

Abstract
The constitutive anabolism of cancer cells supports proliferation but also addicts tumor cells to a steady influx of exogenous nutrients. Limiting access to metabolic substrates could be an effective and selective means to block cancer growth. In this review, we define the pathways by which cancer cells acquire the raw materials for anabolism, highlight the actionable proteins in each pathway, and discuss the status of therapeutic interventions that disrupt nutrient acquisition. Critical open questions to be answered before apical metabolic inhibitors can be successfully and safely deployed in the clinic are also outlined. In summary, recent studies provide strong support that substrate limitation is a powerful therapeutic strategy to effectively, and safely, starve cancer cells to death.
Keywords: autophagy, cancer, macropinocytosis, metabolism, nutrient transporter, sphingolipid
INTRODUCTION
Developing novel and selective therapies that exploit the metabolic differences between normal and transformed cells is a central goal of the cancer metabolism community. Indeed, many small molecule inhibitors of anabolic enzymes that are conditionally essential in cancer cells have been evaluated as anti-neoplastic agents (reviewed in [1–5]). Some of these compounds have shown promising pre-clinical activity, and a few have been evaluated in clinical trials. However, tumor heterogeneity can limit the efficacy of these targeted agents [6–9]. Anabolic enzyme inhibitors will only be effective in the fraction of tumor cells that carry the oncogenic mutation that creates dependency. Pre-existing insensitive sub-clones that have a distinct mutational landscape can produce resistance. Moreover, agents that are merely cytostatic can lead to the adaptive up-regulation of parallel pathways in the cancer cells that they fail to kill. Acquired compensatory mutations can also contribute to the emergence of refractory disease. In summary, metabolic inhibitors suffer from the same limitations as therapies targeting oncogenic signal transduction cascades [10–13]. Thus, while inhibitors of anabolic enzymes are likely to be valuable therapeutic agents, stable remission or cures will almost certainly require drug combinations that hit both primary and adaptive pathways. A broad palette of anti-metabolic agents with complementary modes of action will be required to design effective combination therapies.
An alternate strategy to anabolic enzyme inhibition is to limit access to the nutrients that supply these enzymes with substrates. Blocking the entry of the nutrients that are conditionally essential in cancer cells would target the metabolic pyramid at its apex, inhibiting downstream anabolic enzymes by limiting their access to substrates. Substrate limitation could be effective alone, but should also increase the effectiveness of drugs that reduce enzymatic activity given their distinct mechanisms of action. Could there be an oncological dependency on nutrients that would provide a therapeutic index for the substrate limitation approach? All rapidly proliferating cells import lipids, sugars, and amino acids from the extracellular space to supply energy and raw materials for the biosynthesis of membranes, nucleic acids, and proteins. Normal cells tolerate periods of reduced nutrient availability by becoming quiescent and catabolic similar to bears that hibernate through the winter. Oncogenic mutations limit the metabolic choices a tumor cell can make, leaving cancer cells at a disadvantage when nutrients become scarce. An example of how effective substrate limitation can be against cancer cells is the success of L-asparaginase, a frontline therapy for acute lymphoblastic leukemia (ALL) [14]. L-asparginase is a bacterial enzyme that de-aminates circulating asparagine, depleting this non-essential amino acid. ALL cells are asparagine auxotrophs because they cannot synthesize sufficient asparagine to meet their anabolic demands. Pediatric ALL outcomes have been vastly improved by the introduction of L-asparaginase with minimal toxicity to normal tissues. However, L-asparaginase is not effective against other tumor types, highlighting that the nutritional preferences of cancer cells vary with tissue of origin and mutational status. Many cancer cells are hypersensitive to glucose and/or glutamine depletion [5,8], but recent pre-clinical studies have revealed that other cancers require an ample supply of exogenous serine [15], glycine [16], arginine [17], or low density lipoprotein (LDL) particles that deliver cholesterol and fatty acids [18]. Thus, drugs that limit nutrient uptake could be valuable weapons to add to the cancer metabolism armamentarium.
As mentioned above, many excellent recent reviews describe how metabolic pathways are re-wired in cancer cells and the efforts to drug key enzymes in these pathways [1–5,8,19]. However, these works include only a cursory discussion of approaches that would restrict access to nutrients. In this review, we will define the pathways cancer cells use to acquire the raw materials for biosynthesis, highlight studies that suggest how these access points might be safely targeted, and discuss the benefits and challenges of developing “apical” metabolic inhibitors that inhibit cancer metabolism well upstream.
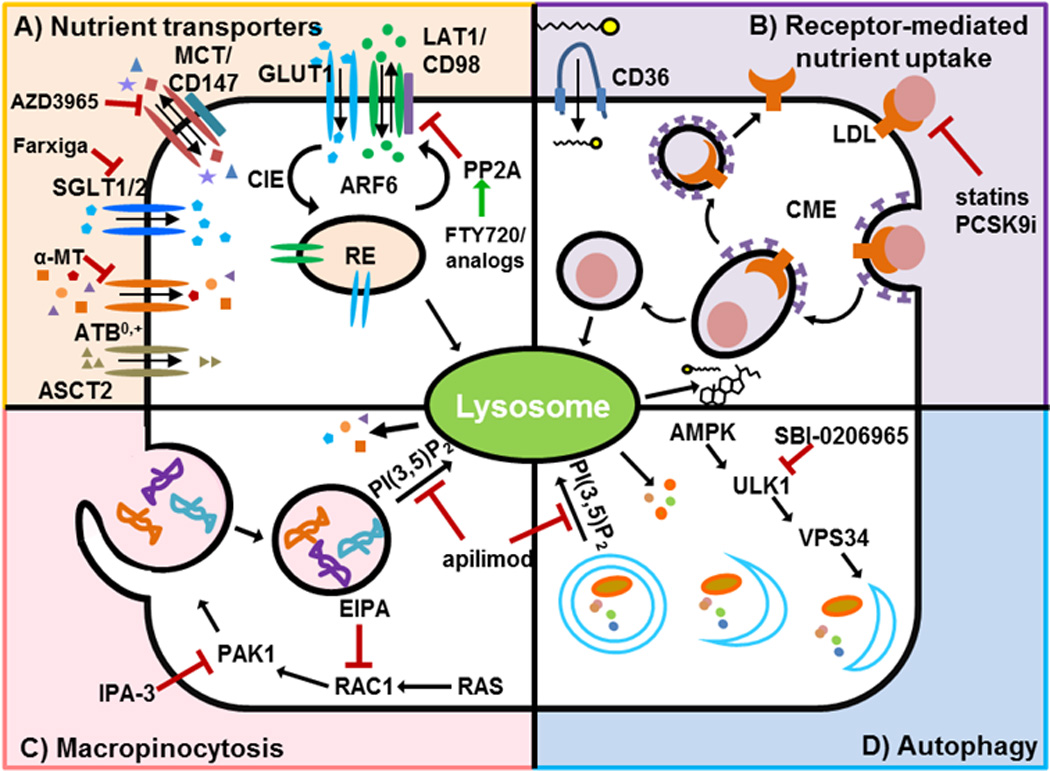
Cell surface nutrient transporters: competitive inhibitors and beyond
Many oncogenic mutations drive nutrient transporter gene expression [20–26]. The post-transcriptional and post-translational mechanisms by which oncogenic mutations orchestrate enhanced nutrient uptake are not as well-studied, but could provide tractable drug targets. Nutrient transporters are internalized through both clathrin-dependent and clathrin-independent endocytosis pathways (CDE and CIE, respectively, Figure 1)[27]. Endocytic vesicles that enter through either pathway can be directed back to the membrane (fast-recycling), routed through the trans-Golgi network for ‘slow’ recycling, or sent to the lysosome for degradation. Endocytic coat proteins, their adaptors, and small GTPase ‘molecular switches’ such as Rab/Arf family members control the destiny of the cargos in these vesicles [28]. Decisions about nutrient transporter endocytosis, recycling, and lysosomal degradation are not static, but rather regulated by signal transduction pathways that respond to growth factors and environmental signals. Understanding how these trafficking decisions are made and de-regulated in cancer cells could expose new ways to target cancer metabolism by attacking the supply wagons. Below, we summarize for each class of surface nutrient transporter with a role in cancer anabolism what is known about the post-transcriptional regulation of their expression and, focusing on recent studies, potential strategies to limit their expression or activity.
Figure 1.
Glucose Transporters
Many cancers and rapidly proliferating cells increase glycolytic flux to provide ATP and biosynthetic intermediates for growth [2,29]. Glucose is a polar compound and therefore requires plasma membrane transporters to enter cells. To date 14 isoforms of the GLUT family of facilitative glucose transporters have been identified that differ in tissue distribution and substrate affinity [30]. GLUT1 (SLC2A1) is often present at high levels on tumor cells, and multiple studies show that it presents a valuable target. For example, although they express a range of glucose transporters, GLUT1 deletion from B-ALL cells is sufficient to block leukemic progression and sensitizes leukemic cells to the ABL kinase inhibitor dasatinib [31]. GLUT1 is also required in solid tumors. Reducing or eliminating GLUT1 expression slows the growth of MMTV-c-ErbB2-driven mammary tumors in vivo, while GLUT1 over-expression accelerates the growth of tumors generated from cells with low levels of this transporter [32]. As partially reducing GLUT1 levels with shRNA reduces both mammary isograft growth [32], head and neck squamous cell carcinoma xenograft growth [33], and murine B16 melanoma metastasis [34], partial inhibition of GLUT1 may be sufficient to limit tumor growth as long as unacceptable toxicity to non-transformed cells that also rely on GLUT1-mediated glucose uptake can be avoided.
Consistent with its central role in glucose uptake and homeostasis, GLUT1 surface expression is regulated post-transcriptionally. Protein kinase C (PKC) activation by phorbol esters leads to phosphorylation of GLUT1 Ser226, promoting both GLUT1 surface expression and glucose uptake [35]. This study is somewhat difficult to reconcile with PKC’s recently described role as a tumor suppressor, but different PKC isoforms or cellular contexts may be important [36]. A tumor suppressor that affects GLUT1 levels in the expected manner is thioredoxin interacting protein (TXNIP) [37]. TXNIP is a member of the α-arrestin family of adaptors that route vesicular traffic [38]. TXNIP interacts directly with GLUT1 to promote internalization [39]. AMPK-dependent TXNIP phosphorylation leads to TXNIP degradation, increasing surface GLUT1 levels. Interestingly, c-Myc binds to and represses the TXNIP promoter in triple-negative breast cancer cells thereby, indirectly, post-transcriptionally increasing GLUT1 surface levels [40]. Her2 expression in breast cancer also reduces TXNIP levels, potentially increasing GLUT1 surface expression [41]. Tumor suppressors are challenging drug targets, but TXNIP might be stabilized in cancer cells by inhibiting AMPK [39]. AMPK has complex roles in tumor cells, but promotes tumor growth in certain contexts [42], perhaps in part through effects on TXNIP. It is possible that, similar to what has been observed with autophagy [43], AMPK has opposing roles in tumor initiation and maintenance and AMPK inhibitors could be useful anti-cancer agents [44].
Other glucose transporters are also up-regulated in tumors. GLUT3 (SLC2A3), normally expressed primarily in neuronal tissues, is over-expressed in glioblastoma [45]. GLUT3 has a higher affinity for glucose than GLUT1 and supports growth in low glucose conditions. Moreover, GLUT3 shRNA reduced brain tumor initiating cell self-renewal and tumorigenic potential in orthotopic xenograft assays, validating that GLUT3 inhibition could be a promising strategy in glioblastoma multiforme, a disease with few treatment options and a poor prognosis. Consistent with this study, nanoparticle-mediated delivery of GLUT3 siRNA reduced GLUT3 mRNA and protein levels and U87MG glioma xenograft growth in a dose-dependent manner [46]. GLUT3 is also ectopically expressed in liver and colorectal cancer where its expression is promoted by AMPK and YAP, a transcription factor that is negatively regulated by the Hippo tumor suppressor pathway [47]. GLUT3 expression is also induced by epithelial mesenchymal transition in non-small cell lung cancer (NSCLC) cells [48]. Interestingly, GLUT3 over-expression in non-malignant human breast cancer epithelial cells conferred malignant phenotypes specifically in 3D culture systems, up-regulating oncogenic signaling, while GLUT3 siRNA caused a phenotypic reversion of malignant breast cancer cells grown in 3D [49]. These studies indicate that GLUT3 may be as important in cancer anabolism as GLUT1.
The glucose analogue 2-deoxy-D-glucose (2-DG) lacks a 2-hydroxyl group and cannot be further metabolized by glycolytic enzymes after it is phosphorylated and trapped in the cell by hexokinase. 2-DG is taken up by GLUTs and is often used to evaluate glucose uptake, both in cancer cells in vitro and in patient tumors in vivo via (18F)-2-deoxy-2-(18F)fluoro-D-glucose positron emission tomography ((18F)-FDG-PET). 2-DG has broad anti-proliferative effects on cancer cells in vitro and in vivo and was evaluated in a number of clinical trials ([31], reviewed in [50]). To be effective, 2-DG must out-compete glucose which is present at millimolar concentrations in the blood; at tolerated doses, 2-DG had no measurable anti-tumor activity [4]. Thus, while GLUTs play a key role in supporting malignant growth and pre-clinical studies strongly suggest that GLUT1 and GLUT3 will be excellent targets for anti-anabolic cancer therapies, new small molecules or gene delivery approaches will be required before GLUTs can be targeted successfully in the clinic. Structures are available for several GLUT family members, including GLUT1 and GLUT3, which could facilitate drug development ([51–53], reviewed in [54,55]). As GLUT1 deficiency leads to a spectrum of neurological symptoms, toxicity may impede the development of GLUT1 inhibitors for cancer patients. However, cancers with gross overexpression of GLUTs and mutations that addict them to glucose metabolism may be exquisitely sensitive to inhibition producing an acceptable therapeutic index.
In addition to the facilitative GLUT family of glucose transporters, the sodium-coupled glucose transporters (SLC5A family, SGLTs) that are normally expressed in the intestine and kidney are also up-regulated in some cancers. Because the Na+ gradient generated by the Na+/K+ ATPase is used to transport glucose, SGLTs can transport glucose against its concentration gradient at the cost of ATP hydrolysis. SGLT1 and SGLT2 expression is elevated prostate and pancreatic tumors where it increases uptake of Me4FDG (Me4FDG is a SGLT but not GLUT substrate, while 2-FDG commonly used for (18F)-FDG-PET is a GLUT but not a SGLT substrate) [56]. An SGLT2 inhibitor FDA-approved for type 2 diabetes, dapagliflozin, inhibited Me4FDG uptake by prostate and pancreatic xenografts in vivo, increasing tumor central necrosis without affecting tumor volume. A second FDA-approved SGLT2 inhibitor, canagliflozin, also increased tumor necrosis and caused a moderate decrease in tumor growth. EGFR associates with and stabilizes surface SGLT1 in prostate cancer cells, and this interaction is sufficient to increase glucose uptake [57]. A follow-up study by the same group found that inhibition of SGLT1 with the non-selective SGLT1 inhibitor phlorizin sensitized prostate cancer cells to EGFR kinase inhibition in vitro [58]. It is possible that the SGLTs play a critical role in poorly perfused areas of solid tumors where glucose levels are low and GLUTs ineffective because active transport is required to import glucose.
Monocarboxylate transporters that import lactate and acetate
The monocarboxylate transporters MCT1 (SLC16A1) and MCT4 (SLC16A3) also contribute to oncogenic anabolism. MCT1 and MCT4 perform proton-coupled transport of pyruvate, lactate, and acetate. MCT1 has a wide tissue distribution, whereas MCT4 expression prevents lactic acid build-up in highly glycolytic cells [59,60] (reviewed in [61]). While MCT4 supports cancer anabolism by exporting lactate to maintain lactate dehydrogenase activity and NAD+ production for glycolysis, MCTs also import anabolic substrates. Although often considered only as a toxic by-product of glycolysis, lactate is an energy-rich molecule that is used as fuel by MCT1-expressing cancer cells [62–64]. Indeed, lactate shuttling coordinated by the expression of different MCTs has been described between tumor cells with glycolytic (export) and oxidative (import) profiles. For example, oxygenated tumor cells that express MCT1 can spare glucose for use by hypoxic cells by oxidizing lactate, creating a symbiotic relationship between tumor cells in perfused and hypoxic areas [64]. Notably, this metabolic symbiosis is also observed between tumor and cancer-associated fibroblasts (CAFs) reprogrammed toward aerobic glycolysis by factors secreted by tumors, a phenomenon coined the “reverse Warburg effect” [65]. Feedback signals from the stroma lead to a reciprocal metabolic shift in the tumor, notably an up-regulation of MCT1, allowing lactate exported by CAFs to be used to support tumor growth in tumor cells [66]. Breaking this lactate cycle via inhibition of MCT1, MCT4, or both may be another way to starve cancer cells for fuel provided that normal cells that rely on MCTs (e.g. red blood cells) can tolerate their inhibition.
While under standard in vitro growth conditions 90% of acetyl CoA is derived from glucose or glutamine, a series of recent studies show that hypoxia and low nutrient conditions increase utilization of exogenous acetate for lipid synthesis and tumor growth [67–70]. Acetate is imported via MCTs, and thus tumors using acetate as a metabolic fuel would also be sensitive to MCT inhibitors. Oxidation of acetate occurs in both orthotopic glioma xenografts and brain metastases of other tumor types with a variety of oncogenic mutations [70]. Given that the primary tumors from which the metastases were derived are not generally 11C-acetate-PET positive, the brain microenvironment may favor the use of acetate as a metabolic fuel. Acetate oxidation was confirmed in glioma patients using isotopic tracing demonstrating that acetate is an important anabolic fuel under physiologic conditions, consistent with 11C-acetate-PET labeling of brain tumors [70,71]. Liver, renal, and prostate tumors as well as some multiple myelomas have been found to be 11C-acetate consumers [72–77]. In summary, acetate is a previously under-appreciated anabolic fuel for cancer cells, particularly under suboptimal growth conditions, and inhibiting acetate import could be a valuable starvation strategy.
MCTs require the chaperone CD147 (basigin) for cell surface expression, and both MCT and CD147 expression correlate with poor patient outcomes in many different cancers [78]. Thus, both MCTs and CD147 could be actionable targets to limit the use of acetate and lactate to fuel cancer growth. Genetic depletion of MCT1, MCT4, and/or CD147 significantly retards growth of a number of different tumor models at least in part by inhibiting glycolysis [63,79–82]. Strategies for targeting tumor lactate metabolism, including uptake, have been recently reviewed [78]. Notably, an MCT1 inhibitor, AZD3965, is currently in clinical trials [NCT01791595, phase 1 for advanced cancers]. Future studies should clarify the tumor types and contexts where acetate and lactate import through MCTs supports biosynthesis, but the clear prediction is that MCT inhibitors will have value as inhibitors of tumor anabolism.
Amino acid transporters that supply anabolism
Like glucose, amino acids cannot cross the lipid bilayer without transporter proteins, and amino acid transporter expression is positively correlated with growth and cancer (reviewed in [55,83]). Given these recent reviews, this section will focus on the most recent reports concerning the amino acid transporters with close connections to cancer anabolism (Figure 1). ASCT2 (SLC1A5) co-transports alanine, serine, cysteine or glutamine and Na+ into the cell, but import is coupled to the export of Na+ and one of these amino acids. ASCT2 thus alters the complement rather than the concentration of amino acids in the cell. As mentioned earlier, the non-essential amino acids serine and glutamine are conditionally essential in some cancers [15,20]. Because glutamine is exchanged for leucine, an activator of mTORC1, ASCT2 can also be required for mTORC1 activation in normal [84] and transformed cells [85]. Consistent with this, elevated ASCT2 protein expression has been documented in leukemias, prostate cancer, breast cancer, melanomas, non-small cell lung cancer and clear-cell renal carcinoma where it is a negative prognostic indicator [86–91]. ASCT2 shRNA inhibits the growth of prostate [91], breast [86], and two neuroblastoma xenografts [92] suggesting that ASCT2 inhibitors [93,94] could be effective in a subset of tumors. Mice with deletions in ACST2 are viable and fertile suggesting that ASCT2 could be targeted in cancer patients with minimal toxicity [67].
Post-translational mechanisms controlling ASCT2 localization and activity have also been uncovered. N-glycosylation is critical for ASCT2 surface localization but does not modulate transport activity [96]. Protein glycosylation is altered in tumor cells [97], in part due to changes in metabolic wiring and glucose availability [98]. As amino acid and other nutrient transporters are glycoproteins, glucose uptake may support the surface expression of other transporters that provide nutrients for growth. Monoubiquitination increases both internalization and lysosomal trafficking of surface proteins [99]. RNF5, a key component of the ER-associated degradation (ERAD) pathway was recently identified ubiquitin ligase for both ASCT2 and the amino acid transporter SNAT2 (SLC38A2) [100]. Eliminating RNF5 promotes ASCT2 expression and reduces differentiation in the MMTV-PyMT mammary tumor model, while RNF5 shRNA accelerates MDA-MB-231 xenograft growth. This study suggests that activation of RNF5 by inducing ER stress, for example with paclitaxel, could be used to decrease ASCT2 expression and starve tumors for glutamine. This work also highlights that interfering with transporter trafficking may actually contribute to the efficacy of drugs already in clinical use.
CD98 (SLC3A2/4F2hc) is also over-expressed in many different cancers, and surface levels are inversely correlated with prognosis [55,101]. Unraveling the role of CD98 in tumor anabolism is somewhat complex due to its multiple roles in cancer cells. Although epidermis-specific deletion of CD98 causes regression of chemically-induced, Ras-dependent tumors, CD98 loss likely limits tumor growth through multiple mechanisms [102]. Surface expression of CD98 is, like ASCT2, negatively regulated by ubiquitination [103], and interfering with CD98 ubiquitination can increase cell proliferation [104]. CD98 functions as a chaperone that is required for surface expression of several integral membrane proteins including LAT1 (SLC7A5), an amino acid transporter that is also over-expressed in cancer cells and negatively correlated with prognosis [105–108]. LAT1 is an obligatory amino acid exchanger and actually exports glutamine, exchanging glutamine imported through ASCT2 for other amino acids including leucine, a key activator of the mTORC1 kinase complex that promotes growth and proliferation [85]. ASCT2/LAT1 targeting makes particular sense in glutamine-dependent tumors such as B-Raf inhibitor-resistant melanomas [88,109]. As LAT1 supplies mTORC1 with amino acids when recruited to lysosomes by LAPTM4b [110], understanding the signals that regulate LAT1 intracellular trafficking will be important to fully appreciate LAT1’s role in tumor anabolism and mTOR activation. LAT1 knockdown slowed prostate cancer cell line growth particularly in combination with enzalutamide (MDV3100), a recently approved anti-androgen therapy [108]. LAT1 shRNA failed to affect NSCLC line NCI-H1299 growth on plastic, but impaired growth on extracellular matrix and in a xenograft model [111]. Both this study and the work with GLUT3 in breast cancer cells demonstrate that evaluating nutrient uptake and dependencies in 3D cultures is important [49]. JPH203, proposed as a more selective inhibitor of LAT1 than BCH, limited lymphoma xenograft growth [112]. Given that JPH203 activates the unfolded protein response and ASCT2 and LAT1 mediated transport are coupled [85], the role of RNF5 [112] in the anti-cancer effects of JPH203 is worth evaluating. Knock-out animals [113] and the capacity to generate knockout cell lines with CRISPR-Cas9 should be a true boon in evaluating the specificity of the multiple agents that decrease amino acid import. LAT1 mediates substrate exchange across the blood brain barrier (BBB) [114]. Thus, disturbing amino acid transport at the BBB may have deleterious effects. Mice conditionally deficient in LAT1 would be valuable in order to assess whether LAT1 could be safely targeted in cancer therapy as deletion of LAT1 in mice results in embryonic lethality [113]. The success of imatinib as a therapy for BCR-Abl-driven leukemias despite the embryonic lethality of Abl deletion [115] supports that LAT1 should not yet be discarded as a potential target.
Like ASCT2, LAT1 and other CD98-associated amino acid transporters are exchangers: rather than concentrating amino acids in cells, they modulate their relative concentrations. ATB0,+ (SLC6A14) in contrast, imports all essential amino acids and glutamine and is highly concentrative due to coupling to transmembrane ion gradients rather than the export of another amino acid [83]. SLC6A14 is expressed at high levels in a subset of tumors including colon, breast, and pancreatic cancer but is only expressed at low levels in normal cells [116]. Indeed, SLC6A14 null mice are viable and fertile, implying that therapeutics interfering with SLC6A14-mediated amino acid transport will be minimally toxic [95]. The SLC6A14-directed inhibitor α-methyl-DL-tryptophan (α-MT) inhibited the growth of an estrogen receptor-positive breast cancer xenograft in vivo [117], while deletion of SLC6A14 dramatically reduced spontaneous tumorigenesis in two different genetically engineered mouse models of breast cancer [95]. SLC6A14 is a particularly promising target due to its broad substrate specificity, concentrating ability, tumor-restricted expression, and demonstrated role in breast cancer models in vivo. Thus, α-MT is another inhibitor of amino acid transport that will benefit from specificity evaluation in knockout models that are now available [95].
Lipid Transport
Rapid proliferation requires the synthesis of membranes, signaling molecules, and lipid modifications required for the activity of many GTPases, including Ras [118]. Lipid requirements can be satisfied by de novo synthesis or via the uptake of exogenous lipoprotein complexes [119]. Fatty acid synthesis in adults is mainly a specialized function of the liver and adipose, tissues that produce and secrete lipid-bound protein complexes for uptake by other organs and tissues. Most normal cells acquire lipids through this mechanism. Some cancer cells, however, synthesize lipids from glucose, glutamine, and acetate [73,77,119]. Metabolic flux experiments with isotopically labeled palmitate demonstrated that cancer cell lines classified as aggressive based on their increased motility, invasiveness, and in vivo growth rates incorporated more exogenous palmitate into structural and signaling lipids than less aggressive cancer cell lines that diverted palmitate towards β-oxidation [120]. This lipid flux analysis suggests that fatty acids may be preferentially used for anabolism in more advanced tumors.
How fatty acids enter cells is incompletely understood [119,121,122]. Fatty acids likely enter by passive diffusion, but a host of surface proteins including CD36, a scavenger receptor with multiple functions, facilitate fatty acid uptake [123]. CD36 contributes to fatty acid import in many cell types but is relatively under-studied in cancer. Insulin signaling promotes CD36 surface localization and long-chain fatty acid uptake [124]. Insulin/IGF-1 signaling has been associated with increased cancer risk, and fatty acid uptake via CD36 could be relevant in certain cancers, particularly those with elevated PI3 kinase signaling. Some studies do link CD36 to cancer progression. In hepatocellular carcinoma, exogenous palmitic acid activated an EMT-like program and induced migration that was decreased by the CD36 inhibitor, sulfo-N-succinimidyl oleate [125]. CD36 expression was also enhanced in a self-renewing, stem-like cell population of glioblastoma, but not in more differentiated counterparts [126]. Glioblastoma cancer stem cells treated with CD36 siRNA formed intracranial tumors in mice more slowly, and low CD36 expression correlated with a slightly better prognosis in patients. While these studies imply a pro-tumorigenic role for CD36 in hepatocellular carcinoma, glioblastoma, and potentially other cancers, interpreting these results is complicated owing to the many functions and wide ligand specificity of CD36. Additional work to determine the extent to which CD36 contributes to cellular fatty acid metabolism and anabolism in cancer is merited.
Growing cells also require cholesterol to generate new cell membranes. By controlling membrane fluidity and lipid raft-mediated signaling, cholesterol can also promote cancer anabolism indirectly. Cholesterol is emulsified into lipoprotein complexes for transport through the blood. Most cholesterol is contained in low-density lipoprotein particles (LDL) and taken up by target cells expressing the LDL receptor (LDLr) (reviewed in [127,128], Figure 1). Ligand binding triggers LDL receptor internalization via CME. Progress through the endosomal system leads to a decline in pH that releases LDL from its receptor, permitting LDL and the LDLr to be diverted into distinct compartments. LDL is directed to the lysosome where it digested releasing cholesterol and fatty acids [129], while the LDLr is usually recycled. Alternatively, surface LDLr that is bound by PCKS9, a serine protease that is secreted by hepatocytes [130], is targeted to the lysosome for degradation. Drugs targeting LDL uptake are widely used clinically to treat hypercholesterolemia [131]. Statins inhibit 3-hydroxy-3-methylgutaryl (HMG)-CoA reductase, the enzyme that catalyzes the rate limiting step of cholesterol synthesis, increasing hepatic LDLr levels and thereby reducing circulating LDL [132]. Consistent with an important role for cholesterol in cancer anabolism, long-term statin use is associated with reduced cancer incidence and improved patient outcomes [131]. PCSK9 inhibitors that have been recently FDA-approved for hypercholesterolemia also work by increasing hepatic LDLr expression [132]. These agents dramatically reduce circulating LDL levels and it will be interesting to see how they affect cancer risk. While cancer cells can synthesize cholesterol, many up-regulate the LDLr and LDL is essential for some cancer cells [18,133]. For example, LDLr expression driven by SREBP1, a transcription factor that is a master regulator of lipid metabolism downstream of PI3 kinase pathway signaling, contributes to glioblastoma growth [134].
Recent work indicates that reducing LDL uptake may be a particularly valuable strategy in prostate cancer, a tumor type often characterized by PI3 kinase pathway activation. Prostate cancer does not exhibit the aerobic glycolysis characteristic of many more rapidly growing cancer classes [73,135,136]. Utilizing label-free spectromicroscopy methods, Yue and colleagues documented a dramatic increase in the accumulation of cholesteryl esters in lipid droplets in malignant prostate relative to normal tissue [18]. Moreover, cholesteryl ester accumulation was tightly correlated with aggressive disease and thus may have prognostic value. Cholesteryl ester accumulation depended on up-regulation of the LDLr following PTEN loss and PI3 kinase activation. Reducing cholesteryl ester accumulation by limiting LDL uptake was sufficient to slow malignant growth in vitro and in vivo, suggesting a potential starvation strategy for prostate cancer. Subsequent studies in PDAC have revealed a similar dependency on LDLr-mediated LDL uptake. shRNA-mediated knockdown of the LDLr disrupts cholesteryl-ester stores and sensitizes PDAC tumors to cytotoxic drugs, while high ldlr expression is negatively correlated with survival in PDAC patients [137]. These recent findings demonstrate that restricting access to exogenous cholesterol by interfering with LDLr-mediated uptake could be effective in multiple difficult to treat cancer classes, particularly in combination with other agents.
Stopping the smorgasbord: is simultaneously blocking multiple nutrient uptake pathways feasible?
As discussed above and shown in Figure 1, cancers depend on transporter-mediated uptake of glucose, acetate, lactate, amino acids, fatty acids, and cholesterol. While inhibition of each of these uptake pathways in isolation slows cancer growth, simultaneously compromising nutrient entry through multiple transporters is likely to be much more effective. Competitive inhibitors of these transporters are generally not very potent, and toxicities limit the use of such agents as exemplified by the poor performance of 2-DG in clinical trials [138]. Innovative strategies to limit transporter-mediated nutrient uptake will be required for combination approaches to be a viable option. One approach would be to target molecules that control the expression of more than one transporter. Similar to unicellular organisms and consistent with requirement for multiple nutrients to grow, nutrient access pathways are coordinately regulated in mammalian cells. For example, Akt or mTOR activation suppresses the lysosomal degradation of glucose and amino acid transporters, LDL receptors, and the transferrin receptor [139]. Precisely how these kinases promote transporter recycling over degradation remains to be defined. As inhibition of the PI3 kinase/Akt/mTOR pathway is generally not sufficient to reduce surface transporter levels (Figure 2), understanding how oncogenic signaling modulates nutrient transporter trafficking could suggest new targeting strategies. Another example of transporter co-regulation comes from a study identifying cargo for the Arf6 GTPase that controls traffic through the CIE pathway [140]. Using a proteomics to identify proteins trapped in endosomes by a constitutively-active Arf6 mutant, the amino acid transporter complex LAT1/CD98, the glucose transporter GLUT1, and the chaperone for monocarboxylate transporters CD147 were all identified as Arf6 cargo. While GTPases like Arf6 are challenging drug targets, drugs targeting GTPases have been developed [141–143]. Whether transporter loss contributes to the anti-neoplastic effects of an existing Arf6 inhibitor, SecinH3, is an open question [144–146].
Figure 2.
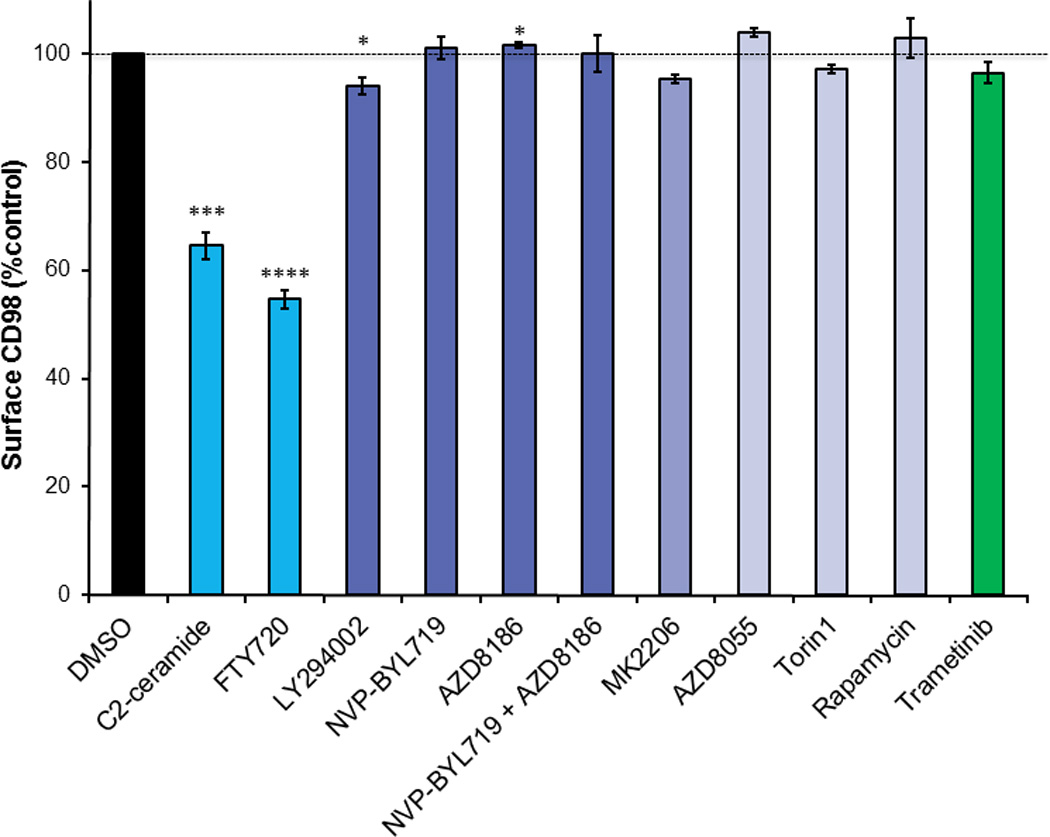
Sphingolipids offer an attractive alternative strategy for coordinate down-regulation of multiple nutrient transporters. These lipids, named for their “enigmatic nature,” reduce surface levels of multiple nutrient transporters by tapping into an evolutionarily conserved mechanism for growth control. In heat stressed yeast, adaptive quiescence is achieved by down-regulating multiple amino acid permeases [147,148]. The mediator of this pro-survival forced starvation is the fungal sphingolipid phytosphingosine [148]. Phytosphingosine produces a transcriptional response that is highly overlapping with nutrient limitation, and preventing permease down-regulation confers resistance to phytosphingosine induced growth inhibition, validating that this sphingolipid phenocopies starvation [149]. While mammalian cells do not produce phytosphingosine, the related sphingolipid ceramide produces a similar response, triggering GLUT1 and CD98 internalization and selectively killing cancer cells through a starvation-like mechanism [150]. Ceramide triggers nutrient transporter loss by directly activating protein phosphatase 2A (PP2A) [150–152]. PP2A is an established tumor suppressor that limits proliferation suggesting that ceramide might have evolved to be an allosteric activator of PP2A–mediated growth arrest in mammalian cells under stress similar to the role of phytosphingosine in yeast [153].
Ceramide itself is extremely hydrophobic and rapidly metabolized in cells making it a poor drug candidate. However, the FDA-approved sphingolipid drug FTY720 phenocopies the effect of phytosphingosine in yeast [149] and ceramide [150] in mammalian cells [154]. FTY720 has excellent pharmacological properties: it is water-soluble and orally bioavailable. Ceramide and FTY720 kill cells at least in part through substrate limitation as the cell-permeable nutrients methyl pyruvate, dimethyl α-ketoglutarate, and dimethyl succinate each protect treated cells from death [149,154]. FTY720-induced nutrient transporter loss is also mediated by protein phosphatase 2A (PP2A) activation [154]. Indeed, other PP2A activators like perphenazine [155] also down-regulate these nutrient transporters although not as potently as FTY720 (unpublished observations). Given that FTY720 induces a starvation-like crisis by down-regulating multiple nutrient transporter proteins, it should be a highly effective apical inhibitor of cancer anabolism. Indeed, FTY720 limits both leukemia and solid tumor growth and metastasis in multiple model systems including breast cancer, bladder cancer, colon cancer, and prostate cancer [156–159]. Unfortunately, although FDA-approved, FTY720 cannot simply be re-purposed for use in cancer patients. At the elevated dose required for anti-cancer activity, FTY720 profoundly reduces heart rate by activating sphingosine-1-phosphate receptors [159–163]. Importantly, FTY720 analogues that do not activate S1P receptors still down-regulate nutrient transporters and kill tumor cells [154,159,164]. The potential of these compounds as cancer cell starvation agents is under evaluation.
Cancer’s big bite: Macropinocytosis supplies nutrients in bulk
As described above, growing cells acquire extracellular nutrients by using nutrient-specific transporter proteins and receptors to move them across the plasma membrane. However, cancer cells can also engulf extracellular fluid relatively non-specifically through macropinocytosis (Figure 1). While a positive correlation between nutrient transporter expression level and cancer progression has long been recognized, the anabolic role of macropinocytosis in cancer cells has only been recently established [165,166]. Activated mutants of the GTPase Ras and the cytosolic tyrosine kinase Src have long been known to trigger macropinocytosis, an actin-dependent process by which cells engulf extracellular material forming large (0.5 – 5.0 µm) vesicles that lack a protein coat [167–171]. The trafficking proteins associated with macropinosomes change rapidly, and there are currently no macropinosome-specific markers. Rather, macropinosomes are detected functionally by measuring the uptake of fluorescently labeled albumin, 70 kD dextran, or 70 kD Ficoll [172,173]. At physiologically relevant concentrations, albumin supports macropinocytosis-dependent proliferation in cells with oncogenic KRAS mutations [165]. Importantly, isotopic tracing experiments confirmed that amino acids derived from extracellular protein were incorporated into central metabolic pathways [165]. As albumin is a fatty acid binding protein, macropinocytosis may also supply Ras-driven cancers with fatty acids for anabolism [129,174]. One caveat of the experiments defining a role for macropinocytosis in cancer cells is that the commonly employed macropinocytosis inhibitor, EIPA, has broad effects on cancer cells apart from its ability to limit macropinocytosis [175]. EIPA inhibits macropinocytosis by blocking the activity of Na+/H+ exchangers, causing a decrease in pH at the plasma membrane. The drop in pH suppresses the Rac1/Cdc42 activation required for membrane ruffling and extension to form macropinosomes [176]. However, given that EIPA blocks cell proliferation through multiple mechanisms unrelated to macropinocytosis and stops cell division even in nutrient replete conditions and in cells not exhibiting macropinocytosis, experiments to conclusively demonstrate that macropinocytosis promotes cancer cell growth will require more specific reagents. While not specific to the macropinocytosis pathway, the Rac1 GTPase and PAK serine threonine kinase are required for the Ras-dependent formation of macropinosomes, and inhibitors of both molecules are available [142,143,177]. In addition, studies thus far have been confined largely to pancreatic cancer cells. Determining how broadly macropinocytosis is used to support cancer anabolism will be an important goal.
The lysosome as a metabolic hub with both anabolic and catabolic functions
The lysosome is commonly thought of as the trash can or recycling bin of the cell. However, the lysosome is required for release of exogenous nutrients from LDL particles and thus plays a key role in anabolism in cancer cells that are dependent on LDL uptake [18,137] (Figure 1). The production of amino acids from extracellular proteins delivered by macropinocytosis also depends on lysosomal degradation [166]. However, oncogenic mutations often reduce lysosomal function [178]. For example, mTORC1 suppresses lysosomal degradation by suppressing the activity of the MiT/TFEB family of transcription factors that promote lysosomal biogenesis and function [179–181]. In keeping with this, mTORC1 inhibitors caused a paradoxical increase in the macropinocytosis-dependent proliferation of Ras-driven pancreatic cancer cells under nutrient stress both in vitro and in vivo [166]. Thus, the disappointing results with rapamycin in human cancer patients may stem in part from its ability to promote lysosomal nutrient generation under certain conditions.
The catabolic functions of the lysosome are much better studied. Autophagy is the adaptive response to nutrient stress, and production of nutrients via autophagy also requires the lysosome (Figure 1). Autophagosome degradation generally promotes survival by promoting the recycling of proteins and organelles into nutrients, also preventing the build-up of damaging reactive oxygen species [43]. In contrast to macropinosome degradation whereby extracellular material is degraded in the lysosome, autophagosome degradation does not increase cell mass but rather recycles cellular constituents that can be used for new purposes, for example sustaining ATP production in starving cells. Consistent with this key role for autophagy in nutrient stressed cells, the MiT/TFE transcription factors that coordinate autophagy–lysosome dynamics are required to maintain intracellular amino acid pools in pancreatic cancer cells [182]. As endocytic nutrient acquisition pathways (LDL degradation and macropinocytosis) and autophagy converge at the lysosome, targeting this organelle would be potent starvation strategy. The FDA-approved anti-malarial agent chloroquine is commonly used as a non-specific inhibitor of autophagy because it blocks lysosomal acidification and thus autophagosome degradation. Although CQ and its derivative HCQ have shown promise in combination with chemotherapies in pre-clinical studies, clinical trials have thus far shown marginal to no benefit in patients, and more refined patient selection strategies or potent inhibitors may be required [183–185]. Moreover, the chemosensitizing effects of CQ in vitro and in pre-clinical models may be at least partially independent of autophagy, as knocking down essential autophagy proteins did not phenocopy the effects of CQ treatment [186]. Thus, the tumor suppressive effects of CQ may stem from the broader role of the lysosome in nutrient generation rather than an oncologic dependency on autophagy (Figure 1). Development of more selective autophagy inhibitors will be critical to clarify the therapeutic potential of autophagy inhibitors in cancer.
The ULK1/2 kinases that initiate autophagosome formation are excellent candidates for such an autophagy-specific inhibitor [187,188]. Compound SBI-0206965 was directly compared to CQ and found to better potentiate the effect of mTORC inhibitors suggesting that ULK1/2 inhibitors may have greater therapeutic value [188]. However, ULK1/2 inhibitors are ill-suited to be apical inhibitors of anabolism as they only impede nutrient recycling via autophagy while leaving nutrient uptake via macropinocytosis and receptor-mediated endocytosis of LDL intact (Figure 1). This may limit their anti-tumor efficacy, particularly in Ras-driven cancers that sustain themselves through LDL uptake [137], macropinocytosis [165,166], and, in some contexts, autophagy (reviewed in [189]). Agents that inhibit all of these lysosomal nutrient generation pathways may already exist. The PIKfyve inhibitor apilimod is being evaluated in clinical trials for auto-immune and inflammatory diseases such as rheumatoid arthritis [190]. PIKfyve is a PI(3)P 5-kinase that modulates lysosomal fusion reactions and Ca2+ release [191]. Although generally well tolerated, apilimod has no demonstrated benefits for rheumatoid arthritis patients [192]. Because PIKfyve is essential for the fusion of both macropinosomes and autophagosomes with the lysosome [193,194] (Figure 1), it could provide a valuable target in cancer patients particularly if combined with other agents that induce nutrient stress. Whether PIKfyve is necessary for LDL degradation has not been tested. Selective inhibitors for the class III PI3 kinase VPS34 also inhibit autophagy and may have similar effects to apilimod as PI3P produced by VPS34 is the substrate for PIKfyve [195]. In summary, evaluating small molecule inhibitors of lysosomal degradation either alone or in combination with other agents merits further investigation as a means to starve cancer cells of both extracellular and intracellular nutrients,
Adapt or die: blocking metabolic adaptations to increase death in nutrient-restricted cancer cells
As mentioned in the introduction, lessons learned from the clinical use of targeted inhibitors of oncogenic signaling make it clear that stable remission or tumor regression are unlikely to be achieved by using targeted metabolic therapies as single agents. Tumor heterogeneity will limit the efficacy of all targeted therapies, and blocking primary and compensatory nutrient access pathways will be required for lasting and broad effects [10–13]. The anti-diabetic biguanide metformin is often discussed as a candidate component of metabolic combination therapies [196–198]. Retrospective studies in cancer patients with diabetes demonstrate a correlation between long-term metformin use and slower disease progression. The anti-cancer effects of metformin have been attributed indirect organismal effects such as reducing circulating insulin levels and also to cancer cell intrinsic effects such as inhibition of mitochondrial complex I and activation of AMPK, a kinase that promotes catabolism over anabolism thereby limiting tumor growth. It is noteworthy that the adaptive increase in oxidative phosphorylation with starvation [199] would be inhibited by metformin. Indeed, limiting glucose is sufficient to sensitize some cancer cells to metformin [200] suggesting that metformin would be useful in combination with starvation agents that block nutrient uptake. As alluded to above, autophagy inhibitors would also block adaptive pathways and be useful in combinations designed to produce nutrient limitation. For example, depletion of the essential autophagy proteins Atg5 or Beclin1 sensitizes leukemia cells to L-asparaginase [90], and an FTY720 analogue that induced transporter loss synergized with the chloroquine to kill patient-derived leukemia cells while leaving normal peripheral blood mononuclear cells unharmed [154]. Indeed, autophagy inhibitors may be a necessary component of any therapeutic strategy to limit exogenous nutrient uptake if tumor regression is the desired endpoint. Combining apical inhibitors that block nutrient access with agents targeting down-stream anabolic enzymes may also be of value.
Food for thought: outstanding questions
Will cancer stem cells have the same metabolic dependencies as “differentiated” cancer cells?
The cancer stem cell (CSC) model hypothesizes that tumors arise from a stem-like population of self-renewing, pluripotent malignant cells [201,202]. CSCs display distinct growth phenotypes and drug sensitivities relative to the bulk tumor and are a potential source of resistant cells. How CSCs will respond to metabolic therapies is an important question to consider. In one recent study, acute myelogenous leukemia initiating cells (LICs) could be sensitized to dietary restriction by AMPK inhibition [203]. AMPK blockade prevented adaptive GLUT1 up-regulation suggesting that targeting GLUT1 could deplete LICs. In another study, leukemia stem cells were found to rely preferentially on oxidative phosphorylation and were unable to switch to glycolysis suggesting that metabolic agents targeting glycolysis would have little effect in this population [204]. In stark contrast, glioblastoma CSCs (termed brain tumor initiating cells, or BTICs) preferentially utilized oxidative metabolism but could readily switch to glycolytic metabolism when mitochondrial function was compromised [205]. Pancreatic CSCs also exhibited an increased dependence on oxidative phosphorylation [206]. Thus, CSCs may rely more on oxidative phosphorylation than their non-stem counterparts in a manner sensitive to tumor context. As metabolic reprogramming regulates stem cell fate and differentiation, CSCs might be forced to differentiate by metabolic therapies. α-ketoglutarate can maintain the undifferentiated state of embryonic stem cells in vitro [207], and alterations in metabolism can block differentiation [208]. Lineage commitment and epigenetic remodeling are also influenced by transporter expression levels in normal stem cells [111,209]. Given that CSC can be rare populations, it will be important to develop tools that can measure metabolic state in few or even in individual cells to test these possibilities. One such technique, NADH FLIM, has been used to monitor the balance of glycolysis and oxidative phosphorylation of stem cells in intestinal crypts [210] and in neural stem cells [211]. NADH FLIM can be performed in vivo, and if stem cells can be marked [212], techniques like NADH FLIM could be used to determine how their metabolism differs from their more differentiated neighbors. Clearly, it will be important to continue to evaluate the effect of starvation-like therapies on both stem and non-stem cancer cells as well as normal stem cell populations.
Will drugs that restrict nutrient access mimic dietary restriction?
Dietary restriction (DR) increases life- and health-span, by conferring protection against cellular stress and inflammation [213]. These benefits of DR may contribute to its ability to prevent cancer initiation, slow metastasis, and promote tumor regression [214,215]. Intermittent fasting schedules may be easier to adhere to than DR regimens yet produce many of the same benefits [216]. A pharmacologic agent that recapitulated these beneficial effects without restricting food intake would be in great demand. Agents that limit nutrient access and thereby increase the effectiveness of less severe DR regimens would also be of value. Therapies that block access to multiple nutrients such as sphingolipids are most likely to mimic DR. Indeed, phytosphingosine, FTY720, and nutrient deprivation produce overlapping transcriptional responses in yeast [149]. The molecular mechanisms underlying the tumor suppressive effects of DR are incompletely defined, but likely include increased resistance to oxidative stress and reductions in circulating growth factors such as IGF-1 and insulin that attenuate PI3 kinase pathway signaling [213,215,217–220]. DR does not suppress the growth of tumors with hyperactive PI3K signaling consistent with the finding that the proliferation of these cells was IGF-1 independent [221]. However, PI3 kinase pathway activation would be unlikely to confer resistance to starvation on a cellular level as PI3 kinase and PTEN loss drive aerobic glycolysis and anabolism that sensitizes cells to nutrient depletion [5,222]. Short term periods of starvation also reduce circulating IGF-1 levels, slow tumor progression [223,224], and protect normal cells from the toxic effects of chemotherapy [225]. As ablation of AMPK was sufficient to sensitize resistant AML leukemia stem cells residing in the hypoxic bone marrow niche to DR [203], suppressing AMPK may enhance the effects of both DR and cellular nutrient uptake inhibitors in cancer patients. As mentioned earlier, AMPK may have a biphasic role in cancer progression and AMPK inhibition may be of value in stressed tumor cells [44].
Will therapies mimicking starvation be safe in cachexic patients?
Cachexia describes the severe loss of muscle and adipose that occurs in 50–80% of late stage cancer patients [226]. This multifactorial syndrome is characterized by inflammatory cytokines and tumor-derived peptides that accelerate substrate mobilization, leading to undesirable weight loss [227,228]. Cancer cachexia is a severe problem that compromises the use of cytotoxic therapy and negatively impacts patient quality of life and survival [229]. There are currently no drugs that reverse the conditions that produce cachexia. Rather, current interventions manage the symptoms rather than neutralize the cause. [229].
At first blush, apical nutrient restriction strategies might seem incompatible with or even likely to induce cachexia. Indeed, clinicians generally encourage patients to increase caloric intake during chemotherapy [215]. However as mentioned above, cachexia is a systemic syndrome driven by inflammation and metabolic imbalances that is not caused by reduced food intake, and does not phenocopy dietary restriction [226]. Reducing glycolysis, and thus lactate production, by substrate limiting tumors may even benefit cachexic patients by interfering with the futile Cori Cycle by which the liver converts tumor lactate into glucose that feeds tumor glycolysis. Given its important role in morbidity and mortality in cancer patients, the effect of therapies that restrict nutrient uptake on whole body metabolism and cancer cachexia will be important to evaluate.
Can agents that affect endocytic trafficking act as single-agent combination therapies?
Thus far, most attempts at targeting cancer metabolism have centered on anabolic enzymes that lie downstream of specific oncogenes. Identifying targets in trafficking pathways might permit simultaneous inactivation of multiple nutrient access pathways (Figure 1). Surprisingly little is known about the proteins that regulate the intracellular trafficking of transporters for key anabolic nutrients. Additional studies in this area could provide exciting new targets – cell biologists likely have much to add to the cancer metabolism field that has so far been dominated by molecular biologists and biochemists. Identifying molecules that are specific to macropinocytosis would be of particular value. This knowledge would provide tools to more accurately define the significance of macropinocytosis in cancer anabolism but could also lead to the development of therapeutic agents that specifically target this nutrient acquisition pathway that is activated in difficult to treat human tumors bearing activated Ras.
How can the therapeutic index of apical metabolic inhibitors be maximized?
DR and intermittent fasting are safe and effective when properly administered. However, reducing food intake too severely or fasting that is too prolonged will have negative effects on the patient as well as the tumor. Apical metabolic inhibitors will need to be titrated to maximize clinical benefit while minimizing negative effects on normal tissues. A better understanding of the degree to which cellular starvation mimics DR in vivo may provide benchmarks that would facilitate optimization of dosing schedules for cellular starvation agents. Moreover, whether intermittent severe nutrient stress (a bad winter) provides a higher therapeutic index than continuous treatment with lower doses (metronomic therapy, chronic lack of a food supply) will be important to test empirically. Intermittent profound starvation seems likely to have the greatest therapeutic index. The hibernation strategy that protects normal cells may only be effective for short periods, while the less robust adaptive strategies available to cancer cells may be sufficient to resist chronic moderate nutrient deprivation of a degree that is tolerated by normal cells.
Concluding Remarks
Many studies cited here demonstrate both the value and feasibility of limiting nutrient access as an alternate approach to blocking anabolism in cancer cells. It is now clear that tumor heterogeneity is a very real obstacle for targeted therapies including those targeting metabolic pathways [6,7]. Compounds that block anabolism at the level of nutrient uptake may limit recurrence if they limit the number of downstream adaptive strategies tumor cells can use to become resistant to therapy. This class of agents may also exhibit broader activity: even if different oncogenic mutations shunt nutrients in different directions, blocking nutrient uptake would still be effective. For example, regardless of whether the carbons and nitrogen in exogenous glutamine end up in α-KG, nucleotides, fatty acids, or pyruvate, blocking glutamine import will curtail the growth of any glutamine-dependent tumor. Of course, understanding how glutamine is utilized in cancers with different oncogenic mutations will be important to develop drug combinations that hit multiple steps in the same pathway, a strategy validated in B-Raf mutant melanomas treated with vemurafenib and MEK or ERK inhibitors and EGFR driven lung tumors [11,109,230]. It is also intriguing to consider applying the concepts of polypharmacology to the cancer metabolism arena [231,232]. Identification of metabolic agents that, like imatinib, hit multiple targets could enhance and broaden anti-neoplastic effects. The ideal starvation agent would inhibit all four nutrient acquisition pathways outlined in Figure 1. By blocking both primary and adaptive pathways, such ideal agents or combinations of agents may produce the sustained responses and tumor regressions that are the goal of all those engaged in developing new cancer therapy regimens.
Table 1.
Selected agents that interfere with nutrient acquisition pathways
| Therapeutic Agent |
Target | Clinical Utility | Cancer | Ref |
|---|---|---|---|---|
| Nutrient Transporters | ||||
| Nano-particle siRNA delivery |
GLUT3 (SLC2A3) |
May be broadly applicable to all transporters |
Glioma | [46] |
| Dapagliflozin (Farxiga) |
SGLT2 (SLC5A2) |
FDA-approved to treat type 2 diabetes | Pancreatic cancer | [56] |
| AZD3965 | MCT1/2 (SLC16A1/ SLC16A7) |
Currently in clinical trials to treat advanced-stage solid tumors (NCT01791595) |
Various cancers | [78, 79, 80] |
| α-methyl-DL- tryptophan |
ATB0,+ (SLC6A14) |
Blocks transport but may not be selective |
ER+ breast cancer | [115] |
| Paclitaxel | ASCT2 (SLC1A5), SNAT2(SLC38A2) |
Widely used cytotoxic agent; induces proteasomal degradation of transporters as a secondary effect |
Breast cancer | [100] |
| FTY720 analogs |
Various transporters |
Down-regulates amino acid and glucose transporters in a PP2A- dependent manner |
Breast cancer Prostate cancer Bladder cancer |
[146, 148, 150– 153] |
| Nutrient Levels | ||||
| L-asparaginase | Asparagine | Frontline therapy for acute lymphoblastic leukemia |
Acute lymphoblastic leukemia |
[14] |
| Arginase/Arginine Deiminase |
Arginine | Pegylated forms of both enzymes have been evaluated in phase I/II trials |
Hepatocellular carcinoma, Melanoma |
[17] |
| Statins | HMG-CoA reductase |
Have anti-proliferative effects in vitro, modest inhibition in vivo In vivo efficacy may depend on reduction of LDL-c |
Breast Cancer | [129] |
| Alirocumab (Praluent) |
PCSK9 | FDA approved PCSK9-neutralizing antibody; increases hepatic surface LDLr to lower LDL-c |
Has not been evaluated |
[128, 130] |
| Macropinocytosis | ||||
| EIPA | NHE-1 | Non-specific inhibition of actin remodeling and membrane extension |
Pancreatic cancer | [159, 169, 170] |
| IPA-3 | Pak1 | Inhibits macropinocytic uptake of certain pathogens |
Has not been evaluated | [172] |
| EHT-1864 | Rac1 | Inhibits Rac1 downstream signaling events including macropinocytosis |
Has not been evaluated |
[171] |
| Autophagy | ||||
| SBI-0206965 | ULK1 | Enhanced starvation and mTOR inhibitor-induced death |
Lung cancer | [182] |
| Lysosomally-Derived Nutients | ||||
| Chloroquine/ Hydroxy- chloroquine |
Lysosomotropic agent |
Disrupts lysosomal fusion degradation; is being evaluated in a number of cancer clinical trials as a single or adjuvant agent |
Various cancers | [177– 179] |
| Apilimod | PIKfyve | Inhibits PI(3,5)P2 formation - required for vesicle-lysosome fusion |
Has not been evaluated |
[187, 188] |
| SAR405 | Vps34 | Selective inhibitor of Vps34 over other PI3 kinases |
Renal cell carcinoma |
[189] |
Acknowledgments
This work was supported by grants to ALE from the NIH (R01 GM089919, R21 CA178230), CDMRP (W81XWH-15-1-0010), the American Cancer Society (RSG-11-111-01-CDD), and the William Lawrence and Blanche Hughes Foundation. ES is supported by Grant Number T32CA009054 from the National Cancer Institute (NCI). SMK was supported by GAANN P200A120207.
REFERENCES
- 1.Tennant DA, Durán RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer. 2010;10:267–277. doi: 10.1038/nrc2817. [DOI] [PubMed] [Google Scholar]
- 2.Hirschey MD, DeBerardinis RJ, Diehl AME, Drew JE, Frezza C, Green MF, Jones LW, Ko YH, Le A, Lea MA, Locasale JW, Longo VD, Lyssiotis CA, McDonnell E, Mehrmohamadi M, Michelotti G, Muralidhar V, Murphy MP, Pedersen PL, Poore B, Raffaghello L, Rathmell JC, Sivanand S, Heiden MG, Vander, Wellen KE, Team TV. Dysregulated metabolism contributes to oncogenesis. Semin. Cancer Biol. 2015;35:S129–S150. doi: 10.1016/j.semcancer.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat. Rev. Drug Discov. 2013;12:829–846. doi: 10.1038/nrd4145. [DOI] [PubMed] [Google Scholar]
- 4.Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat. Rev. Drug Discov. 2011;10:671–684. doi: 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- 5.DeNicola GM, Cantley LC. Cancer’s Fuel Choice: New Flavors for a Picky Eater. Mol. Cell. 2015;60:514–523. doi: 10.1016/j.molcel.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McGranahan N, Swanton C. Biological and Therapeutic Impact of Intratumor Heterogeneity in Cancer Evolution. Cancer Cell. 2015;27:15–26. doi: 10.1016/j.ccell.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Hu J, Locasale JW, Bielas JH, O’Sullivan J, Sheahan K, Cantley LC, Vander Heiden MG, Vitkup D. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat. Biotechnol. 2013;31:522–529. doi: 10.1038/nbt.2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015;17:351–359. doi: 10.1038/ncb3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suvà ML, Regev A, Bernstein BE. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garraway LA, Jänne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012;2:214–226. doi: 10.1158/2159-8290.CD-12-0012. [DOI] [PubMed] [Google Scholar]
- 11.Chong CR, Jänne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013;19:1389–1400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2013;19:1401–1409. doi: 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- 13.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 14.Hunger SP, Mullighan CG. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015;373:1541–1552. doi: 10.1056/NEJMra1400972. [DOI] [PubMed] [Google Scholar]
- 15.Maddocks ODK, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, Vousden KH. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature. 2013;493:542–546. doi: 10.1038/nature11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, Mootha VK. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040–1044. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feun LG, Kuo MT, Savaraj N. Arginine deprivation in cancer therapy. Curr. Opin. Clin. Nutr. Metab. Care. 2015;18:78–82. doi: 10.1097/MCO.0000000000000122. [DOI] [PubMed] [Google Scholar]
- 18.Yue S, Li J, Lee S-Y, Lee HJ, Shao T, Song B, Cheng L, Masterson TA, Liu X, Ratliff TL, Cheng J-X. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014;19:393–406. doi: 10.1016/j.cmet.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang X-Y, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. U. S. A. 2008;105:18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JKV, Markowitz S, Zhou S, Diaz LA, Velculescu VE, Lengauer C, Kinzler KW, Vogelstein B, Papadopoulos N. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–1559. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Rourke JF, Pugh CW, Bartlett SM, Ratcliffe PJ. Identification of hypoxically inducible mRNAs in HeLa cells using differential-display PCR. Role of hypoxia-inducible factor-1. Eur. J. Biochem. 1996;241:403–410. doi: 10.1111/j.1432-1033.1996.00403.x. [DOI] [PubMed] [Google Scholar]
- 23.Flier J, Mueckler M, Usher P, Lodish H. Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science (80−.) 1987;235:1492–1495. doi: 10.1126/science.3103217. [DOI] [PubMed] [Google Scholar]
- 24.Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, de Boer VCJ, Anastasiou D, Ito K, Sasaki AT, Rameh L, Carracedo A, Vander Heiden MG, Cantley LC, Pinton P, Haigis MC, Pandolfi PP. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell. 2012;149:49–62. doi: 10.1016/j.cell.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shroff EH, Eberlin LS, Dang VM, Gouw AM, Gabay M, Adam SJ, Bellovin DI, Tran PT, Philbrick WM, Garcia-Ocana A, Casey SC, Li Y, Dang CV, Zare RN, Felsher DW. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl. Acad. Sci. U. S. A. 2015;112:6539–6544. doi: 10.1073/pnas.1507228112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, Viale A, Zheng H, Paik J, Lim C, Guimaraes AR, Martin ES, Chang J, Hezel AF, Perry SR, Hu J, Gan B, Xiao Y, Asara JM, Weissleder R, Wang YA, Chin L, Cantley LC, DePinho RA. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009;10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maldonado-Báez L, Williamson C, Donaldson JG. Clathrin-independent endocytosis: a cargo-centric view. Exp. Cell Res. 2013;319:2759–2769. doi: 10.1016/j.yexcr.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol. Aspects Med. 34:121–138. doi: 10.1016/j.mam.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu T, Kishton RJ, Macintyre AN, Gerriets VA, Xiang H, Liu X, Abel ED, Rizzieri D, Locasale JW, Rathmell JC. Glucose transporter 1-mediated glucose uptake is limiting for B-cell acute lymphoblastic leukemia anabolic metabolism and resistance to apoptosis. Cell Death Dis. 2014;5:e1470. doi: 10.1038/cddis.2014.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Young CD, Lewis AS, Rudolph MC, Ruehle MD, Jackman MR, Yun UJ, Ilkun O, Pereira R, Abel ED, Anderson SM. Modulation of glucose transporter 1 (GLUT1) expression levels alters mouse mammary tumor cell growth in vitro and in vivo. PLoS One. 2011;6:e23205. doi: 10.1371/journal.pone.0023205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li S, Yang X, Wang P, Ran X. The effects of GLUT1 on the survival of head and neck squamous cell carcinoma. Cell. Physiol. Biochem. 2013;32:624–634. doi: 10.1159/000354466. [DOI] [PubMed] [Google Scholar]
- 34.Koch A, Lang SA, Wild PJ, Gantner S, Mahli A, Spanier G, Berneburg M, Müller M, Bosserhoff AK, Hellerbrand C. Glucose transporter isoform 1 expression enhances metastasis of malignant melanoma cells. Oncotarget. 2015;6:32748–32760. doi: 10.18632/oncotarget.4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee EE, Ma J, Sacharidou A, Mi W, Salato VK, Nguyen N, Jiang Y, Pascual JM, North PE, Shaul PW, Mettlen M, Wang RC. A Protein Kinase C Phosphorylation Motif in GLUT1 Affects Glucose Transport and is Mutated in GLUT1 Deficiency Syndrome. Mol. Cell. 2015;58:845–853. doi: 10.1016/j.molcel.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, Trotter EW, Gallegos LL, Miller CJ, Furnari FB, Hunter T, Brognard J, Newton AC. Cancer-associated protein kinase C mutations reveal kinase’s role as tumor suppressor. Cell. 2015;160:489–502. doi: 10.1016/j.cell.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morrison JA, Pike LA, Sams SB, Sharma V, Zhou Q, Severson JJ, Tan A-C, Wood WM, Haugen BR. Thioredoxin interacting protein (TXNIP) is a novel tumor suppressor in thyroid cancer. Mol. Cancer. 2014;13:62. doi: 10.1186/1476-4598-13-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puca L, Brou C. Α-arrestins - new players in Notch and GPCR signaling pathways in mammals. J. Cell Sci. 2014;127:1359–1367. doi: 10.1242/jcs.142539. [DOI] [PubMed] [Google Scholar]
- 39.Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, Shen C-H, Wen J, Asara J, McGraw TE, Kahn BB, Cantley LC. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell. 2013;49:1167–1175. doi: 10.1016/j.molcel.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen L, O’Shea JM, Kaadige MR, Cunha S, Wilde BR, Cohen AL, Welm AL, Ayer DE. Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proc. Natl. Acad. Sci. U. S. A. 2015;112:5425–5430. doi: 10.1073/pnas.1501555112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nie W, Huang W, Zhang W, Xu J, Song W, Wang Y, Zhu A, Luo J, Huang G, Wang Y, Guan X. TXNIP interaction with the Her-1/2 pathway contributes to overall survival in breast cancer. Oncotarget. 2015;6:3003–3012. doi: 10.18632/oncotarget.3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zadra G, Batista JL, Loda M. Dissecting the Dual Role of AMPK in Cancer: From Experimental to Human Studies. Mol. Cancer Res. 2015;13:1059–1072. doi: 10.1158/1541-7786.MCR-15-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.White E. The role for autophagy in cancer. J. Clin. Invest. 2015;125:42–46. doi: 10.1172/JCI73941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Faubert B, Vincent EE, Poffenberger MC, Jones RG. The AMP-activated protein kinase (AMPK) and cancer: many faces of a metabolic regulator. Cancer Lett. 2015;356:165–170. doi: 10.1016/j.canlet.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 45.Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, Weil RJ, Nakano I, Sarkaria JN, Stringer BW, Day BW, Li M, Lathia JD, Rich JN, Hjelmeland AB. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013;16:1373–1382. doi: 10.1038/nn.3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu C-F, Liu Y, Shen S, Zhu Y-H, Wang J. Targeting glucose uptake with siRNA-based nanomedicine for cancer therapy. Biomaterials. 2015;51:1–11. doi: 10.1016/j.biomaterials.2015.01.068. [DOI] [PubMed] [Google Scholar]
- 47.Wang W, Xiao Z-D, Li X, Aziz KE, Gan B, Johnson RL, Chen J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 2015;17:490–499. doi: 10.1038/ncb3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masin M, Vazquez J, Rossi S, Groeneveld S, Samson N, Schwalie PC, Deplancke B, Frawley LE, Gouttenoire J, Moradpour D, Oliver TG, Meylan E. GLUT3 is induced during epithelial-mesenchymal transition and promotes tumor cell proliferation in non-small cell lung cancer. Cancer Metab. 2014;2:11. doi: 10.1186/2049-3002-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Onodera Y, Nam J-M, Bissell MJ. Increased sugar uptake promotes oncogenesis via EPAC/RAP1 and O-GlcNAc pathways. J. Clin. Invest. 2014;124:367–384. doi: 10.1172/JCI63146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pelicano H, Martin DS, Xu R-H, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25:4633–4646. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- 51.Nomura N, Verdon G, Kang HJ, Shimamura T, Nomura Y, Sonoda Y, Hussien SA, Qureshi AA, Coincon M, Sato Y, Abe H, Nakada-Nakura Y, Hino T, Arakawa T, Kusano-Arai O, Iwanari H, Murata T, Kobayashi T, Hamakubo T, Kasahara M, Iwata S, Drew D. Structure and mechanism of the mammalian fructose transporter GLUT5. Nature. 2015;526:397–401. doi: 10.1038/nature14909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deng D, Xu C, Sun P, Wu J, Yan C, Hu M, Yan N. Crystal structure of the human glucose transporter GLUT1. Nature. 2014;510:121–125. doi: 10.1038/nature13306. [DOI] [PubMed] [Google Scholar]
- 53.Deng D, Sun P, Yan C, Ke M, Jiang X, Xiong L, Ren W, Hirata K, Yamamoto M, Fan S, Yan N. Molecular basis of ligand recognition and transport by glucose transporters. Nature. 2015;526:391–396. doi: 10.1038/nature14655. [DOI] [PubMed] [Google Scholar]
- 54.Deng D, Yan N. GLUT, SGLT, and SWEET: Structural and mechanistic investigations of the glucose transporters. Protein Sci. 2015 doi: 10.1002/pro.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCracken AN, Edinger AL. Nutrient transporters: the Achilles’ heel of anabolism. Trends Endocrinol. Metab. 2013;24:200–208. doi: 10.1016/j.tem.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scafoglio C, Hirayama BA, Kepe V, Liu J, Ghezzi C, Satyamurthy N, Moatamed NA, Huang J, Koepsell H, Barrio JR, Wright EM. Functional expression of sodium-glucose transporters in cancer. Proc. Natl. Acad. Sci. U. S. A. 2015;112:E4111–E4119. doi: 10.1073/pnas.1511698112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weihua Z, Tsan R, Huang W-C, Wu Q, Chiu C-H, Fidler IJ, Hung M-C. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell. 2008;13:385–393. doi: 10.1016/j.ccr.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ren J, Bollu LR, Su F, Gao G, Xu L, Huang W-C, Hung M-C, Weihua Z. EGFR-SGLT1 interaction does not respond to EGFR modulators, but inhibition of SGLT1 sensitizes prostate cancer cells to EGFR tyrosine kinase inhibitors. Prostate. 2013;73:1453–1461. doi: 10.1002/pros.22692. [DOI] [PubMed] [Google Scholar]
- 59.Ullah MS, Davies AJ, Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006;281:9030–9037. doi: 10.1074/jbc.M511397200. [DOI] [PubMed] [Google Scholar]
- 60.Baek G, Tse YF, Hu Z, Cox D, Buboltz N, McCue P, Yeo CJ, White MA, DeBerardinis RJ, Knudsen ES, Witkiewicz AK. MCT4 defines a glycolytic subtype of pancreatic cancer with poor prognosis and unique metabolic dependencies. Cell Rep. 2014;9:2233–2249. doi: 10.1016/j.celrep.2014.11.025. [DOI] [PubMed] [Google Scholar]
- 61.Pinheiro C, Longatto-Filho A, Azevedo-Silva J, Casal M, Schmitt FC, Baltazar F. Role of monocarboxylate transporters in human cancers: state of the art. J. Bioenerg. Biomembr. 2012;44:127–139. doi: 10.1007/s10863-012-9428-1. [DOI] [PubMed] [Google Scholar]
- 62.Semenza GL. Tumor metabolism: cancer cells give and take lactate. J. Clin. Invest. 2008;118:3835–3837. doi: 10.1172/JCI37373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kennedy KM, Scarbrough PM, Ribeiro A, Richardson R, Yuan H, Sonveaux P, Landon CD, Chi J-T, Pizzo S, Schroeder T, Dewhirst MW. Catabolism of exogenous lactate reveals it as a legitimate metabolic substrate in breast cancer. PLoS One. 2013;8:e75154. doi: 10.1371/journal.pone.0075154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, Wahl ML, Feron O, Dewhirst MW. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Invest. 2008;118:3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 66.Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, Lanciotti M, Serni S, Cirri P, Chiarugi P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012;72:5130–5140. doi: 10.1158/0008-5472.CAN-12-1949. [DOI] [PubMed] [Google Scholar]
- 67.Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K, McGarry L, James D, Shanks E, Kalna G, Saunders RE, Jiang M, Howell M, Lassailly F, Thin MZ, Spencer-Dene B, Stamp G, van den Broek NJF, Mackay G, Bulusu V, Kamphorst JJ, Tardito S, Strachan D, Harris AL, Aboagye EO, Critchlow SE, Wakelam MJO, Schulze A, Gottlieb E. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 2015;27:57–71. doi: 10.1016/j.ccell.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kamphorst JJ, Chung MK, Fan J, Rabinowitz JD. Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab. 2014;2:23. doi: 10.1186/2049-3002-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, Walters H, Tantawy MN, Fu A, Manning HC, Horton JD, Hammer RE, McKnight SL, Tu BP. Acetate dependence of tumors. Cell. 2014;159:1591–1602. doi: 10.1016/j.cell.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, Huang Z, Barnett S, Mickey BE, DeBerardinis RJ, Tu BP, Maher EA, Bachoo RM. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell. 2014;159:1603–1614. doi: 10.1016/j.cell.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yamamoto Y, Nishiyama Y, Kimura N, Kameyama R, Kawai N, Hatakeyama T, Kaji M, Ohkawa M. 11C-acetate PET in the evaluation of brain glioma: comparison with 11C-methionine and 18F-FDG-PET. Mol. Imaging Biol. 2008;10:281–287. doi: 10.1007/s11307-008-0152-5. [DOI] [PubMed] [Google Scholar]
- 72.Grassi I, Nanni C, Allegri V, Morigi JJ, Montini GC, Castellucci P, Fanti S. The clinical use of PET with (11)C-acetate. Am. J. Nucl. Med. Mol. Imaging. 2012;2:33–47. [PMC free article] [PubMed] [Google Scholar]
- 73.Zadra G, Photopoulos C, Loda M. The fat side of prostate cancer. Biochim. Biophys. Acta. 2013;1831:1518–1532. doi: 10.1016/j.bbalip.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ho C, Chen S, Leung YL, Cheng T, Wong K, Cheung SK, Liang R, Chim CS. 11C-acetate PET/CT for metabolic characterization of multiple myeloma: a comparative study with 18F-FDG PET/CT. J. Nucl. Med. 2014;55:749–752. doi: 10.2967/jnumed.113.131169. [DOI] [PubMed] [Google Scholar]
- 75.Oyama N, Akino H, Kanamaru H, Suzuki Y, Muramoto S, Yonekura Y, Sadato N, Yamamoto K, Okada K. 11C-Acetate PET Imaging of Prostate Cancer. J. Nucl. Med. 2002;43:181–186. [PubMed] [Google Scholar]
- 76.Ho C-L, Yu SCH, Yeung DWC. 11C-Acetate PET Imaging in Hepatocellular Carcinoma and Other Liver Masses. J. Nucl. Med. 2003;44:213–221. [PubMed] [Google Scholar]
- 77.Yoshii Y, Furukawa T, Saga T, Fujibayashi Y. Acetate/acetyl-CoA metabolism associated with cancer fatty acid synthesis: overview and application. Cancer Lett. 2015;356:211–216. doi: 10.1016/j.canlet.2014.02.019. [DOI] [PubMed] [Google Scholar]
- 78.Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J. Clin. Invest. 2013;123:3685–3692. doi: 10.1172/JCI69741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Marchiq I, Le Floch R, Roux D, Simon M-P, Pouyssegur J. Genetic disruption of lactate/H+ symporters (MCTs) and their subunit CD147/BASIGIN sensitizes glycolytic tumor cells to phenformin. Cancer Res. 2015;75:171–180. doi: 10.1158/0008-5472.CAN-14-2260. [DOI] [PubMed] [Google Scholar]
- 80.Polański R, Hodgkinson CL, Fusi A, Nonaka D, Priest L, Kelly P, Trapani F, Bishop PW, White A, Critchlow SE, Smith PD, Blackhall F, Dive C, Morrow CJ. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin. Cancer Res. 2014;20:926–937. doi: 10.1158/1078-0432.CCR-13-2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Le Floch R, Chiche J, Marchiq I, Naiken T, Naïken T, Ilc K, Ilk K, Murray CM, Critchlow SE, Roux D, Simon M-P, Pouysségur J. CD147 subunit of lactate/H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proc. Natl. Acad. Sci. U. S. A. 2011;108:16663–16668. doi: 10.1073/pnas.1106123108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lim KS, Lim KJ, Price AC, Orr BA, Eberhart CG, Bar EE. Inhibition of monocarboxylate transporter-4 depletes stem-like glioblastoma cells and inhibits HIF transcriptional response in a lactate-independent manner. Oncogene. 2014;33:4433–4441. doi: 10.1038/onc.2013.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bhutia YD, Babu E, Ramachandran S, Ganapathy V. Amino Acid transporters in cancer and their relevance to “glutamine addiction”: novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015;75:1782–1788. doi: 10.1158/0008-5472.CAN-14-3745. [DOI] [PubMed] [Google Scholar]
- 84.Nakaya M, Xiao Y, Zhou X, Chang J-H, Chang M, Cheng X, Blonska M, Lin X, Sun S-C. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. 2014;40:692–705. doi: 10.1016/j.immuni.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, MacKeigan JP, Porter JA, Wang YK, Cantley LC, Finan PM, Murphy LO. Bidirectional Transport of Amino Acids Regulates mTOR and Autophagy. Cell. 2009;136:521–534. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van Geldermalsen M, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D, Ritchie W, Feng Y, Bailey CG, Deng N, Harvey K, Beith JM, Selinger CI, O’Toole SA, Rasko JEJ, Holst J. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene. 2015 doi: 10.1038/onc.2015.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu Y, Yang L, An H, Chang Y, Zhang W, Zhu Y, Xu L, Xu J. High expression of Solute Carrier Family 1, member 5 (SLC1A5) is associated with poor prognosis in clear-cell renal cell carcinoma. Sci. Rep. 2015;5:16954. doi: 10.1038/srep16954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang Q, Beaumont KA, Otte NJ, Font J, Bailey CG, van Geldermalsen M, Sharp DM, Tiffen JC, Ryan RM, Jormakka M, Haass NK, Rasko JEJ, Holst J. Targeting glutamine transport to suppress melanoma cell growth. Int. J. Cancer. 2014;135:1060–1071. doi: 10.1002/ijc.28749. [DOI] [PubMed] [Google Scholar]
- 89.Shimizu K, Kaira K, Tomizawa Y, Sunaga N, Kawashima O, Oriuchi N, Tominaga H, Nagamori S, Kanai Y, Yamada M, Oyama T, Takeyoshi I. ASC amino-acid transporter 2 (ASCT2) as a novel prognostic marker in non-small cell lung cancer. Br. J. Cancer. 2014;110:2030–2039. doi: 10.1038/bjc.2014.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Willems L, Jacque N, Jacquel A, Neveux N, Maciel TT, Lambert M, Schmitt A, Poulain L, Green AS, Uzunov M, Kosmider O, Radford-Weiss I, Moura IC, Auberger P, Ifrah N, Bardet V, Chapuis N, Lacombe C, Mayeux P, Tamburini J, Bouscary D. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood. 2013;122:3521–3532. doi: 10.1182/blood-2013-03-493163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang Q, Hardie R-A, Hoy AJ, van Geldermalsen M, Gao D, Fazli L, Sadowski MC, Balaban S, Schreuder M, Nagarajah R, Wong JJ-L, Metierre C, Pinello N, Otte NJ, Lehman ML, Gleave M, Nelson CC, Bailey CG, Ritchie W, Rasko JEJ, Holst J. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015;236:278–289. doi: 10.1002/path.4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ren P, Yue M, Xiao D, Xiu R, Gan L, Liu H, Qing G. ATF4 and N-Myc coordinate glutamine metabolism in MYCN-amplified neuroblastoma cells through ASCT2 activation. J. Pathol. 2015;235:90–100. doi: 10.1002/path.4429. [DOI] [PubMed] [Google Scholar]
- 93.Colas C, Grewer C, Otte NJ, Gameiro A, Albers T, Singh K, Shere H, Bonomi M, Holst J, Schlessinger A. Ligand Discovery for the Alanine-Serine-Cysteine Transporter (ASCT2, SLC1A5) from Homology Modeling and Virtual Screening. PLoS Comput. Biol. 2015;11:e1004477. doi: 10.1371/journal.pcbi.1004477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schulte ML, Dawson ES, Saleh SA, Cuthbertson ML, Manning HC. 2-Substituted Nγ-glutamylanilides as novel probes of ASCT2 with improved potency. Bioorg. Med. Chem. Lett. 2015;25:113–116. doi: 10.1016/j.bmcl.2014.10.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Babu E, Bhutia YD, Ramachandran S, Gnanaprakasam JP, Prasad PD, Thangaraju M, Ganapathy V. Deletion of the amino acid transporter Slc6a14 suppresses tumour growth in spontaneous mouse models of breast cancer. Biochem. J. 2015;469:17–23. doi: 10.1042/BJ20150437. [DOI] [PubMed] [Google Scholar]
- 96.Console L, Scalise M, Tarmakova Z, Coe IR, Indiveri C. N-linked glycosylation of human SLC1A5 (ASCT2) transporter is critical for trafficking to membrane. Biochim. Biophys. Acta. 2015;1853:1636–1645. doi: 10.1016/j.bbamcr.2015.03.017. [DOI] [PubMed] [Google Scholar]
- 97.Christiansen MN, Chik J, Lee L, Anugraham M, Abrahams JL, Packer NH. Cell surface protein glycosylation in cancer. Proteomics. 2014;14:525–546. doi: 10.1002/pmic.201300387. [DOI] [PubMed] [Google Scholar]
- 98.Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nat. Rev. Mol. Cell Biol. 2012;13:270–276. doi: 10.1038/nrm3305. [DOI] [PubMed] [Google Scholar]
- 99.Hicke L, Dunn R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu. Rev. Cell Dev. Biol. 2003;19:141–172. doi: 10.1146/annurev.cellbio.19.110701.154617. [DOI] [PubMed] [Google Scholar]
- 100.Jeon YJ, Khelifa S, Ratnikov B, Scott DA, Feng Y, Parisi F, Ruller C, Lau E, Kim H, Brill LM, Jiang T, Rimm DL, Cardiff RD, Mills GB, Smith JW, Osterman AL, Kluger Y, Ronai ZA. Regulation of glutamine carrier proteins by RNF5 determines breast cancer response to ER stress-inducing chemotherapies. Cancer Cell. 2015;27:354–369. doi: 10.1016/j.ccell.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Edinger AL. Controlling cell growth and survival through regulated nutrient transporter expression. Biochem. J. 2007;406:1–12. doi: 10.1042/BJ20070490. [DOI] [PubMed] [Google Scholar]
- 102.Estrach S, Lee S-A, Boulter E, Pisano S, Errante A, Tissot FS, Cailleteau L, Pons C, Ginsberg MH, Féral CC. CD98hc (SLC3A2) loss protects against ras-driven tumorigenesis by modulating integrin-mediated mechanotransduction. Cancer Res. 2014;74:6878–6889. doi: 10.1158/0008-5472.CAN-14-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Eyster CA, Cole NB, Petersen S, Viswanathan K, Früh K, Donaldson JG. MARCH ubiquitin ligases alter the itinerary of clathrin-independent cargo from recycling to degradation. Mol. Biol. Cell. 2011;22:3218–3230. doi: 10.1091/mbc.E10-11-0874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ablack JNG, Metz PJ, Chang JT, Cantor JM, Ginsberg MH. Ubiquitylation of CD98 limits cell proliferation and clonal expansion. J. Cell Sci. 2015;128:4273–4278. doi: 10.1242/jcs.178129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Isoda A, Kaira K, Iwashina M, Oriuchi N, Tominaga H, Nagamori S, Kanai Y, Oyama T, Asao T, Matsumoto M, Sawamura M. Expression of L-type amino acid transporter 1 (LAT1) as a prognostic and therapeutic indicator in multiple myeloma. Cancer Sci. 2014;105:1496–1502. doi: 10.1111/cas.12529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yazawa T, Shimizu K, Kaira K, Nagashima T, Ohtaki Y, Atsumi J, Obayashi K, Nagamori S, Kanai Y, Oyama T, Takeyoshi I. Clinical significance of coexpression of L-type amino acid transporter 1 (LAT1) and ASC amino acid transporter 2 (ASCT2) in lung adenocarcinoma. Am. J. Transl. Res. 2015;7:1126–1139. [PMC free article] [PubMed] [Google Scholar]
- 107.Kaira K, Nakamura K, Hirakawa T, Imai H, Tominaga H, Oriuchi N, Nagamori S, Kanai Y, Tsukamoto N, Oyama T, Asao T, Minegishi T. Prognostic significance of L-type amino acid transporter 1 (LAT1) expression in patients with ovarian tumors. Am. J. Transl. Res. 2015;7:1161–1171. [PMC free article] [PubMed] [Google Scholar]
- 108.Xu M, Sakamoto S, Matsushima J, Kimura T, Ueda T, Mizokami A, Kanai Y, Ichikawa T. Upregulation of LAT1 during anti-androgen therapy contribute to progression in prostate cancer cells. J. Urol. 2015 doi: 10.1016/j.juro.2015.11.071. [DOI] [PubMed] [Google Scholar]
- 109.Hernandez-Davies JE, Tran TQ, Reid MA, Rosales KR, Lowman XH, Pan M, Moriceau G, Yang Y, Wu J, Lo RS, Kong M. Vemurafenib resistance reprograms melanoma cells towards glutamine dependence. J. Transl. Med. 2015;13:210. doi: 10.1186/s12967-015-0581-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Milkereit R, Persaud A, Vanoaica L, Guetg A, Verrey F, Rotin D. LAPTM4b recruits the LAT1-4F2hc Leu transporter to lysosomes and promotes mTORC1 activation. Nat. Commun. 2015;6:7250. doi: 10.1038/ncomms8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dann SG, Ryskin M, Barsotti AM, Golas J, Shi C, Miranda M, Hosselet C, Lemon L, Lucas J, Karnoub M, Wang F, Myers JS, Garza SJ, Follettie MT, Geles KG, Klippel A, Rollins RA, Fantin VR. Reciprocal regulation of amino acid import and epigenetic state through Lat1 and EZH2. EMBO J. 2015;34:1773–1785. doi: 10.15252/embj.201488166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rosilio C, Nebout M, Imbert V, Griessinger E, Neffati Z, Benadiba J, Hagenbeek T, Spits H, Reverso J, Ambrosetti D, Michiels J-F, Bailly-Maitre B, Endou H, Wempe MF, Peyron J-F. L-type amino-acid transporter 1 (LAT1): a therapeutic target supporting growth and survival of T-cell lymphoblastic lymphoma/T-cell acute lymphoblastic leukemia. Leukemia. 2015;29:1253–1266. doi: 10.1038/leu.2014.338. [DOI] [PubMed] [Google Scholar]
- 113.Poncet N, Mitchell FE, Ibrahim AFM, McGuire VA, English G, Arthur JSC, Shi Y-B, Taylor PM. The catalytic subunit of the system L1 amino acid transporter (slc7a5) facilitates nutrient signalling in mouse skeletal muscle. PLoS One. 2014;9:e89547. doi: 10.1371/journal.pone.0089547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Christensen HN. Role of amino acid transport and countertransport in nutrition and metabolism. Physiol Rev. 1990;70:43–77. doi: 10.1152/physrev.1990.70.1.43. [DOI] [PubMed] [Google Scholar]
- 115.Wang JYJ. The capable ABL: what is its biological function? Mol. Cell. Biol. 2014;34:1188–1197. doi: 10.1128/MCB.01454-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bhutia YD, Babu E, Prasad PD, Ganapathy V. The amino acid transporter SLC6A14 in cancer and its potential use in chemotherapy. Asian J. Pharm. Sci. 2014;9:293–303. [Google Scholar]
- 117.Karunakaran S, Ramachandran S, Coothankandaswamy V, Elangovan S, Babu E, Periyasamy-Thandavan S, Gurav A, Gnanaprakasam JP, Singh N, Schoenlein PV, Prasad PD, Thangaraju M, Ganapathy V. SLC6A14 (ATB0,+) protein, a highly concentrative and broad specific amino acid transporter, is a novel and effective drug target for treatment of estrogen receptor-positive breast cancer. J. Biol. Chem. 2011;286:31830–31838. doi: 10.1074/jbc.M111.229518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hancock JF, Magee AI, Childs JE, Marshall CJ. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell. 1989;57:1167–1177. doi: 10.1016/0092-8674(89)90054-8. [DOI] [PubMed] [Google Scholar]
- 119.Currie E, Schulze A, Zechner R, Walther TC, Farese RV. Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18:153–161. doi: 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Louie SM, Roberts LS, Mulvihill MM, Luo K, Nomura DK. Cancer cells incorporate and remodel exogenous palmitate into structural and oncogenic signaling lipids. Biochim. Biophys. Acta. 2013;1831:1566–1572. doi: 10.1016/j.bbalip.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stremmel W, Pohl L, Ring A, Herrmann T. A new concept of cellular uptake and intracellular trafficking of long-chain fatty acids. Lipids. 2001;36:981–989. doi: 10.1007/s11745-001-0809-2. [DOI] [PubMed] [Google Scholar]
- 122.Kamp F, Hamilton JA. How fatty acids of different chain length enter and leave cells by free diffusion. Prostaglandins. Leukot. Essent. Fatty Acids. 2006;75:149–159. doi: 10.1016/j.plefa.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 123.Glatz JFC, Luiken JJFP, Bonen A. Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol. Rev. 2010;90:367–417. doi: 10.1152/physrev.00003.2009. [DOI] [PubMed] [Google Scholar]
- 124.van Oort MM, van Doorn JM, Bonen A, Glatz JFC, van der Horst DJ, Rodenburg KW, Luiken JJFP. Insulin-induced translocation of CD36 to the plasma membrane is reversible and shows similarity to that of GLUT4. Biochim. Biophys. Acta. 2008;1781:61–71. doi: 10.1016/j.bbalip.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 125.Nath A, Li I, Roberts LR, Chan C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci. Rep. 2015;5:14752. doi: 10.1038/srep14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hale JS, Otvos B, Sinyuk M, Alvarado AG, Hitomi M, Stoltz K, Wu Q, Flavahan W, Levison B, Johansen ML, Schmitt D, Neltner JM, Huang P, Ren B, Sloan AE, Silverstein RL, Gladson CL, DiDonato JA, Brown JM, McIntyre T, Hazen SL, Horbinski C, Rich JN, Lathia JD. Cancer stem cell-specific scavenger receptor 36 drives glioblastoma progression. Stem Cells. 2014;32:1746–1758. doi: 10.1002/stem.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wijers M, Kuivenhoven JA, van de Sluis B. The life cycle of the low-density lipoprotein receptor: insights from cellular and in-vivo studies. Curr. Opin. Lipidol. 2015;26:82–87. doi: 10.1097/MOL.0000000000000157. [DOI] [PubMed] [Google Scholar]
- 128.Goldstein JL, Brown MS. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009;29:431–438. doi: 10.1161/ATVBAHA.108.179564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Edwards IJ, Berquin IM, Sun H, O’flaherty JT, Daniel LW, Thomas MJ, Rudel LL, Wykle RL, Chen YQ. Differential effects of delivery of omega-3 fatty acids to human cancer cells by low-density lipoproteins versus albumin. Clin. Cancer Res. 2004;10:8275–8283. doi: 10.1158/1078-0432.CCR-04-1357. [DOI] [PubMed] [Google Scholar]
- 130.Zhang D-W, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD, Cohen JC, Hobbs HH. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 2007;282:18602–18612. doi: 10.1074/jbc.M702027200. [DOI] [PubMed] [Google Scholar]
- 131.Demierre M-F, Higgins PDR, Gruber SB, Hawk E, Lippman SM. Statins and cancer prevention. Nat. Rev. Cancer. 2005;5:930–942. doi: 10.1038/nrc1751. [DOI] [PubMed] [Google Scholar]
- 132.Everett BM, Smith RJ, Hiatt WR. Reducing LDL with PCSK9 Inhibitors--The Clinical Benefit of Lipid Drugs. N. Engl. J. Med. 2015;373:1588–1591. doi: 10.1056/NEJMp1508120. [DOI] [PubMed] [Google Scholar]
- 133.Rudling MJ, Angelin B, Peterson CO, Collins VP. Low Density Lipoprotein Receptor Activity in Human Intracranial Tumors and Its Relation to the Cholesterol Requirement. Cancer Res. 1990;50:483–487. [PubMed] [Google Scholar]
- 134.Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D, Kuga D, Amzajerdi AN, Soto H, Zhu S, Babic I, Tanaka K, Dang J, Iwanami A, Gini B, Dejesus J, Lisiero DD, Huang TT, Prins RM, Wen PY, Robins HI, Prados MD, Deangelis LM, Mellinghoff IK, Mehta MP, James CD, Chakravarti A, Cloughesy TF, Tontonoz P, Mischel PS. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011;1:442–456. doi: 10.1158/2159-8290.CD-11-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liu Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006;9:230–234. doi: 10.1038/sj.pcan.4500879. [DOI] [PubMed] [Google Scholar]
- 136.Wu X, Daniels G, Lee P, Monaco ME. Lipid metabolism in prostate cancer. Am. J. Clin. Exp. Urol. 2014;2:111–120. [PMC free article] [PubMed] [Google Scholar]
- 137.Guillaumond F, Bidaut G, Ouaissi M, Servais S, Gouirand V, Olivares O, Lac S, Borge L, Roques J, Gayet O, Pinault M, Guimaraes C, Nigri J, Loncle C, Lavaut M-N, Garcia S, Tailleux A, Staels B, Calvo E, Tomasini R, Iovanna JL, Vasseur S. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. U. S. A. 2015;112:2473–2478. doi: 10.1073/pnas.1421601112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dwarakanath BS, Singh D, Banerji AK, Sarin R, Venkataramana NK, Jalali R, Vishwanath PN, Mohanti BK, Tripathi RP, Kalia VK, Jain V. Clinical studies for improving radiotherapy with 2-deoxy-D-glucose: present status and future prospects. J. Cancer Res. Ther. 2009;5(Suppl. 1):S21–S26. doi: 10.4103/0973-1482.55136. [DOI] [PubMed] [Google Scholar]
- 139.Edinger AL, Thompson CB. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol. Biol. Cell. 2002;13:2276–2288. doi: 10.1091/mbc.01-12-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Eyster CA, Higginson JD, Huebner R, Porat-Shliom N, Weigert R, Wu WW, Shen R-F, Donaldson JG. Discovery of new cargo proteins that enter cells through clathrin-independent endocytosis. Traffic. 2009;10:590–599. doi: 10.1111/j.1600-0854.2009.00894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Agola JO, Hong L, Surviladze Z, Ursu O, Waller A, Strouse JJ, Simpson DS, Schroeder CE, Oprea TI, Golden JE, Aubé J, Buranda T, Sklar LA, Wandinger-Ness A. A competitive nucleotide binding inhibitor: in vitro characterization of Rab7 GTPase inhibition. ACS Chem. Biol. 2012;7:1095–1108. doi: 10.1021/cb3001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shutes A, Onesto C, Picard V, Leblond B, Schweighoffer F, Der CJ. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J. Biol. Chem. 2007;282:35666–35678. doi: 10.1074/jbc.M703571200. [DOI] [PubMed] [Google Scholar]
- 143.Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2004;101:7618–7623. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Grossmann AH, Yoo JH, Clancy J, Sorensen LK, Sedgwick A, Tong Z, Ostanin K, Rogers A, Grossmann KF, Tripp SR, Thomas KR, D’Souza-Schorey C, Odelberg SJ, Li DY. The small GTPase ARF6 stimulates β-catenin transcriptional activity during WNT5A-mediated melanoma invasion and metastasis. Sci. Signal. 2013;6:ra14. doi: 10.1126/scisignal.2003398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bill A, Schmitz A, Albertoni B, Song J-N, Heukamp LC, Walrafen D, Thorwirth F, Verveer PJ, Zimmer S, Meffert L, Schreiber A, Chatterjee S, Thomas RK, Ullrich RT, Lang T, Famulok M. Cytohesins are cytoplasmic ErbB receptor activators. Cell. 2010;143:201–211. doi: 10.1016/j.cell.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 146.Bill A, Schmitz A, König K, Heukamp LC, Hannam JS, Famulok M. Anti-proliferative effect of cytohesin inhibition in gefitinib-resistant lung cancer cells. PLoS One. 2012;7:e41179. doi: 10.1371/journal.pone.0041179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chung N, Jenkins G, Hannun YA, Heitman J, Obeid LM. Sphingolipids signal heat stress-induced ubiquitin-dependent proteolysis. J. Biol. Chem. 2000;275:17229–17232. doi: 10.1074/jbc.C000229200. [DOI] [PubMed] [Google Scholar]
- 148.Chung N, Mao C, Heitman J, Hannun YA, Obeid LM. Phytosphingosine as a specific inhibitor of growth and nutrient import in Saccharomyces cerevisiae. J. Biol. Chem. 2001;276:35614–35621. doi: 10.1074/jbc.M105653200. [DOI] [PubMed] [Google Scholar]
- 149.Welsch CA, Roth LWA, Goetschy JF, Movva NR. Genetic, biochemical, and transcriptional responses of Saccharomyces cerevisiae to the novel immunomodulator FTY720 largely mimic those of the natural sphingolipid phytosphingosine. J. Biol. Chem. 2004;279:36720–36731. doi: 10.1074/jbc.M406179200. [DOI] [PubMed] [Google Scholar]
- 150.Guenther GG, Peralta ER, Rosales KR, Wong SY, Siskind LJ, Edinger AL. Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc. Natl. Acad. Sci. U. S. A. 2008;105:17402–17407. doi: 10.1073/pnas.0802781105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dobrowsky RT, Kamibayashi C, Mumby MC, Hannun YA. Ceramide activates heterotrimeric protein phosphatase 2A. J. Biol. Chem. 1993;268:15523–15530. [PubMed] [Google Scholar]
- 152.Chalfant CE, Szulc Z, Roddy P, Bielawska A, Hannun YA. The structural requirements for ceramide activation of serine-threonine protein phosphatases. J. Lipid Res. 2004;45:496–506. doi: 10.1194/jlr.M300347-JLR200. [DOI] [PubMed] [Google Scholar]
- 153.Guenther GG, Edinger AL. A new take on ceramide: starving cells by cutting off the nutrient supply. Cell Cycle. 2009;8:1122–1126. doi: 10.4161/cc.8.8.8161. [DOI] [PubMed] [Google Scholar]
- 154.Romero Rosales K, Singh G, Wu K, Chen J, Janes MR, Lilly MB, Peralta ER, Siskind LJ, Bennett MJ, Fruman DA, Edinger AL. Sphingolipid-based drugs selectively kill cancer cells by down-regulating nutrient transporter proteins. Biochem. J. 2011;439:299–311. doi: 10.1042/BJ20110853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gutierrez A, Pan L, Groen RWJ, Baleydier F, Kentsis A, Marineau J, Grebliunaite R, Kozakewich E, Reed C, Pflumio F, Poglio S, Uzan B, Clemons P, VerPlank L, An F, Burbank J, Norton S, Tolliday N, Steen H, Weng AP, Yuan H, Bradner JE, Mitsiades C, Look AT, Aster JC. Phenothiazines induce PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. J. Clin. Invest. 2014;124:644–655. doi: 10.1172/JCI65093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chua C-W, Lee DT-W, Ling M-T, Zhou C, Man K, Ho J, Chan FL, Wang X, Wong Y-C. FTY720, a fungus metabolite, inhibits in vivo growth of androgen-independent prostate cancer. Int. J. Cancer. 2005;117:1039–1048. doi: 10.1002/ijc.21243. [DOI] [PubMed] [Google Scholar]
- 157.Azuma H, Takahara S, Ichimaru N, Wang JD, Itoh Y, Otsuki Y, Morimoto J, Fukui R, Hoshiga M, Ishihara T, Nonomura N, Suzuki S, Okuyama A, Katsuoka Y. Marked prevention of tumor growth and metastasis by a novel immunosuppressive agent, FTY720, in mouse breast cancer models. Cancer Res. 2002;62:1410–1419. [PubMed] [Google Scholar]
- 158.Azuma H, Takahara S, Horie S, Muto S, Otsuki Y, Katsuoka Y. Induction of apoptosis in human bladder cancer cells in vitro and in vivo caused by FTY720 treatment. J. Urol. 2003;169:2372–2377. doi: 10.1097/01.ju.0000064938.32318.91. [DOI] [PubMed] [Google Scholar]
- 159.Chen B, Roy SG, McMonigle RJ, Keebaugh A, McCracken AN, Selwan E, Fransson R, Fallegger D, Huwiler A, Kleinman MT, Edinger AL, Hanessian S. Azacyclic FTY720 Analogues That Limit Nutrient Transporter Expression but Lack S1P Receptor Activity and Negative Chronotropic Effects Offer a Novel and Effective Strategy to Kill Cancer Cells in Vivo. ACS Chem. Biol. 2015 doi: 10.1021/acschembio.5b00761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Camm J, Hla T, Bakshi R, Brinkmann V. Cardiac and vascular effects of fingolimod: mechanistic basis and clinical implications. Am. Heart J. 2014;168:632–644. doi: 10.1016/j.ahj.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 161.Sanna MG, Liao J, Jo E, Alfonso C, Ahn M-Y, Peterson MS, Webb B, Lefebvre S, Chun J, Gray N, Rosen H. Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J. Biol. Chem. 2004;279:13839–13848. doi: 10.1074/jbc.M311743200. [DOI] [PubMed] [Google Scholar]
- 162.Forrest M, Sun S-Y, Hajdu R, Bergstrom J, Card D, Doherty G, Hale J, Keohane C, Meyers C, Milligan J, Mills S, Nomura N, Rosen H, Rosenbach M, Shei G-J, Singer II, Tian M, West S, White V, Xie J, Proia RL, Mandala S. Immune cell regulation and cardiovascular effects of sphingosine 1-phosphate receptor agonists in rodents are mediated via distinct receptor subtypes. J. Pharmacol. Exp. Ther. 2004;309:758–768. doi: 10.1124/jpet.103.062828. [DOI] [PubMed] [Google Scholar]
- 163.Koyrakh L, Roman MI, Brinkmann V, Wickman K. The heart rate decrease caused by acute FTY720 administration is mediated by the G protein-gated potassium channel I. Am. J. Transplant. 2005;5:529–536. doi: 10.1111/j.1600-6143.2005.00754.x. [DOI] [PubMed] [Google Scholar]
- 164.Fransson R, McCracken AN, Chen B, McMonigle RJ, Edinger AL, Hanessian S. Design, Synthesis, and Anti-leukemic Activity of Stereochemically Defined Constrained Analogs of FTY720 (Gilenya) ACS Med. Chem. Lett. 2013;4:969–973. doi: 10.1021/ml4002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, Rabinowitz JD, Metallo CM, Vander Heiden MG, Bar-Sagi D. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497:633–637. doi: 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, Thompson CB. The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell. 2015;162:259–270. doi: 10.1016/j.cell.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Swanson JA. Shaping cups into phagosomes and macropinosomes. Nat. Rev. Mol. Cell Biol. 2008;9:639–649. doi: 10.1038/nrm2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mercer J, Helenius A. Gulping rather than sipping: macropinocytosis as a way of virus entry. Curr. Opin. Microbiol. 2012;15:490–499. doi: 10.1016/j.mib.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 169.Levin R, Grinstein S, Schlam D. Phosphoinositides in phagocytosis and macropinocytosis. Biochim. Biophys. Acta. 2015;1851:805–823. doi: 10.1016/j.bbalip.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 170.Egami Y, Taguchi T, Maekawa M, Arai H, Araki N. Small GTPases and phosphoinositides in the regulatory mechanisms of macropinosome formation and maturation. Front. Physiol. 2014;5:374. doi: 10.3389/fphys.2014.00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Maltese WA, Overmeyer JH. Non-apoptotic cell death associated with perturbations of macropinocytosis. Front. Physiol. 2015;6:38. doi: 10.3389/fphys.2015.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chung J-J, Huber TB, Gödel M, Jarad G, Hartleben B, Kwoh C, Keil A, Karpitskiy A, Hu J, Huh CJ, Cella M, Gross RW, Miner JH, Shaw AS. Albumin-associated free fatty acids induce macropinocytosis in podocytes. J. Clin. Invest. 2015;125:2307–2316. doi: 10.1172/JCI79641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Commisso C, Flinn RJ, Bar-Sagi D. Determining the macropinocytic index of cells through a quantitative image-based assay. Nat. Protoc. 2014;9:182–192. doi: 10.1038/nprot.2014.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, Thompson CB, Rabinowitz JD. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. U. S. A. 2013;110:8882–8887. doi: 10.1073/pnas.1307237110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Matthews H, Ranson M, Kelso MJ. Anti-tumour/metastasis effects of the potassium-sparing diuretic amiloride: an orally active anti-cancer drug waiting for its call-of-duty? Int. J. Cancer. 2011;129:2051–2061. doi: 10.1002/ijc.26156. [DOI] [PubMed] [Google Scholar]
- 176.Koivusalo M, Welch C, Hayashi H, Scott CC, Kim M, Alexander T, Touret N, Hahn KM, Grinstein S. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J. Cell Biol. 2010;188:547–563. doi: 10.1083/jcb.200908086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Redelman-Sidi G, Iyer G, Solit DB, Glickman MS. Oncogenic activation of Pak1-dependent pathway of macropinocytosis determines BCG entry into bladder cancer cells. Cancer Res. 2013;73:1156–1167. doi: 10.1158/0008-5472.CAN-12-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kroemer G, Jäättelä M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer. 2005;5:886–897. doi: 10.1038/nrc1738. [DOI] [PubMed] [Google Scholar]
- 179.Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–914. doi: 10.4161/auto.19653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–1108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012;5:ra42. doi: 10.1126/scisignal.2002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, Settleman J, Stephanopoulos G, Dyson NJ, Zoncu R, Ramaswamy S, Haas W, Bardeesy N. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361–365. doi: 10.1038/nature14587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhi X, Zhong Q. Autophagy in cancer. F1000Prime Rep. 2015;7:18. doi: 10.12703/P7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Thorburn A, Thamm DH, Gustafson DL. Autophagy and cancer therapy. Mol. Pharmacol. 2014;85:830–838. doi: 10.1124/mol.114.091850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Rebecca VW, Amaravadi RK. Emerging strategies to effectively target autophagy in cancer. Oncogene. 2015 doi: 10.1038/onc.2015.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8:200–212. doi: 10.4161/auto.8.2.18554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Petherick KJ, Conway OJL, Mpamhanga C, Osborne SA, Kamal A, Saxty B, Ganley IG. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem. 2015;290:11376–11383. doi: 10.1074/jbc.C114.627778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Egan DF, Chun MGH, Vamos M, Zou H, Rong J, Miller CJ, Lou HJ, Raveendra-Panickar D, Yang C-C, Sheffler DJ, Teriete P, Asara JM, Turk BE, Cosford NDP, Shaw RJ. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell. 2015;59:285–297. doi: 10.1016/j.molcel.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.White E. Exploiting the bad eating habits of Ras-driven cancers. Genes Dev. 2013;27:2065–2071. doi: 10.1101/gad.228122.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Cai X, Xu Y, Cheung AK, Tomlinson RC, Alcázar-Román A, Murphy L, Billich A, Zhang B, Feng Y, Klumpp M, Rondeau J-M, Fazal AN, Wilson CJ, Myer V, Joberty G, Bouwmeester T, Labow MA, Finan PM, Porter JA, Ploegh HL, Baird D, De Camilli P, Tallarico JA, Huang Q. PIKfyve, a class III PI kinase, is the target of the small molecular IL-12/IL-23 inhibitor apilimod and a player in Toll-like receptor signaling. Chem. Biol. 2013;20:912–921. doi: 10.1016/j.chembiol.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.McCartney AJ, Zhang Y, Weisman LS. Phosphatidylinositol 3,5-bisphosphate: low abundance, high significance. Bioessays. 2014;36:52–64. doi: 10.1002/bies.201300012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Krausz S, Boumans MJH, Gerlag DM, Lufkin J, van Kuijk AWR, Bakker A, de Boer M, Lodde BM, Reedquist KA, Jacobson EW, O’Meara M, Tak PP. Brief report: a phase IIa, randomized, double-blind, placebo-controlled trial of apilimod mesylate, an interleukin-12/interleukin-23 inhibitor, in patients with rheumatoid arthritis. Arthritis Rheum. 2012;64:1750–1755. doi: 10.1002/art.34339. [DOI] [PubMed] [Google Scholar]
- 193.Kerr MC, Wang JTH, Castro NA, Hamilton NA, Town L, Brown DL, Meunier FA, Brown NF, Stow JL, Teasdale RD. Inhibition of the PtdIns(5) kinase PIKfyve disrupts intracellular replication of Salmonella. EMBO J. 2010;29:1331–1347. doi: 10.1038/emboj.2010.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.de Lartigue J, Polson H, Feldman M, Shokat K, Tooze SA, Urbé S, Clague MJ. PIKfyve Regulation of Endosome-Linked Pathways. Traffic. 2009;10:883–893. doi: 10.1111/j.1600-0854.2009.00915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ronan B, Flamand O, Vescovi L, Dureuil C, Durand L, Fassy F, Bachelot M-F, Lamberton A, Mathieu M, Bertrand T, Marquette J-P, El-Ahmad Y, Filoche-Romme B, Schio L, Garcia-Echeverria C, Goulaouic H, Pasquier B. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat. Chem. Biol. 2014;10:1013–1019. doi: 10.1038/nchembio.1681. [DOI] [PubMed] [Google Scholar]
- 196.Hardie DG. AMPK: a target for drugs and natural products with effects on both diabetes and cancer. Diabetes. 2013;62:2164–2172. doi: 10.2337/db13-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Morales DR, Morris AD. Metformin in cancer treatment and prevention. Annu. Rev. Med. 2015;66:17–29. doi: 10.1146/annurev-med-062613-093128. [DOI] [PubMed] [Google Scholar]
- 198.Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab. 2014;20:953–966. doi: 10.1016/j.cmet.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 199.Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, Sabatini DM. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. 2014;508:108–112. doi: 10.1038/nature13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, Chandel NS. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. 2014;3:e02242. doi: 10.7554/eLife.02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells - what challenges do they pose? Nat. Rev. Drug Discov. 2014;13:497–512. doi: 10.1038/nrd4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat. Rev. Cancer. 2013;13:727–738. doi: 10.1038/nrc3597. [DOI] [PubMed] [Google Scholar]
- 203.Saito Y, Chapple RH, Lin A, Kitano A, Nakada D. AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell Stem Cell. 2015;17:585–596. doi: 10.1016/j.stem.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O’Dwyer KM, Liesveld JL, Brookes PS, Becker MW, Jordan CT. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12:329–341. doi: 10.1016/j.stem.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, Popescu R, Della Donna L, Evers P, Dekmezian C, Reue K, Christofk H, Mischel PS, Pajonk F. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. U. S. A. 2011;108:16062–16067. doi: 10.1073/pnas.1106704108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, Barneda D, Sellers K, Campos-Olivas R, Graña O, Viera CR, Yuneva M, Sainz B, Heeschen C. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015;22:590–605. doi: 10.1016/j.cmet.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 207.Carey BW, Finley LWS, Cross JR, Allis CD, Thompson CB. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. 2015;518:413–416. doi: 10.1038/nature13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, Travins J, Weiss S, Looper R, Ligon KL, Verhaak RGW, Yan H, Kaelin WG., Jr Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Oburoglu L, Tardito S, Fritz V, de Barros SC, Merida P, Craveiro M, Mamede J, Cretenet G, Mongellaz C, An X, Klysz D, Touhami J, Boyer-Clavel M, Battini J-L, Dardalhon V, Zimmermann VS, Mohandas N, Gottlieb E, Sitbon M, Kinet S, Taylor N. Glucose and glutamine metabolism regulate human hematopoietic stem cell lineage specification. Cell Stem Cell. 2014;15:169–184. doi: 10.1016/j.stem.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 210.Stringari C, Edwards RA, Pate KT, Waterman ML, Donovan PJ, Gratton E. Metabolic trajectory of cellular differentiation in small intestine by Phasor Fluorescence Lifetime Microscopy of NADH. Sci. Rep. 2012;2:568. doi: 10.1038/srep00568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Stringari C, Nourse JL, Flanagan LA, Gratton E. Phasor fluorescence lifetime microscopy of free and protein-bound NADH reveals neural stem cell differentiation potential. PLoS One. 2012;7:e48014. doi: 10.1371/journal.pone.0048014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Blanpain C, Simons BD. Unravelling stem cell dynamics by lineage tracing. Nat. Rev. Mol. Cell Biol. 2013;14:489–502. doi: 10.1038/nrm3625. [DOI] [PubMed] [Google Scholar]
- 213.Hursting SD, Dunlap SM, Ford NA, Hursting MJ, Lashinger LM. Calorie restriction and cancer prevention: a mechanistic perspective. Cancer Metab. 2013;1:10. doi: 10.1186/2049-3002-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Mirzaei H, Suarez JA, Longo VD. Protein and amino acid restriction, aging and disease: from yeast to humans. Trends Endocrinol. Metab. 2014;25:558–566. doi: 10.1016/j.tem.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Meynet O, Ricci J-E. Caloric restriction and cancer: molecular mechanisms and clinical implications. Trends Mol. Med. 2014;20:419–427. doi: 10.1016/j.molmed.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 216.Longo VD, Mattson MP. Fasting: molecular mechanisms and clinical applications. Cell Metab. 2014;19:181–192. doi: 10.1016/j.cmet.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Longo VD, Fontana L. Calorie restriction and cancer prevention: metabolic and molecular mechanisms. Trends Pharmacol. Sci. 2010;31:89–98. doi: 10.1016/j.tips.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Lv M, Zhu X, Wang H, Wang F, Guan W. Roles of caloric restriction, ketogenic diet and intermittent fasting during initiation, progression and metastasis of cancer in animal models: a systematic review and meta-analysis. PLoS One. 2014;9:e115147. doi: 10.1371/journal.pone.0115147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Pallavi R, Giorgio M, Pelicci PG. Insights into the beneficial effect of caloric/ dietary restriction for a healthy and prolonged life. Front. Physiol. 2012;3:318. doi: 10.3389/fphys.2012.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Saleh AD, Simone BA, Palazzo J, Savage JE, Sano Y, Dan T, Jin L, Champ CE, Zhao S, Lim M, Sotgia F, Camphausen K, Pestell RG, Mitchell JB, Lisanti MP, Simone NL. Caloric restriction augments radiation efficacy in breast cancer. Cell Cycle. 2013;12:1955–1963. doi: 10.4161/cc.25016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kalaany NY, Sabatini DM. Tumours with PI3K activation are resistant to dietary restriction. Nature. 2009;458:725–731. doi: 10.1038/nature07782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 223.Lee C, Raffaghello L, Brandhorst S, Safdie FM, Bianchi G, Martin-Montalvo A, Pistoia V, Wei M, Hwang S, Merlino A, Emionite L, de Cabo R, Longo VD. Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci. Transl. Med. 2012;4:124ra27. doi: 10.1126/scitranslmed.3003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.De Lorenzo MS, Baljinnyam E, Vatner DE, Abarzúa P, Vatner SF, Rabson AB. Caloric restriction reduces growth of mammary tumors and metastases. Carcinogenesis. 2011;32:1381–1387. doi: 10.1093/carcin/bgr107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Raffaghello L, Lee C, Safdie FM, Wei M, Madia F, Bianchi G, Longo VD. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc. Natl. Acad. Sci. 2008;105:8215–8220. doi: 10.1073/pnas.0708100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Argilés JM, Busquets S, Stemmler B, López-Soriano FJ. Cancer cachexia: understanding the molecular basis. Nat. Rev. Cancer. 2014;14:754–762. doi: 10.1038/nrc3829. [DOI] [PubMed] [Google Scholar]
- 227.Petruzzelli M, Schweiger M, Schreiber R, Campos-Olivas R, Tsoli M, Allen J, Swarbrick M, Rose-John S, Rincon M, Robertson G, Zechner R, Wagner EF. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 2014;20:433–447. doi: 10.1016/j.cmet.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 228.Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE, Spiegelman BM. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature. 2014;513:100–104. doi: 10.1038/nature13528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Fearon K, Arends J, Baracos V. Understanding the mechanisms and treatment options in cancer cachexia. Nat. Rev. Clin. Oncol. 2013;10:90–99. doi: 10.1038/nrclinonc.2012.209. [DOI] [PubMed] [Google Scholar]
- 230.Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat. Rev. Clin. Oncol. 2014;11:473–481. doi: 10.1038/nrclinonc.2014.104. [DOI] [PubMed] [Google Scholar]
- 231.Peters J-U. Polypharmacology - foe or friend? J. Med. Chem. 2013;56:8955–8971. doi: 10.1021/jm400856t. [DOI] [PubMed] [Google Scholar]
- 232.Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat. Rev. Cancer. 2010;10:130–137. doi: 10.1038/nrc2787. [DOI] [PMC free article] [PubMed] [Google Scholar]