

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is driven by the inactivation of the tumor suppressor genes (TSGs), CDKN2A (P16) and SMAD4 (DPC4), commonly by homozygous deletions (HDs). Using a combination of high density single-nucleotide polymorphism (SNP) microarray and whole genome sequencing (WGS), we fine-mapped novel breakpoints surrounding deletions of CDKN2A and SMAD4 and characterized them by their underlying structural variants (SVs). Only one third of CDKN2A and SMAD4 deletions (6 of 18) were simple interstitial deletions, rather, the majority of deletions were caused by complex rearrangements, specifically, a translocation on one side of the TSG in combination with an inversion on the other side. We designate these as “TransFlip” mutations. Characteristics of TransFlip mutations are: (1) a propensity to target the TSGs CDKN2A and SMAD4 (P < 0.005), (2) not present in the germline of the examined samples, (3) non-recurrent breakpoints, (4) relatively small (47 bp to 3.4 kb) inversions, (5) inversions can be either telomeric or centromeric to the TSG, and (6) non-reciprocal, and non-recurrent translocations. TransFlip mutations are novel complex genomic rearrangements with unique breakpoint signatures in pancreatic cancer. We hypothesize that they are a common but poorly understood mechanism of TSG inactivation in human cancer.
INTRODUCTION
Homozygous deletions (HD) are a common mechanism by which tumor suppressor genes (TSGs) are inactivated in cancer, and are a common mapping tool for the discovery of TSGs (Lee et al., 1987; Hahn et al., 1996). HDs can range in size from kilobases to megabases, and are enriched at fragile sites and have been associated with chromothripsis (Bignell et al., 2010; Stephens et al., 2011). HDs are often the result of double-stranded DNA breaks (DSB), which can be repaired through a variety of mechanisms including homologous, microhomologous, and non-homologous pathways (Supporting Information Table 1). Non-homologous end joining (NHEJ), microhomology-mediated end-joining (MMEJ), and microhomology-mediated break-induced replication (MMBIR, also known as fork stalling and template switching, FoSTeS) are the dominant pathways by which DSB can be repaired, and each of these mechanisms manifests differently in the repaired DNA (Chiang et al., 2012). Recent research has highlighted the additional mechanisms of chromoplexy, LINE1 (L1) 3′ transduction, and breakage-fusion-bridge cycles (BFB) (Campbell et al., 2010; Baca et al., 2013; Tubio et al., 2014).
Cyclin-dependent kinase inhibitor 2A (CDKN2A) and deleted in pancreatic carcinoma (SMAD4) are two of the most commonly homozygously deleted TSGs in cancer (Cox et al., 2005). In pancreatic ductal adenocarcinoma (PDAC), CDKN2A and SMAD4 homozygous deletions are seen in about 40 and 30% of cases, respectively (Caldas et al., 1994; Hahn et al., 1996; Jones et al., 2008; Biankin et al., 2012). The CDKN2A locus, at 9p21, encodes 2 distinct proteins, P16 (INK4A) and P14 (ARF1), which regulate cyclin-dependent kinases and the tumor suppressor TP53, respectively. The SMAD4 locus, at 18q21.1, encodes a transcription factor in the TGF-beta signaling pathway.
In this study, we first identified somatic HDs in PDAC cancer cell lines using high density single-nucleotide polymorphism (SNP) microarrays. We then determined the precise breakpoints and characterized the underlying complex structural rearrangements (SV; interstitial deletion, translocation, inversion) using paired-end whole genome sequencing (WGS). We further characterized all deletions (heterozygous and homozygous) involving the PDAC TSGs, CDKN2A, and SMAD4, and confirmed rearrangements by PCR amplification and bidirectional Sanger sequencing across the breakpoints. We report two distinct patterns of TSG deletions: the first is simple interstitial deletion, and second, are deletions that result from the combination of an inter-chromosomal translocation and inversion flanking the HD of the TSG, designated here as TransFlip mutations. For somatic HDs in other regions of the genome, TransFlip mutations are rare but are an important mechanism by which TSGs are inactivated in PDAC, where TransFlip mutations occurred more frequently than simple interstitial deletions.
MATERIALS AND METHODS
Preparation of Genomic DNA and Karyotyping
Cancer cell lines were generated and matched normal DNA were obtained from eight patients with familial pancreatic adenocarcinoma (FPC, defined as a patient with PDAC who is from a kindred in which at least two family members were diagnosed with PDAC) as previously described (Kamiyama et al., 2013; Norris et al., 2015). STR analysis using ABI Profiler kit (Life Technologies, Carlsbad, CA) and 3130xL Genetic Analyzer (Life Technologies) confirmed tumor-normal pair matching, and xenografting in nude mice confirmed that the cell line isolated was the neoplastic component. Genomic DNA was extracted from early passage (<20 passages) cell lines and matched normal EBV-transformed lymphoblasts or frozen normal tissue using QIAamp DNA mini kit (Qiagen, Valencia, CA), per manufacturer’s instruction. Cell lines were karyotyped using methodology previously described (Griffin et al., 1994). For comparison, DNA from the tumors of sporadic PDAC patients was obtained, as part of the rapid autopsy program previously reported (Embuscado et al., 2005).
High Density SNP Microarray
The Omni2.5 array (Illumina, San Diego, CA) was used to analyze cancer cell lines and matched normal samples, as previously described (Norris et al., 2015). GenomeStudio and KaryoStudio (Illumina) were used to identify regions of homozygous deletion, heterozygous deletion, and loss of heterozygosity (LOH), and matched normal was used to remove germline deletions. These approximate coordinates were used to inform breakpoint analysis of WGS data.
Whole Genome Sequencing (WGS)
Sequencing on an Illumina HiSeq 2000 (Illumina) was carried out at 60× coverage by Personal Genome Diagnostics (PGDx, Baltimore, MD) using 3 μg of genomic DNA and generating 200 bp (2 × 100 bp reads) sequence per fragment (median total size ~500 bp). Paired reads were aligned to human genome (hg19) with Eland v.2 algorithm using the CASAVA 1.7 software (Illumina).
Breakpoint Characterization and Sanger Sequencing Confirmation
The exact genomic coordinates of CDKN2A and SMAD4 deletion breakpoints were determined by visual inspection of aligned WGS reads in IGV (Broad Institute, Boston, MA). The approximate genomic coordinates from SNP microarray data were used as a starting point. The reads were visually scanned in IGV for evidence of a deletion breakpoint, specifically: (1) drop in coverage, (2) mismatched bases at read ends, (3) reads with aberrant insert size, (4) reads with aberrant pair orientation, (5) reads with pair mapped to another chromosome, or (6) flanking reads with unmapped pairs. Tumor DNA and matched normal were amplified with M13-tagged primers (Integrated DNA Technologies, Coralville, IA), designed with Primer3 (Untergasser et al., 2007), using Platinum PCR Super Mix (Life Technologies), per manufacturer’s protocol. Amplicons were size-separated using polyacrylamide gel electrophoresis (PAGE) and ethidium bromide staining, to confirm the presence of novel junctions in the tumor (cancer cell line and available primary tumor DNA for Pa227C, Pa228C, Pa229C, and Pa230C) and the absence in the normal DNA. Amplicons from tumor DNA were Sanger sequenced on the 3730xL DNA Analyzer (Life Technologies) and analyzed with Sequencher software (Gene Codes, Ann Arbor, MI). To eliminate the possibility of the TransFlip mutations being specific to familial PDAC, we confirmed the presence of both an interstitial deletion and a TransFlip mutation in CDKN2A deletions in a set of four sporadic PDAC tumors’ WGS data.
RESULTS
Nucleotide Resolution of HDs
During our attempts to identify FPC predisposition genes, we closely examined HD regions identified by high density SNP microarrays in a series of well-characterized FPC cell lines (Norris et al., 2015). Visual inspection of WGS data in IGV revealed abrupt loss of coverage at both breakpoints corresponding to regions first identified by the SNP microarray. Aberrantly mapped pairs and reads with “rainbow ends” (where half of the read IGV perfectly matches the reference and the other half has highlighted mispaired bases) flank both breakpoints (Fig. 1). As expected, the mispaired bases of the “rainbow end” map to the other side of the HD for a simple interstitial deletion, the definition of split reads. Aberrantly mapped read pairs are colored by the IGV software to indicate a deletion, insertion, inversion, or translocation (Supporting Information Fig. 1).
Figure 1.
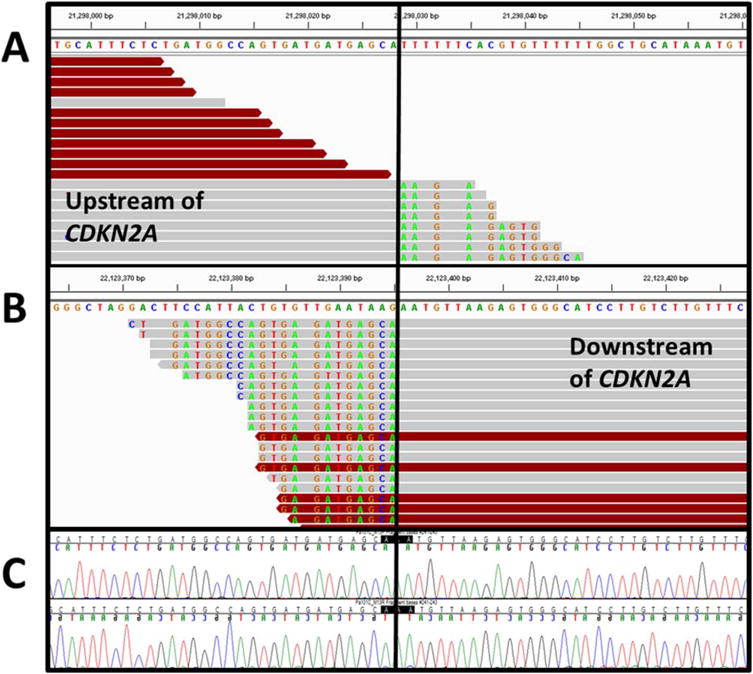
Signature of a HD in IGV. Shown is an interstitial deletion (0.8Mb) of CDKN2A in Pa101C (hg19: chr9:21,298,029; chr9:22,123,396), visualized with IGV and the reference sequence shown above each side of the breakpoint. The reads upstream (A) and downstream (B) of the CDKN2A HD are aligned, with the solid black line indicating the HD breakpoint. Colored bases indicate mismatched bases from reference, since they align to the other side of the breakpoint. Red-colored reads have an aberrant insert size with their pair, indicative of a HD. For this HD, there is no microhomology at the breakpoint and no non-templated bases inserted at the breakpoint. (C) Bidirectional Sanger sequencing confirms the novel junction. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Simple Interstitial Deletions Underlie Most Somatic HDs
Using this approach, we identified a total of 85 somatic HDs in our PDAC cancer cell lines from 8 FPC patients. The number of somatic HDs varied by sample, from 3 (Pa101C) to 18 (Pa229C and Pa231C), with a median of 9.5. The majority (69/85, 81%) of all somatic HDs were simple, interstitial deletions, in which a segment of a chromosome arm was deleted and the ends joined (Fig. 2A, Supporting Information Tables 2 and 3). The HDs associated with complex rearrangements (16/85, 19%) were categorized as inversion at one breakpoint and translocation at the other (Trans-Flip mutations, 7/16, 44%), inversion at both breakpoints (INV1INV, 6/16, 38%), intra-chromosomal translocation at both breakpoints (ICT1ICT, 1/16, 6%), or inter-chromosomal translocation at both breakpoints (TRANS-TRANS, 2/16, 13%). Deletions associated with complex rearrangements were significantly larger than interstitial HDs, with medians of 153 and 9 kb deleted chromosome material respectively (P < 0.05 by unpaired, two-tailed t test) (Supporting Information Table 2). Of the complex rearrangements, inversions produced the smallest HDs (median of 57 kb), while SVs involving translocations (TRANS+TRANS, ICT1ICT, and TransFlip mutations) produced significantly larger HDs (P < 0.05 by unpaired, two-tailed t test).
Figure 2.
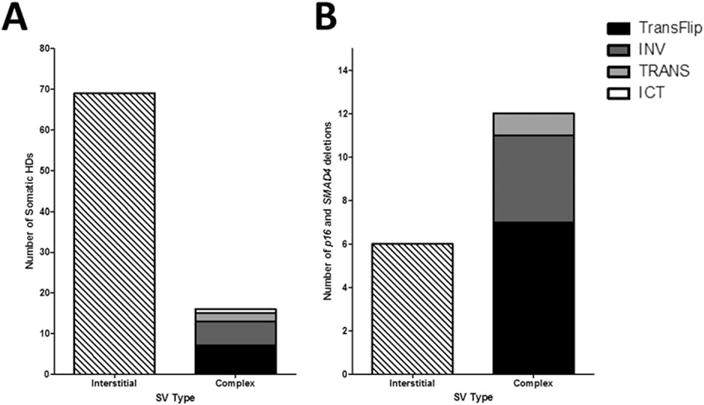
Most somatic HDs are simple interstitial deletions, but TransFlip mutations are more prevalent in CDKN2A and SMAD4 deletions. (A) Somatic HDs were categorized as interstitial, where breakpoints were directly end-joined, or complex structural variants (SV). The majority (69/85) of somatic HDs were interstitial. The 16 complex SVs were further characterized as TransFlip mutation (translocation at one breakpoint and inversion at the other breakpoint, 7/16), INV (inversion at both breakpoints, 6/16), TRANS (inter-chromosomal translocation at both breakpoints, 2/16), or ICT (intra-chromosomal translocation at both breakpoints, 1/16). (B) When looking at just CDKN2A and SMAD4 deletions, the majority (12/18) are complex rearrangements, and most frequently TransFlip mutations (7/18). There are no ICT SVs underlying CDKN2A and SMAD4 deletions.
While most interstitial deletions were intergenic (44/69, 64%), most (12/16, 75%) complex rearrangements resulted in the deletion of one or multiple gene(s) (Supporting Information Table 3). Specifically, TransFlip mutations were enriched at the PDAC TSGs CDKN2A and SMAD4 HDs, compared to interstitials (4 of 7 vs. 5 of 69, P < 0.005 by Fisher’s exact test).
Complex TransFlip Mutations Underlie many CDKN2A and SMAD4 Deletions in Pancreatic Cancer
We then performed a comprehensive analysis of all CDKN2A and SMAD4 deletions, including both homozygous and heterozygous deletions, in our PDAC cancer cell lines from eight FPC patients. Deletions of CDKN2A and SMAD4 were present in seven samples each, with 18 total deletions producing the 2 heterozygous CDKN2A deletions (Pa102C, Pa230C), 5 homozygous CDKN2A deletions (Pa101C, Pa222C, Pa228C, Pa229C, Pa231C), 3 heterozygous SMAD4 deletions (Pa101C, Pa228C, Pa230C), and 4 homozygous SMAD4 deletions (Pa102C, Pa227C, Pa229C, Pa231C). These generated a total of 26 novel junctions (where interstitial deletions produce one novel junction and TransFlip mutations produce at least two novel junctions). All 26 novel junctions were confirmed to be present in the cancer cell line by PCR and bidirectional Sanger sequencing, and absent in the matched normal sample. For 4 cases, the resected primary tumor was available (Pa227C, Pa228C, Pa229C, and Pa230C), and all corresponding novel junctions were present, confirming that these TransFlip mutations are not an artifact of cell culture.
Complex SVs were found to underlie the majority (12/18, 66%) of CDKN2A and SMAD4 deletions (Fig. 2B). Most samples (5/8, 63%) had CDKN2A and/or SMAD4 inactivated by a TransFlip mutation (Tables 1 and 2, Supporting Information Table 4). TransFlip mutations were coupled with LOH (CDKN2A, Pa229C, and Pa231C), with an interstitial deletion (CDKN2A, Pa222C), or with another TransFlip mutation (SMAD4, Pa102C, and Pa227C), to produce a HD of the TSG. TransFlip mutations and inactivation of the other allele of the TSGs by point mutation were mutually exclusive. In five samples with a point mutation in combination with a deletion of the TSG, 4 were the result of an INV rearrangement centromeric to the TSG and a terminal deletion (CDKN2A: 1/2; SMAD4: 3/3). The remaining one CDKN2A heterozygous deletion was a simple interstitial deletion (Pa102C).
TABLE 1.
Summary of CDKN2A and SMAD4 Deletions
| Gene | Sample | HD Size (Mb) | CNV | SV(s) |
|---|---|---|---|---|
| CDKN2A | Pa101C | 0.8 | 2Xa-HD-2Xa | ID |
| Pa102C | . | 3X-1X-3X | ID | |
| Pa222C | 0.1 | 1X-HD-1X | ID+TransFlip | |
| Pa227Cb | . | . | . | |
| Pa228C | 0.5 | 1X-HD-1X | IDc | |
| Pa229C | 4 | 2Xa-HD-2Xa | TransFlipc | |
| Pa230C | . | 1X-2X | INVc | |
| Pa231C | 0.7 | 1X-HD-2Xa | TransFlip | |
| SMAD4 | Pa101C | . | 3X-2Xa | INV |
| Pa102C | 6 | 1X-HD-1X | TransFlip+TransFlip | |
| Pa222Cb | . | . | . | |
| Pa227C | 5 | 2Xa-HD-1X | TransFlipc+TransFlipc | |
| Pa228C | . | 3X-1X to qtel | INVc | |
| Pa229C | 1.4 | 1X-HD-1X | IDc | |
| Pa230C | . | 1X to qtel | INVc | |
| Pa231C | 0.6 | 1X-HD-1X | ID+TRANS |
2X = Copy-Neutral LOH
CDKN2A is not deleted in Pa227C and SMAD4 is not deleted in Pa222C, Dots (.) indicate that the structural or copy number alteration was not detected.
Confirmed in resected primary tumor samples.
TABLE 2.
Structural Variants that Underlie CDKN2A and SMAD4 Deletions
| Tumor suppressor gene | Interstitial deletion | TransFlip (TRANS+INV) | TRANS alone | INV alone |
|---|---|---|---|---|
| CDKN2A (p16) | 4 | 3 | 0 | 0 |
| SMAD4 (DPC4) | 2 | 4 | 1 | 3 |
Characteristics of TransFlip Mutations in Pancreatic Cancer
All translocations were non-reciprocal and conventional karyotyping only detected one of the translocation junctions (1/11, 9%, Pa231C’s t(4;9)(p15.1;p21.3)) (Supporting Information Table 5). Inversion sizes ranged from 47 bp to 3.3 Mb for CDKN2A, and 53 bp to 35.3 Mb for SMAD4, and inversions associated with TransFlip mutations were the shortest (47 bp to 3.4 kb). The inversion half of a TransFlip mutation was either centromeric or telomeric to the TSG deletion (Fig. 3). In addition to the characteristics above, TransFlip mutations were (1) confirmed in the primary tumors to exclude the possibility of cell culture artifact, and (2) were not present in any of the germline HDs in our samples. Distinct TransFlip mutations were also identified in sporadic PDAC samples. The fundamental features of TransFlip mutations are summarized in Table 3.
Figure 3.
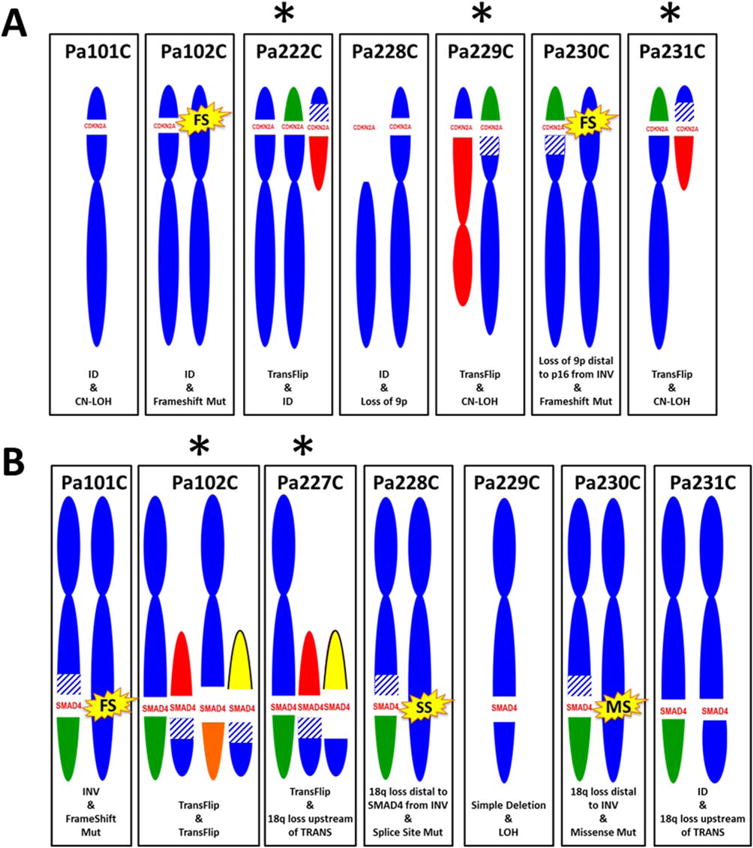
Derivative chromosomes for CDKN2A and SMAD4 deletions. Shown are the derivative chromosomes 9 (A) and 18 (B). Deletions are indicated by gap of blue chromosome material, on the short arm of chromosome 9 for CDKN2A and the long arm of chromosome 18 for SMAD4. Translocations (TRANS) are indicated by non-blue-colored chromosome partners (all are non-recurrent), inversions (INV) are indicated by blue striping, point mutations (FS: frameshift, SS: splice site, MS: missense) are indicated by yellow starbursts. TransFlip mutations (*) underlie 3 CDKN2A deletions (Pa222C, Pa229C, and Pa231C), and 2 SMAD4 deletions (Pa102C and Pa227C). Simple interstitial deletions (ID) and loss of heterozygosity (LOH, including copy-neutral events, or CN-LOH) also underlie CDKN2A and SMAD4 deletions in our cell lines. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
TABLE 3.
TransFlip Mutations
| Characteristics of TransFlip Mutations |
|---|
| Non-recurrent breakpoints |
| Not present in the germline of these PDAC patients |
| Enriched at TSGs |
| Inversion size ranges from 47bp to 3.4kb |
| Fold-back inversions are uncommon |
| Inversions can be telomeric or centromeric to the affected TSG |
| Translocations are inter-chromosomal and non-reciprocal |
| Non-recurrent chromosome partners in translocations |
| Can occur with LOH, a second TransFlip mutation, or an interstitial deletion to produce a TSG HD |
Lack of Alternate Mechanisms of CDKN2A and SMAD4 Deletion
For the simple interstitial deletions and the complex rearrangements targeting CDKN2A and SMAD4, only 1 case had non-templated base insertion at the junction, which argues against NHEJ as the dominant mechanism (Supporting Information Table 6). Microhomology was seen at the breakpoint in 12 of 14 (86%) CDKN2A novel junctions and 7/12 (58%) SMAD4 novel junctions. However, the median microhomology was only 1 base for both CDKN2A and SMAD4 (range of 0–7 and 0–3 bases, respectively). Therefore MMEJ and FoSteS/MMBIR are unlikely to be a dominant mechanism of the CDKN2A and SMAD4 rearrangements in our cohort. Only one inversion of a TransFlip mutation (Pa222C SMAD4) had a relative copy number increase, indicative of a fold-back inversion, a manifestation of BFB cycles (Fig. 4). BFB cycles do not explain the majority of TransFlip mutations.
Figure 4.

Fold-back inversion seen in one TransFlip mutation. High density SNP microarray of 18q21 in Pa222C reveals a copy number increase (green arrow) associated with the inversion side of the Trans-Flip mutation that results in the homozygous deletion of SMAD4 (black box). This is indicative of a fold-back inversion, and was not seen in any of the other inversions. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Chromothripsis of chromosomes 9 and 18 was seen in 1 and 2 cases, respectively (Supporting Information Fig. 2). Pa102C exhibited chromothripsis for both chromosomes and Pa227C exhibited chromothripsis for chromosome 18 (it did not have a CDKN2A deletion). Chromothripsis-associated genome remodeling has been shown to be mostly intra-chromosomal translocations (rearrangements within the same chromosome), compared to the exclusively inter-chromosomal translocations of TransFlip mutations (Stephens et al., 2011). HDs have been associated with fragile sites, but fragile sites do not account for the spectrum of CDKN2A and SMAD4 rearrangements, since the breakpoints ranged over megabases and the translocation partners were not always in fragile sites themselves (Bignell et al., 2010).
In most cases (CDKN2A: 13/14, 93% and SMAD4: 9/12, 75%), the regions involved in the rearrangement are not actively transcribed regions (here defined as having genes within 10 kb of both sides of the breakpoint), and so chromoplexy is not a dominant mechanism. Only one case (CDKN2A interstitial HD in Pa101C) had repetitive elements of the same family (in this case, LINE-1) flanking both ends of the breakpoint (Supporting Information Table 6). A recent report by Startek and colleagues highlights the requirement of an absolute minimum of 96% homology between LINE elements for mediating NAHR (Startek et al., 2015). The LINE-1 elements flanking the interstitial deletion of CDKN2A in Pa101C (L1PA4, chr9:21,297,268-21,300,617 and L1MA5A, chr9:22,121,352-22,123,446), have only 72% homology, arguing against a LINE-LINE-mediated NAHR event. L1 3′ transduction was not obvious at any CDKN2A or SMAD4 rearrangements, since all deletion junctions lacked the signatures of a non-templated polyA insertion and TT/AAA ORF2p endonuclease recognition sequence.
DISCUSSION
TransFlip mutations are more prevalent in CDKN2A and SMAD4 deletions than simple interstitial deletions, in contrast to deletions elsewhere where the opposite is the case. The rearrangement breakpoints in TransFlip mutations are often in gene-free regions (they do not produce fusion transcripts), but TransFlip mutations always result in the deletion of one or more genes, with enrichment at TSGs. All translocations in TransFlip mutations were non-reciprocal, which is typical of epithelial tumors (Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer, http://cgap.nci.nih.gov/Chromosomes/Mitelman). The inversions associated with TransFlip mutations could be either centromeric or telomeric to the TSG, ranged from 47 bp to 3.4 kb in length, and only one was a fold-back inversion. Fold-back inversions have been reported in PDAC, but not in combination with translocations (Campbell et al., 2010).
To the best of our knowledge, TransFlip mutations have not been described in a somatic disease setting, but the combination of an inversion and translocation at a deletion breakpoint has been reported in recurrent translocations of renal cell carcinoma cell lines (Ali et al., 2013). Unlike TransFlip mutations, the inversions described in these lines were centromeric and the genetic material distal to the breakpoint was deleted on the derivative chromosome (in TransFlip mutations, the distal sequence participates in a separate translocation event). It is of interest to note that there are a couple of reports of translocations associated with TSG HDs, but these studies were done at the cytogenetic level and did not report inversions (Misawa et al., 2004; Herholz et al., 2007). It is possible that these represent unidentified TransFlip mutations that would have been recognized if the investigators had more detailed information, such as that provided by WGS.
Previous analyses of CDKN2A deletion breakpoints found that the likely mechanism of repair was tissue-specific. Illegitimate V(D)J recombination was implicated in the majority of CDKN2A deletions in lymphoid leukemia, while evidence of NHEJ and MMEJ were seen in lung and other non-lymphoid cancers (Cayuela et al., 1997; Sasaki et al., 2003; Raschke et al., 2005). While microhomology was present at most breakpoints of both TransFlip mutations and interstitial deletions, the significance of the few bases involved seems insufficient to explain the mechanism, given the expected microhomology length of 1.35 bases genome-wide by chance alone. A microhomology length of less than 7 bases is statistically insignificant, based on a geometric distribution and accounting for GC content. Only one junction of a TransFlip mutation (the inversion half of the CDKN2A TransFlip mutation in Pa231C) had 7 bases of microhomology at the junction, but the corresponding translocation half of the TransFlip mutation had only 1 base of microhomology. No one mechanism (NHEJ, MMEJ, chromothripsis, chromoplexy, LINE-LINE-mediated NAHR, L1 3′ transduction, or BFB cycles) explains the prevalence of complex rearrangements, specifically TransFlip mutations, as a mechanism of CDKN2A and SMAD4 inactivation in our cohort.
We propose that TransFlip mutations occur by a novel mechanism, given their unique signature (Table 3). It is possible that the translocation and inversion occur simultaneously to resolve breaks in the DNA strand, or occur sequentially (Fig. 5). It remains to be determined if TransFlip mutations are the result of an inversion that requires a telomere (possibly via translocation) to survive (or vice versa), or if TransFlip mutations are a manifestation of a specific DNA repair pathway defect currently unknown. What is clear is that TransFlip mutations are associated with the inactivation of TSGs, not just HDs.
Figure 5.
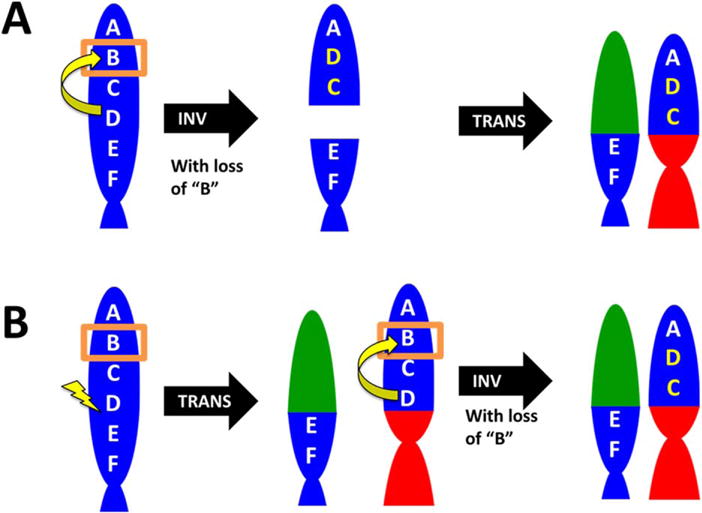
Proposed sequential mechanisms for TransFlip mutations. It is possible that the translocation and inversion occur simultaneously to resolve the deletion of B (boxed). Here we propose two stepwise mechanisms, with the TSG indicated by “B” on an example chromosome (blue). (A) An inversion of “BCD” (yellow) results in a DNA break and deletion of B, which is then resolved by a non-reciprocal translocation (red, green). (B) A DNA break between “D” and “E” (lightning bolt) is repaired by a non-reciprocal translocation (red, green) and followed by an inversion of “BCD” (yellow) and a deletion of “B”. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
It is possible that TransFlip mutations are unique to pancreatic cancer. In 2007, in their seminal work, Griffin and colleagues revealed the particularly high level of genomic instability in pancreatic cancer cell lines, which was also evident by the complex karyotypes of these distinct pancreatic cancer cell lines (Supporting Information Table 5) (Griffin et al., 2007). Genomic instability is a main driver of complex rearrangements in cancer, and often leads to the inactivation of tumor suppressor genes, including CDKN2A and SMAD4, which were also recurrently lost in the cell lines reported by Griffin in 2007. Future studies should explore the prevalence of TransFlip mutations in other cancers, to determine if Trans-Flips are a manifestation of a pancreatic cancer specific type of genomic instability.
Given the lack of TransFlip mutations associated with germline HDs in our cohort, we are keenly interested in whether TransFlip mutations underlie any human genetic disease, or whether they are completely unique to cancer. TransFlip mutations could be the result of an endogenous process possessed by all cells that are selected for in cancer because of TSG deletions. Alternatively, TransFlip mutations could manifest in cancer because of a fundamental defect in cancer cells, such as DNA repair, cell cycle regulation, or chromatin remodeling, or even the manifestation of the reaction of cells to a specific mutagen.
Complex rearrangements like TransFlip mutations will impact PARE (personalized analysis of rearranged ends) analysis for early detection and minimal residual disease testing (Leary et al., 2010). For monitoring minimal residual disease in a patient using PARE, it is imperative to use tumor-specific rearrangements that are under positive selection. TransFlip mutations are ideal PARE molecules since they often delete TSGs, thus providing selective advantage to the tumor cell, and have two novel junctions that can be assessed in the testing. One challenge of Trans-Flip mutations and other complex rearrangement is reliably detecting them from WGS data, and it remains to be seen if SV calling algorithms detect them as well as they do interstitial deletions, especially in primary tumors where DNA from stromal cells may obscure the detection of structural variants. We emphasize the utility of high density SNP microarray to first identify the approximate breakpoints of deletions that aid in locating the junction in WGS data.
The most exciting implication of TransFlip mutations is that they are potentially targetable via synthetic lethality, similar to BRCA mutations conferring sensitivity to poly ADP ribose polymerase (PARP) inhibitors or by exploiting genes in the HD, like in the co-deletion of MTAP with CDKN2A (Farmer et al., 2005; Hustinx et al., 2005).
Our study underscores the high complexity of structural rearrangements that can occur and which may not be fully appreciated without the combined use of multiple techniques, including conventional cytogenetics, high density SNP microarray, and WGS. Further work is needed to elucidate the mechanisms that underlie these complex rearrangements and determine the prevalence of TransFlip mutations as a mechanism of TSG inactivation.
Supplementary Material
Acknowledgments
The authors acknowledge Drs. Scott Kern, Yi Ning, Kathleen Burns, Victor Velculescu, Michael Goggins, Mihoko Kamiyama, Ken Kinzler, Bert Vogelstein, and Sian Jones for helpful discussions. They thank Stacy Riel, Kerry Powell, Isabelle Newkirk, Adam Lessing, Michael Borges, Michael Mullendore, Dante Truste, Roxanne Ashworth, Chrisanthe Papapavlou, Danielle Hagan, and Hong Liang for outstanding technical support.
Supported by: The Sol Goldman Pancreatic Cancer Research Center (pilot project); Grant number: R21CA164592;
PanCan/AACR Innovation Award; Grant number: SPORE P50-CA62924-13 (Kern);
The Stringer Foundation, and Michael Rolfe Pancreatic Cancer Foundation.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- Ali H, Daser A, Dear P, Wood H, Rabbitts P, Rabbitts T. Nonreciprocal chromosomal translocations in renal cancer involve multiple DSBs and NHEJ associated with breakpoint inversion but not necessarily with transcription. Genes Chromosomes Cancer. 2013;52:402–409. doi: 10.1002/gcc.22038. [DOI] [PubMed] [Google Scholar]
- Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M, Van Allen E, Kryukov GV, Sboner A, Theurillat JP, Soong TD, Nickerson E, Auclair D, Tewari A, Beltran H, Onofrio RC, Boysen G, Guiducci C, Barbieri CE, Cibulskis K, Sivachenko A, Carter SL, Saksena G, Voet D, Ramos AH, Winckler W, Cipicchio M, Ardlie K, Kantoff PW, Berger MF, Gabriel SB, Golub TR, Meyerson M, Lander ES, Elemento O, Getz G, Demichelis F, Rubin MA, Garraway LA. Punctuated evolution of prostate cancer genomes. Cell. 2013;153:666–677. doi: 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, Chang DK, Cowley MJ, Gardiner BB, Song S, Harliwong I, Idrisoglu S, Nourse C, Nourbakhsh E, Manning S, Wani S, Gongora M, Pajic M, Scarlett CJ, Gill AJ, Pinho AV, Rooman I, Anderson M, Holmes O, Leonard C, Taylor D, Wood S, Xu Q, Nones K, Fink JL, Christ A, Bruxner T, Cloonan N, Kolle G, Newell F, Pinese M, Mead RS, Humphris JL, Kaplan W, Jones MD, Colvin EK, Nagrial AM, Humphrey ES, Chou A, Chin VT, Chantrill LA, Mawson A, Samra JS, Kench JG, Lovell JA, Daly RJ, Merrett ND, Toon C, Epari K, Nguyen NQ, Barbour A, Zeps N, Australian Pancreatic Cancer Genome I. Kakkar N, Zhao F, Wu YQ, Wang M, Muzny DM, Fisher WE, Brunicardi FC, Hodges SE, Reid JG, Drummond J, Chang K, Han Y, Lewis LR, Dinh H, Buhay CJ, Beck T, Timms L, Sam M, Begley K, Brown A, Pai D, Panchal A, Buchner N, De Borja R, Denroche RE, Yung CK, Serra S, Onetto N, Mukhopadhyay D, Tsao MS, Shaw PA, Petersen GM, Gallinger S, Hruban RH, Maitra A, Iacobuzio-Donahue CA, Schulick RD, Wolfgang CL, Morgan RA, Lawlor RT, Capelli P, Corbo V, Scardoni M, Tortora G, Tempero MA, Mann KM, Jenkins NA, Perez-Mancera PA, Adams DJ, Largaespada DA, Wessels LF, Rust AG, Stein LD, Tuveson DA, Copeland NG, Musgrove EA, Scarpa A, Eshleman JR, Hudson TJ, Sutherland RL, Wheeler DA, Pearson JV, McPherson JD, Gibbs RA, Grimmond SM. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, Buck G, Chen L, Beare D, Latimer C, Widaa S, Hinton J, Fahey C, Fu B, Swamy S, Dalgliesh GL, Teh BT, Deloukas P, Yang F, Campbell PJ, Futreal PA, Stratton MR. Signatures of mutation and selection in the cancer genome. Nature. 2010;463:893–898. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, Weinstein CL, Hruban RH, Yeo CJ, Kern SE. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994;8:27–32. doi: 10.1038/ng0994-27. [DOI] [PubMed] [Google Scholar]
- Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal SA, Leroy C, Jia M, Menzies A, Butler AP, Teague JW, Griffin CA, Burton J, Swerdlow H, Quail MA, Stratton MR, Iacobuzio-Donahue C, Futreal PA. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–1113. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayuela JM, Gardie B, Sigaux F. Disruption of the multiple tumor suppressor gene MTS1/p16(INK4a)/CDKN2 by illegitimate V(D)J recombinase activity in T-cell acute lymphoblastic leukemias. Blood. 1997;90:3720–3726. [PubMed] [Google Scholar]
- Chiang C, Jacobsen JC, Ernst C, Hanscom C, Heilbut A, Blumenthal I, Mills RE, Kirby A, Lindgren AM, Rudiger SR, McLaughlan CJ, Bawden CS, Reid SJ, Faull RL, Snell RG, Hall IM, Shen Y, Ohsumi TK, Borowsky ML, Daly MJ, Lee C, Morton CC, MacDonald ME, Gusella JF, Talkowski ME. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat Genet. 2012;44:390–397, S391. doi: 10.1038/ng.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox C, Bignell G, Greenman C, Stabenau A, Warren W, Stephens P, Davies H, Watt S, Teague J, Edkins S, Birney E, Easton DF, Wooster R, Futreal PA, Stratton MR. A survey of homozygous deletions in human cancer genomes. Proc Natl Acad Sci USA. 2005;102:4542–4547. doi: 10.1073/pnas.0408593102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embuscado EE, Laheru D, Ricci F, Yun KJ, de Boom Witzel S, Seigel A, Flickinger K, Hidalgo M, Bova GS, Iacobuzio-Donahue CA. Immortalizing the complexity of cancer metastasis: Genetic features of lethal metastatic pancreatic cancer obtained from rapid autopsy. Cancer Biol Ther. 2005;4:548–554. doi: 10.4161/cbt.4.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Griffin CA, Hruban RH, Long PP, Morsberger LA, Douna-Issa F, Yeo CJ. Chromosome abnormalities in pancreatic adenocarcinoma. Genes Chromosomes Cancer. 1994;9:93–100. doi: 10.1002/gcc.2870090204. [DOI] [PubMed] [Google Scholar]
- Griffin CA, Morsberger L, Hawkins AL, Haddadin M, Patel A, Ried T, Schrock E, Perlman EJ, Jaffee E. Molecular cytogenetic characterization of pancreas cancer cell lines reveals high complexity chromosomal alterations. Cytogenet Genome Res. 2007;118:148–156. doi: 10.1159/000108295. [DOI] [PubMed] [Google Scholar]
- Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- Herholz H, Kern W, Schnittger S, Haferlach T, Dicker F, Haferlach C. Translocations as a mechanism for homozygous deletion of 13q14 and loss of the ATM gene in a patient with B-cell chronic lymphocytic leukemia. Cancer Genet Cytogenet. 2007;174:57–60. doi: 10.1016/j.cancergencyto.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Hustinx SR, Hruban RH, Leoni LM, Iacobuzio-Donahue C, Cameron JL, Yeo CJ, Brown PN, Argani P, Ashfaq R, Fukushima N, Goggins M, Kern SE, Maitra A. Homozygous deletion of the MTAP gene in invasive adenocarcinoma of the pancreas and in periampullary cancer: A potential new target for therapy. Cancer Biol Ther. 2005;4:83–86. doi: 10.4161/cbt.4.1.1380. [DOI] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiyama H, Rauenzahn S, Shim JS, Karikari CA, Feldmann G, Hua L, Kamiyama M, Schuler FW, Lin MT, Beaty RM, Karanam B, Liang H, Mullendore ME, Mo G, Hidalgo M, Jaffee E, Hruban RH, Jinnah HA, Roden RB, Jimeno A, Liu JO, Maitra A, Eshleman JR. Personalized chemotherapy profiling using cancer cell lines from selectable mice. Clin Cancer Res. 2013;19:1139–1146. doi: 10.1158/1078-0432.CCR-12-2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leary RJ, Kinde I, Diehl F, Schmidt K, Clouser C, Duncan C, Antipova A, Lee C, McKernan K, De La Vega FM, Kinzler KW, Vogelstein B, Diaz LA, Jr, Velculescu VE. Development of personalized tumor biomarkers using massively parallel sequencing. Sci Transl Med. 2010;2:20ra14. doi: 10.1126/scitranslmed.3000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WH, Bookstein R, Hong F, Young LJ, Shew JY, Lee EY. Human retinoblastoma susceptibility gene: Cloning, identification, and sequence. Science. 1987;235:1394–1399. doi: 10.1126/science.3823889. [DOI] [PubMed] [Google Scholar]
- Misawa A, Hosoi H, Imoto I, Iehara T, Sugimoto T, Inazawa J. Translocation (1;22)(p36;q11.2) with concurrent del(22)(q11.2) resulted in homozygous deletion of SNF5/INI1 in a newly established cell line derived from extrarenal rhabdoid tumor. J Hum Genet. 2004;49:586–589. doi: 10.1007/s10038-004-0191-y. [DOI] [PubMed] [Google Scholar]
- Norris AL, Roberts NJ, Jones S, Wheelan SJ, Papadopoulos N, Vogelstein B, Kinzler KW, Hruban RH, Klein AP, Eshleman JR. Familial and sporadic pancreatic cancer share the same molecular pathogenesis. Fam Cancer. 2015;14:95–103. doi: 10.1007/s10689-014-9755-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschke S, Balz V, Efferth T, Schulz WA, Florl AR. Homozygous deletions of CDKN2A caused by alternative mechanisms in various human cancer cell lines. Genes Chromosomes Cancer. 2005;42:58–67. doi: 10.1002/gcc.20119. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Kitagawa Y, Sekido Y, Minna JD, Kuwano H, Yokota J, Kohno T. Molecular processes of chromosome 9p21 deletions in human cancers. Oncogene. 2003;22:3792–3798. doi: 10.1038/sj.onc.1206589. [DOI] [PubMed] [Google Scholar]
- Startek M, Szafranski P, Gambin T, Campbell IM, Hixson P, Shaw CA, Stankiewicz P, Gambin A. Genome-wide analyses of LINE-LINE-mediated nonallelic homologous recombination. Nucleic Acids Res. 2015;43:2188–2198. doi: 10.1093/nar/gku1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal S, Leroy C, Jia M, Menzies A, Butler AP, Teague JW, Quail MA, Burton J, Swerdlow H, Carter NP, Morsberger LA, Iacobuzio-Donahue C, Follows GA, Green AR, Flanagan AM, Stratton MR, Futreal PA, Campbell PJ. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubio JM, Li Y, Ju YS, Martincorena I, Cooke SL, Tojo M, Gundem G, Pipinikas CP, Zamora J, Raine K, Menzies A, Roman-Garcia P, Fullam A, Gerstung M, Shlien A, Tarpey PS, Papaemmanuil E, Knappskog S, Van Loo P, Ramakrishna M, Davies HR, Marshall J, Wedge DC, Teague JW, Butler AP, Nik-Zainal S, Alexandrov L, Behjati S, Yates LR, Bolli N, Mudie L, Hardy C, Martin S, McLaren S, O’Meara S, Anderson E, Maddison M, Gamble S, Group IBC, Group IBC, Group IPC, Foster C, Warren AY, Whitaker H, Brewer D, Eeles R, Cooper C, Neal D, Lynch AG, Visakorpi T, Isaacs WB, van’t Veer L, Caldas C, Desmedt C, Sotiriou C, Aparicio S, Foekens JA, Eyfjord JE, Lakhani SR, Thomas G, Myklebost O, Span PN, Borresen-Dale AL, Richardson AL, Van de Vijver M, Vincent-Salomon A, Van den Eynden GG, Flanagan AM, Futreal PA, Janes SM, Bova GS, Stratton MR, McDermott U, Campbell PJ. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 2014;345:1251343. doi: 10.1126/science.1251343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JA. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007;35:W71–W74. doi: 10.1093/nar/gkm306. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.