

Abstract
Background:
The evolution of antibiotic-resistant bacteria (ARB) and antibiotic-resistance genes (ARGs) has been accelerated recently by the indiscriminate application of antibiotics. Antibiotic resistance has challenged the success of medical interventions and therefore is considered a hazardous threat to human health.
Objectives:
The present study aimed to describe the use of zinc finger nuclease (ZFN) technology to target and disrupt a plasmid-encoded β-lactamase, which prevents horizontal gene transfer-mediated evolution of ARBs.
Materials and Methods:
An engineered ZFN was designed to target a specific sequence in the ampicillin resistance gene (ampR) of the pTZ57R plasmid. The Escherichia coli bacteria already contained the pZFN kanamycin-resistant (kanaR) plasmid as the case or the pP15A, kanaR empty vector as the control, were transformed with the pTZ57R; the ability of the designed ZFN to disrupt the β-lactamase gene was evaluated with the subsequent disturbed ability of the bacteria to grow on ampicillin (amp) and ampicillin-kanamycin (amp-kana)-containing media. The effect of mild hypothermia on the ZFN gene targeting efficiency was also evaluated.
Results:
The growth of bacteria in the case group on the amp and amp-kana-containing media was significantly lower compared with the control group at 37°C (P < 0.001). Despite being more efficient in hypothermic conditions at 30°C (P < 0.001), there were no significant associations between the incubation temperature and the ZFN gene targeting efficiency.
Conclusions:
Our findings revealed that the ZFN technology could be employed to overcome ampicillin resistance by the targeted disruption of the ampicillin resistance gene, which leads to inactivation of β-lactam synthesis. Therefore, ZFN technology could be engaged to decrease the antibiotic resistance issue with the construction of a ZFN archive against different ARGs. To tackle the resistance issue at the environmental level, recombinant phages expressing ZFNs against different ARGs could be constructed and released into both hospital and urban wastewater systems.
Keywords: Ampicillin Resistance, Microbial Drug Resistance, Beta-Lactamases, Gene Targeting, Genome Editing, Zinc Finger Nuclease
1. Background
Antibiotic-resistant bacteria (ARB) were described as the greatest risk to human health in the recent annual report of the World Economic Forum on global risks (1). Today, antibiotics are widely used not only in medicine but also in aquaculture, agriculture and livestock, growth enhancers and protective agents (2-4). A direct result of the vast applications of these antibiotics is their constant release into the natural ecosystems (5), which subsequently leads to the evolution of ARBs and antibiotic resistance genes (ARGs) (2) due to the selective pressure applied in bacterial populations (6). Antibiotic-resistant bacteria and ARGs are capable of being transmitted to humans via direct or indirect contact (7), which supports evidence that clinical resistance is intimately correlated with environmental resistance (8, 9). Antibiotic resistance has made the treatment approaches of bacterial infections insufficient and therefore leads to increasing rates of morbidity and mortality (7, 10).
The evolution of ARBs takes place via acquisition of spontaneous mutation in the bacterial chromosome or horizontal gene transfer (HGT) of resistance genes through conjugation, transformation, or transduction. HGT is performed through mobile DNA elements (11), and plasmid-mediated transmission is an important mechanism (12). Out of the five major classes of antibiotics, β-lactam antibiotics are the most widely used; thus, resistance to β-lactam antibiotics is a serious threat to human health (13, 14). Synthesis of β-lactamase enzymes is the most prevalent and important mechanism employed by bacteria to resist beta-lactam antibiotics (15). The integration of β-lactamase genes in plasmids has been a key factor in the development and transmission of resistance to β-lactam antibiotics among bacteria (16, 17).
Since the measures taken to deal with antibiotic resistance have only focused on the control of the resistance process, there is a pressing need to develop new approaches for solving the problem. Our proposed solution is based on the utilization of one of the gene editing tools, “ZFNs,” to target ARGS and either pathologic or virulent genes in mobile DNA elements or the bacterial genome, thereby knocking these genes out and preventing HGT between bacteria. A ZFN is an engineered endonuclease composed of a DNA-binding domain, referred to as a zinc finger protein (ZFP) combined with the non-specific cleavage domain of the type IIS Fok I restriction endonuclease, which is covalently linked to the carboxy-terminal of the ZFP via a flexible peptide linker. Owing to the capability of being manipulated for their recognition specificities, ZFNs offer highly sequence-specific gene-targeting tools to induce double-strand breaks (DSBs) in a wide variety of DNA sites (18, 19). After the recognition of half target sites by ZFPs, successful dimerization of FOKI cleavage domains induces a DSB at the site of the spacer, which subsequently results in the activation of cellular DNA repair systems. Double-strand breaks can be repaired by homologous recombination (HR) or non-homologous end joining (NHEJ) mechanisms (20). In the presence or absence of donor DNA, HR and NHEJ phenomena respectively occur to repair the DNA lesions. NHEJ is an error-prone mechanism that introduces insertion/deletion mutations at the DSB site, leading to mutations that change the open reading frame (ORF) or terminate the translation of the gene product (21). A reliable strategy to disrupt the gene of interest is based on applying two pairs of ZFNs that target two different sites of the desired gene, which results in the deletion of a large DNA fragment between the sites and disrupts the gene efficiently (22).
2. Objectives
The present study was designed to investigate the ability of an engineered ZFN to disrupt the β-lactamase encoding gene of an ampR plasmid, pTZ57R, as an antibiotic resistance gene model. ZFNs are powerful tools for targeted genome manipulation and have been used extensively for different purposes in many other organisms, such as plants, rats, drosophila, and zebra fish (21, 23-25); however, ZFNs have never been used for targeting ARGs.
3. Materials and Methods
3.1. Preparation of Recombinant pTZ57R
In order to achieve more efficient resistance disruption, it was scheduled to target two different regions of ampR gene by two pairs of ZFNs. Additionally, the context-dependent assembly (CoDA) approach was used to engineer ZFNs because it is an efficient and simple method compared with the other strategies. However, as there was only one potential target site identified by the CoDA platform in the β-lactamase gene of the pTZ57R plasmid (Val 258-Ala 266), the ZFN target site (Figure 1) was cloned in the pTZ57R (Thermo Scientific, Waltham, MA, USA) into the EcoRI and BamHI restriction sites. To this end, the ZFN target site was synthesized as two oligos (Bioneer Co., Korea); such that their hybridization created the double-stranded DNA target site, with EcoRI and BamHI overhangs at the two ends. This strategy made it possible for only one pair of ZFNs to target two parts of the pTZ57R plasmid.
Figure 1. The ZFN Target Site Identified by the CoDA Platform in the β-lactamase Gene of the pTZ57R Plasmid.

A mixture of two oligos was heat-denatured to single-stranded DNA and then cooled to allow the strands to renaturate. The pTZ57R plasmid was extracted using a GeneJET™ Plasmid Miniprep Kit (Thermo Scientific, Waltham, MA, USA) and was digested using EcoRI and BamHI restriction enzymes (Thermo Scientific, Waltham, MA, USA). The ligation reaction was carried out between the synthetic ZFN target site and the linearized plasmid using T4 DNA ligase (Thermo Scientific, Waltham, MA, USA), and then the ligation product was transformed into a chemically competent Escherichia coli strain TOP10 cells. The recombinant clones were selected by colony polymerase chain reaction (PCR) using a ZFN target site forward primer (5′ - GATCCAGTTATCTACACGACGGGGAGTCAGGCG - 3′) and M13 reverse sequencing primer (5′- CAGGAAACAGCTATGA - 3′) (Bioneer Co., Korea). To confirm the recombinant pTZ57R plasmid, the positive clones selected by colony PCR were subjected to plasmid extraction; the PCR reaction was also performed on the purified plasmid DNA with the same condition. The PCR products were detected on 1.5% agarose gel.
3.2. pP15A, kanaR Vector Construction
To construct an appropriate vector for ZFN expression, an M13KO7 helper phage (containing the P15A origin of replication and the kanamycin resistance gene (kanaR)) was applied for transduction in the E.coli TG1 strain. Selection of the M13KO7 helper phage was based on having a compatible origin of replication with pTZ57R. The M13KO7 double-strand plasmid was extracted, and a 2592-bp DNA fragment, including the P15A origin of replication and the kanaR, was then PCR amplified using the following primers: 5′ AGAATTCTCAAAGCAACCATAGTACGC 3′ (forward) containing an EcoRI cut site and 5′AGGATCCTCTAGACTCTGATGTTACATTGCACAAG 3′ (reverse) containing BamHI and XbaI cut sites at the 5′ ends (Bioneer Co., Korea). The PCR products were digested with the EcoRI restriction enzyme, purified, and then treated with Pfu DNA polymerase (Thermo Scientific, Waltham, MA, USA) to create a phosphorylated blunt end. After gel purification (Bioneer Co., Korea), the PCR product was self-ligated and transformed into chemically competent E. coli TOP10 cells. A single positive clone containing the self-ligated kanaR replicon, named “pP15A, kanaR” (Figure 2), was grown for plasmid extraction (Thermo Scientific, Waltham, MA, USA). The purified plasmid was confirmed by single and double-digestion reactions by BamHI and XbaI restriction enzymes (Thermo Scientific, Waltham, MA, USA) and after confirmation was used to clone and express the left and right ZFN arrays.
Figure 2. pP15A, kanaR vector Constructed for ZFN Cloning and Expression.

3.3. ZFN Cloning in pP15A, kanaR
The nucleotide sequences coding for the left and right ZFP arrays were downloaded from http://zifit.partners.org/ZiFiT. Furthermore, the obligate heterodimeric FokI variants, ELD/KKR, were obtained from Addgene: pCLR2070 and Addgene: pCLR2068 (https://www.addgene.org/). The left and right ZFN arrays and linker peptide coding sequences were codon optimized based on the codon frequencies in E. coli and linked to the FOKI KKR and ELDs, respectively. The linker peptide “TGPNRGVTK” was used to join the ZFPs to the FOKI cleavage domains, which is the known linker for ZFNs that target two half sites separated by a seven-nucleotide spacer. Two E. coli lacuv5 promoters were used to express the ZFN arrays (Figure 3). The described construct with other essential elements for protein expression, which is 2208 bp in length, was synthesized and cloned in the pP15A, kanaR vector into the BamHI and XbaI restriction sites (GeneCust, Dudelange, Luxembourg). The ZFN expression vector was 4800 bp in length and is abbreviated as “pZFN”.
Figure 3. This Figure Illustrates the pZFN, Containing ZFN Expression Cassette Cloned into the pP15A, kanaR Vector, Which is 4800 bp in Length.

3.4. Evaluation of Zinc Finger Nuclease Efficiency
To evaluate the ZFN efficiency required to disrupt the ampR, the E. coli TOP10 cells containing pZFN as the case or pP15A, kanaR as the control were transformed with recombinant pTZ57R. To this end, these two bacteria were inoculated in LB broth supplemented with kanamycin (30 µg/ml). In order to normalize the conditions, the optical densities at 600 nm (OD600) were measured and the transformants were diluted with fresh broth medium into an equal optical density at 600 nm (OD600). These bacteria were made chemically competent by CaCl2 (100 mM) and utilized as the transformation host for equal amounts of recombinant pTZ57R plasmid (3 µl). The transformation products were dispersed on three different Luria-Bertani (LB) (Merck, NJ, USA) plates containing: amp (100 µg/ml), kana (30 µg/ml), and amp (100 µg/ml); plus kana (30 µg/ml) separately. All media were supplemented with a final concentration of 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) (Sigma-Aldrich, MI, USA) to induce protein expression from the Lacuv5 promoters and 100 µM ZnCl2 (Merck, NJ, USA). Furthermore, to evaluate the effect of the temperature on ZFN gene targeting efficiency, all of the above samples were duplicated and incubated at two different temperatures: 30°C and 37°C. The experiments were also performed in triplicate.
3.4.1. Colony Counting
After 16 hours of transformation, the plates were observed to evaluate the rate of bacterial growth on all of the media in both the case and control groups. The growth of bacteria on LB agar containing kanamycin ensures that the bacterial hosts have not lost their kanaR plasmid (pZFN or pP15A, kanaR). The comparison of colony numbers on amp and amp plus kana LB plates between the two groups was used to indicate the ZFN efficiency required to disrupt amp resistance. Therefore, the colony counting on the amp and amp-kana LB plates in all experiments was performed manually.
3.4.2. Spectrophotometry Analysis
To examine and compare the bacterial growth on amp and amp-kana LB plates between the case and control groups, the bacterial colonies on the LB plates were washed in an equal volume of fresh broth medium, and then the optical densities at 600 nm (OD600) were measured using spectrophotometry (Eppendorf Spectrometer, Hamburg, Germany) based on previous studies (26).
3.5. Statistical Analysis
The comparisons of the colony counting and OD600 measurement results for amp and amp-kana-containing media between the case and control groups were performed using the Mann-Whitney U test. The statistical analyses were carried out using the Statistical Package for the Social Sciences (SPSS) software, version 20 (SPSS Inc., IL, USA). Graphs were produced with GraphPad Prism version 6.0 (GraphPad Software, San Diego, CA, USA, available at http://www.graphpad.com/).
3.6. ZFN Expression Assay by SDS-PAGE and Western Blot
To evaluate the ZFN expression, a single positive clone of E. coli TOP10 containing pZFN was inoculated in LB medium containing kanamycin (30 µg/ml), and the bacteria were shaken overnight at 37°C. A 1:50 dilution ratio of the overnight culture was prepared with the LB medium supplemented with 30 µg/ml kanamycin and 100 µM ZnCl2, and the bacteria were grown with shaking at 30°C to an OD600 of 0.8. To induce the protein expression, 1 mM IPTG was added into the growth media; a 1-ml sample was collected 1, 2, and 4 h after induction. Then, the bacteria were harvested by centrifugation at 14,000 rpm for 5 minute at 4°C. The pellets were resuspended in a sample loading buffer and lysed by boiling. The cell lysates were analysed using 15% SDS-polyacrylamide gel electrophoresis (PAGE) gel. Then the proteins were transferred onto a nitrocellulose membrane at 70 V for 90 min. The blotted membrane was blocked with 5% skim milk in Tris buffered saline (TBS) containing 0.05% Tween-20 (TBS-T) overnight, washed with TBS-T, and incubated with horseradish peroxidase-conjugated anti-his antibody (Sigma-Aldrich, MI, USA), diluted in TBS-T for 2 h. After washing with TBS-T to remove the detached antibodies, detection was developed using 3, 3’-diaminobenzidine (DAB) or 3, 3, 5, 5-tetramethylbenzidine (TMB) in various experiments. The E. coli TOP10, E. coli TOP10 containing pP15A, kanaR and the noninduced sample of E. coli TOP10 containing pZFN cell lysates were also analyzed by SDS-PAGE and western blot as the negative controls. The same western blot analysis was performed by anti-HA antibody (Cell Signaling Technology, USA) for the right ZFN expression assay.
4. Results
4.1. Cloning of ZFN target site in pTZ57R
The PCR amplification of the recombinant pTZ57R containing the ZFN target site into the EcoRI-BamHI restriction sites resulted in a 130-bp PCR product, as shown in Figure 4.
Figure 4. Confirmation of the ZFN Target Site Cloning in pTZ57R.
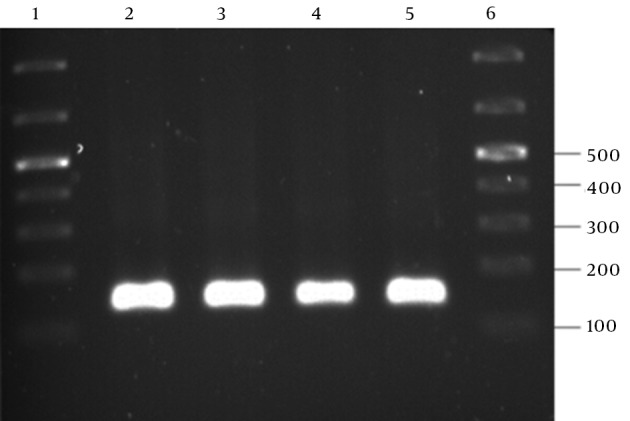
Lanes 2–5, reveal amplification of a 130-bp PCR product; Lanes 1 and 6, GeneRuler 100-bp DNA ladder (Thermo Scientific, Waltham, MA, USA)
4.2. pP15A, kanaR Vector Confirmation
PCR amplification of the M13K07 plasmid DNA fragment containing P15A origin of replication and kanaR gene resulted in a 2592-bp band on agarose gel electrophoresis (Figure 5). The plasmid DNA purified from a single positive clone containing self-ligated pP15A, kanaR was confirmed by both single and double digestion reactions with BamHI and XbaI restriction enzymes (Figure 6).
Figure 5. PCR Amplification of p15A Origin of Replication and kanaR Gene.
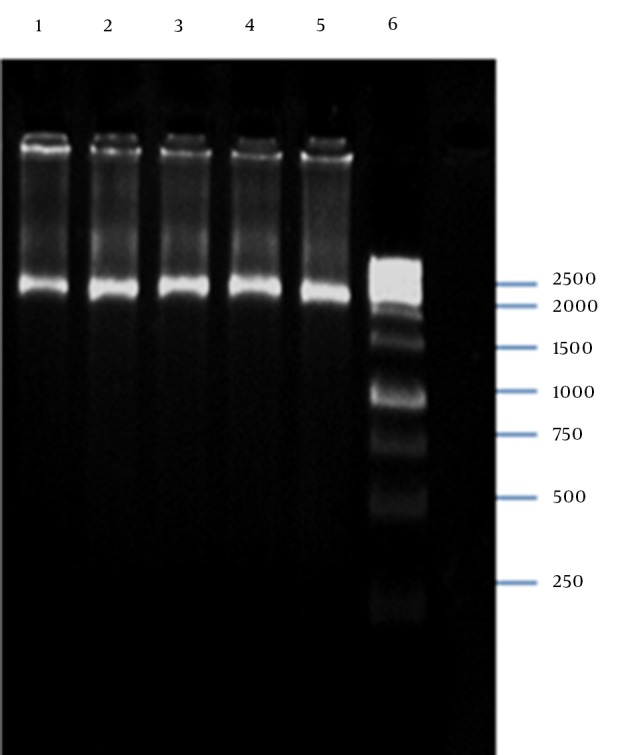
Lanes 1–5 Show the PCR Product 2592 bp in Length as the Sequence Containing p15A Origin of Replication and kanaR gene; Lane 6, GeneRuler 1 kb DNA Ladder (Thermo Scientific, Waltham, MA, USA)
Figure 6. Confirmation of Self-Ligated pP15A, kanaR by Digestion.
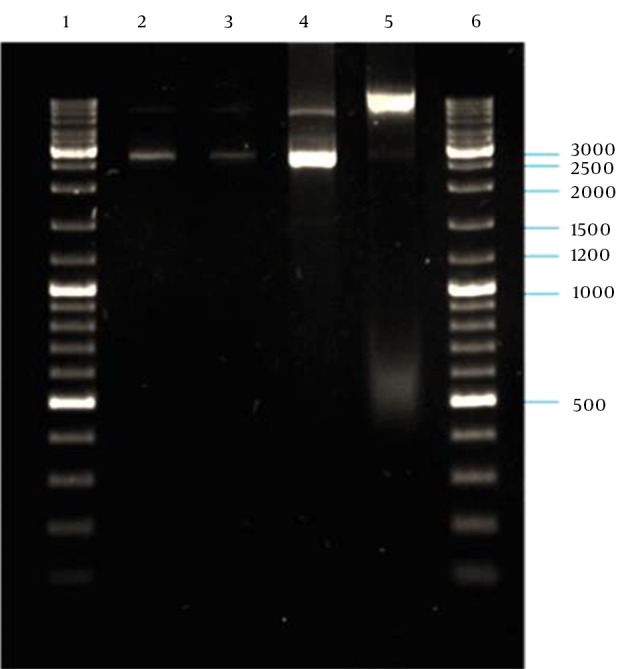
Lane 2: digestion of pP15A, kanaR vector with BamHI; lane 3, xbaI; Lane 4, double digestion with BamHI-XbaI; Lane 5, undigested pP15A, kanaR vector; Lanes 1 and 6, GeneRuler DNA ladder mix (Thermo Scientific, Waltham, MA, USA).
4.3. ZFN Cloning in pP15A, kanaR
Digestion of pZFN with BamHI-XbaI resulted in two restricted DNA fragments (Figure 7), which confirmed cloning of ZFN expression cassette in the pP15A,KanaR vector.
Figure 7. Confirmation of pZFN by Digestion.
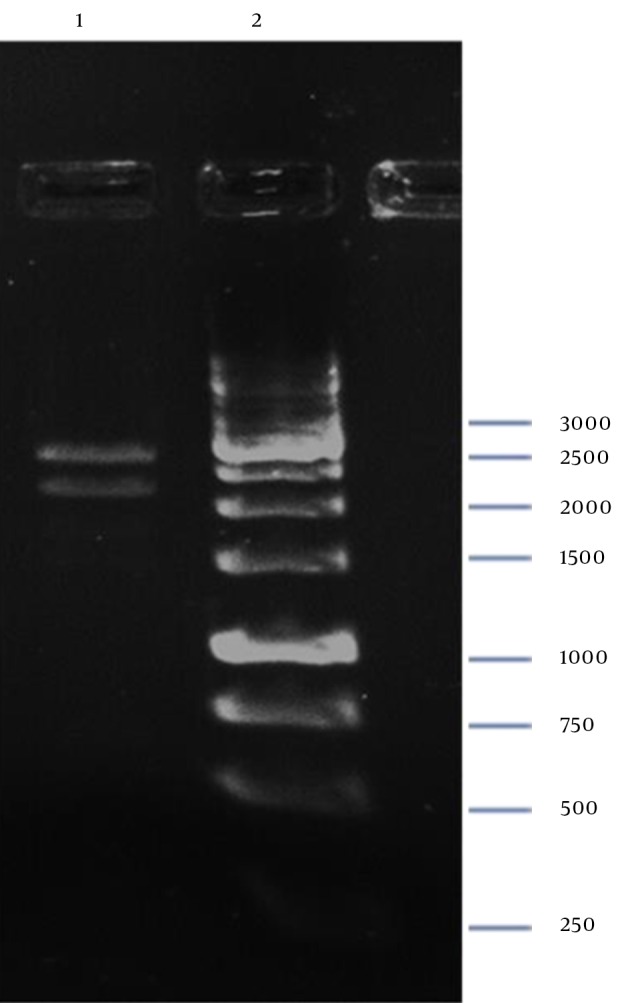
Lane 1, 2208-bp and 2592-bp Fragments; Lane 2, GeneRuler 1kb DNA Ladder (Thermo Scientific, Waltham, MA, USA).
4.4. Evaluation of ZFN Efficiency
4.4.1. Colony Counting
According to the experimental data shown in Figures 8 and 9A, the number of E. coli TOP10 colonies containing pZFN transformed with the pTZ57R ampR plasmid as the case group was significantly lower compared to the E. coli TOP10 colonies containing pP15A, kanaR, transformed with the pTZ57R as the control group on amp-kana (P < 0.001) and amp (P < 0.001) containing LB media at 37ºC.
Figure 8. The Transformation Results From Experiment 1 at 37°C.

Figures A – C, show the transformation products of E. coli TOP10 containing pZFN, transformed with pTZ57R, on amp-kana, kana and amp-Containing Media; figures D – F, Display the Transformation Products of E. coli TOP10 containing the pP15A, kanaR vector transformed with pTZ57R on the same media; the number of colonies on amp-kana and amp in the case group significantly decreased compared to the control group. The High rate of bacterial growth on kana-containing media ensures that the transformants did not lose their kanaR plasmids.
Figure 9. Graphic Comparison of Colony Counting and OD600 Measurement Results Between Case and Control Groups.

The dot plot graph of colony counting results; A, and OD600 measurement results; B from the three transformation procedures performed at two different incubation temperatures: 30 and 37°C. The data shown in the graphs reveal a significant reduction in colony numbers and bacterial growth in the E. coli TOP10 containing pZFN, transformed with pTZ57R, compared to the E. coli TOP10 containing the pP15A, kanaR vector transformed with pTZ57R, on amp and amp-kana-containing media at both temperatures. A comparison of two case groups regarding colony counting and OD600 results did not show any statistically significant association among incubation temperature or ZFN gene targeting efficiency.
Regarding the colony counting results provided in Figures 9A and 10, there was also a significant difference between the case and control groups on amp-kana-containing LB plates (P < 0.001) and amp-containing LB plates (P < 0.001) incubated at 30°C, although there was no significant association between the incubation temperature and the ZFN gene-targeting efficiency. Furthermore, the growth of bacteria on LB agar containing kanamycin was extremely high in both the case and control groups in all experiments (Figure 8).
Figure 10. The Transformation Results From Experiment 1 at 30°C.

Figures A and B, show the transformation products of E. coli TOP10 Containing pZFN, Transformed With pTZ57R, as the case group on amp-kana and amp containing media; Figures C and D, show the transformation products of E. coli TOP10 containing pP15A, kanaR vector transformed with pTZ57R, as the control group, on amp-kana and amp containing media. The plates were incubated overnight at 30°C. A significantly lower level of bacterial growth in the case group compared to the control group was also observed at this temperature.
4.4.2. Spectrophotometry Analysis
The optical density measurement results provided in Figure 9B also demonstrated a significant reduction in bacterial growth in the case compared with the control groups (P <0.001) at both temperatures: 37°C and 30°C, which were consistent with the colony counting results. The spectrophotometry results also confirmed that there was no significant association between the incubation temperature and the ZFN gene-targeting efficiency, which was also in agreement with the colony counting results.
4.5. SDS-PAGE and Western Blot Results
As a result of low copy numbers of pZFN, a low expression level of ZFN was achieved, which was not detectable by SDS-PAGE analysis (Figure 11). The ZFN expression assay performed by western blotting in various experiments resulted in a faint 34-kd protein band, which was visible in about 5 minutes of detection, but it was not capable to be captured (Figure 12).
Figure 11. A Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) Analysis Performed to Detect Zinc Finger Nuclease (ZFN) Expression.

Lanes 1 – 3, The Total Cell Protein (Tcp) of Escherichia coli top10 containing pZFN induced with 1 Mm Isopropyl Β-D-1-thiogalactopyranoside 4, 2, And 1 hour After Induction, Respectively; lane 4, the non-induced E. coli TOP10 containing pZFN; lane 5: the prestained protein ladder (Thermo Scientific, Waltham, MA, USA); lane 6, the TCP of the E. coli TOP10 containing pP15a, kanamycin-resistant ; lane 7 the TCP of E. coli TOP10 as negative controls. The ZFN protein band was not visible on SDS-PAGE due to the low levels of expression; B, thermo scientific prestained protein ladder size.
Figure 12. Detection of Zinc Finger Nuclease Expression by Western Blot Using HRP-Conjugated Anti-His Antibody Resulted in a 34-kd Faint Protein Band.

A, 3, 3, 5, 5-Tetramethylbenzidine and; B, 3,3’-Diaminobenzidine Substrates. However, due to low levels of expression, the band was not captured. the referred bands are his-tagged positive controls
5. Discussion
Antibiotic resistance as a public health threat is associated with high rates of morbidity and mortality. Since the present approaches are only capable of controlling the resistance process, development of a new approach to solve the antibiotic resistance problem is highly required. Identification of ZFNs as the first widely utilized gene editing tool has opened new insights into efficient, targeted manipulation of genomes at any desired region. In the present study, we demonstrated that targeting the β-lactam resistance gene with ZFNs leads to inactivation of β-lactamase synthesis and therefore ampR disruption. In the present study, the CoDA platform was used to design and construct the desired ZFN, while other strategies, including modular assembly (MA) and oligomerized pool engineering (OPEN), also exist to construct the proposed ZFN. In contrast to the MA and OPEN strategies, which are either associated with some technical challenges or are time and labor-intensive, the CoDA approach is an efficient and simple method to engineer and construct ZFNs in a short period of time (27-30). Consistent with the present study, the CoDA platform was also used in previous studies regarding ZFN construction (30-32). Efficient disruption of antibiotic resistance could be achieved by targeting two separate regions of the β-lactamase encoding gene. As the CoDA approach had only explored one potential target site in the β-lactamase gene, the ZFN target site identified in the ampR gene was first cloned in the pTZ57R vector, which natively contained one target site. In agreement with the present experiment, previous studies have also used two separate target sites for more efficient gene disruption (22, 33). In contrast, in other studies, ZFN gene targeting was achieved using only one target site (34, 35). Moreover, according to the plasmid incompatibility rule, if two plasmid vectors are to exist in one bacterial cell simultaneously, the plasmids need to have compatible origins of replication and two different selection markers are required to identify the cells that contain both vectors (36). Therefore, to construct a suitable vector for ZFN cloning and expression to be compatible with the pTZ57R plasmid, both the P15A origin of replication and the Kanamycin resistance gene were employed.
The colony counting data revealed a significant reduction of bacterial growth on amp-kana and amp-containing LB plates in the ZFN-treated bacteria compared to the control group, which was treated with only pP15A, kanaR. The colony counting data were also verified by spectrophotometric analysis through the measurement of OD600. These findings imply the role of ZFN in the disruption of ampR through targeted deletion of the β-lactamase encoding gene of the pTZ57R ampR plasmid, which has led to a reduction in bacterial growth. Regarding β-lactamase encoding gene deletion, no bacterial growth on amp and amp-kana containing media in the ZFN-treated group was expected. Limited bacterial growth was observed on these media, which is illustrative of insufficient ZFN gene disruption efficiency. Consistent with the present data, the ZFN gene-targeting efficiency in previous studies was reported to be incomplete in various species and cell types (37). Another explanation was obtained through a comparison of the present model with a restriction-modification system as one of the natural defense mechanisms against invader DNA (e.g., from the bacteriophage resource) in bacteria.
The efficiency of restriction-modification systems to disrupt the invader DNA is not complete, and only the phage titer would be decreased after the restriction. Another possibility is that β-lactamase gene expression occurs before the cleavage of the gene; thus, the remaining resistance gene product could be responsible for the bacterial growth observed on amp-kana and amp-containing LB plates. Moreover, the lower copy numbers of the p15A origin of replication (15–20 copies per cell) in pZFN compared to the pMB1 origin of replication (> 1000) in pTZ57R might be a logical explanation for the imbalance of ZFNs and the target site copy numbers, which lead to insufficiency of ZFN and disrupt the gene of interest. Another possible deduction is that despite the existence of pZFN in these bacteria, which could be inferred from their ability to grow on kanamycin-containing media, ZFN expression or proper folding or ZFN cleavage activity did not occur. It is also possible that the mutations induced in the resistance gene did not confer the protein function to resist against β-lactam antibiotics. Furthermore, the growth of bacteria on amp-containing media could also be explained by the fact that some of the colonies observed here might have lost their pZFN, kanaR plasmid and only maintained the pTZ57R.
Therefore, the high rate of bacterial growth on kana-containing LB medium in both the case and control groups ensures that the transformants in both groups have maintained kanaR plasmids (pZFN or pP15A, kanaR) and, consequently, the ability to grow on kanamycin-containing media. The findings of the present report suggested that ZFNs tend to be more efficient in hypothermic conditions in comparison with incubation at 37ºC, although the difference was not statistically significant. Doyon and colleagues (38) also demonstrated the positive effect of transient hypothermia (30ºC) on targeted gene disruption in mammalian cells. Despite being expressed at low levels, the engineered ZFN was able to decrease the rate of bacterial growth on amp and amp-kana-containing media by targeted disruption of the pTZ57R β-lactamase gene. This finding could be interpreted by the enzymatic nature of ZFNs as molecular scissors, which, like every enzyme molecule, could target and cleave many DNA molecules that contain the ZFN target site as the substrate. The present study was performed to clarify the ability of ZFNs to overcome antibiotic resistance by targeted disruption of ARGs. However, there are a few points that could have improved the results. First of all, developing an advanced ZFN archive to target a broader range of DNA sequences would allow ZFNs that specifically target the catalytic domain encoding region of the ARGs to be chosen, which could result in more efficient resistance disruption. Moreover, strategies resulting in higher expression levels of ZFN could be employed by making use of the pRSFDuet vector containing RSF1030 origin of replication with a copy number greater than 100, which is compatible with the PMB1 origin of replication in pTZ57R and also carries the kanaR gene. Additionally, this vector contains two T7 promoters, which are suitable for coexpression of both left and right ZFNs in E. coli BL21 (DE3), an optimized expression strain.
Other gene editing tools, including transcription activator-like effector nucleases (TALENs) and the clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated (Cas) system, could also be engaged to this end. Recently, Citorik and colleagues engineered the CRISPR/Cas system to target a few ARGs, including extended spectrum β-lactamases blaSHV-18 and blaNDM-1 and DNA gyrase gyrA, which are responsible for resistance to quinolones, and reported the efficiency of these systems in reducing the number of viable E. coli transformants harboring the resistance genes; they announced this system as a novel generation of antimicrobials, which could act in a sequence-specific manner due to their recognition specificities (39). To tackle the antibiotic resistance issue at the environmental level, phages that are able to transduce the bacterial species with resistance genes can be employed. These phages should be engineered to express ZFNs or other gene editing tools, including TALEN and the CRISPR/Cas system, against conserved sequences among different resistance genes. Due to the diversity and dissemination of resistance genes, several recombinant phages will be required to put this idea into practice. Consequently, a recombinant phage library could be constructed for different bacteria with different types of antibiotic resistance. The recombinant phages can ultimately be released into the hospital and urban wastewater systems to reduce antibiotic resistance. In practice, this approach could be useful for most resistant bacteria, especially mycobacterium species, as it is one of the most clinically resistant bacterial strains (40). Moreover, there are several pathogenic microorganisms with dispersed or compact virulence genes (pathologic islands), which can be converted to nonpathogenic species by employing the gene editing technology to make these genes knockout, which could make the pathogenic species suitable candidates to be used as live attenuated vaccines.
Acknowledgments
The authors gratefully acknowledge all their colleagues in the Genetics and Molecular Biology Department.
Footnotes
Authors’ Contribution:The original idea was developed by Dr. Hossein Khanahmad. Mansoureh Shahbazi Dastjerdeh and Dr. Hossein Khanahmad performed the study concepts and design, developed the protocol, conducted the analysis, and interpreted the data. Mansoureh Shahbazi Dastjerdeh, Shirin Kouhpayeh, Zahra mohammadi, and Laleh Shariati wrote the manuscript. The laboratory tasks were mainly performed by Mansoureh Shahbazi Dastjerdeh. Faezeh Sabzehei, Zahra Hejazi, Parisa Rabiei, and Mostafa Manian collaborated on the laboratory work. Data acquisition was primarily performed by Mansoureh Shahbazi. Statistical analyses were carried out by Mansoureh Shahbazi, Dr. Hossein Khanahmad, and Shirin Kouhpayeh. Critical revision of the manuscript was performed by Shirin Kouhpayeh and Dr. Hossein Khanahmad. Dr. Hossein Khanahmad was the study supervisor, and Dr. Mansour Salehi was the advisor of the project.
Funding/Support:This work was supported by Grant 393358 from the Isfahan University of Medical Sciences.
References
- 1.Spellberg B, Bartlett JG, Gilbert DN. The future of antibiotics and resistance. N Engl J Med. 2013;368(4):299–302. doi: 10.1056/NEJMp1215093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kummerer K. Antibiotics in the aquatic environment--a review--part II. Chemosphere. 2009;75(4):435–41. doi: 10.1016/j.chemosphere.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 3.Singer RS, Finch R, Wegener HC, Bywater R, Walters J, Lipsitch M. Antibiotic resistance—the interplay between antibiotic use in animals and human beings. Lance Infect Dise. 2003;3(1):47–51. doi: 10.1016/s1473-3099(03)00490-0. [DOI] [PubMed] [Google Scholar]
- 4.Cabello FC. Heavy use of prophylactic antibiotics in aquaculture: a growing problem for human and animal health and for the environment. Environ Microbiol. 2006;8(7):1137–44. doi: 10.1111/j.1462-2920.2006.01054.x. [DOI] [PubMed] [Google Scholar]
- 5.Sarmah AK, Meyer MT, Boxall AB. A global perspective on the use, sales, exposure pathways, occurrence, fate and effects of veterinary antibiotics (VAs) in the environment. Chemosphere. 2006;65(5):725–59. doi: 10.1016/j.chemosphere.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 6.Hoa PT, Managaki S, Nakada N, Takada H, Shimizu A, Anh DH, et al. Antibiotic contamination and occurrence of antibiotic-resistant bacteria in aquatic environments of northern Vietnam. Sci Total Environ. 2011;409(15):2894–901. doi: 10.1016/j.scitotenv.2011.04.030. [DOI] [PubMed] [Google Scholar]
- 7.Grundmann H, Klugman KP, Walsh T, Ramon-Pardo P, Sigauque B, Khan W, et al. A framework for global surveillance of antibiotic resistance. Drug Resist Updat. 2011;14(2):79–87. doi: 10.1016/j.drup.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 8.Tatavarthy A, Peak K, Veguilla W, Reeves F, Cannons A, Amuso P, et al. Comparison of antibiotic susceptibility profiles and molecular typing patterns of clinical and environmental Salmonella enterica serotype Newport. J Food Prot. 2006;69(4):749–56. doi: 10.4315/0362-028x-69.4.749. [DOI] [PubMed] [Google Scholar]
- 9.Abriouel H, Omar NB, Molinos AC, Lopez RL, Grande MJ, Martinez-Viedma P, et al. Comparative analysis of genetic diversity and incidence of virulence factors and antibiotic resistance among enterococcal populations from raw fruit and vegetable foods, water and soil, and clinical samples. Int J Food Microbiol. 2008;123(1-2):38–49. doi: 10.1016/j.ijfoodmicro.2007.11.067. [DOI] [PubMed] [Google Scholar]
- 10.Kemper N. Veterinary antibiotics in the aquatic and terrestrial environment. Ecolog Indicat. 2008;8(1):1–13. [Google Scholar]
- 11.Lavilla Lerma L, Benomar N, Knapp CW, Correa Galeote D, Galvez A, Abriouel H. Diversity, distribution and quantification of antibiotic resistance genes in goat and lamb slaughterhouse surfaces and meat products. PLoS One. 2014;9(12):e29384. doi: 10.1371/journal.pone.0114252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Norman A, Hansen LH, Sorensen SJ. Conjugative plasmids: vessels of the communal gene pool. Philos Trans R Soc Lond B Biol Sci. 2009;364(1527):2275–89. doi: 10.1098/rstb.2009.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andes D, Craig WA. Treatment of infections with ESBL-producing organisms: pharmacokinetic and pharmacodynamic considerations. Clin Microbiol Infect. 2005;11 Suppl 6:10–7. doi: 10.1111/j.1469-0691.2005.01265.x. [DOI] [PubMed] [Google Scholar]
- 14.Livermore DM, Brown DF. Detection of beta-lactamase-mediated resistance. J Antimicrob Chemother. 2001;48 Suppl 1:59–64. doi: 10.1093/jac/48.suppl_1.59. [DOI] [PubMed] [Google Scholar]
- 15.Drawz SM, Bonomo RA. Three decades of beta-lactamase inhibitors. Clin Microbiol Rev. 2010;23(1):160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilke MS, Lovering AL, Strynadka NC. Beta-lactam antibiotic resistance: a current structural perspective. Curr Opin Microbiol. 2005;8(5):525–33. doi: 10.1016/j.mib.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 17.Horrevorts AM, Borst J, Puyk RJ, De Ridder R, Dzoljicdanilovic G, Degener JE, et al. Ecology of Pseudomonas aeruginosa in patients with cystic fibrosis. J Med Microbiol. 1990;31(2):119–24. doi: 10.1099/00222615-31-2-119. [DOI] [PubMed] [Google Scholar]
- 18.Wu J, Kandavelou K, Chandrasegaran S. Custom-designed zinc finger nucleases: what is next? Cell Mol Life Sci. 2007;64(22):2933–44. doi: 10.1007/s00018-007-7206-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sander JD, Maeder ML, Joung JK. Engineering Designer Nucleases with Customized Cleavage Specificities. Curr Protoc Mol Biol. 2011;12(3):1–3. doi: 10.1002/0471142727.mb1213s96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis D, Stokoe D. Zinc finger nucleases as tools to understand and treat human diseases. BMC Med. 2010;8:42. doi: 10.1186/1741-7015-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Remy S, Tesson L, Menoret S, Usal C, Scharenberg AM, Anegon I. Zinc-finger nucleases: a powerful tool for genetic engineering of animals. Transgenic Res. 2010;19(3):363–71. doi: 10.1007/s11248-009-9323-7. [DOI] [PubMed] [Google Scholar]
- 22.McMahon MA, Rahdar M, Porteus M. Gene editing: not just for translation anymore. Nat Methods. 2012;9(1):28–31. doi: 10.1038/nmeth.1811. [DOI] [PubMed] [Google Scholar]
- 23.Hauschild J, Petersen B, Santiago Y, Queisser AL, Carnwath JW, Lucas-Hahn A, et al. Efficient generation of a biallelic knockout in pigs using zinc-finger nucleases. Proc Natl Acad Sci U S A. 2011;108(29):12013–7. doi: 10.1073/pnas.1106422108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cui X, Ji D, Fisher DA, Wu Y, Briner DM, Weinstein EJ. Targeted integration in rat and mouse embryos with zinc-finger nucleases. Nat Biotechnol. 2011;29(1):64–7. doi: 10.1038/nbt.1731. [DOI] [PubMed] [Google Scholar]
- 25.Meyer M, de Angelis MH, Wurst W, Kuhn R. Gene targeting by homologous recombination in mouse zygotes mediated by zinc-finger nucleases. Proc Natl Acad Sci U S A. 2010;107(34):15022–6. doi: 10.1073/pnas.1009424107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sondi I, Salopek-Sondi B. Silver nanoparticles as antimicrobial agent: a case study on E. coli as a model for Gram-negative bacteria. J Colloid Interface Sci. 2004;275(1):177–82. doi: 10.1016/j.jcis.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 27.Wright DA, Thibodeau-Beganny S, Sander JD, Winfrey RJ, Hirsh AS, Eichtinger M, et al. Standardized reagents and protocols for engineering zinc finger nucleases by modular assembly. Nat Protoc. 2006;1(3):1637–52. doi: 10.1038/nprot.2006.259. [DOI] [PubMed] [Google Scholar]
- 28.Maeder ML, Thibodeau-Beganny S, Sander JD, Voytas DF, Joung JK. Oligomerized pool engineering (OPEN): an 'open-source' protocol for making customized zinc-finger arrays. Nat Protoc. 2009;4(10):1471–501. doi: 10.1038/nprot.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JS, Lee HJ, Carroll D. Genome editing with modularly assembled zinc-finger nucleases. Nat Methods. 2010;7(2):91. doi: 10.1038/nmeth0210-91a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sander JD, Dahlborg EJ, Goodwin MJ, Cade L, Zhang F, Cifuentes D, et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat Methods. 2011;8(1):67–9. doi: 10.1038/nmeth.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Osborn MJ, DeFeo AP, Blazar BR, Tolar J. Synthetic zinc finger nuclease design and rapid assembly. Hum Gene Ther. 2011;22(9):1155–65. doi: 10.1089/hum.2011.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson KA, McEwen AE, Pruett-Miller SM, Zhang J, Kildebeck EJ, Porteus MH. Expanding the Repertoire of Target Sites for Zinc Finger Nuclease-mediated Genome Modification. Mol Ther Nucleic Acids. 2013;2:e29384. doi: 10.1038/mtna.2013.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee HJ, Kweon J, Kim E, Kim S, Kim JS. Targeted chromosomal duplications and inversions in the human genome using zinc finger nucleases. Genome Res. 2012;22(3):539–48. doi: 10.1101/gr.129635.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qu X, Wang P, Ding D, Li L, Wang H, Ma L, et al. Zinc-finger-nucleases mediate specific and efficient excision of HIV-1 proviral DNA from infected and latently infected human T cells. Nucleic Acids Res. 2013;41(16):7771–82. doi: 10.1093/nar/gkt571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qu X, Wang P, Ding D, Wang X, Zhang G, Zhou X, et al. Zinc finger nuclease: a new approach for excising HIV-1 proviral DNA from infected human T cells. Mol Biol Rep. 2014;41(9):5819–27. doi: 10.1007/s11033-014-3456-3. [DOI] [PubMed] [Google Scholar]
- 36.Velappan N, Sblattero D, Chasteen L, Pavlik P, Bradbury AR. Plasmid incompatibility: more compatible than previously thought? Protein Eng Des Sel. 2007;20(7):309–13. doi: 10.1093/protein/gzm005. [DOI] [PubMed] [Google Scholar]
- 37.Guschin DY, Waite AJ, Katibah GE, Miller JC, Holmes MC, Rebar EJ. A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol. 2010;649:247–56. doi: 10.1007/978-1-60761-753-2_15. [DOI] [PubMed] [Google Scholar]
- 38.Doyon Y, Choi VM, Xia DF, Vo TD, Gregory PD, Holmes MC. Transient cold shock enhances zinc-finger nuclease-mediated gene disruption. Nat Methods. 2010;7(6):459–60. doi: 10.1038/nmeth.1456. [DOI] [PubMed] [Google Scholar]
- 39.Citorik RJ, Mimee M, Lu TK. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat Biotechnol. 2014;32(11):1141–5. doi: 10.1038/nbt.3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hatfult GF, Sarkis GJ. DNA sequence, structure and gene expression of mycobacteriophage L5: a phage system for mycobacterial genetics. Molecul Microbiol. 1993;7(3):395–405. doi: 10.1111/j.1365-2958.1993.tb01131.x. [DOI] [PubMed] [Google Scholar]