

Abstract
Background:
Collapsin response mediator protein-2 (CRMP2), a multifunctional cytosolic protein highly expressed in the brain, is degraded by calpain following traumatic brain injury (TBI), possibly inhibiting posttraumatic neurite regeneration. Lipid peroxidation (LP) is involved in triggering postinjury CRMP2 proteolysis. We examined the hypothesis that propofol could attenuate LP, calpain-induced CRMP2 degradation, and brain injury after TBI.
Methods:
A unilateral moderate controlled cortical impact injury was induced in adult male Sprague-Dawley rats. The animals were randomly divided into seven groups: Sham control group, TBI group, TBI + propofol groups (including propofol 1 h, 2 h, and 4 h groups), TBI + U83836E group and TBI + fat emulsion group. The LP inhibitor U83836E was used as a control to identify that antioxidation partially accounts for the potential neuroprotective effects of propofol. The solvent of propofol, fat emulsion, was used as the vehicle control. Ipsilateral cortex tissues were harvested at 24 h post-TBI. Immunofluorescent staining, Western blot analysis, and terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling were used to evaluate LP, calpain activity, CRMP2 proteolysis and programmed cell death. The data were statistically analyzed using one-way analysis of variance and a paired t-test.
Results:
Propofol and U83836E significantly ameliorated the CRMP2 proteolysis. In addition, both propofol and U83836E significantly decreased the ratio of 145-kDa αII-spectrin breakdown products to intact 270-kDa spectrin, the 4-hydroxynonenal expression and programmed cell death in the pericontusional cortex at 24 h after TBI. There was no difference between the TBI group and the fat emulsion group.
Conclusions:
These results demonstrate that propofol postconditioning alleviates calpain-mediated CRMP2 proteolysis and provides neuroprotective effects following moderate TBI potentially by counteracting LP and reducing calpain activation.
Keywords: Collapsin Response Mediator Protein-2, Lipid Peroxidation, Neuroprotection, Propofol, Traumatic Brain Injury
INTRODUCTION
Traumatic brain injury (TBI) is a critical worldwide public health problem with high morbidity and mortality.[1] Following the primary brain injury resulting from the direct mechanical insult, the brain damage is aggravated by the secondary injury caused by a series of pathophysiological events continuing for hours to days after TBI.[2] The current experimental and clinical studies focused on the neuroprotection following TBI, with the purpose of preventing the secondary insults and improve functional outcome.[3] Timely and effective therapeutic interventions after trauma would improve the long-term prognosis of patients with TBI;[4] therefore, the therapeutic window would also be an issue of great importance.
Substantial evidence indicates that free radical production and oxidative damage play an important role in the secondary injury after TBI.[2,5] Lipid peroxidation (LP)-mediated oxidative injury after TBI, initiated by reactive oxygen species and reactive nitrogen species, results in the increased permeability of membranes, decreased membrane adenosine triphosphatase activity, mitochondrial dysfunction, and cell damage.[5,6] As a consequence of LP-mediated membrane injury, aldehydic breakdown products like 4-hydroxynonenal (4-HNE) are generated during the first hour following TBI.[5] The reactive aldehyde 4-HNE acts as a key mediator of oxidative injury by binding critical cellular proteins, resulting in impaired mitochondrial bioenergetic function.[7]
Posttraumatic oxidative damage may lead to mitochondrial dysfunction, consequently exacerbating neuronal calcium dysregulation.[5] Calpains, a family of calcium-activated neutral cysteine proteases that may be activated within minutes to hours following TBI in animals, play a vital role in TBI-induced neuronal damage.[6,8] Transient calpain activation may trigger multiple cell signaling and remodeling pathways.[8] Trauma-induced sustained calpain activation contributes to the degradation of extensive cellular proteins, including components of the cytoskeleton such as αII-spectrin, eventually causing neurodegeneration, neurological dysfunction, and neuronal death following TBI.[6,9] Collapsin response mediator protein-2 (CRMP2), a cytosolic protein highly expressed in the brain, plays a major role in axonal guidance, specification, elongation and branching, neurotransmitter release, resistance to glutamate toxicity and neuronal cell death.[10] The proteolysis of CRMP2 mediated by calpain following TBI may be a potential inhibitory factor for posttraumatic neurite regeneration.[11]
Propofol is a widely used intravenous (i.v.) anesthetic similar in structure to the natural antioxidant Vitamin E; it is commonly used in the sedation of TBI patients in the intensive care unit or during the intraoperative period. Propofol has been demonstrated to have potential protective effects against oxidative injury in animals and humans, but its signaling pathways are poorly elucidated.[12]
In this study, we hypothesized that propofol attenuates LP, calpain-induced CRMP2 degradation and programmed cell death, providing neuroprotection during the early period after TBI. To test this hypothesis, we investigated the effects of postinjury propofol on oxidative stress, calpain-mediated CRMP2 proteolysis and programmed cell death and the therapeutic window in a controlled cortical impact (CCI) model in rats. The levels of 4-HNE-modified proteins were determined to evaluate the LP-induced injury. The integrity of αII-spectrin and CRMP2 were detected to examine calpain activation and CRMP2 proteolysis. Histological evaluations were also performed to estimate TBI-induced programmed cell death.
METHODS
Animals
The present study employed 70 adult male Sprague-Dawley rats (Vital River Laboratory Animal Technology Co., Ltd., China) weighing 220–270 g. All animals were housed in plastic cages in an animal room with a controlled 12 h light-dark cycle. Water and food were provided ad libitum. Appropriate experimental protocols were maintained to meet the guidelines established by the Animal Care and Use Committee of Capital Medical University, which conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. We have made every effort to relieve animal suffering and reduce the numbers of animals used for this study.
Rat model of focal (controlled cortical impact) traumatic brain injury
A unilateral CCI injury was induced using a controlled cortical impactor (Impact One™ Stereotaxic Impactor for CCI, Richmond, IL, USA). The animals were anesthetized with 10% chloral hydrate (Sigma-Aldrich, St Louis, MO, USA) (400 mg/kg, i.p.). The core body temperature was maintained at 37 ± 1°C by a localized temperature therapy system with a warming pad beneath the rats. Catheters were inserted into the tail vein, and artery for i.v. injection and arterial blood gas analysis, respectively. The animals were mounted on a stereotaxic instrument (Narishige Scientific Instrument Lab, Japan) in a prone position and fixed by auxiliary ear and incisor bars. The animals received a midline cranial skin incision and unilateral (on the right side) craniotomy (4 mm in diameter) lateral and 0.5 mm anterior to the bregma, directly over the forelimb sensorimotor cortex.[13] The CCI was accomplished with a rounded-tip impact probe (3 mm in diameter) connected to an electromechanical actuator. The cortical impact was delivered at a velocity of 6.0 m/s with a depth of 1.0 mm below the cortical surface, remaining for 150 ms, which caused moderate TBI.[14] The impactor rod was angled 18° away from vertical to maintain the impact probe perpendicular to the cortical surface. After the CCI, the wound was sutured, and the animals were kept warm until recovery from anesthesia. We excluded five animals that survived <24 h postoperatively and two animals with dural laceration after the impact.
Experimental groups
The animals were divided randomly into seven experimental groups by using a table of random number (n = 9 per group after exclusion): Sham control group, TBI group, TBI + propofol 1 h group, TBI + propofol 2 h group, TBI + propofol 4 h group, TBI + U83836E group, and TBI + fat emulsion group. The LP inhibitor U83836E was used as a control to identify that antioxidation partially accounts for the potential neuroprotective effects of propofol. The solvent of propofol, fat emulsion, was used as the vehicle control. Sham-injured control animals underwent all surgical procedures except the impact injury.
Preparation and dosing of propofol, U83836E, and fat emulsion
Propofol was purchased from Fresenius Kabi (Bad Homburg, Germany, Lot No. 16EL0259). Propofol 12.5 mg/kg was i.v. injected within 5 min at 1 h, 2 h and 4 h after TBI in the propofol 1 h group, propofol 2 h group and propofol 4 h group, respectively, followed by propofol 40 mg·kg−1·h−1 i.v. infusion for 2 h. The present study used a clinically relevant dosage of propofol based on the preliminary experiment and pharmacological calculation according to the different body surface areas.
U83836E (Enzo Life Sciences, Inc., Farmingdale, NY, USA) was freshly diluted in normal saline (Baxter International Inc., IL, USA) to 0.75 mg/ml. The dilutions were made to deliver the initial 2 mg/kg i.v. injection at 15 min after TBI followed by a 7 mg/kg intraperitoneal (i.p.) injection. The employed dose was based on a previously published study on the effects of U83836E in the mouse CCI model.[2,9] The dose used in that study was converted to the dose for rats according to the different body surface areas.
The fat emulsion was obtained from Fresenius Kabi. The fat emulsion was i.v. injected within 5 min at 1 h after TBI, followed by a continuous infusion for 2 h. The dose and infusion rates in the vehicle control (fat emulsion) group were the same as those for propofol.
Brain tissue collection and preparation
At 24 h after CCI, the animals were anesthetized and sacrificed by decapitation (n = 6 per group for Western blot analysis; n = 3 per group for immunofluorescent staining and terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling [TUNEL]). The brains were removed rapidly and rinsed with ice-cold normal saline. The pericontusional cortex was removed from the right hemisphere [Figure 1a], quick-frozen in liquid nitrogen and kept frozen at −80°C until use in the laboratory (Laboratory of Clinical Medical Research, Beijing Tian tan Hospital, China). For Western blot analysis, the brain tissues were homogenized in ice-cold lysis buffer using a motor-driven pellet pestle (Pellet Pestle Cordless Motor, Kimble Chase, Vineland, NJ, USA). The lysis buffer contained 50 mmol/L Tris-HCl (Sigma-Aldrich, USA) (pH 7.4), 1 mmol/L ethylene diamine tetraacetic acid (Sigma-Aldrich), 1 mmol/L sodium orthovanadate (Sigma-Aldrich), 1 mmol/L sodium fluoride (Sigma-Aldrich), 1% NP-40 (Sigma-Aldrich), 0.5% sodium deoxycholate (Sigma-Aldrich) and freshly added protease inhibitor cocktail (Applygen Technologies Inc., Beijing, China). The homogenized brain samples were then lysed for 60 min at 4°C. The homogenates were then centrifuged at 12,000 r/min for 20 min at 4°C. The supernatant was collected, and the total protein concentration was determined by a BCA Protein Assay Kit (Pierce Company, Rockford, IL, USA).
Figure 1.
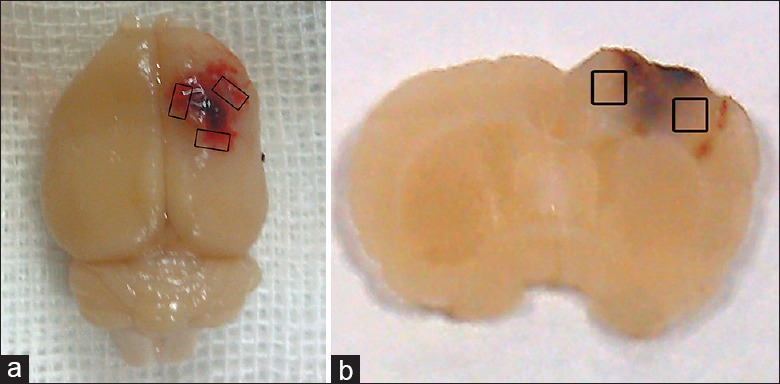
Schematic representation of the sample area for detection. (a) Image of a representative TBI brain showing cortex contusion and the pericontusional region surrounding the injured cortex. The sample area for detection is marked with the black boxes. TBI: Traumatic brain injury; (b) a coronal section of a rat brain used for immunofluorescent staining and TUNEL analysis. The black boxes indicate the detected region. TUNEL: Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling.
Western blot analysis
Balanced protein samples (30 μg per lane) were heated for 5 min at 95°C and then loaded on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. After electrophoresis, the gels were transferred to a polyvinylidene difluoride (PVDF) membrane (Immobilon-P, Millipore Corporation, Billerica, MA, USA) at 4°C by the wet transfer method (200 mA, 3 h). Following several rinses with phosphate buffer solution-Tween (PBST, containing 0.1% Tween-20), the transferred PVDF membrane was blocked with 5% nonfat milk in PBST for 1 h at room temperature. The membrane was then incubated for 3 h at room temperature with a rabbit polyclonal antibody against CRMP2 (Cell Signaling Technology, Danvers, MA, USA) and a mouse monoclonal antibody against αII-spectrin (Merck Millipore, Darmstadt, Germany). A mouse monoclonal antibody against β-actin (Santa Cruz Biotechnology, Paso Robles, CA, USA) was employed to verify equal loading of the samples. Horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG (Santa Cruz Biotechnology, Paso Robles, CA, USA) was used as the secondary antibody. After incubation with the primary and secondary antibodies, a chemiluminescent detection kit (Immobilon-P, Millipore Corporation, Billerica, MA, USA) was used to visualize the protein bands. The relative band intensities were tested by densitometry using FluorChem FC2 software (ProteinSimple, CA, USA).
Immunofluorescent staining
The animals were anesthetized with 10% chloral hydrate (400 mg/kg, i.p.) and perfusion-fixed with 4% paraformaldehyde (PFA) (Sinopharm Chemical Reagent Co., Ltd., China) in 0.1 mol/L phosphate buffer (PB, pH 7.4). The brains were then removed and immersed in the same PFA solution for 1 day and subsequently shifted into 30% sucrose in 0.1 mol/L PB at 4°C until further processing. The brains were embedded in an optimum cutting temperature compound (Sakura Finetek USA, Torrance, CA, USA). Coronal sections, 20 μm thick, were then cut with a cryostat (Leica CM1950, Leica Biosystems Nussloch GmbH, Nussloch, Germany) and mounted onto polylysine-coated glass slides. Prior to immunofluorescent staining, frozen sections were immersed in 0.01 mol/L phosphate buffer saline (PBS) solution and rinsed for 10 min. After permeabilizing in 0.01 mol/L PBS containing 0.2% Triton X-100 for 30 min, the brain sections were blocked in 10% goat serum for 1 h at room temperature; the sections were incubated overnight at room temperature with a mixture of primary antibodies consisting of the rabbit anti-4-HNE antibody (1:100, Abcam, Cambridge, UK) and mouse anti-neurofilament (NF) antibody (1:100, Cell Signaling Technology, Danvers, MA, USA) as a neuronal cell marker. The sections were then washed with 0.01 mol/L PBS 3 times (10 min each). For double immunofluorescent labeling, immunoreactivity for 4-HNE and NF was detected by incubation with Alexa Fluor 555-conjugated goat antirabbit IgG antibody and Alexa Fluor 488-conjugated goat antimouse IgG antibody (Cell Signaling Technology), respectively, for 1 h in the dark at room temperature. Negative controls were performed by replacing the primary antibodies with 10% goat serum. The sections were subsequently washed in 0.01 mol/L PBS as described above and mounted on slides with UltraCruz mounting medium containing 6-diamidine-2-phenylindole (DAPI) (Santa Cruz Biotechnology, Paso Robles, CA, USA). The fluorescent signals were detected with fluorescent microscopy (DMI4000 B, Leica Microsystems Ltd., Wetzlar, Germany).
Assessment of brain injury (transferase-mediated dUTP nick-end labeling, TUNEL)
The in situ DNA fragmentation of brain sections was detected by TUNEL labeling using an in situ Cell Death Detection Kit, Fluorescein (Roche Diagnostics GmbH, Mannheim, Germany), in accordance with the manufacturer's instructions. In brief, frozen brain sections were fixed for 20 min using 4% PFA in PB (pH 7.4) at room temperature and subsequently washed in PBS. The sections were then incubated with 0.1% Triton X-100 in 0.1% sodium citrate for 2 min on ice and rinsed with PBS. The sections were incubated with a TUNEL reaction mixture in a humidified chamber for 60 min at 37°C to add fluorescein-dUTP to the 3’-OH termini of the fragmented DNA. The sections were then rinsed in PBS and mounted on slides with UltraCruz mounting medium containing DAPI. The positive cells were detected by fluorescence microscopy.
Cell counting
Immunofluorescent staining and TUNEL labeling were performed on three successive coronal cortex sections obtained from the center of the injured area on the right lateral forelimb sensorimotor cortex which is 0.5 mm anterior to the bregma[13] (n = 3 per group), and the images were acquired from three separate fields surrounding the contusional region in each section. Positive cells in the pericontusional area [Figure 1b] were counted and analyzed using ImageJ software (National Institutes of Health, USA). The 4-HNE-positive cells were considered to be all those cells expressing 4-HNE-modified proteins in both the nucleus and the cytoplasm. The severity of the brain injury was assessed by calculating the average number of TUNEL-positive cells per 100 cells.
Statistical analysis
All experiments were performed at least in triplicate. The results are presented as the means ± standard deviation. Differences among these groups were determined by one-way analysis of variance (ANOVA) with a Bonferroni post-hoc comparison using SPSS version 13.0 (IBM Corporation, USA). Differences within groups were determined with a paired t-test. A value of P < 0.05 was considered statistically significant.
RESULTS
Hemodynamics and arterial blood gas analysis
All the animals except the excluded seven ones survived during the study period. As shown in Table 1, the heart rate (HR), mean arterial pressure (MAP), pH, arterial partial pressure of oxygen (PaO2), and partial pressure of carbon dioxide in the artery (PaCO2) in all groups were stable before the craniotomy (baseline values) and at the end of the experimental treatment. There were no significant differences in hemodynamics and blood gases in the experimental groups compared with the Sham group before the craniotomy or at the end of the experiment. The PaO2 decreased at the end of the infusion in the propofol 1 h group, and the PaCO2 decreased at the end of the U83836E i.p. injection in the U83836E group (P < 0.05) within an acceptable range. Although these changes were statistically significant, there was no potential clinical significance. No significant differences were found in the HR, MAP, pH, PaO2, and PaCO2 between the 2 time points in the other groups. This observation suggested that propofol, U83836E and fat emulsion did not significantly influence the animals’ circulation and respiration [Table 1].
Table 1.
Hemodynamic and arterial blood gas data of rats before the craniotomy and after the experimental treatment (n = 6)
| Group | Time points | HR (bpm) | MAP (mmHg) | PaO2 (mmHg) | PaCO2 (mmHg) |
|---|---|---|---|---|---|
| Sham | Before | 290 ± 5 | 80 ± 15 | 106 ± 27 | 46 ± 7 |
| After | 287 ± 7 | 79 ± 9 | 111 ± 19 | 44 ± 7 | |
| TBI | Before | 277 ± 16 | 77 ± 9 | 109 ± 11 | 42 ± 11 |
| After | 277 ± 10 | 78 ± 7 | 109 ± 11 | 41 ± 8 | |
| TBI + | Before | 286 ± 6 | 81 ± 8 | 113 ± 18 | 41 ± 9 |
| propofol 1 h | After | 274 ± 16 | 86 ± 12 | 102 ± 11* | 40 ± 4 |
| TBI + propofol 2 h | Before | 273 ± 14 | 77 ± 8 | 102 ± 19 | 43 ± 10 |
| After | 265 ± 13 | 83 ± 7 | 110 ± 19 | 38 ± 8 | |
| TBI + propofol 4 h | Before | 274 ± 26 | 80 ± 10 | 99 ± 12 | 46 ± 5 |
| After | 277 ± 16 | 85 ± 8 | 112 ± 12 | 39 ± 9 | |
| TBI + U83836E | Before | 276 ± 22 | 75 ± 4 | 104 ± 8 | 43 ± 7 |
| After | 277 ± 13 | 81 ± 8 | 113 ± 7 | 38 ± 5 | |
| TBI + FE | Before | 267 ± 27 | 75 ± 4 | 98 ± 9 | 42 ± 8 |
| After | 284 ± 13 | 82 ± 7 | 108 ± 15 | 38 ± 7* |
The data are presented as the means ± SD. There were no significant differences among all groups before the craniotomy or at the end of the experimental treatment. The “before” and “after” time points are referred to as “before the craniotomy” and “at the end of experimental treatment,” respectively: For example, “after TBI” in the TBI group and “at the end of drug infusion or injection” in the three propofol groups, the U83836E group and the fat emulsion group. HR: Heart rate; MAP: Mean arterial pressure; PaO2: Arterial partial pressure of oxygen; PaCO2: Partial pressure of carbon dioxide in the artery; FE: Fat emulsion; bpm: Beat per minute; TBI: Traumatic brain injury. Significant differences (one-way ANOVA and paired t-test), *P<0.05 versus base values.
Propofol attenuates posttraumatic oxidative damage
4-hydroxynonenal, one of the major products of LP, contributes to oxidative stress-induced cell injury. The effects of propofol on oxidative damage at 24 h after TBI were examined by immunofluorescent staining for 4-HNE-modified proteins. 4-HNE-positive cells were not observed in the Sham control group. 4-HNE-positive cells in the region surrounding the impacted cortex were significantly increased in all experimental groups (P < 0.01) [Figure 2]. There were fewer 4-HNE staining cells in the three propofol groups and the U83836E group compared with the TBI group (P < 0.01) [Figure 2]. Fewer 4-HNE positive cells were detected in three propofol groups and the U83836E group than the fat emulsion group [Figure 2]. No significant difference was found between the TBI group and the vehicle control group (the fat emulsion group) (P > 0.05) [Figure 2]. There was no significant difference among the three propofol groups (P > 0.05) [Figure 2]. 4-HNE-modified proteins were detected in both the cytoplasm and the nucleus. Cells with 4-HNE-modified proteins were expressed in the cytoplasm of neurons (cells staining positive for NFs) and nonneuronal cells [Figure 2]. We counted all the 4-HNE-positive cells.
Figure 2.
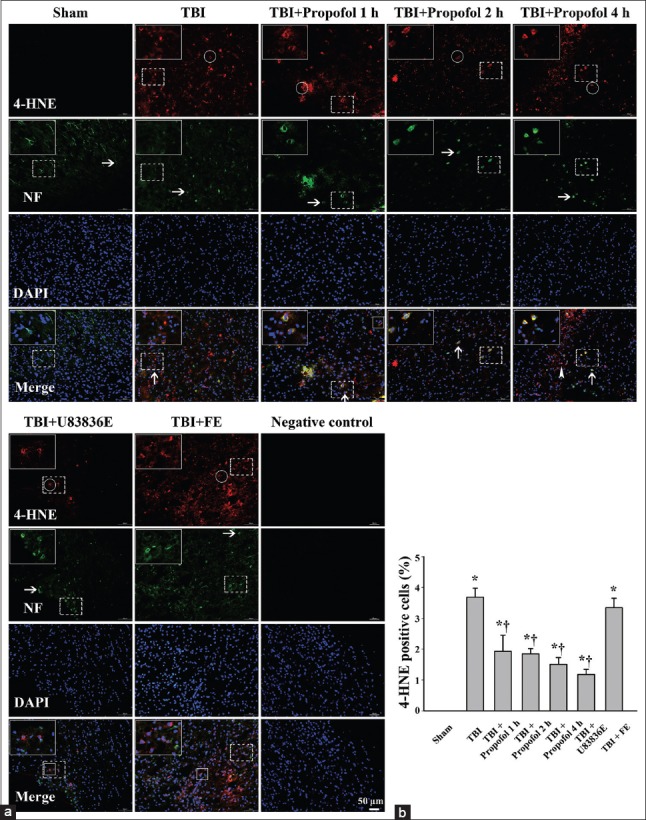
Immunofluorescence staining of 4-hydroxynonenal positive cells in the pericontusional cortex at 24 h following moderate TBI in the Sham group (Sham), TBI group (TBI), three groups treated with propofol at different time points after TBI (TBI + Propofol 1 h, TBI + Propofol 2 h, TBI + Propofol 4 h), the lipid peroxidation inhibitor U83836E group (TBI + U83836E) and the vehicle control fat emulsion group (TBI + FE). TBI: Traumatic brain injury; (a) Horizontal arrows indicate representative neurofilaments (NF; green). Round rings and arrowheads indicate representative 4-HNE-modified proteins (red) expressed in the cytoplasm and the nucleus, respectively. Vertical arrows and boxes represent 4-HNE-positive neurons and non-neuronal cells, respectively; (b) quantitation of 4-HNE-positive cells. The data are presented as the mean ± standard deviation (SD) (n = 3). *P < 0.01 vs. Sham group; †P < 0.01 vs. TBI group. Bar = 50 μm.
The effect of propofol on calpain-mediated αII-spectrin breakdown
To provide insight into whether post-TBI treatment with propofol could attenuate calpain activation, Western blot analysis was performed to measure the levels of the calpain-mediated proteolysis product of αII-spectrin in the pericontusional cortex at 24 h following TBI. The calpain-mediated spectrin breakdown product (SBDP, 145 kDa) levels increased markedly in all experimental groups following TBI [Figure 3]. The ratio of calpain-cleaved SBDP to intact αII-spectrin was lower in the three propofol groups and the U83836E group (P < 0.01) than in the TBI group [Figure 3]. The 145/270 kDa spectrin ratio in the vehicle control (fat emulsion) group was similar to that of the TBI group (P > 0.05) [Figure 3].
Figure 3.
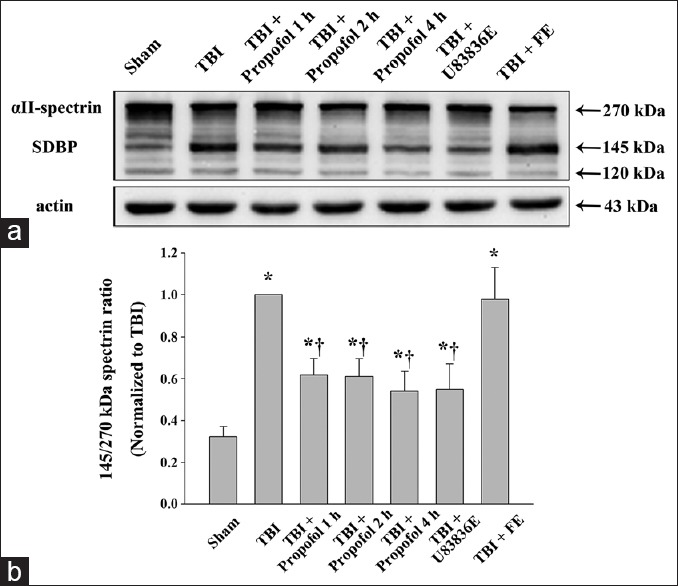
Western blot analysis of αII-spectrin in the pericontusional cortex at 24 h following moderate TBI. (a) Brain tissue lysates were immunoblotted with anti-αII-spectrin (top) and anti-β-actin (bottom) antibodies, respectively. The lanes were loaded with protein from the Sham group (Sham), the TBI group (TBI), the propofol 1 h group (TBI + Propofol 1 h), the propofol 2 h group (TBI + Propofol 2 h), the propofol 4 h group (TBI + Propofol 4 h), the lipid peroxidation inhibitor U83836E group (TBI + U83836E) and the vehicle control fat emulsion group (TBI + FE). TBI: Traumatic brain injury; (b) quantitative analysis for the ratio of the calpain-mediated 145-kDa spectrin breakdown product (SBDP) to intact 270-kDa αII-spectrin. The densitometric ratio was normalized against the TBI group. The results were expressed as the means ± standard deviation (SD) (n = 6). *P < 0.01 vs. Sham group; †P < 0.01 vs. TBI group.
Propofol reduces the proteolysis of collapsin response mediator protein-2 after traumatic brain injury
We examined whether the propofol treatment could attenuate the proteolysis of CRMP2 in vivo following TBI. We examined the pericontusional cortex tissues harvested 24 h post-TBI because CRMP2 degradation is most significant at this time point.[11] CRMP2 breakdown products (55 kDa) were observed in all experimental groups [Figure 4]. An ANOVA demonstrated that the ratio of the 55-kDa breakdown product to intact 62-kDa CRMP2 was lower in the three propofol groups and in the U83836E group (P < 0.01) than in the TBI group [Figure 4]. There was no significant difference between the TBI group and the vehicle control (fat emulsion) group (P > 0.05) [Figure 4].
Figure 4.
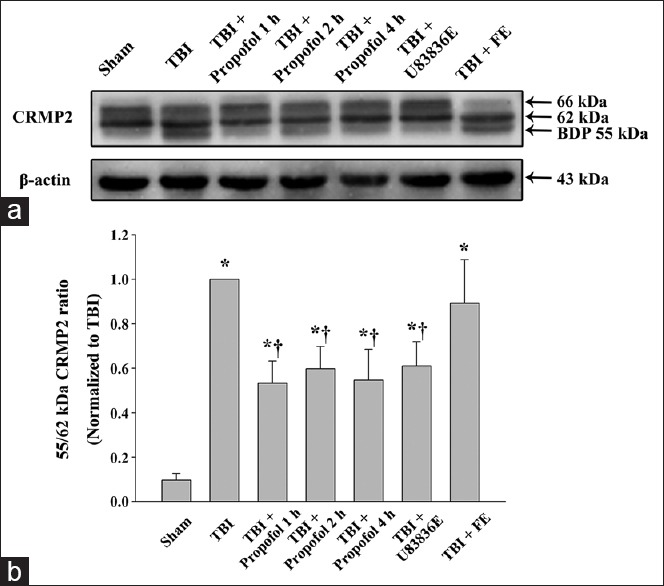
Western blot analysis of collapsin response mediator protein-2 (CRMP2) proteolysis in the ipsilateral cortex surrounding the impacted area at 24 h following moderate TBI. (a) Brain tissue lysates were immunoblotted with anti-CRMP2 (top) and anti-β-actin (bottom) antibodies. The lanes were loaded with protein from the Sham group (Sham), the TBI group (TBI), the propofol 1 h group (TBI + Propofol 1 h), the propofol 2 h group (TBI + Propofol 2 h), the propofol 4 h group (TBI + Propofol 4 h), the lipid peroxidation inhibitor U83836E group (TBI + U83836E) and the vehicle control fat emulsion group (TBI + FE). TBI: Traumatic brain injury; (b) densitometric analysis for the ratio of the 55-kDa breakdown product to intact 62-kDa CRMP2. The densitometric ratio was normalized against the TBI group. The data are expressed as the means ± standard deviation (SD) (n = 6). *P < 0.01 vs. Sham group; †P < 0.01 vs. TBI group.
Propofol protects neurons from traumatic brain injury-induced programmed cell death
Transferase-mediated dUTP nick-end labeling was performed in the pericontusional region at 24 h after TBI. No TUNEL-positive cells were observed in the Sham group [Figure 5]. The ratio of TUNEL-positive cells in the area surrounding the impacted cortex was increased compared with the Sham group (P < 0.01) [Figure 5]. In the three propofol groups and the U83836E group, the ratio of TUNEL-positive cells in the same area was significantly decreased compared with the TBI group (P < 0.01) [Figure 5]. This result demonstrated that the propofol and the antioxidant U83836E treatments after TBI could inhibit programmed cell death in the cortex surrounding the injured region. There was no significant difference between the TBI group and the vehicle control (fat emulsion) group (P > 0.05) [Figure 5], indicating that the solvent of propofol did not provide neuroprotective effects in TBI rats.
Figure 5.
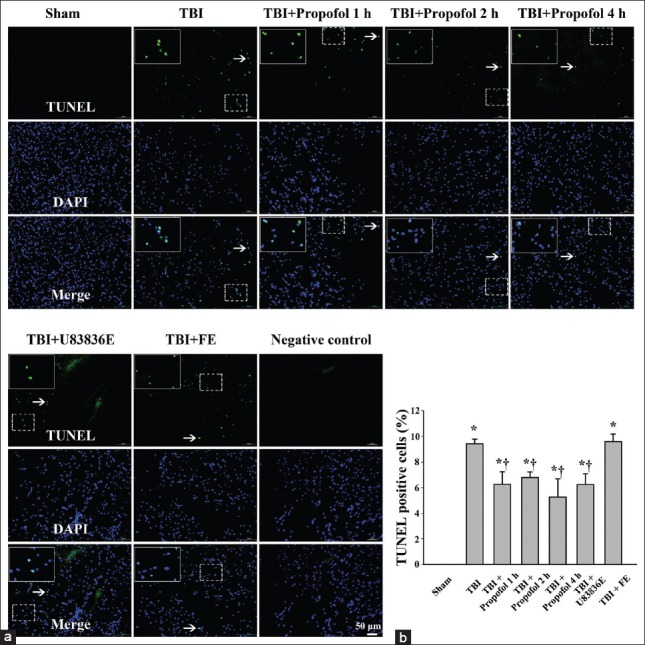
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) of programmed cell death in the pericontusional cortex at 24 h following moderate TBI in the Sham group (Sham), TBI group (TBI), three propofol groups (TBI + Propofol 1 h, TBI + Propofol 2 h, TBI + Propofol 4 h), lipid peroxidation (LP) inhibitor U83836E group (TBI + U83836E) and the vehicle control fat emulsion group (TBI + FE). TUNEL labeling demonstrated the reduction in programmed cell death after the administration of propofol and U83836E following TBI. TBI: Traumatic brain injury; TUNEL: Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling. (a) The white arrows indicate the TUNEL-positive cells, which represent programmed cell death; (b) quantitation of TUNEL-positive cells. The results are presented as the means ± standard deviation (SD) (n = 3). *P < 0.01 vs. Sham group; †P < 0.01 vs. TBI group. Bar = 50 μm.
DISCUSSION
This study utilized the rat CCI injury model to investigate changes in oxidative stress and the calpain-CRMP2 pathway, as well as the neuroprotective effects of propofol following TBI injury. The results showed that propofol attenuated oxidative stress, reduced calpain activation and CRMP2 proteolysis and improved cell survival at an early stage in the pericontusional cortex after TBI. The fat emulsion was used as the vehicle control to prove that propofol rather than its solvent provided the neuroprotective effects. In addition, we demonstrated that propofol can afford neuroprotection in the extended therapeutic window up to at least 4 h following TBI.
Oxidative stress is known to play an important role in the pathology of secondary brain injury following TBI. The brain is extremely sensitive to LP toxicity because it contains a large amount of LP-susceptible unsaturated fatty acids, like arachidonic acid.[5] The levels of LP increased significantly at 3 h and continued to increase, with peak levels occurring at 48 h post trauma.[15] Propofol (2,6-diisopropylphenol) is a rapidly acting i.v. anesthetic that has been proposed as a neuroprotective agent due to its ability to reduce the cerebral metabolic rate of oxygen and preserve cerebral autoregulation.[16] The antioxidant potential of propofol at a therapeutic concentration has been demonstrated in many disease models and clinical settings.[17,18] Propofol provides significant neuroprotection by alleviating LP and mitochondrial dysfunction in a fetal cerebral ischemia-reperfusion model.[19] The antioxidant U83836E has been proved to attenuate posttraumatic LP in the mouse CCI model.[2,9] Since both propofol and U83836E are effective LP inhibitors, the U83836E group was used as a control group in this study. The present experiment employed a more clinically relevant regimen using i.v. bolus of propofol followed by a continuous infusion for 2 h. Our results indicate that either early treatment (i.e., 1 h) or delayed treatment (i.e., 4 h) with propofol after TBI results in a significant reduction in LP, providing similar effects as the antioxidant U83836E on the inhibition of oxidative stress. Nonetheless, even with early treatment, the LP levels are elevated significantly compared with the Sham control group, indicating that secondary brain injury cascades, like oxidative stress, are triggered rapidly by TBI but can be attenuated by propofol intervention.
Calpains, a family of Ca2+-dependent cysteine proteases, exist as inactive proenzymes under physiological conditions.[20] Once activated by cytoplasmic Ca2+ overload following TBI, calpains hydrolyze numerous intracellular proteins including cytoskeletal proteins (e.g., αII-spectrin), which may result in neurological dysfunction and neurodegeneration.[6,11] Calpains may also contribute to the triggering of apoptotic cascades due to their ability to activate caspases.[21] U83836E, a potent LP inhibitor, produces a dose-related attenuation of calpain-mediated αII-spectrin degradation and provides neuroprotection following TBI.[9] CRMP2 is a multifunctional adaptor protein distributed in the central nervous system.[22] CRMP2 has been shown to play an important role in regulating cytosolic calcium, neuronal plasticity, and excitotoxicity.[23] CRMP2 breakdown products mediated by calpain are observed in both TBI and middle cerebral artery occlusion models.[11,24] Recent evidence has suggested that CRMP2 can be pharmacologically manipulated, offering new opportunities to preserve neural function in certain neurological diseases.[22] The present study shows that propofol rather than its solvent administered in the early period post-TBI can inhibit calpain activation and attenuate the calpain-mediated degradation of CRMP2 in moderate TBI in rats, which may be a conceivable mechanism of its neuroprotective effects.
Programmed cell death includes the apoptosis and nonapoptotic death which is termed necroptosis and plays a vital role in tissue homeostasis.[25] The apoptosis of neurons and glia is considered to be a vital factor in the secondary brain injury following TBI.[26] Pharmacological strategies have been investigated to alleviate apoptosis.[26] Propofol provides protection and reduces the apoptotic cell numbers in vitro.[27] Propofol has been demonstrated to reduce neuronal apoptosis and ameliorate cerebral ischemia-reperfusion injury.[28] The neuroprotective effects of propofol against apoptosis may be mediated by inhibiting caspase-3 expression, increasing Bcl-2 expression and suppressing a series of oxidative events.[29] Our results suggest that postinjury treatment with propofol in the acute posttraumatic period (initiated within 4 h after TBI) significantly attenuates TBI-induced programmed cell death, potentially due to the reduction in calpain-mediated CRMP2 proteolysis, while the fat emulsion alone was without protective effects.
Propofol is a widely used anesthetic for patients with TBI in the clinical setting. The possibility that propofol could provide a significant effect even when the treatment onset is postponed to an extended time after injury is important. To explore the therapeutic window of propofol posttreatment following TBI, the onset of propofol was initiated at different time points posttrauma. There was no significant difference among the propofol 1 h, 2 h, and 4 h groups. Our results indicated that propofol provides evident neuroprotection even when the onset is delayed up to 4 h posttrauma. Although propofol has been demonstrated to improve the neurobehavioral outcomes after cerebral ischemia,[30] the experimental behavioral studies in TBI are controversial and less encouraging.[31] The protective action of propofol and functional improvement after propofol therapy have been shown in the rat models of intracerebral hemorrhage injury and TBI.[3] However, a recently published study indicated that propofol impaired neurologic recovery and decreased the survival rate after TBI.[31] Different experimental design may possibly account for the disparate results. In addition, a multicenter randomized controlled trial (RCT) proved that high-dose propofol resulted in better neurological outcomes in patients with moderate and severe TBI.[32] A recently reported meta-analysis demonstrated that sedation with propofol and midazolam have similar effects in patients with severe TBI on the mortality, Glasgow Outcome Scale score, and intracranial pressure.[33] Consequently, further well-designed experimental studies and RCTs are urgently needed to elucidate the precise effects of propofol and other sedatives on post-TBI cascades and outcomes.
There are a few limitations in this study that still need further clarification. First, although we have demonstrated that propofol ameliorates oxidative stress, suppresses calpain activation and CRMP2 proteolysis and reduces apoptosis, propofol likely may interact with other signaling pathways that may or may not be involved in post-TBI peroxidation, calpain activation and CRMP2 degradation. Second, given that we harvested the samples at 24 h following TBI, we can only make conclusions regarding the transient neuroprotective effects of propofol postconditioning in TBI rats. However, the long-term effects and dose-dependent effects of propofol must be further investigated. Additional time points are also needed to detect therapeutic time window of propofol administration. Third, rats differ markedly from humans in many respects, making rat experiments difficult to extrapolate to humans. Consequently, extrapolating the therapeutic regimen of propofol for TBI rats to patients with moderate TBI should be performed cautiously and requires further clinical research.
Despite these limitations, our study demonstrates that propofol posttreatment can alleviate calpain-mediated CRMP2 proteolysis and provide neuroprotection in a model of moderate TBI in vivo. These effects may occur by potentially counteracting LP and reducing calpain activation, even when propofol administration is delayed up to 4 h after TBI.
ACKNOWLEDGMENTS
We thank Dr. Yuan-Li Zhao for providing the controlled cortical impactor and Ya-Jie Wang, An-Chen Guo, Hao-Wen Li, Li Liu and Yuan Ren for technical assistance.
Footnotes
Edited by: Li-Min Chen
Source of Support: This study was supported by grants from the Beijing Nova Program (No. 2007B074), the Beijing Municipal Health System “215” High-level Medical Personnel Culturing Project (No. 2009-3-19) and the Youth Foundation of Beijing Tian Tan Hospital (No. KY2011-03).
Conflict of Interest: None declared.
REFERENCES
- 1.Georgiou AP, Manara AR. Role of therapeutic hypothermia in improving outcome after traumatic brain injury: A systematic review. Br J Anaesth. 2013;110:357–67. doi: 10.1093/bja/aes500. [DOI] [PubMed] [Google Scholar]
- 2.Mustafa AG, Singh IN, Wang J, Carrico KM, Hall ED. Mitochondrial protection after traumatic brain injury by scavenging lipid peroxyl radicals. J Neurochem. 2010;114:271–80. doi: 10.1111/j.1471-4159.2010.06749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo T, Wu J, Kabadi SV, Sabirzhanov B, Guanciale K, Hanscom M, et al. Propofol limits microglial activation after experimental brain trauma through inhibition of nicotinamide adenine dinucleotide phosphate oxidase. Anesthesiology. 2013;119:1370–88. doi: 10.1097/ALN.0000000000000020. [DOI] [PubMed] [Google Scholar]
- 4.Boer C, Franschman G, Loer SA. Prehospital management of severe traumatic brain injury: Concepts and ongoing controversies. Curr Opin Anaesthesiol. 2012;25:556–62. doi: 10.1097/ACO.0b013e328357225c. [DOI] [PubMed] [Google Scholar]
- 5.Hall ED, Vaishnav RA, Mustafa AG. Antioxidant therapies for traumatic brain injury. Neurotherapeutics. 2010;7:51–61. doi: 10.1016/j.nurt.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng Y, Thompson BM, Gao X, Hall ED. Temporal relationship of peroxynitrite-induced oxidative damage, calpain-mediated cytoskeletal degradation and neurodegeneration after traumatic brain injury. Exp Neurol. 2007;205:154–65. doi: 10.1016/j.expneurol.2007.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh IN, Gilmer LK, Miller DM, Cebak JE, Wang JA, Hall ED. Phenelzine mitochondrial functional preservation and neuroprotection after traumatic brain injury related to scavenging of the lipid peroxidation-derived aldehyde 4-hydroxy-2-nonenal. J Cereb Blood Flow Metab. 2013;33:593–9. doi: 10.1038/jcbfm.2012.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kampfl A, Posmantur R, Nixon R, Grynspan F, Zhao X, Liu SJ, et al. mu-calpain activation and calpain-mediated cytoskeletal proteolysis following traumatic brain injury. J Neurochem. 1996;67:1575–83. doi: 10.1046/j.1471-4159.1996.67041575.x. [DOI] [PubMed] [Google Scholar]
- 9.Mustafa AG, Wang JA, Carrico KM, Hall ED. Pharmacological inhibition of lipid peroxidation attenuates calpain-mediated cytoskeletal degradation after traumatic brain injury. J Neurochem. 2011;117:579–88. doi: 10.1111/j.1471-4159.2011.07228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taghian K, Lee JY, Petratos S. Phosphorylation and cleavage of the family of collapsin response mediator proteins may play a central role in neurodegeneration after CNS trauma. J Neurotrauma. 2012;29:1728–35. doi: 10.1089/neu.2011.2063. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Z, Ottens AK, Sadasivan S, Kobeissy FH, Fang T, Hayes RL, et al. Calpain-mediated collapsin response mediator protein-1,-2, and-4 proteolysis after neurotoxic and traumatic brain injury. J Neurotrauma. 2007;24:460–72. doi: 10.1089/neu.2006.0078. [DOI] [PubMed] [Google Scholar]
- 12.Vasileiou I, Xanthos T, Koudouna E, Perrea D, Klonaris C, Katsargyris A, et al. Propofol: A review of its non-anaesthetic effects. Eur J Pharmacol. 2009;605:1–8. doi: 10.1016/j.ejphar.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 13.Jones TA, Liput DJ, Maresh EL, Donlan N, Parikh TJ, Marlowe D, et al. Use-dependent dendritic regrowth is limited after unilateral controlled cortical impact to the forelimb sensorimotor cortex. J Neurotrauma. 2012;29:1455–68. doi: 10.1089/neu.2011.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu S, Kaneko Y, Bae E, Stahl CE, Wang Y, van Loveren H, et al. Severity of controlled cortical impact traumatic brain injury in rats and mice dictates degree of behavioral deficits. Brain Res. 2009;1287:157–63. doi: 10.1016/j.brainres.2009.06.067. [DOI] [PubMed] [Google Scholar]
- 15.Ansari MA, Roberts KN, Scheff SW. A time course of contusion-induced oxidative stress and synaptic proteins in cortex in a rat model of TBI. J Neurotrauma. 2008;25:513–26. doi: 10.1089/neu.2007.0451. [DOI] [PubMed] [Google Scholar]
- 16.Tawfeeq NA, Halawani MM, Al-Faridi K, Aal-Shaya WA, Taha WS. Traumatic brain injury: Neuroprotective anaesthetic techniques, an update. Injury. 2009;40(Suppl 4):S75–81. doi: 10.1016/j.injury.2009.10.040. [DOI] [PubMed] [Google Scholar]
- 17.Kalimeris K, Kouni S, Kostopanagiotou G, Nomikos T, Fragopoulou E, Kakisis J, et al. Cognitive function and oxidative stress after carotid endarterectomy: Comparison of propofol to sevoflurane anesthesia. J Cardiothorac Vasc Anesth. 2013;27:1246–52. doi: 10.1053/j.jvca.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 18.Chen RM, Tai YT, Chen TG, Lin TH, Chang HC, Chen TL, et al. Propofol protects against nitrosative stress-induced apoptotic insults to cerebrovascular endothelial cells via an intrinsic mitochondrial mechanism. Surgery. 2013;154:58–68. doi: 10.1016/j.surg.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 19.Harman F, Hasturk AE, Yaman M, Arca T, Kilinc K, Sargon MF, et al. Neuroprotective effects of propofol, thiopental, etomidate, and midazolam in fetal rat brain in ischemia-reperfusion model. Childs Nerv Syst. 2012;28:1055–62. doi: 10.1007/s00381-012-1782-0. [DOI] [PubMed] [Google Scholar]
- 20.Kawasaki H, Kawashima S. Regulation of the calpain-calpastatin system by membranes (review) Mol Membr Biol. 1996;13:217–24. doi: 10.3109/09687689609160599. [DOI] [PubMed] [Google Scholar]
- 21.Stefanis L. Caspase - dependent and-Independent neuronal death: Two distinct pathways to neuronal injury. Neuroscientist. 2005;11:50–62. doi: 10.1177/1073858404271087. [DOI] [PubMed] [Google Scholar]
- 22.Hensley K, Venkova K, Christov A, Gunning W, Park J. Collapsin response mediator protein-2: An emerging pathologic feature and therapeutic target for neurodisease indications. Mol Neurobiol. 2011;43:180–91. doi: 10.1007/s12035-011-8166-4. [DOI] [PubMed] [Google Scholar]
- 23.Hou ST, Jiang SX, Aylsworth A, Ferguson G, Slinn J, Hu H, et al. CaMKII phosphorylates collapsin response mediator protein 2 and modulates axonal damage during glutamate excitotoxicity. J Neurochem. 2009;111:870–81. doi: 10.1111/j.1471-4159.2009.06375.x. [DOI] [PubMed] [Google Scholar]
- 24.Bu X, Zhang N, Yang X, Liu Y, Du J, Liang J, et al. Proteomic analysis of cPKCßII-interacting proteins involved in HPC-induced neuroprotection against cerebral ischemia of mice. J Neurochem. 2011;117:346–56. doi: 10.1111/j.1471-4159.2011.07209.x. [DOI] [PubMed] [Google Scholar]
- 25.Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;11(147):742–58. doi: 10.1016/j.cell.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raghupathi R, Graham DI, McIntosh TK. Apoptosis after traumatic brain injury. J Neurotrauma. 2000;17:927–38. doi: 10.1089/neu.2000.17.927. [DOI] [PubMed] [Google Scholar]
- 27.Holownia A, Mroz RM, Wielgat P, Skiepko A, Sitko E, Jakubow P, et al. Propofol protects rat astroglial cells against tert-butyl hydroperoxide-induced cytotoxicity; the effect on histone and cAMP-response-element-binding protein (CREB) signalling. J Physiol Pharmacol. 2009;60:63–9. [PubMed] [Google Scholar]
- 28.Cui DR, Wang L, Jiang W, Qi AH, Zhou QH, Zhang XL. Propofol prevents cerebral ischemia-triggered autophagy activation and cell death in the rat hippocampus through the NF-kB/p53 signaling pathway. Neuroscience. 2013;246:117–32. doi: 10.1016/j.neuroscience.2013.04.054. [DOI] [PubMed] [Google Scholar]
- 29.Li J, Han B, Ma X, Qi S. The effects of propofol on hippocampal caspase-3 and Bcl-2 expression following forebrain ischemia-reperfusion in rats. Brain Res. 2010;1356:11–23. doi: 10.1016/j.brainres.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 30.Xi HJ, Zhang TH, Tao T, Song CY, Lu SJ, Cui XG, et al. Propofol improved neurobehavioral outcome of cerebral ischemia-reperfusion rats by regulating Bcl-2 and Bax expression. Brain Res. 2011;1410:24–32. doi: 10.1016/j.brainres.2011.06.060. [DOI] [PubMed] [Google Scholar]
- 31.Thal SC, Timaru-Kast R, Wilde F, Merk P, Johnson F, Frauenknecht K, et al. Propofol impairs neurogenesis and neurologic recovery and increases mortality rate in adult rats after traumatic brain injury. Crit Care Med. 2014;42:129–41. doi: 10.1097/CCM.0b013e3182a639fd. [DOI] [PubMed] [Google Scholar]
- 32.Kelly DF, Goodale DB, Williams J, Herr DL, Chappell ET, Rosner MJ, et al. Propofol in the treatment of moderate and severe head injury: A randomized, prospective double-blinded pilot trial. J Neurosurg. 1999;90:1042–52. doi: 10.3171/jns.1999.90.6.1042. [DOI] [PubMed] [Google Scholar]
- 33.Gu JW, Yang T, Kuang YQ, Huang HD, Kong B, Shu HF, et al. Comparison of the safety and efficacy of propofol with midazolam for sedation of patients with severe traumatic brain injury: A meta-analysis. J Crit Care. 2014;29:287–90. doi: 10.1016/j.jcrc.2013.10.021. [DOI] [PubMed] [Google Scholar]