

Abstract
The composition and function of the central nervous system (CNS) is extremely complex. In addition to hundreds of subtypes of neurons, other cell types, including glia (astrocytes, oligodendrocytes, and microglia) and vascular cells (endothelial cells and pericytes) also play important roles in CNS function. Such heterogeneity makes the study of gene transcription in CNS challenging. Transcriptomic studies, namely the analyses of the expression levels and structures of all genes, are essential for interpreting the functional elements and understanding the molecular constituents of the CNS. Microarray has been a predominant method for large-scale gene expression profiling in the past. However, RNA-sequencing (RNA-Seq) technology developed in recent years has many advantages over microarrays, and has enabled building more quantitative, accurate, and comprehensive transcriptomes of the CNS and other systems. The discovery of novel genes, diverse alternative splicing events, and noncoding RNAs has remarkably expanded the complexity of gene expression profiles and will help us to understand intricate neural circuits. Here, we discuss the procedures and advantages of RNA-Seq technology in mammalian CNS transcriptome construction, and review the approaches of sample collection as well as recent progress in building RNA-Seq-based transcriptomes from tissue samples and specific cell types.
Keywords: central nervous system, RNA-sequencing, gene expression, alternative splicing, noncoding RNA, transcriptome complexity
Introduction
The mammalian central nervous system (CNS) is of enormous structural complexity as well as cellular diversity. The abundance of gene expression varies dramatically by neuroanatomical location and cell class in the CNS (Cahoy and others 2008; Hawrylycz and others 2012; Zhang and others 2014). Understanding CNS functions and diseases requires a thorough understanding of the interaction of various components and cell classes within CNS tissues. A comprehensive analysis of the genes and RNA isoforms that are expressed in different anatomical subdivisions and cell types is therefore crucial for understanding CNS mechanisms and providing therapeutic solutions for neurological disorders, such as spinal cord injury, stroke, autism, or Alzheimer’s disease.
Prior to the development of high-throughput profiling approaches, gene expression and function studies had previously been restricted to a small number of genes at a time. The advances in high-throughput technologies and computational analyses have promoted a significant expansion from single-gene to genome-wide transcriptional analyses. Recent large-scale studies have revealed that the transcriptome is much more complex than the “one gene, one protein” concept initially proposed in the central dogma of genetics in the 1950s (Mattick and Gagen 2001; Wu 2011). The mammalian transcriptome consists of all transcripts synthesized in an organism including protein-coding, noncoding, alternatively spliced, sense, antisense, and edited RNAs (Snyder and Gerstein 2003). Previously, the majority of transcriptome studies were performed using microarrays (Cahoy and others 2008; Hawrylycz and others 2012; Lein and others 2007). While the use of microarray technology has provided valuable insights into CNS functions and disorders, microarray technology suffers from limitations in resolution, dynamic range, and accuracy (Bareyre and Schwab 2003; Carmel and others 2001; Miller and others 2013; Velardo and others 2004). The past decade has witnessed a revolution in next- generation sequencing (NGS) technology, which greatly affected the field of gene transcription (Wang and others 2009). RNA-sequencing (RNA-Seq) represents an innovative method for both mapping and quantifying a large number of transcribed RNAs in a given sample in a single experiment. RNA-Seq approaches particularly enable the discovery of novel transcripts, alternative splicing events, and genetic variations.
Therefore, an increasing number of studies have used RNA-Seq approaches to sequence CNS transcriptomes and characterize their layers of transcriptional complexity (Puthanveettil and others 2013; Wu and others 2010; Zhang and others 2014). Rapid progress has been made in understanding the orchestrated gene expression under normal and pathological conditions. Notably, the recent advancements in network analyses enable researchers to investigate the relationships between genes in the context of network and to identify key determinants in important processes (Chen and others 2013; Luo and others 2015; Voineagu and others 2011). The CNS transcriptome has been studied not only at the levels of tissues and organs but also at the level of their constituent cell types. The systematic dissection of CNS into its primary cell types has provided unprecedented insight into the transcriptional dynamics and functions of CNS development, organization, and diseases (Ng and others 2013). Here, we review some recent progress in building RNA-Seq–based transcriptomes of the CNS, with a focus on brain and spinal cord. First, we introduce and compare the commonly used technologies for building transcriptome.
Microarray
In the 1990s, DNA microarray technology was invented that enabled researchers to study gene expression at a genome-wide scale. Microarray experiments are based on the hybridization of cDNA molecules to specific nucleic acid probes immobilized on slides or chips. Based on the intensities of fluorescence signals at each position on the array, the gene expression level can be calculated (Brazma and others 2001). Microarray studies have led to important advances in the understanding of a wide range of biological problems, from the identification of differentially expressed genes between healthy and diseased tissues to drug discovery, and have greatly facilitated progress in understanding the molecular mechanisms of CNS functions and diseases (Bareyre and Schwab 2003; Cahoy and others 2008; Debouck and Goodfellow 1999; Dugas and others 2006; Kim and others 2012; Shen and others 2012; Thompson and others 2014; Zamanian and others 2012).
RNA-Sequencing
Next-generation sequencing (NGS) refers to a variety of massively parallel sequencing technology that arose after the introduction of first NGS platform (Roche 454) in 2004. Since then, other high-throughput sequencing platforms have been developed, such as Illumina, Applied Biosystems SOLiD, Complete Genomics, Pacific Biosciences (PacBio), and others. In RNA-sequencing (RNA-Seq), cDNAs generated from RNA samples are sequenced using NGS, producing millions of short sequence reads. The number of reads mapped within a gene or an exon can then be used as a measure of its abundance in the analyzed sample.
Typical RNA-Seq experimental procedures are as follows. The first step is to isolate and purify total RNA. The choice of ribosomal RNA (rRNA)–depleted or poly (A)–selected RNA as the input for constructing an RNA-Seq library depends on the experimental purpose. The oligo-d(T) hybridization method is the most widely used method to select polyadenylated RNAs. This approach is very effective for enriching mRNA, but also loses many noncoding RNAs (ncRNAs). In contrast, rRNA-depleted samples have the advantage of including other RNA species. The next step is library preparation. Briefly, RNAs are converted into a library of cDNA fragments with adaptors attached to one or both ends of each fragment. Subsequently, the molecules in the library, with or without amplification, are sequenced and short sequences from one end (single-end) or both ends (paired-end) are obtained. These reads are typically 30 to 500 bp in length, depending on the DNA-sequencing technology used. The reads obtained after sequencing are either aligned to a reference genome or transcriptome, or assembled de novo without genomic sequence guidance, to create a genome-scale transcription map providing both transcriptional structure and expression level for each gene.
Microarray or RNA-Sequencing?
Although microarray technology has facilitated valuable insights into CNS functions and disorders throughout the past decade, it also suffers from major limitations. We, and others, have found that RNA-Seq has several advantages over microarrays (Wang and others 2009; Zhang and others 2014). First, sequencing technology is much more sensitive and quantitative than arrays. Second, RNA-Seq provides a larger dynamic range for detection of transcripts (>9000-folds) compared with standard arrays. Third, sequence data are more specific and have less background. Furthermore, sequencing experiments do not depend on the limited probes present on tiled microarrays and can therefore be used to interrogate any location in the genome, including unannotated genes. Finally, sequencing is not limited by array hybridization chemistry, such as melting temperature, cross-hybridization, and secondary structure concerns. Importantly, RNA-Seq also enables identification of splicing isoforms and estimation of expression levels by providing splice junction reads. Table 1 lists the major differences between microarray and RNA-Seq approaches.
Table 1.
Comparison of Microarray and RNA-Sequencing (RNA-Seq) for Studying the Transcriptome.
| Technology | Microarray | RNA-Seq |
|---|---|---|
| Basis | Hybridization | High-throughput sequencing |
| Resolution | From several to hundreds of bases | Single-base |
| Required amount of input RNA | Nanograms (ng) to micrograms (μg) | Picograms (pg) to nanograms (ng) |
| High throughput | Yes | Yes |
| Reliance on reference sequence | Yes | Not necessarily |
| Ability to multiplex | No | Yes |
| Background noise | High | Low |
| Dynamic range | 100-folds | >9000-folds |
| Splicing isoform detection | Limited to custom-designed microarrays | Yes |
| Allelic expression | Limited to custom-designed microarrays | Yes |
We compared the transcriptome datasets of the major brain cell classes by RNA-Seq and microarray using the same set of RNA samples. Both RNA-Seq and microarray technologies revealed consistent gene expression signatures and demonstrated a high correlation of gene expression across cell classes (Fig. 1A). Importantly, RNA-Seq not only detected the majority of differentially expressed genes revealed by microarray but also identified many more cell-type–specific genes than did the microarray platform in astrocytes, neurons, oligodendrocytes, and microglia (ranging from 1471 [oligodendrocytes] to 3529 [microglia] genes at a fourfold enrichment or depletion cutoff; Fig. 1B–E) (Zhang and others 2014).
Figure 1.

Comparison of RNA-Seq and microarray analyses of differentially expressed genes across eight neural cell types. (A) High Spearman’s rank correlations between gene expression data from RNA-Seq and microarray platforms was observed across different cell types. (B–E) Comparing differentially expressed genes identified by RNA-Seq and microarray in astrocytes, neurons, oligodendrocytes, and microglia. Adapted from Zhang and others (2014) with permission and modifications.
Approaches of Sample Collection for CNS Transcriptome Characterization
The mammalian CNS is a very complex system; for example, the brain is a highly heterogeneous organ and the transcriptomes of different neural cell types differ dramatically from each other. In addition, even in the same type of cells, the transcriptome could dramatically change in response to stimuli (Stevens 1998). Therefore, treatment parameters, sample collection, and cellular heterogeneity are major challenges for building an accurate and complete CNS transcriptome.
Several approaches have been established to help reduce the cellular heterogeneity of mammalian CNS. Microdissection has been used for this purpose for decades. Traditional microdissection requires that a highly skilled and experienced researcher conducts the assay to ensure accuracy. In 1986, laser microbeam microdissection was first developed (Monajembashi and others 1986). In 1996, the first study employing laser-captured microdissection (LCM) appeared in Science (Emmert-Buck and others 1996). The LCM technique uses an inverted light microscope with a laser device that greatly facilitated the visualization and capture of the homogeneous cells of interest. LCM technology was commercialized and rapidly adopted by scientific communities internationally. LCM can be performed on both frozen and formalin-fixed paraffin-embedded tissues (Coudry and others 2007). However, the major limitation of LCM is similar to traditional microdissection: the identification of cell populations is based on morphological characteristics, so it requires a skilled histologist to perform this technique.
Although microdissection approaches can be used to reduce cellular heterogeneity, it is still difficult to collect cell populations of high purity. Immunoassays, such as immunopanning and fluorescence-activated cell sorting (FACS), which are based on the presence of specific cell-surface markers, have been developed to separate pure cell populations. FACS is now a popular and powerful technique for separating pure cell populations based on cell surface markers. FACS also allows the purification of individual cells based on size, granularity, and fluorescence (Herzenberg and others 2002). This technique has been applied to thousands of neural studies (Guez-Barber and others 2012; Moller and others 2012). Another useful technique, immunopanning, uses antibodies against specific cell-surface proteins to select certain cell types from a suspension of heterogeneous cells. Unlike FACS, immunopanning does not rely on fluorescent signals to detect specific cell populations, so it may be used to purify unlabeled cells (Barres and others 1992). In addition, genetic tagging methods such as BacTrap method (artificial chromosomes transgenic lines generated for Translating Ribosome Affinity Purification) permits in situ profiling of mRNA translation of specific cell types without the concern that the procedure of purification could alter their gene expression (Heiman and others 2008).
The aforementioned methods can use only one or several molecule markers to label a cell population; however, this is sometimes not sufficient to completely define a cell type. To date, single-cell RNA-Seq has the highest degree of specificity for transcriptome studies. Earlier studies were not able to obtain a large number of reads from single-cell RNA-Seq because of the limited amount of RNA in a single cell (5–10 pg). Recently, advancements in sequencing technology and microfluidics devices have helped researchers profile thousands of genes at once in single cells (Chen and others 2015; Tang and others 2010). In Figure 2, different approaches to sample collection for CNS transcriptome characterization are described.
Figure 2.

Different approaches for reducing cellular heterogeneity for CNS transcriptome characterization.
Building RNA Sequencing Transcriptome Using Tissue Samples from the CNS
The nervous system has the highest number of alternative splicing (AS) events in comparison to other organs. However, before NGS technology became available, it was challenging to characterize complex AS and its role in the nervous system. RNA-Seq can allow more efficient de novo identification of genes and splicing isoforms, and permit characterization of AS events at greater depth and coverage than can analyses based on microarray platforms. For instance, Mills and others (2013) conducted RNA-Seq of the frontal gray matter (GM) and white matter (WM) and identified 1652 genes and 882 splicing isoforms that were differentially expressed between GM and WM. One example is the human G protein-coupled receptor 123 (GPR123) gene that has four isoforms (GPR123-002, GPR123-003, GPR123-004, GPR123-005). The expression of the GPR123-004 isoform cannot be detected in GM; while in WM, the GPR123-004 isoform is the major isoform. The expressions of the other isoforms are lower than that of the GPR123-004 isoform in WM (Mills and others 2013). In another study, transcriptome analyses of prefrontal cortex and cerebellar tissue samples were carried out from 35 individuals ranging in age from 2 days to 98 years. This study demonstrated that approximately 40% of genes in the human brain undergo dramatic splicing changes in both brain regions across the lifespan. Thousands of these splicing changes were previously uncharacterized. About 15% of the differential splicing events were observed between the prefrontal cortex and cerebellum. About 30% of age-related splicing changes were detected during 25 to 98 years of age, while the other 70% occurred from 0 to 14 years (Mazin and others 2013).
As only ~2% of the human genome encodes protein sequences, the majority of transcribed sequences are non-coding (Birney and others 2007; Guttman and others 2010; Lee 2012; Okazaki and others 2002; Wu and others 2008). microRNAs have been extensively reviewed previously, thus we focus on long noncoding RNAs (lncRNAs) here. lncRNAs (>200 bp in length) play crucial roles in transcriptional regulation during various biological processes, including brain development and cell fate determination (Mercer and others 2008; Mercer and others 2010; Pastori and Wahlestedt 2012). For example, Hu and others (2014) used the aforementioned RNA-Seq data set for human brain development to reconstruct transcripts. They discovered that around 40% of the transcripts were novel, including 4.2% new exons and new exon extensions, 3.6% antisense transcripts, and 29.7% potential noncoding RNAs. Moreover, that study identified antisense lncRNAs located upstream of protein-coding genes that were associated with neuronal functions and those lncRNAs were driven by bidirectional promoters enriched in neuron-related transcription factor binding sites (Hu and others 2014). Another study characterized the expression of lncRNAs in 38 human and 40 macaque prefrontal cortex (PFC) samples in a developmental time series (He and others 2014). 5835 GENCODE annotated lncRNAs and 1888 currently unannotated ones were identified. Importantly, 409 lncRNAs are development-associated lncRNAs and showed dramatic expression changes at different developmental time points. Overexpression of these development-associated lncRNAs affected gene expression in human SH-SY5Y cells, suggesting their potential roles in human PFC development.
In addition to AS and noncoding RNA, RNA editing is another RNA processing factor that expands the molecular complexity of the CNS transcriptome. RNA modifications such as adenosine-to-inosine (A-to-I) editing may occur in vertebrates at the post-transcriptional level and are functionally involved in the regulation of gene expression. Using RNA-Seq and an inosine chemical erasing approach, one study analyzed A-to-I editing in the human adult brain and identified 66 new A-to-I editing sites in CDS (coding sequence), and 19,725 new A-to-I editing sites in non-CDS regions. Moreover, the transcripts of 2114 genes were found to be edited with inosines. These edited transcripts were enriched for GO (gene ontology) terms associated with nerve impulses and energy metabolism, and so on (Sakurai and others 2014). In a separate study, a new computational methodology was developed to identify potential A-to-G(I) editing events with only RNA-Seq data and no prior knowledge of the RNA-editing characteristics or genomic sequences from the donor individual (Picardi and others 2012). In human spinal cord, this strategy detected 15 RNA-editing candidates with only A-to-G transitions in protein-coding gene sequences, and three of them were known editing sites located in SRP9, CCNI, and FLNA coding regions. Six of the remaining 12 RNA-editing candidates could be validated with whole-exome sequence data. In addition to RNA-editing candidates in protein-coding regions, 156 RNA-editing candidates in untranslated regions (UTR) and 21 in alternatively spliced exons were also detected. However, only a small number of these events were validated with whole-exome sequence data.
Importantly, RNA-Seq technology also facilitates analyses of the mammalian CNS transcriptome under pathological conditions. For example, Autism spectrum disorder (ASD) is a heritable neurodevelopmental disease characterized by abnormalities in social interaction and language development (Klauck 2006). Gene co-expression network analysis showed abnormalities in cortical patterning in the ASD brain compared with healthy brain. The modules of coexpressed genes associated with autism included a neuronal module enriched for known autism susceptibility genes, such as the neuronal specific splicing factor A2BP1 (also known as FOX1) (Martin and others 2007). In order to investigate the A2BP1-dependent splicing events in ASD, RNA-Seq for three ASD samples with reduced expression of A2BP1 and three healthy samples was carried out. 212 alternative splicing events were differentially detected between ASD (low A2BP1) and normal samples. Moreover, 36 of 176 predicted targets of A2BP1 were differentially expressed. These results suggested the involvement of alternative splicing dysfunction in ASD pathogenesis (Voineagu and others 2011). Spinal cord injury (SCI) is another devastating neurological disorder with no effective treatment. To generate a comprehensive view of the mechanisms involved in SCI pathology, we sequenced mouse spinal cord samples from contusive models in the acute (2 days after injury) and subacute (7 days after injury) phases, and systematically characterized the transcriptomic changes to identify genes and pathways critical in SCI pathology (Chen and others 2013). Totals of 2832 and 4207 differentially expressed genes were identified in the acute and subacute stages, respectively. Comparing RNA-Seq and microarray data side-by-side showed that RNA-Seq data exhibits larger dynamic range and allows more differentially expressed genes to be detected than did microarray data. Importantly, we developed a systems-based analysis framework to identify potentially crucial genes in the global gene network by combining information regarding molecular interactions and cellular localization with our differential expression data (Fig. 3). Some candidate genes identified (such as Hmox1, Lcn2, Tlr4, Ccl11, and Ccr1) have been shown by other groups to play important roles in SCI, which demonstrates the validity of our approach (Freria and others 2012; Kanno and others 2009; Knerlich-Lukoschus and others 2011; Rathore and others 2011). Many genes whose functions in SCI have not been well studied, such as genes in the LXR/RXR pathway, were also identified and can be further investigated in future experiments (Myers and others 2014). In addition, pharmacogenomic information was incorporated into our analyses. Among the genes identified, those with existing drug information can be readily tested in animal models of SCI. Therefore, this study exemplified how global gene profiling can translate into identifying genes of interest for performing functional tests and generating new hypotheses. Furthermore, the identification of splicing isoforms and estimation of expression levels provide useful information for enhancing the specificity of drug design and reducing potential side effects. We are in the process of testing some potential therapeutic targets and drugs in animal models of SCI. Characterizing gene expression and regulatory networks in the CNS can ultimately lead to therapeutic strategies for neurological disorders.
Figure 3.

A systems-based analysis framework for identifying potentially crucial genes in the global gene network. (A) A schematic diagram of gene expression network. The connected edges of a potentially key gene are highlighted in blue. (B) The workflow for the analysis framework in identifying potentially important genes. Adapted from Chen and others (2013) with permission and modifications.
Characterizing the Cell-Type–Specific Transcriptomes of the CNS
Most of the current publicly available gene expression data were generated from tissue homogenates. Gene expression occurring in minor cell types may thus go undetected, as these cell types comprise only a small portion of the total tissue RNA. Also, changes in gene expression in different cell types may occur in opposite directions, thereby appearing static in composite tissue data. To better understand the functions and interactions of the cell types in the brain, neurons, astrocytes, oligodendrocyte precursor cells, newly formed oligodendrocytes, myelinating oligodendrocytes, microglia, endothelial cells, and pericytes were purified from mouse cerebral cortex, using immunopanning and FACS (Zhang and others 2014). We generated a high-resolution transcriptome database of >22,000 genes for these eight cell types by RNA sequencing and identified thousands of new cell-type enriched genes and splicing isoforms. These can serve as novel tools for cell identification and genetic manipulation (Fig. 4).
Figure 4.

Validation of cell-type enriched genes from RNA-Seq results by qRT-PCR and in situ hybridization. (A) 34 of the selected 40 newly identified cell-type enriched genes were validated by qRT-PCR using Fluidigm BioMark microfluidic technology. Warmer colors represent higher abundance transcripts, and cooler colors represent lower abundance transcripts. Black indicates no amplification. (B) Fold enrichment plots for Atp13a4, Cpne7, Fam70b, Tmem88b, and Rcsd1 compared to housekeeping gene Gapdh. (C–G) In situ hybridization showing the colocalization of novel cell-type specific genes with classical cell markers. Left, image of the cortex and hippocampus at low magnification. Scale bar, 200 μm. Right, image of the cortex at high magnification. Scale bar, 50 μm. Adapted from Zhang and others (2014) with permission and modifications.
Studying alternative splicing in purified cell types can provide new insights into the molecular composition, function and diseases of the CNS. But most analyses to date have been carried out in tissues comprised of mixed cell types, so the exact cellular sources of spliced iso-forms were not previously clear. The complex profiles of differentially spliced RNAs in glia, neurons, and vascular cells have not been investigated previously. In the study, a high-quality, large-scale splicing dataset for the major cell classes of the brain was generated.
Analyzing the aforementioned RNA-Seq data from eight representative cell classes led to the discovery of 3113 nonredundant cassette exons with cell-type specific splicing (Yan and others 2015). Approximately, 35% of these have resulted from newly identified alternative splicing events (Fig. 5A). In addition, known and new cassette exons are under tissue- or cell-type specific regulation, by for example, RNA binding proteins (RBPs) Rbfox and Ptbp2 (Fig. 5B–D). Furthermore, there were 1014 highly conserved nonsense-mediated decay (NMD) cassette exons that may function directly in controlling gene expression levels. These NMD exons are enriched in RBPs including splicing factors and regulators for other steps in RNA metabolism. Notably, a group of NMD exons reside in chromatin regulator genes. Thus, NMD can modulate chromatin regulators, thereby coupling AS- and epigenetics-based mechanisms (Yan and others 2015).
Figure 5.

Tissue– or cell-type–specific regulation of cassette exons. (A) Comparison of cell-type–specific known and novel cassette exons across different cell types. MO, myelinating oligodendrocyte; NFO, newly formed oligodendrocyte; OPC, oligodendrocyte precursor cells. (B) RNA map correlating Rbfox binding position with Rbfox-dependent neuron-specific exon inclusion (red) and exclusion (blue). (C) The number of known and novel cassette exons in which splicing was changed by conditional depletion of Ptbp2. (D) An RNA map correlating Ptbp2 binding position with Ptbp2-dependent exon inclusion (red) and exclusion (blue). Adapted from Yan and others (2015) with permission and modifications.
The specific enrichment of alternatively spliced RNAs in different cell classes implies that these distinct transcript isoforms may contribute to the distinct functions of each cell type. The purified brain cell data can be used to better understand the differences in glial and neuronal function. For instance, there are two isoforms, Pkm1 and Pkm2, for the gene encoding the glycolytic enzyme pyruvate kinase (Pkm). Pkm1 is exclusively expressed in neurons and maintains cellular energy levels by catalyzing the last step in ATP formation in aerobic glycolysis in neurons, while Pkm2 is exclusively expressed in astrocytes (Fig. 6A and B). Pkm2 contains an inducible nuclear translocation signal important for cells to regulate the glycolytic flux depending on their energy status (Zhang and others 2014). Emerging evidence has demonstrated the essential roles of glia in a variety of neurological diseases, so in recent years, the research community has increasingly focused on glia cells to comprehensively understand the pathophysiology of neurological disorders (Molofsky and others 2012). Thus, the transcriptome database described here will provide valuable resource for insights into the involvement of glia in CNS functions.
Figure 6.
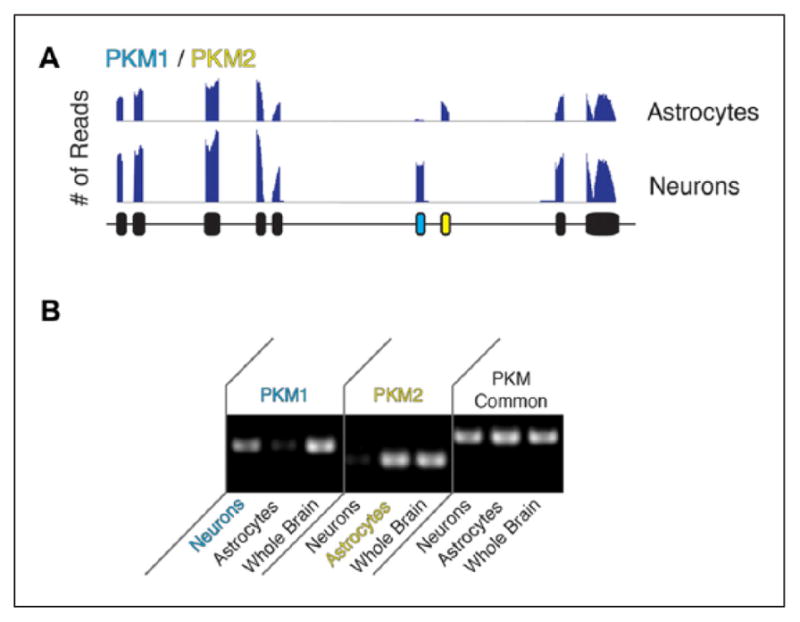
(A) Sequence traces of cell-type specific expression of PKM1 and PKM2 transcripts. The blue exon in neurons and yellow exons in astrocytes are mutually exclusive. (B) Confirmation of cell-type specific expression of PKM1 and PKM2 transcripts by PCR using primers targeting exons unique to PKM1, unique to PKM2, and common exons. Adapted from Zhang and others (2014) with permission and modifications.
Compared with their protein-coding counterparts, lncRNAs are more cell-type specific and generally expressed at lower levels, which makes it difficult to detect and assemble these transcripts, especially if the lncRNAs are expressed in the minor cell types within a tissue (Cabili and others 2011; Guttman and others 2010; Mercer and others 2008). A previous study used targeted capture (using tiling arrays to target selected portions of the transcriptome) followed by RNA-Seq to characterize lncRNA expression for the subventricular zone (SVZ) and neural stem cell lineages. A combination of microdissection and FACS were used to isolate adult mouse tissues from SVZ, olfactory bulb (OB), the dentate gyrus (DG), as well as representing cell populations of activated NSC, transit-amplifying cell, migratory neuroblast, and niche astrocyte. Thousands of currently unannotated lncRNAs were identified. Results indicated that lncRNAs are more temporally and spatially regulated than are mRNAs in the brain. In addition, coexpression analysis of mRNAs and lncRNAs suggested that some lncRNAs are associated with specific neural cell type functions as well as neurological diseases (Ramos and others 2013).
Although targeted capture followed by RNA-Seq can be used to validate transcripts that are expressed at low levels, this approach requires a priori knowledge of the target region. De novo identification of novel lncRNAs from purified cell type RNA-Seq data can further expand the known lncRNA repertoire and elucidate lncRNA functions in the CNS. From the brain cell data set, we reported 811 lncRNAs (FPKM [fragments per kilobase of exon model per million mapped reads] >1) and 227 lncRNAs with a stringent criterion (FPKM >5) in at least one cell type based on GENCODE annotation (Zhang and others 2014). Some lncRNAs are highly expressed: 12 of them have FPKM values >100 in the brain. Astrocytes and neurons express larger numbers of lncRNAs (109 ±2 and 92 ±3, respectively) than do myelinating oligodendrocytes and endothelial cells (44 ±3 and 48 ±3, respectively). We are in the process of broadening the lncRNA catalog by reconstructing the transcriptome ab initio and characterizing the regulation of unannotated lncRNAs in cell fate determination by integrating differential gene expression and transcription factor occupancy information. lncRNAs involved in oligodendrocyte precursor cell formation have been identified in loss-of-function experiments.
Further, the RNA-Seq transcriptomes of the brain cell classes also include previously undetected cell-type specific transcription factors, secreted ligands and membrane receptors, ion channels, cell adhesion molecules, and enzymes (Zhang and others 2014). The complete datasets have been deposited in a database displayed with an interactive web browser (http://web.stanford.edu/group/barres_lab/brain_rnaseq.html; http://jiaqianwulab.org/resource.htm). The research community can easily access this database to search for the transcription and alternative splicing profiles of their genes of interest in various cell classes (Fig. 7).
Figure 7.

Brain Cell RNA-Seq browser (http://jiaqianwulab.org/resource.htm). Sequence traces are shown for various brain cell classes. An example of alternatively spliced exons is surrounded by a dotted line.
Besides various cell types in the brain, cell types of interest from the spinal cord have also been purified and subjected to transcriptome analysis. Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease caused by progressive loss of motor neurons. Chiu and others (2013) specifically isolated CD11b-positive microglia from spinal cords of the SOD1G93A mouse model of ALS, lipo-polysaccharide (LPS)-injected, or healthy mice to investigate microglia activation. In ALS microglia, a neurodegeneration-specific gene expression profile differed greatly from that of LPS-induced microglia and M1 or M2 macrophages. Analysis of differentially expressed genes in those data indicated that ALS microglia coex-pressed both potentially neuroprotective and neurotoxic factors, including Insulin-like growth factor1 and pro-granulin as neuroprotective factors, and matrix metallo-proteinase 12 and optineurin as neurotoxic factors.
In addition to studying purified cell types from animals, analysis of gene expression during stem cell neural differentiation is another powerful approach to gain insights into the mechanisms and pathways involved in early cell-fate specification in the CNS, which may ultimately be very useful for pharmacological screening and developing therapies for neurodegenerative diseases and neurological trauma (Chen and others 2015; Lin and others 2014; Wu and others 2010). In order to examine the fundamental mechanisms governing the acquisition of neurogenic potential and the transition to gliogenic potential from stem cells, the transcriptome changes that occur during the differentiation of human embryonic stem cells (hESCs) into the neural lineage have been characterized (Wu and others 2010). Undifferentiated hESCs, and cells at three stages of early neural differentiation, including N1 (early initiation), N2 (neural progenitor), and N3 (early glial-like), were analyzed using a combination of RNA-Seq technologies (Illumina single-end, paired-end, and Roche 454). The results revealed enormous complexity in gene transcription and splicing dynamics during neural cell differentiation. Previously unannotated transcripts and spliced isoforms specific for each stage of differentiation were found. Interestingly, splicing isoform diversity is the greatest in undifferentiated hESCs, but then decreases on differentiation, a phenomenon we named as isoform specialization. During neural differentiation, differential expression was observed for many types of genes, including those involved in key signaling pathways and those encoding many extracellular receptors exhibiting stage-specific regulation. Another group performed loss function study of lncRNA to test their roles in the maintenance of mouse ESCs pluripotency and differentiation (Lin and others 2014). Tcl1 Upstream Neuron-Associated lncRNA (TUNA) (also known as Megamind) was found to bind to three RNA-binding proteins, polypyrimidine tract- binding protein (PTBP1), heterogeneous nuclear ribonucleoprotein K (hnRNP-K), and nucleolin (NCL). Inhibition of neural differentiation of mouse ESCs was observed on depletion of TUNA or the individual RNA-binding proteins, suggesting the important role of lncRNAs in stem cell neural differentiation. These results provided valuable information for the mechanisms underlying early neural differentiation.
Furthermore, recent advancements in sequencing technology and microfluidics devices have made transcriptome analysis at single-cell resolution possible. Single-cell sequencing will be instrumental for characterizing CNS cell-type heterogeneity and finding novel markers for subtypes of neuron and glia. In one study, the transcriptome of a collection of 3005 individual cells from the mouse somatosensory cortex and region I of hippocampus proper (CA1) were sequenced. A divisive biclustering method (BackSPIN) was developed to clustering these genes and cell types and successfully identified nine major classes of cells. These transcriptome datasets revealed not only well-known cell markers, such as Tbr1 for S1 pyramidal cells, but also novel markers for each cell class, such as Spink8 for hippocampal pyramidal cells. Moreover, 47 subclasses of cells were revealed when performing biclustering of data from each of the nine major cell classes (Zeisel and others 2015). In humans, a profiling study of single-cell gene expression was performed by sequencing RNAs from single outer radial glia (ORG) of human cortex that were isolated by FACS. This study revealed ORG-enriched genes, such as lncRNAs and targets of neurogenin. Moreover, characterizing the diverse co-expression patterns of classic progenitor markers, proneural transcription factors, and ORG-enriched genes in single-cell sequencing data indicated that there were more progenitors expressing pro-neural neurogenin targets in human than in ferret and mouse, suggesting higher transcriptional heterogeneity in human than in other species (Johnson and others 2015). Another single-cell transcriptome study used 466 cells from cortical tissue of eight human adults and four fetuses for single-cell RNA sequencing. This study successfully classified individual cells into all of the major cell types of the brain, categorized neuron subtypes, and identified genes that are differentially expressed between fetal and adult neurons. Interestingly, the discovery of gradients of gene expression between quiescent and replicating fetal neuronal populations reflected the developmental transition between these two populations (Darmanis and others 2015).
Overall, characterization of the CNS transcriptome by RNA-Seq approaches can provide more comprehensive and accurate gene transcription information, including details about the products of alternative splicing, noncoding RNAs, novel transcripts, and of other kinds of RNA processing. Increasingly available data in RNA-Seq databases will help the neuroscience community to make new discoveries at unprecedented speed and depth. However, many challenges still remain. For example, the majority of the read counts are produced by very highly expressed genes. Many genes of interest with low expression levels may have gone undetected due to very low read counts. These low read counts can also be quite variable, which can cause biases in differential expression analyses. Additionally, RNA-Seq-based transcriptome studies often relied on an existing annotation for quantification of transcript. As current gene annotations are still far from accurate and complete, the RNA-Seq–based CNS transcriptome may still only reflect partial gene expression profiles. Thus, the development and improvement of annotation-agnostic analysis approaches are required to strengthen the power of RNA-Seq. For example, Jaffe et al. recently implemented an annotation-agnostic differential expression analysis to characterize the transcriptomes of 72 human prefrontal cortex samples covering six life stages and revealed 50,650 differentially expression regions (DERs) across development and aging. These DERs are conserved across 16 human brain regions and the mouse cortex. Interestingly, most of these DERs have the highest expression levels in the fetus and around 41% of DERs are located in nonexonic sequences (Jaffe and others 2015). Furthermore, gene transcription and protein expression levels are not always well correlated. Thus, integrating data obtained from multiple “omic” approaches is critical to reaching comprehensive conclusions. There are many challenges ahead in developing statistical and computational strategies for integrating these data, for improving annotation, and for making transcriptome databases freely available to the scientific community. Eventually, large-scale systematic functional experiments will be required to understand the significance and regulation of the gene expression changes.
Acknowledgments
The authors thank Mary Ann Cushman for critical reading and editing of the manuscript. In addition, the authors would like to acknowledge the authors of the many excellent studies that could not be cited in this review due to space constraints.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: JQW, XD, and YY were supported by grants from the National Institutes of Health R01 NS088353 and R00 HL093213; the Staman Ogilvie Fund—Memorial Hermann Foundation; Mission Connect—a program of the TIRR Foundation; the Senator Lloyd & B. A. Bentsen Center for Stroke Research; UTHealth BRAIN Initiative and CTSA UL1 TR000371; and a grant from the University of Texas System Neuroscience and Neurotechnology Research Institute (Grant No. 362469).
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflict of interest with respect to the research, authorship, and/or publication of this article.
References
- Bareyre FM, Schwab ME. Inflammation, degeneration and regeneration in the injured spinal cord: insights from DNA microarrays. Trends Neurosci. 2003;26(10):555–63. doi: 10.1016/j.tins.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Barres BA, Hart IK, Coles HS, Burne JF, Voyvodic JT, Richardson WD, et al. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70(1):31–46. doi: 10.1016/0092-8674(92)90531-g. [DOI] [PubMed] [Google Scholar]
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, et al. Minimum information about a microarray experiment (MIAME)—toward standards for microarray data. Nat Genet. 2001;29(4):365–71. doi: 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–27. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28(1):264–78. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmel JB, Galante A, Soteropoulos P, Tolias P, Recce M, Young W, et al. Gene expression profiling of acute spinal cord injury reveals spreading inflammatory signals and neuron loss. Physiol Genomics. 2001;7:201–13. doi: 10.1152/physiolgenomics.00074.2001. [DOI] [PubMed] [Google Scholar]
- Chen K, Dai X, Wu J. Alternative splicing: An important mechanism in stem cell biology. World J Stem Cells. 2015;7(1):1–10. doi: 10.4252/wjsc.v7.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Deng S, Lu H, Zheng Y, Yang G, Kim D, et al. RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS One. 2013;8(8):e72567. doi: 10.1371/journal.pone.0072567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KN, Dong XM, Wu JQ. The application of single-cell sequencing in dynamic transcriptomes. In: Wang X, editor. Single cell sequencing and systems immunology. Dordrecht: Springer Netherlands; 2015. pp. 41–63. [Google Scholar]
- Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O’Keeffe S, Phatnani HP, et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013;4(2):385–401. doi: 10.1016/j.celrep.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coudry RA, Meireles SI, Stoyanova R, Cooper HS, Carpino A, Wang X, et al. Successful application of microarray technology to microdissected formalin-fixed, paraffin-embedded tissue. J Mol Diagn. 2007;9(1):70–9. doi: 10.2353/jmoldx.2007.060004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM, et al. A survey of human brain transcrip-tome diversity at the single cell level. Proc Natl Acad Sci U S A. 2015;112(23):7285–90. doi: 10.1073/pnas.1507125112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debouck C, Goodfellow PN. DNA microarrays in drug discovery and development. Nat Genet. 1999;21(1 Suppl):48–50. doi: 10.1038/4475. [DOI] [PubMed] [Google Scholar]
- Dugas JC, Tai YC, Speed TP, Ngai J, Barres BA. Functional genomic analysis of oligodendrocyte differentiation. J Neurosci. 2006;26(43):10967–83. doi: 10.1523/JNEUROSCI.2572-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang Z, Goldstein SR, et al. Laser capture microdissection. Science. 1996;274(5289):998–1001. doi: 10.1126/science.274.5289.998. [DOI] [PubMed] [Google Scholar]
- Freria CM, Velloso LA, Oliveira AL. Opposing effects of Toll-like receptors 2 and 4 on synaptic stability in the spinal cord after peripheral nerve injury. J Neuroinflammation. 2012;9:240. doi: 10.1186/1742-2094-9-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guez-Barber D, Fanous S, Harvey BK, Zhang Y, Lehrmann E, Becker KG, et al. FACS purification of immunolabeled cell types from adult rat brain. J Neurosci Methods. 2012;203(1):10–8. doi: 10.1016/j.jneumeth.2011.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol. 2010;28(5):503–10. doi: 10.1038/nbt.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489(7416):391–9. doi: 10.1038/nature11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Bammann H, Han D, Xie G, Khaitovich P. Conserved expression of lincRNA during human and macaque prefrontal cortex development and maturation. RNA. 2014;20(7):1103–11. doi: 10.1261/rna.043075.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135(4):738–48. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzenberg LA, Parks D, Sahaf B, Perez O, Roederer M. The history and future of the fluorescence activated cell sorter and flow cytometry: a view from Stanford. Clin Chem. 2002;48(10):1819–27. [PubMed] [Google Scholar]
- Hu HY, He L, Khaitovich P. Deep sequencing reveals a novel class of bidirectional promoters associated with neuronal genes. BMC Genomics. 2014;15:457. doi: 10.1186/1471-2164-15-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AE, Shin J, Collado-Torres L, Leek JT, Tao R, Li C, et al. Developmental regulation of human cortex transcription and its clinical relevance at single base resolution. Nat Neurosci. 2015;18(1):154–61. doi: 10.1038/nn.3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MB, Wang PP, Atabay KD, Murphy EA, Doan RN, Hecht JL, et al. Single-cell analysis reveals transcriptional heterogeneity of neural progenitors in human cortex. Nat Neurosci. 2015;18(5):637–46. doi: 10.1038/nn.3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno H, Ozawa H, Dohi Y, Sekiguchi A, Igarashi K, Itoi E. Genetic ablation of transcription repressor Bach1 reduces neural tissue damage and improves locomotor function after spinal cord injury in mice. J Neurotrauma. 2009;26(1):31–9. doi: 10.1089/neu.2008.0667. [DOI] [PubMed] [Google Scholar]
- Kim DS, Li KW, Boroujerdi A, Peter Yu Y, Zhou CY, Deng P, et al. Thrombospondin-4 contributes to spinal sensitization and neuropathic pain states. J Neurosci. 2012;32(26):8977–87. doi: 10.1523/JNEUROSCI.6494-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauck SM. Genetics of autism spectrum disorder. Eur J Hum Genet. 2006;14(6):714–20. doi: 10.1038/sj.ejhg.5201610. [DOI] [PubMed] [Google Scholar]
- Knerlich-Lukoschus F, von der Ropp-Brenner B, Lucius R, Mehdorn HM, Held-Feindt J. Spatiotemporal CCR1, CCL3(MIP-1αalpha), CXCR4, CXCL12(SDF-1α) expression patterns in a rat spinal cord injury model of posttraumatic neuropathic pain. J Neurosurg Spine. 2011;14(5):583–97. doi: 10.3171/2010.12.SPINE10480. [DOI] [PubMed] [Google Scholar]
- Lee JT. Epigenetic regulation by long noncoding RNAs. Science. 2012;338(6113):1435–9. doi: 10.1126/science.1231776. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445(7124):168–76. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Lin N, Chang KY, Li Z, Gates K, Rana ZA, Dang J, et al. An evolutionarily conserved long noncoding RNA TUNA controls pluripotency and neural lineage commitment. Mol Cell. 2014;53(6):1005–19. doi: 10.1016/j.molcel.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Coskun V, Liang A, Yu J, Cheng L, Ge W, et al. Single-cell transcriptome analyses reveal signals to activate dormant neural stem cells. Cell. 2015;161(5):1175–86. doi: 10.1016/j.cell.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CL, Duvall JA, Ilkin Y, Simon JS, Arreaza MG, Wilkes K, et al. Cytogenetic and molecular characterization of A2BP1/FOX1 as a candidate gene for autism. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(7):869–76. doi: 10.1002/ajmg.b.30530. [DOI] [PubMed] [Google Scholar]
- Mattick JS, Gagen MJ. The evolution of controlled multitasked gene networks: the role of introns and other noncoding RNAs in the development of complex organisms. Mol Biol Evol. 2001;18(9):1611–30. doi: 10.1093/oxfordjournals.molbev.a003951. [DOI] [PubMed] [Google Scholar]
- Mazin P, Xiong J, Liu X, Yan Z, Zhang X, Li M, et al. Widespread splicing changes in human brain development and aging. Mol Syst Biol. 2013;9:633. doi: 10.1038/msb.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS. Specific expression of long noncoding RNAs in the mouse brain. Proc Natl Acad Sci U S A. 2008;105(2):716–21. doi: 10.1073/pnas.0706729105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Qureshi IA, Gokhan S, Dinger ME, Li G, Mattick JS, et al. Long noncoding RNAs in neuronalglial fate specification and oligodendrocyte lineage maturation. BMC Neurosci. 2010;11:14. doi: 10.1186/1471-2202-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Woltjer RL, Goodenbour JM, Horvath S, Geschwind DH. Genes and pathways underlying regional and cell type changes in Alzheimer’s disease. Genome Med. 2013;5(5):48. doi: 10.1186/gm452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JD, Kavanagh T, Kim WS, Chen BJ, Kawahara Y, Halliday GM, et al. Unique transcriptome patterns of the white and grey matter corroborate structural and functional heterogeneity in the human frontal lobe. PLoS One. 2013;8(10):e78480. doi: 10.1371/journal.pone.0078480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller K, Stahl T, Boltze J, Wagner DC. Isolation of inflammatory cells from rat brain tissue after stroke. Exp Transl Stroke Med. 2012;4(1):20. doi: 10.1186/2040-7378-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AV, Krencik R, Ullian EM, Tsai HH, Deneen B, Richardson WD, et al. Astrocytes and disease: a neurodevelopmental perspective. Genes Dev. 2012;26(9):891–907. doi: 10.1101/gad.188326.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monajembashi S, Cremer C, Cremer T, Wolfrum J, Greulich KO. Microdissection of human chromosomes by a laser microbeam. Exp Cell Res. 1986;167(1):262–5. doi: 10.1016/0014-4827(86)90223-5. [DOI] [PubMed] [Google Scholar]
- Myers SA, Andres KR, Hagg T, Whittemore SR. CD36 deletion improves recovery from spinal cord injury. Exp Neurol. 2014;256:25–38. doi: 10.1016/j.expneurol.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SY, Lin L, Soh BS, Stanton LW. Long noncoding RNAs in development and disease of the central nervous system. Trends Genet. 2013;29(8):461–8. doi: 10.1016/j.tig.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, et al. Analysis of the mouse transcrip-tome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420(6915):563–73. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- Pastori C, Wahlestedt C. Involvement of long noncoding RNAs in diseases affecting the central nervous system. RNA Biol. 2012;9(6):860–70. doi: 10.4161/rna.20482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picardi E, Gallo A, Galeano F, Tomaselli S, Pesole G. A novel computational strategy to identify A-to-I RNA editing sites by RNA-Seq data: de novo detection in human spinal cord tissue. PLoS One. 2012;7(9):e44184. doi: 10.1371/journal.pone.0044184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthanveettil SV, Antonov I, Kalachikov S, Rajasethupathy P, Choi YB, Kohn AB, et al. A strategy to capture and characterize the synaptic transcriptome. Proc Natl Acad Sci U S A. 2013;110(18):7464–9. doi: 10.1073/pnas.1304422110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos AD, Diaz A, Nellore A, Delgado RN, Park KY, Gonzales-Roybal G, et al. Integration of genome-wide approaches identifies lncRNAs of adult neural stem cells and their progeny in vivo. Cell Stem Cell. 2013;12(5):616–28. doi: 10.1016/j.stem.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathore KI, Berard JL, Redensek A, Chierzi S, Lopez-Vales R, Santos M, et al. Lipocalin 2 plays an immunomodulatory role and has detrimental effects after spinal cord injury. J Neurosci. 2011;31(38):13412–9. doi: 10.1523/JNEUROSCI.0116-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai M, Ueda H, Yano T, Okada S, Terajima H, Mitsuyama T, et al. A biochemical landscape of A-to-I RNA editing in the human brain transcriptome. Genome Res. 2014;24(3):522–34. doi: 10.1101/gr.162537.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen EH, Overly CC, Jones AR. The Allen Human Brain Atlas: comprehensive gene expression mapping of the human brain. Trends Neurosci. 2012;35(12):711–4. doi: 10.1016/j.tins.2012.09.005. [DOI] [PubMed] [Google Scholar]
- Snyder M, Gerstein M. Genomics. Defining genes in the genomics era. Science. 2003;300(5617):258–60. doi: 10.1126/science.1084354. [DOI] [PubMed] [Google Scholar]
- Stevens CF. Neuronal diversity: too many cell types for comfort? Curr Biol. 1998;8(20):R708–10. doi: 10.1016/s0960-9822(98)70454-3. [DOI] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Nordman E, Li B, Xu N, Bashkirov VI, et al. RNA-Seq analysis to capture the transcriptome landscape of a single cell. Nat Protoc. 2010;5(3):516–35. doi: 10.1038/nprot.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CL, Ng L, Menon V, Martinez S, Lee CK, Glattfelder K, et al. A high-resolution spatiotemporal atlas of gene expression of the developing mouse brain. Neuron. 2014;83(2):309–23. doi: 10.1016/j.neuron.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velardo MJ, Burger C, Williams PR, Baker HV, López MC, Mareci TH, et al. Patterns of gene expression reveal a temporally orchestrated wound healing response in the injured spinal cord. J Neurosci. 2004;24(39):8562–76. doi: 10.1523/JNEUROSCI.3316-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474(7351):380–4. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JQ. Characterize mammalian transcriptome complexity. Saarbrücken, Germany: Lambert Academic; 2011. p. 204. [Google Scholar]
- Wu JQ, Du J, Rozowsky J, Zhang Z, Urban AE, Euskirchen G, et al. Systematic analysis of transcribed loci in ENCODE regions using RACE sequencing reveals extensive transcription in the human genome. Genome Biol. 2008;9(1):R3. doi: 10.1186/gb-2008-9-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JQ, Habegger L, Noisa P, Szekely A, Qiu C, Hutchison S, et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proc Natl Acad Sci U S A. 2010;107(11):5254–9. doi: 10.1073/pnas.0914114107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Weyn-Vanhentenryck SM, Wu J, Sloan SA, Zhang Y, Chen K, et al. Systematic discovery of regulated and conserved alternative exons in the mammalian brain reveals NMD modulating chromatin regulators. Proc Natl Acad Sci U S A. 2015;112(11):3445–50. doi: 10.1073/pnas.1502849112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, et al. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32(18):6391–410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Munoz-Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, Jureus A, et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347(6226):1138–42. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34(36):11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]