

Abstract
Background:
Pulmonary hypertension (PH) frequently complicates the course of idiopathic pulmonary fibrosis (IPF) patients and is associated with significantly worse outcomes. The aim of the present study was to investigate the incidence of PH in IPF patients and evaluate the correlation between clinical parameters and systolic pulmonary artery pressure (sPAP).
Methods:
Hospitalized patients with IPF, who were evaluated for sPAP by Doppler echocardiography from January 2004 to December 2011, were enrolled in our study. Patients were defined as PH by an estimated sPAP > 50 mmHg and graded as PH likely, PH possible and PH unlikely, based on the 2009 European Society of Cardiology/European Respiratory Society PH Guidelines. The correlations between clinical parameters and sPAP were analyzed by multiple linear regression.
Results:
Totally, 119 IPF patients were enrolled in our study and 28 (23.5%), 20 (16.8%) and 71 (59.7%) patients were PH likely, PH possible and PH unlikely, respectively. Borg dyspnea score was positively correlated with sPAP, r = 0.467, P < 0.001. Oxygen saturation was negatively correlated with sPAP, r = −0.416, P < 0.001. Diffusing capacity of the lung for carbon monoxide percentage predicted was negatively correlated with sPAP, r = −0.424, P = 0.003. N-terminal fragment of pro-brain natriuretic peptide and pulmonary artery width was positively correlated with sPAP, r = 0.452, P = 0.011 and r = 0.513, P < 0.001, respectively.
Conclusions:
The incidence of PH in IPF patients was 23.5% in a single center of China. PH may worsen the dyspnea, right heart dysfunction and decrease the life quality of the patients with IPF.
Keywords: Idiopathic Pulmonary Fibrosis, Incidence, Pulmonary Hypertension
INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a specific form of chronic, progressive, fibrosing interstitial pneumonia of unknown cause that occurs primarily in older adults.[1] Pulmonary hypertension (PH) describes a group of devastating diseases causing breathlessness, loss of exercise capacity, and death due to right heart failure. PH frequently complicates IPF and relates to the worse outcomes.[2,3,4,5,6,7,8,9,10,11] Data regarding the presence and significance of PH mainly came from IPF patients who were evaluated for lung transplantation.[1] The incidence of PH in IPF patients ranged from 8.1%[5] to 86.4%,[12] due to different measurement time of PH during the course of the disease, diverse diagnosis method and criteria used to define PH. Previous studies demonstrated that reduced diffusing capacity of the lung for carbon monoxide (DLCO) or supplemental oxygen requirement or impaired exercise capacity might better reflect coexistence of PH in IPF patients.[2,3,4,13] Nathan et al.[14] found that pulmonary function parameters such as forced vital capacity (FVC) and total lung capacity (TLC) had a poor relationship with PH. In addition, studies[15,16] showed that plasma brain natriuretic peptide (BNP) correlated with systolic pulmonary artery pressure (sPAP) in IPF patients and could be used as a biomarker to assess the prognosis of IPF patients with PH.[10,17]
All of these studies indicated that PH might not be the rare event in IPF patients and contribute to the pathophysiological and clinical features of IPF. The aim of this study was to investigate the incidence of PH in IPF patients and to evaluate the correlation between sPAP and clinical parameters in a single center in China.
METHODS
Study population
The study population was selected from the database of Beijing Institute of Respiratory Medicine Interstitial Lung Disease Group, Beijing Chao-Yang Hospital, Capital Medical University, China. Hospitalized patients with IPF were enrolled from January 2004 to December 2011. Diagnosis of IPF was based on the criteria of the American Thoracic Society and European Respiratory Society (ATS/ERS) consensus classification of idiopathic interstitial pneumonias (IIPs).[18] All the patients fulfilled the criteria: (1) Compatible clinical manifestations such as progressive dyspnea and bilateral predominantly basal crackles; (2) restrictive lung functional defect and gas exchange impairment; (3) typical abnormalities indicative of usual interstitial pneumonitis (UIP) on chest high resolution computerized tomography (HRCT) including bilateral lung reticular abnormality with predominantly basal/subpleural honeycombing and/or traction bronchiectasis; (4) bronchoalveolar lavage fluid profiles in accordance with UIP or/and pathological features indicative of UIP on surgical lung biopsy; (5) no evidence of known causes of pulmonary fibrosis. Patients who fulfilled the following criteria were excluded: (1) Other types of interstitial lung disease (ILD), such as connective tissue disease-ILD, drug-induced ILD, or other types of IIP; (2) acute pulmonary embolism or chronic thromboembolic PH; (3) left heart function failure and other heart diseases; (4) chest HRCT, pulmonary function, blood gas analysis and echocardiography data were missing. The flow chart of the patient enrollment in the study is represented in Figure 1.
Figure 1.
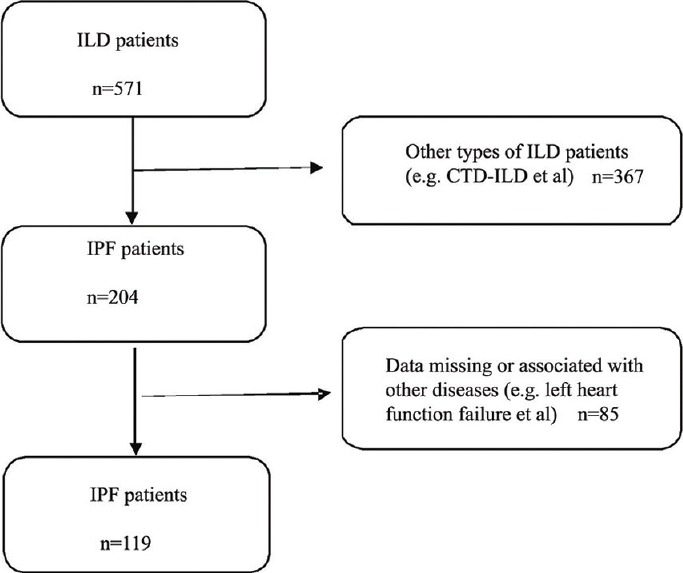
Flow chart of the study population enrollment. ILD: Interstitial lung disease; CTD: Connective tissue disease; IPF: Idiopathic pulmonary fibrosis.
The study protocol was approved by the Human Ethics Review Committee of the Beijing Chao-Yang Hospital, Capital Medical University, China. Written informed consent was obtained from all the patients.
Smoking status definition
Smoking status was defined as current smokers (a minimum of one cigarette a day for a minimum of 1-year without cessation, or stopped smoking <12 months before clinical presentation), ex-smokers (a minimum of one cigarette a day for a minimum of 1-year but had stopped smoking for at least 12 months before clinical presentation) and nonsmokers.
Borg dyspnea scale
Dyspnea was evaluated by self-assessment according to the modified Borg dyspnea scale and graded from 0 (no dyspnea) to 10 (maximal imaginable dyspnea).[19]
Pulmonary function test
Lung volumes and DLCO were measured by Master Screen (Jaeger, Würzburg, Germany) using the routine method.[20] The parameters included the predicted percentage of maximal VC, the predicted percentage of FVC, the predicted percentage of TLC, and predicted percentage of DLCO (DLCO % pred).
Arterial blood gas analysis
Arterial blood gas analysis was measured by ALB 700 (Radiometer Medical Bronshoj, Denmark). Arterial partial pressure of oxygen (PaO2), oxygen saturation (SaO2) and the inspired oxygen concentration (FiO2) were recorded. Oxygen index (OI, PaO2/FiO2) was calculated.
Doppler echocardiography
Doppler transthoracic echocardiography (Philips IE33, Washington, USA) was routinely made according to the diagnostic evaluation protocol of ILD. PAP was estimated based on the peak tricuspid regurgitation velocity (TRV). The tricuspid regurgitation pressure gradient (TRPG) was calculated according to the modified Bernoulli equation: TRPG = 4 × (TRV)2 and sPAP was calculated from the equation: sPAP = TRPG + estimated right atrial pressure. PH was defined as an estimated sPAP >50 mmHg by Doppler echocardiography based on the 2009 European Society of Cardiology (ESC)/ERS PH Guideline[21] and was divided into three grades: (1) PH unlikely: TRV ≤2.8 m/s, sPAP ≤36 mmHg. (2) PH possible: TRV 2.9–3.4 m/s, sPAP 37–50 mmHg. (3) PH likely: TRV >3.4 m/s, sPAP >50 mmHg.
Measurement for plasma N-terminal fragment of pro-brain natriuretic peptide
Plasma N-terminal fragment of pro-BNP (NT-pro BNP) was detected by using rapid detection kit (colloidal gold method) in FIA-8000 immunoassay analyzer (Pulang Nanjing Medical Equipment Limited Company, Jiangsu, China). The range of normal value is 0–228 pg/ml.
Statistical analysis
Statistical analysis was performed with SPSS 17.0 software package (SPSS Inc., Chicago, IL, USA). Categorical data were reported as frequencies and percentages, and continuous variables were expressed as mean ± standard deviation or as median and interquartile range unless stated otherwise. The Kolmogorov-Smirnov test was applied to assess the distribution of continuous variables. Nonparametric test and analysis of variance were used to compare the difference of clinical parameters among groups. The Chi-squared test was used for comparisons of categorical variables. Multiple linear regression analysis was used to find potential factors affecting the outcome of patients. The correlation between clinical parameters and sPAP was analyzed with Pearson correlation if continuous variables were in the normal distribution and with the Spearman rank correlation if continuous variables were not in the normal distribution, respectively. A P < 0.05 was considered statistically significant.
RESULTS
Demographic variables
Of 119 IPF patients involved in this study, 101 were males and 18 females. The average age at initial diagnosis as IPF was 68 (57, 73) years. 89 patients (75%) had a history of smoking, of which 20 patients (17%) were current smokers. Thirty patients (25%) were nonsmokers.
Incidence of pulmonary hypertension in idiopathic pulmonary fibrosis patients
One hundred and nineteen IPF patients were evaluated for sPAP by echocardiography and were diagnosed as: PH likely (n = 28, 23.5%), PH possible (n = 20, 16.8%), PH unlikely (n = 71, 59.7%), respectively. The incidence of PH in IPF was 23.5% (28/119) when only likely PH was definite and the incidence of PH in IPF was 40.3% (48/119) when likely PH and possible PH were taken together.
Relationship of clinical and physiological parameters and systolic pulmonary artery pressure
As shown in Table 1, there was no statistically significant difference in age, gender, disease duration among IPF patients with different sPAP. However, there was statistically significant difference in Borg dyspnea score, SaO2, OI, the width of pulmonary artery and NT-pro BNP (P < 0.05). The width of pulmonary artery and NT-pro BNP would be analyzed additionally, for the reason that they might not be the cause of inducing PH. By using multiple linear regression, there was a correlation between Borg dyspnea score, SaO2 and sPAP (P < 0.05). The regression equation could be got after the confounding factors were adjusted. Y =3.278 + 0.16X1 − 0.023X2. Y is sPAP, X1 is Borg dyspnea score, and X2 is SaO2. In the case of constant SaO2, the risk of sPAP increases by 0.16 when Borg dyspnea score increases by one. When the Borg dyspnea score is constant, the risk of sPAP decreases by 0.023 when the SaO2 increases by 1%.
Table 1.
Clinical and physiological parameters related to PH in IPF patients
| Parameters | PH unlikely (n = 71) | PH possible (n = 20) | PH likely (n = 28) | P1* | P2† |
|---|---|---|---|---|---|
| Age (years) | 65.44 ± 9.25 | 66.10 ± 12.35 | 69.00 ± 9.65 | 0.275 | |
| Male/female | 60/11 | 16/4 | 25/3 | 0.670 | |
| Duration (months) | 12 (2, 48) | 24 (6, 45) | 24 (12, 36) | 0.479 | |
| Borg dyspnea score | 2.63 ± 1.67 | 4.08 ± 1.82 | 4.54 ± 1.71 | 0.001 | 0.001 |
| SaO2 (%) | 95.9 (93.7, 96.8) | 94.5 (88.85, 96.4) | 92.65 (89.05, 95.4) | 0.001 | 0.045 |
| OI (mmHg) | 332.9 ± 94.4 | 284.5 ± 122.9 | 230.5 ± 108.8 | 0.001 | 0.226 |
| FVC (L) | 2.45 ± 0.83 | 2.22 ± 0.70 | 2.16 ± 0.54 | 0.288 | |
| FVC % pred | 71.18 ± 21.97 | 71.98 ± 23.53 | 64.91 ± 14.01 | 0.443 | |
| FEV1 (L) | 2.02 ± 0.64 | 1.83 ± 0.43 | 1.79 ± 0.35 | 0.241 | |
| FEV1% pred | 76.31 ± 21.24 | 76.21 ± 22.26 | 69.23 ± 10.42 | 0.422 | |
| FEV1/FVC (%) | 83.51 ± 8.10 | 84.08 ± 6.87 | 83.82 ± 10.19 | 0.971 | |
| TLC (L) | 4.30 ± 1.14 | 3.92 ± 1.33 | 3.87 ± 1.01 | 0.293 | |
| TLC % pred | 71.28 ± 17.88 | 74.81 ± 30.15 | 62.93 ± 15.77 | 0.205 | |
| RV/TLC (%) | 42.68 ± 10.42 | 40.64 ± 15.06 | 43.01 ± 8.80 | 0.798 | |
| DLCO % pred | 36.87 ± 17.70 | 29.02 ± 15.34 | 21.67 ± 16.88 | 0.006 | 0.002 |
| DLCO-VA %pred | 60.67 ± 21.49 | 48.50 ± 19.67 | 43.12 ± 27.81 | 0.011 | 0.696 |
| Width of PA (mm) | 25.29 ± 2.89 | 27.59 ± 3.61 | 30.18 ± 5.87 | 0.001 | |
| NT-pro BNP‡ (pg/ml) | 260 (55, 510) | 766 (184, 2734) | 2049 (507, 4662) | 0.002 |
Data were presented as mean ± SD/medians and IQR or number (%) unless otherwise indicated. *P1 value was obtained by using different statistical method to compare the difference of clinical parameters among IPF patients with different sPAP; †P2 value was obtained using multiple linear regression; ‡Data on 53 patients were available. P<0.05 was considered to be statistically significant. PH: pulmonary hypertension; SaO2: Oxygen saturation; OI: Oxygen index; FVC: Forced vital capacity; FEV1: Forced expiratory volume in 1 s; TLC: Total lung capacity; RV: Residual volume; DLCO: Diffusing capacity of the lung for carbon monoxide; VA: Alveolar ventilation; PA: Pulmonary artery; NT-pro BNP: N-terminal fragment of pro-brain natriuretic peptide; SD: Standard deviation; IQR: Interquartile range; IPF: Idiopathic pulmonary fibrosis.
Similarly, a statistically significant difference was found in DLCO % pred and DLCO/alveolar ventilation(VA) %pred (P < 0.05). No significant difference existed in FVC, forced expiratory volume in 1 s (FEV1), TLC, FEV1/FVC and residual volume (RV)/TLC. By using multiple linear regression, there was a relationship between DLCO % pred and sPAP (P = 0.002). The regression equation could be got after the confounding factors were adjusted. Y = 2.024 − 0.015X. Y is sPAP, X is DLCO % pred. Decreased DLCO % pred was the risk factors of sPAP increasing. The risk of sPAP decreases by 0.015 when DLCO % pred increases by 1.
Correlation between clinical parameters and systolic pulmonary artery pressure
Borg dyspnea score showed a significantly positive correlation with sPAP (r = 0.467, P < 0.001). Conversely, SaO2 showed a significantly negative correlation with sPAP (r = −0.416, P < 0.001). DLCO % pred also showed a significantly negative correlation with sPAP (r = −0.424, P = 0.003). As shown in Figure 2, the pulmonary artery width and NT-pro BNP showed a significantly positive correlation with sPAP (r = 0.513, P < 0.001; r = 0.452, P = 0.011 [Figure 2a and b]).
Figure 2.
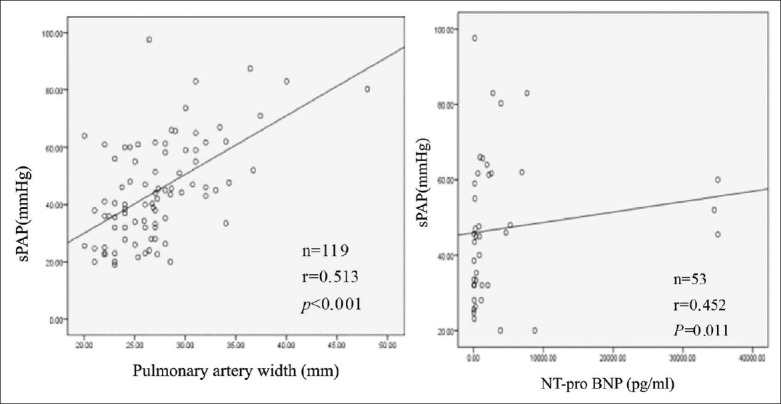
Correlation analysis between the pulmonary artery width and NT-pro BNP with sPAP in IPF patients, respectively. (a) A positive correlation was found between the pulmonary artery width and sPAP in subjects with IPF (r = 0.513, P < 0.001); (b) A positive correlation was found between NT-pro BNP and sPAP in subjects with IPF (r = 0.452, P = 0.011). Analysis was performed with the Spearman rank correlation coefficient and Pearson rank correlation coefficient. sPAP: Systolic pulmonary artery pressure; NT-pro BNP: N-terminal fragment of pro-BNP; IPF: Idiopathic pulmonary fibrosis.
DISCUSSION
Recently, awareness of complications and comorbid conditions associated with IPF is increasing because they can affect disease outcome and survival. The gold standard for diagnosis of PH is mean PAP above 25 mmHg at rest as assessed by right heart catheterization (RHC). It is not routinely performed due to its invasive feature. Noninvasive Doppler transthoracic echocardiography is done clinically as an alternative approach for the detection of PH. However, no consistent and reliable sPAP cut-off value was used for echocardiographic diagnosis of PH until recently. We conducted an investigation of PH in IPF patients by using Doppler echocardiography to evaluate sPAP. PH was diagnosed by sPAP >50 mmHg based on the echocardiographic diagnosis criteria described in 2009 ESC/ERS PH guidelines[21] in a single center of China. The main findings in our study were: (1) The incidence of PH in IPF patients was 23.5%. (2) In IPF patients with PH, Borg dyspnea score was positively while SaO2 and DLCO % pred was negatively correlated with sPAP. (3) NT-pro BNP and the pulmonary artery width were positively correlated with sPAP.
The presence of PH has been associated with increased risk of mortality for patients with IPF.[1] The incidence of PH in IPF patients was variable depending on the measuring methods of sPAP or diagnostic criteria of PH as well as study population. The reported incidence of PH in IPF patients was mostly gained during the end stage recommended for lung transplantation. However, PH is a common complication in patients with IPF assessed by echocardiography and is not limited in patients with advanced disease.[2,13] Agarwal et al.[22] defined PH as sPAP ≥40 mmHg by Doppler echocardiography and showed that the incidence of PH in IPF patients was 36%. Recently, another two studies[11,13] reported a higher incidence of PH (39.7 − 55.0%) in IPF patients, based on sPAP >36 mmHg by echocardiography. The 23.5% incidence of PH in IPF in the current study is lower than the aforementioned report because of the use of more strict criteria of sPAP >50 mmHg. However, the incidence of PH in IPF patients was 40.3% if likely PH and possible PH were pooled up, or PH was defined as sPAP >36 mmHg. Nadrous et al.[2] reported that the incidence of PH in IPF patients was 84.1% when PH was defined as sPAP ≥36 mmHg by Doppler echocardiography, but the incidence was 30.7% if PH was defined as sPAP >50 mmHg, which is still higher than our outcomes. It is possible that the lower incidence of PH in our current study might be due to the fact that every patient was initially diagnosed as IPF. It was a pity that we could not evaluate the extent of the disease by chest HRCT and pulmonary function parameters.
In our study, we found that there was statistically significant difference in Borg dyspnea score, SaO2, and DLCO % pred among IPF patients with different degree of PH. Borg dyspnea score showed a significant positive c orrelation with sPAP and SaO2 or DLCO % pred showed a significant negative correlation with sPAP, which suggests that Borg dyspnea score, SaO2, and DLCO % pred may be predictors of sPAP. Borg dyspnea score, SaO2, and DLCO % pred may reflect hypoxic condition of IPF patients. Hypoxemia may induce vasoconstriction and sPAP elevating further. Therefore, hypoxemia would be one of the mechanisms, which induces the development of PH in IPF patients. Currently, there is no specific therapy recommended for PH associated with IPF. Studies[3,12] reported that sPAP elevations had a significant association with oxygen requirements in IPF patients. Huppmann et al.[23] also confirmed that patients with signs of PH also seemed to benefit from pulmonary rehabilitation (PR). Therefore, long-term oxygen therapy to improve the anoxic condition and PR to improve respiratory function should be recommended to IPF patients with HP.
Survival of patients with PH is closely related to right ventricular (RV) function.[24,25,26,27] Both pulmonary artery width and NT-pro BNP are the markers of reflecting right heart function. Studies confirmed that the plasma level of BNP correlated with the elevation of sPAP[15,16] and was a predictor to prognosis of IPF.[10,17] Moreover, NT-pro BNP correlates with sPAP better than BNP and shows less variability.[28] Our study showed that both NT-pro BNP and pulmonary artery width were positively correlated with sPAP in IPF patients. IPF patients may have poorer right ventricular function accompany the elevation of sPAP. It is very important to initially evaluate sPAP in IPF patients.[11,29] Echocardiography and NT-pro BNP measurement, as the noninvasive, repeatable, and inexpensive methods, can be used widely to detect RV dysfunction in IPF patients.
Our study has some limitations. First, the lack of RHC to confirm the presence and degree of PH might result in false estimation of the incidence of PH. Second, the outcome might be influenced by the potential patients selecting biases from a single center. On the other hand, the protocols for IPF and PH diagnosis were relatively consistent.
In spite of the above-mentioned limitations, the current study has its peculiar clinical value. RHC is not used as a routine tool because it is an invasive procedure and could cause some morbidity (1.1%) and mortality (0.055%) even when performed at experienced centers.[30] Doppler echocardiography is noninvasive, convenient, inexpensive, widely available and even relatively reliable for evaluating PH.[10,16,31] A meta-analysis[32] summarized that the correlation coefficient of sPAP estimated by echocardiography versus RHC was 0.70, and the sensitivity and specificity of echocardiography in diagnosing PH are 83% and 72%, respectively.
In conclusion, the present study showed that the incidence of PH in IPF patients was 23.5% at a tertiary referral center in China. The increased Borg dyspnea score, decreased SaO2, and reduced DLCO % pred were all associated with an elevation of sPAP in IPF patients. IPF patients with higher sPAP may have poorer right ventricular function. Therefore, in patients with IPF, evaluating the presence of PH may be useful in determining disease prognosis and evolution. Well-designed, prospective, longitudinal, multicenter studies are needed in the future.
Footnotes
Edited by: Li-Min Chen
Source of Support: This work was supported by grants from the National Key Technology R&D Program, the Ministry of Science and Technology of China (No. 2012BAI05B02) and the Key Program of Beijing Nature Science Foundation (No. 7131008).
Conflict of Interest: None declared.
REFERENCES
- 1.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nadrous HF, Pellikka PA, Krowka MJ, Swanson KL, Chaowalit N, Decker PA, et al. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Chest. 2005;128:2393–9. doi: 10.1378/chest.128.4.2393. [DOI] [PubMed] [Google Scholar]
- 3.Lettieri CJ, Nathan SD, Barnett SD, Ahmad S, Shorr AF. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2006;129:746–52. doi: 10.1378/chest.129.3.746. [DOI] [PubMed] [Google Scholar]
- 4.Zisman DA, Ross DJ, Belperio JA, Saggar R, Lynch JP, 3rd, Ardehali A, et al. Prediction of pulmonary hypertension in idiopathic pulmonary fibrosis. Respir Med. 2007;101:2153–9. doi: 10.1016/j.rmed.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamada K, Nagai S, Tanaka S, Handa T, Shigematsu M, Nagao T, et al. Significance of pulmonary arterial pressure and diffusion capacity of the lung as prognosticator in patients with idiopathic pulmonary fibrosis. Chest. 2007;131:650–6. doi: 10.1378/chest.06-1466. [DOI] [PubMed] [Google Scholar]
- 6.Shorr AF, Wainright JL, Cors CS, Lettieri CJ, Nathan SD. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur Respir J. 2007;30:715–21. doi: 10.1183/09031936.00107206. [DOI] [PubMed] [Google Scholar]
- 7.Mejý′a M, Carrillo G, Rojas-Serrano J, Estrada A, Sua×rez T, Alonso D, et al. Idiopathic pulmonary fibrosis and emphysema: Decreased survival associated with severe pulmonary arterial hypertension. Chest. 2009;136:10–5. doi: 10.1378/chest.08-2306. [DOI] [PubMed] [Google Scholar]
- 8.Gläser S, Noga O, Koch B, Opitz CF, Schmidt B, Temmesfeld B, et al. Impact of pulmonary hypertension on gas exchange and exercise capacity in patients with pulmonary fibrosis. Respir Med. 2009;103:317–24. doi: 10.1016/j.rmed.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 9.Cai M, Zhu M, Ban C, Su J, Ye Q, Liu Y, et al. Clinical features and outcomes of 210 patients with idiopathic pulmonary fibrosis. Chin Med J (Engl) 2014;127:1868–73. [PubMed] [Google Scholar]
- 10.Song JW, Song JK, Kim DS. Echocardiography and brain natriuretic peptide as prognostic indicators in idiopathic pulmonary fibrosis. Respir Med. 2009;103:180–6. doi: 10.1016/j.rmed.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 11.Castria D, Refini RM, Bargagli E, Mezzasalma F, Pierli C, Rottoli P. Pulmonary hypertension in idiopathic pulmonary fibrosis: Prevalence and clinical progress. Int J Immunopathol Pharmacol. 2012;25:681–9. doi: 10.1177/039463201202500314. [DOI] [PubMed] [Google Scholar]
- 12.Nathan SD, Shlobin OA, Ahmad S, Koch J, Barnett SD, Ad N, et al. Serial development of pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respiration. 2008;76:288–94. doi: 10.1159/000114246. [DOI] [PubMed] [Google Scholar]
- 13.Papakosta D, Pitsiou G, Daniil Z, Dimadi M, Stagaki E, Rapti A, et al. Prevalence of pulmonary hypertension in patients with idiopathic pulmonary fibrosis: Correlation with physiological parameters. Lung. 2011;189:391–9. doi: 10.1007/s00408-011-9304-5. [DOI] [PubMed] [Google Scholar]
- 14.Nathan SD, Shlobin OA, Ahmad S, Urbanek S, Barnett SD. Pulmonary hypertension and pulmonary function testing in idiopathic pulmonary fibrosis. Chest. 2007;131:657–63. doi: 10.1378/chest.06-2485. [DOI] [PubMed] [Google Scholar]
- 15.Leuchte HH, Neurohr C, Baumgartner R, Holzapfel M, Giehrl W, Vogeser M, et al. Brain natriuretic peptide and exercise capacity in lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med. 2004;170:360–5. doi: 10.1164/rccm.200308-1142OC. [DOI] [PubMed] [Google Scholar]
- 16.Goto K, Arai M, Watanabe A, Hasegawa A, Nakano A, Kurabayashi M. Utility of echocardiography versus BNP level for the prediction of pulmonary arterial pressure in patients with pulmonary arterial hypertension. Int Heart J. 2010;51:343–7. doi: 10.1536/ihj.51.343. [DOI] [PubMed] [Google Scholar]
- 17.Leuchte HH, Baumgartner RA, Nounou ME, Vogeser M, Neurohr C, Trautnitz M, et al. Brain natriuretic peptide is a prognostic parameter in chronic lung disease. Am J Respir Crit Care Med. 2006;173:744–50. doi: 10.1164/rccm.200510-1545OC. [DOI] [PubMed] [Google Scholar]
- 18.American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165:277–304. doi: 10.1164/ajrccm.165.2.ats01. [DOI] [PubMed] [Google Scholar]
- 19.Borg GA. Psychophysical bases of perceived exertion. Med Sci Sports Exerc. 1982;14:377–81. [PubMed] [Google Scholar]
- 20.Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, et al. Standarization of spirometry. Eur Respir J. 2005;26:319–38. doi: 10.1183/09031936.05.00034805. [DOI] [PubMed] [Google Scholar]
- 21.Galiè N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT) Eur Heart J. 2009;30:2493–537. doi: 10.1093/eurheartj/ehp297. [DOI] [PubMed] [Google Scholar]
- 22.Agarwal R, Gupta D, Verma JS, Aggarwal AN, Jindal SK. Noninvasive estimation of clinically asymptomatic pulmonary hypertension in idiopathic pulmonary fibrosis. Indian J Chest Dis Allied Sci. 2005;47:267–71. [PubMed] [Google Scholar]
- 23.Huppmann P, Sczepanski B, Boensch M, Winterkamp S, Schönheit-Kenn U, Neurohr C, et al. Effects of inpatient pulmonary rehabilitation in patients with interstitial lung disease. Eur Respir J. 2013;42:444–53. doi: 10.1183/09031936.00081512. [DOI] [PubMed] [Google Scholar]
- 24.Ghio S, Klersy C, Magrini G, D’Armini AM, Scelsi L, Raineri C, et al. Prognostic relevance of the echocardiographic assessment of right ventricular function in patients with idiopathic pulmonary arterial hypertension. Int J Cardiol. 2010;140:272–8. doi: 10.1016/j.ijcard.2008.11.051. [DOI] [PubMed] [Google Scholar]
- 25.Ghio S, Pazzano AS, Klersy C, Scelsi L, Raineri C, Camporotondo R, et al. Clinical and prognostic relevance of echocardiographic evaluation of right ventricular geometry in patients with idiopathic pulmonary arterial hypertension. Am J Cardiol. 2011;107:628–32. doi: 10.1016/j.amjcard.2010.10.027. [DOI] [PubMed] [Google Scholar]
- 26.Sachdev A, Villarraga HR, Frantz RP, McGoon MD, Hsiao JF, Maalouf JF, et al. Right ventricular strain for prediction of survival in patients with pulmonary arterial hypertension. Chest. 2011;139:1299–309. doi: 10.1378/chest.10-2015. [DOI] [PubMed] [Google Scholar]
- 27.Rivera-Lebron BN, Forfia PR, Kreider M, Lee JC, Holmes JH, Kawut SM. Echocardiographic and hemodynamic predictors of mortality in idiopathic pulmonary fibrosis. Chest. 2013;144:564–70. doi: 10.1378/chest.12-2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takatsuki S, Wagner BD, Ivy DD. B-type natriuretic peptide and amino-terminal pro-B-type natriuretic peptide in pediatric patients with pulmonary arterial hypertension. Congenit Heart Dis. 2012;7:259–67. doi: 10.1111/j.1747-0803.2011.00620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimura M, Taniguchi H, Kondoh Y, Kimura T, Kataoka K, Nishiyama O, et al. Pulmonary hypertension as a prognostic indicator at the initial evaluation in idiopathic pulmonary fibrosis. Respiration. 2013;85:456–63. doi: 10.1159/000345221. [DOI] [PubMed] [Google Scholar]
- 30.Hoeper MM, Lee SH, Voswinckel R, Palazzini M, Jais X, Marinelli A, et al. Complications of right heart catheterization procedures in patients with pulmonary hypertension in experienced centers. J Am Coll Cardiol. 2006;48:2546–52. doi: 10.1016/j.jacc.2006.07.061. [DOI] [PubMed] [Google Scholar]
- 31.Dahiya A, Vollbon W, Jellis C, Prior D, Wahi S, Marwick T. Echocardiographic assessment of raised pulmonary vascular resistance: Application to diagnosis and follow-up of pulmonary hypertension. Heart. 2010;96:2005–9. doi: 10.1136/hrt.2010.204834. [DOI] [PubMed] [Google Scholar]
- 32.Janda S, Shahidi N, Gin K, Swiston J. Diagnostic accuracy of echocardiography for pulmonary hypertension: A systematic review and meta-analysis. Heart. 2011;97:612–22. doi: 10.1136/hrt.2010.212084. [DOI] [PubMed] [Google Scholar]