic progenitor mobilization by granulocyte colony-stimulating factor in mice. Exp Hematol. 2015;43:110–4. e1–2. doi: 10.1016/j.exphem.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Charles JF, Aliprantis AO. Osteoclasts: more than ‘bone eaters’. Trends Mol Med. 2014;20:449–59. doi: 10.1016/j.molmed.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cho DI, Kim MR, Jeong HY, Jeong HC, Jeong MH, Yoon SH, Kim YS, Ahn Y. Mesenchymal stem cells reciprocally regulate the M1/M2 balance in mouse bone marrow-derived macrophages. Exp Mol Med. 2014;46:e70. doi: 10.1038/emm.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI, Zhao RC, Shi Y. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. 2008;2:141–50. doi: 10.1016/j.stem.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 123.Nakajima H, Uchida K, Guerrero AR, Watanabe S, Sugita D, Takeura N, Yoshida A, Long G, Wright KT, Johnson WE, Baba H. Transplantation of mesenchymal stem cells promotes an alternative pathway of macrophage activation and functional recovery after spinal cord injury. J Neurotrauma. 2012;29:1614–25. doi: 10.1089/neu.2011.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Maggini J, Mirkin G, Bognanni I, Holmberg J, Piazzon IM, Nepomnaschy I, Costa H, Canones C, Raiden S, Vermeulen M, Geffner JR. Mouse Bone Marrow-Derived Mesenchymal Stromal Cells Turn Activated Macrophages into a Regulatory-Like Profile. PloS One. 2010:5. doi: 10.1371/journal.pone.0009252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Prockop DJ. Concise review: two negative feedback loops place mesenchymal stem/stromal cells at the center of early regulators of inflammation. Stem Cells. 2013;31:2042–6. doi: 10.1002/stem.1400. [DOI] [PubMed] [Google Scholar]
- 126.Gómez-Barrena E, Rosset P, Lozano D, Stanovici J, Ermthaller C, Gerbhard F. Bone fracture healing: Cell therapy in delayed unions and nonunions. Bone. 2015;70:93–101. doi: 10.1016/j.bone.2014.07.033. [DOI] [PubMed] [Google Scholar]
- 127.Goodman SB, Gibon E, Pajarinen J, Lin TH, Keeney M, Ren PG, Nich C, Yao Z, Egashira K, Yang F, Konttinen YT. Novel biological strategies for treatment of wear particle-induced periprosthetic osteolysis of orthopaedic implants for joint replacement. J R Soc Interface. 2014:11. doi: 10.1098/rsif.2013.0962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Córdova LA, Trichet V, Escriou V, Rosset P, Amiaud J, Battaglia S, Charrier C, Berreur M, Brion R, Gouin F, Layrolle P, Passuti N, Heymann D. Inhibition of osteolysis and increase of bone formation after local administration of siRNA-targeting RANK in a polyethylene particle-induced osteolysis model. Acta Biomater. 2015;13:150–158. doi: 10.1016/j.actbio.2014.10.042. [DOI] [PubMed] [Google Scholar]
- 129.Heymann D. Bisphosphonates and Bone Diseases: Past, Present and Future. Curr Pharm Des. 2010;16:2948–2949. doi: 10.2174/138161210793563572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nardi A, Ventura L, Cozzi L, Tonini G, Zennaro R, Celi M, Ramazzina E. The bone anabolic therapy. Aging Clin Exp Res. 2013;25:121–124. doi: 10.1007/s40520-013-0133-7. [DOI] [PubMed] [Google Scholar]
- 131.Andrews EB, Gilsenan AW, Midkiff K, Sherrill B, Wu Y, Mann BH, Masica D. The US postmarketing surveillance study of adult osteosarcoma and teriparatide: Study design and findings from the first 7 years. J Bone Miner Res. 2012;27:2429–2437. doi: 10.1002/jbmr.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Allen MR. Bisphosphonates and osteonecrosis of the jaw: moving from the bedside to the bench. Cells Tissues Organs. 2009;189:289–294. doi: 10.1159/000151371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Carvalho NNC, Voss LA, Almeida MOP, Salgado CL, Bandeira F. Atypical femoral fractures during prolonged use of bisphosphonates: short-term responses to strontium ranelate and teriparatide. J Clin Endocrinol Metab. 2011;96:2675–2680. doi: 10.1210/jc.2011-0593. [DOI] [PubMed] [Google Scholar]
- 134.Simon LS. Osteoporosis. Rheum Dis Clin North Am. 2007;33:149–76. doi: 10.1016/j.rdc.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 135.Simpson AH, Murray IR. Osteoporotic fracture models. Curr Osteoporos Rep. 2015;13:9–15. doi: 10.1007/s11914-014-0246-8. [DOI] [PubMed] [Google Scholar]
- 136.Silverman SL, Siris E, Kendler DL, Belazi D, Brown JP, Gold DT, Lewiecki EM, Papaioannou A, Simonelli C, Ferreira I, Balasubramanian A, Dakin P, Ho P, Siddhanti S, Stolshek B, Recknor C. Persistence at 12 months with denosumab in postmenopausal women with osteoporosis: interim results from a prospective observational study. Osteoporos Int. 2015;26:361–72. doi: 10.1007/s00198-014-2871-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pietschmann P, Mechtcheriakova D, Meshcheryakova A, Foger-Samwald U, Ellinger I. Immunology of Osteoporosis: A Mini-Review. Gerontology. 2015 doi: 10.1159/000431091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- 139.Lencel P, Magne D. Inflammaging: the driving force in osteoporosis? Med Hypotheses. 2011;76:317–21. doi: 10.1016/j.mehy.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 140.Topini C, Topini D, Cerica G, Nardocci F, Topini G. Osteoporosis and risk of fracture, analysis on a population admitted in rehabilitation post-acute. Clin Cases Miner Bone Metab. 2014;11:129–31. [PMC free article] [PubMed] [Google Scholar]
- 141.Perren SM. Fracture healing: fracture healing understood as the result of a fascinating cascade of physical and biological interactions. Part I. An Attempt to Integrate Observations from 30 Years AO Research. Acta Chir Orthop Traumatol Cech. 2014;81:355–64. [PubMed] [Google Scholar]
- 142.Donoso O, Pino AM, Seitz G, Osses N, Rodriguez JP. Osteoporosis-associated alteration in the signalling status of BMP-2 in human MSCs under adipogenic conditions. J Cell Biochem. 2015 doi: 10.1002/jcb.25082. [DOI] [PubMed] [Google Scholar]
- 143.Coughlan T, Dockery F. Osteoporosis and fracture risk in older people. Clin Med. 2014;14:187–91. doi: 10.7861/clinmedicine.14-2-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ibrahim N, Khamis MF, Mod Yunoh MF, Abdullah S, Mohamed N, Shuid AN. Targeted delivery of lovastatin and tocotrienol to fracture site promotes fracture healing in osteoporosis model: micro-computed tomography and biomechanical evaluation. PLoS One. 2014;9:e115595. doi: 10.1371/journal.pone.0115595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nikolaou VS, Efstathopoulos N, Kontakis G, Kanakaris NK, Giannoudis PV. The influence of osteoporosis in femoral fracture healing time. Injury. 2009;40:663–8. doi: 10.1016/j.injury.2008.10.035. [DOI] [PubMed] [Google Scholar]
- 146.Panteli M, Pountos I, Jones E, Giannoudis PV. Biological and molecular profile of fracture non-union tissue: current insights. J Cell Mol Med. 2015;19:685–713. doi: 10.1111/jcmm.12532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Copuroglu C, Calori GM, Giannoudis PV. Fracture non-union: who is at risk? Injury. 2013;44:1379–82. doi: 10.1016/j.injury.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 148.Behrens A, van Deursen JM, Rudolph KL, Schumacher B. Impact of genomic damage and ageing on stem cell function. Nat Cell Biol. 2014;16:201–7. doi: 10.1038/ncb2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Iwakura T, Miwa M, Sakai Y, Niikura T, Lee SY, Oe K, Hasegawa T, Kuroda R, Fujioka H, Doita M, Kurosaka M. Human hypertrophic nonunion tissue contains mesenchymal progenitor cells with multilineage capacity in vitro. J Orthop Res. 2009;27:208–15. doi: 10.1002/jor.20739. [DOI] [PubMed] [Google Scholar]
- 150.Kloen P, Doty SB, Gordon E, Rubel IF, Goumans MJ, Helfet DL. Expression and activation of the BMP-signaling components in human fracture nonunions. J Bone Joint Surg Am. 2002;84-A:1909–18. doi: 10.2106/00004623-200211000-00001. [DOI] [PubMed] [Google Scholar]
- 151.Qu G, von Schroeder HP. The osteogenic potential of pseudoarthrosis tissue and bone from human scaphoid non-unions. J Hand Surg Eur Vol. 2008;33:449–56. doi: 10.1177/1753193408090122. [DOI] [PubMed] [Google Scholar]
- 152.Fernandez-Bances I, Perez-Basterrechea M, Perez-Lopez S, Nunez Batalla D, Fernandez Rodriguez MA, Alvarez-Viejo M, Ferrero-Gutierrez A, Menendez-Menendez Y, Garcia-Gala JM, Escudero D, Paz Aparicio J, Carnero Lopez S, Lopez Fernandez P, Gonzalez Suarez D, Otero Hernandez J. Repair of long-bone pseudoarthrosis with autologous bone marrow mononuclear cells combined with allogenic bone graft. Cytotherapy. 2013;15:571–7. doi: 10.1016/j.jcyt.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 153.Hisatome T, Yasunaga Y, Yanada S, Tabata Y, Ikada Y, Ochi M. Neovascularization and bone regeneration by implantation of autologous bone marrow mononuclear cells. Biomaterials. 2005;26:4550–6. doi: 10.1016/j.biomaterials.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 154.Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med. 2010;2:640–53. doi: 10.1002/wsbm.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hernigou P, Poignard A, Manicom O, Mathieu G, Rouard H. The use of percutaneous autologous bone marrow transplantation in nonunion and avascular necrosis of bone. J Bone Joint Surg Br. 2005;87:896–902. doi: 10.1302/0301-620X.87B7.16289. [DOI] [PubMed] [Google Scholar]
- 156.Patterson TE, Kumagai K, Griffith L, Muschler GF. Cellular strategies for enhancement of fracture repair. J Bone Joint Surg Am. 2008;90(Suppl 1):111–9. doi: 10.2106/JBJS.G.01572. [DOI] [PubMed] [Google Scholar]
- 157.Lin TH, Tamaki Y, Pajarinen J, Waters HA, Woo DK, Yao Z, Goodman SB. Chronic inflammation in biomaterial-induced periprosthetic osteolysis: NF-kappaB as a therapeutic target. Acta Biomater. 2014;10:1–10. doi: 10.1016/j.actbio.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Branemark R, Branemark PI, Rydevik B, Myers RR. Osseointegration in skeletal reconstruction and rehabilitation: a review. J Rehabil Res Dev. 2001;38:175–81. [PubMed] [Google Scholar]
- 159.Anderson JM, Rodriguez A, Chang DT. Foreign body reaction to biomaterials. Semin Immunol. 2008;20:86–100. doi: 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Trindade R, Albrektsson T, Tengvall P, Wennerberg A. Foreign Body Reaction to Biomaterials: On Mechanisms for Buildup and Breakdown of Osseointegration. Clin Implant Dent Relat Res. 2014 doi: 10.1111/cid.12274. in press. [DOI] [PubMed] [Google Scholar]
- 161.Albrektsson T, Dahlin C, Jemt T, Sennerby L, Turri A, Wennerberg A. Is marginal bone loss around oral implants the result of a provoked foreign body reaction? Clin Implant Dent Relat Res. 2014;16:155–65. doi: 10.1111/cid.12142. [DOI] [PubMed] [Google Scholar]
- 162.Sundfeldt M, Carlsson LV, Johansson CB, Thomsen P, Gretzer C. Aseptic loosening, not only a question of wear: a review of different theories. Acta Orthop. 2006;77:177–97. doi: 10.1080/17453670610045902. [DOI] [PubMed] [Google Scholar]
- 163.Albrektsson T, Dahl E, Enbom L, Engevall S, Engquist B, Eriksson AR, Feldmann G, Freiberg N, Glantz PO, Kjellman O, et al. Osseointegrated oral implants. A Swedish multicenter study of 8139 consecutively inserted Nobelpharma implants. J Periodontol. 1988;59:287–96. doi: 10.1902/jop.1988.59.5.287. [DOI] [PubMed] [Google Scholar]
- 164.Papaspyridakos P, Mokti M, Chen CJ, Benic GI, Gallucci GO, Chronopoulos V. Implant and prosthodontic survival rates with implant fixed complete dental prostheses in the edentulous mandible after at least 5 years: a systematic review. Clin Implant Dent Relat Res. 2014;16:705–17. doi: 10.1111/cid.12036. [DOI] [PubMed] [Google Scholar]
- 165.Zwartele RE, Witjes S, Doets HC, Stijnen T, Poll RG. Cementless total hip arthroplasty in rheumatoid arthritis: a systematic review of the literature. Arch Orthop Trauma Surg. 2012;132:535–46. doi: 10.1007/s00402-011-1432-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gallo J, Raska M, Konttinen YT, Nich C, Goodman SB. Innate immunity sensors participating in pathophysiology of joint diseases: a brief overview. J Long Term Eff Med Implants. 2014;24:297–317. doi: 10.1615/jlongtermeffmedimplants.2014010825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gallo J, Vaculova J, Goodman SB, Konttinen YT, Thyssen JP. Contributions of human tissue analysis to understanding the mechanisms of loosening and osteolysis in total hip replacement. Acta Biomater. 2014;10:2354–66. doi: 10.1016/j.actbio.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pajarinen J, Jamsen E, Konttinen YT, Goodman SB. Innate immune reactions in septic and aseptic osteolysis around hip implants. J Long Term Eff Med Implants. 2014;24:283–96. doi: 10.1615/jlongtermeffmedimplants.2014010564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhang X, Morham SG, Langenbach R, Young DA, Xing L, Boyce BF, Puzas EJ, Rosier RN, O’Keefe RJ, Schwarz EM. Evidence for a direct role of cyclo-oxygenase 2 in implant wear debris-induced osteolysis. J Bone Miner Res. 2001;16:660–70. doi: 10.1359/jbmr.2001.16.4.660. [DOI] [PubMed] [Google Scholar]
- 170.Childs LM, Goater JJ, O’Keefe RJ, Schwarz EM. Efficacy of etanercept for wear debris-induced osteolysis. J Bone Miner Res. 2001;16:338–47. doi: 10.1359/jbmr.2001.16.2.338. [DOI] [PubMed] [Google Scholar]
- 171.Gibon E, Ma T, Ren PG, Fritton K, Biswal S, Yao Z, Smith L, Goodman SB. Selective inhibition of the MCP-1-CCR2 ligand-receptor axis decreases systemic trafficking of macrophages in the presence of UHMWPE particles. J Orthop Res. 2012;30:547–53. doi: 10.1002/jor.21548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Keeney M, Waters H, Barcay K, Jiang X, Yao Z, Pajarinen J, Egashira K, Goodman SB, Yang F. Mutant MCP-1 protein delivery from layer-by-layer coatings on orthopedic implants to modulate inflammatory response. Biomaterials. 2013;34:10287–95. doi: 10.1016/j.biomaterials.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lin TH, Sato T, Barcay KR, Waters H, Loi F, Zhang R, Pajarinen J, Egashira K, Yao Z, Goodman SB. NF-kappaB decoy oligodeoxynucleotide enhanced osteogenesis in mesenchymal stem cells exposed to polyethylene particle. Tissue Eng Part A. 2015;21:875–83. doi: 10.1089/ten.tea.2014.0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Beane OS, Fonseca VC, Cooper LL, Koren G, Darling EM. Impact of aging on the regenerative properties of bone marrow-, muscle-, and adipose-derived mesenchymal stem/stromal cells. PLoS One. 2014;9:e115963. doi: 10.1371/journal.pone.0115963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Rao AJ, Nich C, Dhulipala LS, Gibon E, Valladares R, Zwingenberger S, Smith RL, Goodman SB. Local effect of IL-4 delivery on polyethylene particle induced osteolysis in the murine calvarium. J Biomed Mater Res A. 2013;101:1926–34. doi: 10.1002/jbm.a.34486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Pajarinen J, Kouri VP, Jamsen E, Li TF, Mandelin J, Konttinen YT. The response of macrophages to titanium particles is determined by macrophage polarization. Acta Biomater. 2013;9:9229–40. doi: 10.1016/j.actbio.2013.06.027. [DOI] [PubMed] [Google Scholar]
- 177.Pajarinen J, Tamaki Y, Antonios JK, Lin TH, Sato T, Yao Z, Takagi M, Konttinen YT, Goodman SB. Modulation of mouse macrophage polarization in vitro using IL-4 delivery by osmotic pumps. J Biomed Mater Res A. 2015;103:1339–45. doi: 10.1002/jbm.a.35278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Pirris SM, Nottmeier EW, Kimes S, O’Brien M, Rahmathulla G. A retrospective study of iliac crest bone grafting techniques with allograft reconstruction: do patients even know which iliac crest was harvested? Clinical article J Neurosurg Spine. 2014;21:595–600. doi: 10.3171/2014.6.SPINE13902. [DOI] [PubMed] [Google Scholar]
- 179.Grayson WL, Frohlich M, Yeager K, Bhumiratana S, Chan ME, Cannizzaro C, Wan LQ, Liu XS, Guo XE, Vunjak-Novakovic G. Engineering anatomically shaped human bone grafts. Proc Natl Acad Sci U S A. 2010;107:3299–304. doi: 10.1073/pnas.0905439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jing W, Smith AA, Liu B, Li J, Hunter DJ, Dhamdhere G, Salmon B, Jiang J, Cheng D, Johnson CA, Chen S, Lee K, Singh G, Helms JA. Reengineering autologous bone grafts with the stem cell activator WNT3A. Biomaterials. 2015;47:29–40. doi: 10.1016/j.biomaterials.2014.12.014. [DOI] [PubMed] [Google Scholar]
- 181.Kanczler JM, Oreffo RO. Osteogenesis and angiogenesis: the potential for engineering bone. Eur Cell Mater. 2008;15:100–14. doi: 10.22203/ecm.v015a08. [DOI] [PubMed] [Google Scholar]
- 182.Wang M, Zhang G, Wang Y, Liu T, Zhang Y, An Y, Li Y. Crosstalk of mesenchymal stem cells and macrophages promotes cardiac muscle repair. Int J Biochem Cell Biol. 2015;58:53–61. doi: 10.1016/j.biocel.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 183.Spiller KL, Anfang RR, Spiller KJ, Ng J, Nakazawa KR, Daulton JW, Vunjak-Novakovic G. The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials. 2014;35:4477–4488. doi: 10.1016/j.biomaterials.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Spiller KL, Vunjak-Novakovic G. Clinical translation of controlled protein delivery systems for tissue engineering. Drug Deliv Transl Res. 2015;5:101–15. doi: 10.1007/s13346-013-0135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wernike E, Montjovent MO, Liu Y, Wismeijer D, Hunziker EB, Siebenrock KA, Hofstetter W, Klenke FM. VEGF incorporated into calcium phosphate ceramics promotes vascularisation and bone formation in vivo. Eur Cell Mater. 2010;19:30–40. doi: 10.22203/ecm.v019a04. [DOI] [PubMed] [Google Scholar]
- 186.Aro HT, Govender S, Patel AD, Hernigou P, Perera de Gregorio A, Popescu GI, Golden JD, Christensen J, Valentin A. Recombinant human bone morphogenetic protein-2: a randomized trial in open tibial fractures treated with reamed nail fixation. J Bone Joint Surg Am. 2011;93:801–8. doi: 10.2106/JBJS.I.01763. [DOI] [PubMed] [Google Scholar]
- 187.Das A, Tanner S, Barker DA, Green D, Botchwey EA. Delivery of S1P receptor-targeted drugs via biodegradable polymer scaffolds enhances bone regeneration in a critical size cranial defect. J Biomed Mater Res A. 2014;102:1210–8. doi: 10.1002/jbm.a.34779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kaplan JM, Youd ME, Lodie TA. Immunomodulatory activity of mesenchymal stem cells. Curr Stem Cell Res Ther. 2011;6:297–316. doi: 10.2174/157488811797904353. [DOI] [PubMed] [Google Scholar]
- 189.Mikita J, Dubourdieu-Cassagno N, Deloire MS, Vekris A, Biran M, Raffard G, Brochet B, Canron MH, Franconi JM, Boiziau C, Petry KG. Altered M1/M2 activation patterns of monocytes in severe relapsing experimental rat model of multiple sclerosis. Amelioration of clinical status by M2 activated monocyte administration. Mult Scler. 2011;17:2–15. doi: 10.1177/1352458510379243. [DOI] [PubMed] [Google Scholar]