

Abstract
Background:
Although the onset of anemia during infectious disease is commonly correlated with production of inflammatory cytokines, the mechanisms by which cytokines induce anemia are poorly defined. This study focused on the mechanism research.
Methods:
Different types of mice were infected perorally with Toxoplasma gondii strain ME49. At the indicated times, samples from each mouse were harvested, processed, and analyzed individually. Blood samples were analyzed using a Coulter Counter and red blood cell (RBC) survival was measured by biotinylation. Levels of tumor necrosis factor-α (TNF-α), inducible nitric oxide synthase (iNOS), and inducible protein 10 (IP-10) mRNA in liver tissue were measured by real-time polymerase chain reaction.
Results:
T. gondii-infected mice exhibited anemia due to a decrease in both erythropoiesis and survival time of RBC in the circulation (P < 0.02). In addition, infection-stimulated anemia was associated with fecal occult, supporting previous literature that hemorrhage is a consequence of T. gondii infection in mice. Infection-induced anemia was abolished in interferon gamma (IFNγ) and IFNγ receptor deficient mice (P < 0.05) but was still evident in mice lacking TNF-α, iNOS, phagocyte NADPH oxidase or IP-10 (P < 0.02). Neither signal transducer and activator of transcription 1 (STAT1) deficient mice nor 129S6 controls exhibited decreased erythropoiesis, but rather suffered from an anemia resulting solely from increased loss of circulating RBC.
Conclusions:
Infection-stimulated decrease in erythropoiesis and losses of RBC have distinct mechanistic bases. These results show that during T. gondii infection, IFNγ is responsible for an anemia that results from both a decrease in erythropoiesis and a STAT1 independent loss of circulating RBC.
Keywords: Anemia, Hemorrhage, Signal Transducer and Activator of Transcription 1, Toxoplasma gondii
INTRODUCTION
Anemia accompanies a variety of chronic illnesses including inflammatory disorders, neoplasia, and infection.[1] In some infection diseases such as Mycoplasma pneumoniae, anemia correlates with increased risk of early death.[2] Correlative clinical reports, supported by in-vitro studies, suggest a significant role for the inflammatory cytokines interferon gamma (IFNγ) and tumor necrosis factor-α (TNF-α) in the onset of anemia.[3,4] However, direct in-vivo studies of the causes of anemia during infectious disease have not been performed, and the underlying mechanisms remain poorly defined.
Interferon gamma critically mediates host resistance to numerous infections. Deficiencies in the production of IFNγ or the IFNγ receptor (IFNγ-R) result in impaired resistance to bacterial,[5] viral,[6] and parasitic infections.[7] IFNγ initiates protection from infection by inducing the expression of genes that enhance immunity and exert antimicrobial functions.[8] In addition to its well-recognized protective effects, pathological effects have also been attributed to IFNγ production during viral,[6] helminth,[9] and parasitic[10,11] infections. Specifically, production of IFNγ has been correlated to the onset of anemia during both autoimmune and infectious disease.[1] In vitro evidence has suggested an inhibitory role of IFNγ in the maturation of erythroid progenitors.[12,13] However, direct in-vivo mechanisms for IFNγ-induced anemia during infection remain to be fully elucidated.
The binding of IFNγ to cellular receptors stimulates recruitment and activation of members of the Janus kinase and signal transducer and activator of transcription (STAT) family. Phosphorylation of STAT1 stimulates the transcription of most IFNγ-inducible genes.[14] Specifically, STAT1 is involved in the transcriptional regulation of IFNγ-inducible genes phagocyte NADPH oxidase (Phox), inducible nitric oxide synthase (iNOS) and inducible protein 10 (IP-10),[15,16,17] all of which are involved in host resistance to infection. These antimicrobial genes also are implicated in the induction of pathology during a variety of infections.[18,19]
Toxoplasma gondii is an obligate intracellular, protozoan parasite that causes significant disease in immunocompromised patients and upon transplacental transmission to the developing fetus. Host defense against T. gondii infection has been extensively studied in murine models, where it has been found that protection is mediated by a robust type 1 immune response characterized by the production of large quantities of IFNγ.[20] In this setting, however, IFNγ also functions pathologically. Liesenfeld et al. demonstrated that intestinal pathology accompanying acute T. gondii infection was abrogated by suppression of IFNγ.[11] In addition, we demonstrated that mice infected with T. gondii exhibit anemia that is characterized by decreased hematocrits and is dependent upon production of IFNγ.[21] Mice lacking hemostatic function (i.e., fibrin[ogen]−/− or warfarin-treated mice) displayed exacerbated IFNγ-induced anemia, suggesting that hemorrhage is responsible for the anemia. However, previous literature suggests that IFNγ may also suppress erythropoiesis during infection. Therefore, in this study, we further characterize the IFNγ-dependent anemia during T. gondii infection in mice. We demonstrate that IFNγ production during T. gondii infection both decreases red blood cell (RBC) production as evidenced by reduced numbers of circulating reticulocytes, and simultaneously increases the loss of circulating RBC. However, analyses of erythropoiesis and RBC loss in fibrin (ogen)−/−, IFNγ−/−, IFNγ-R−/−, and STAT1−/− mice revealed that infection-stimulated anemia primarily results from IFNγ-dependent, STAT1-independent RBC loss. These findings will lead to a better understanding of the pathways that result in IFNγ-mediated protection versus IFNγ-mediated pathology, and should thereby enable the development of therapies that alleviate pathology while maintaining protection.
METHODS
Mice
Six to ten weeks old male mice were used for all experiments. C57BL/6, 129S6, STAT1−/−, IFNγ−/−, IFN-γR−/−, TNF−/−, and NOS2−/− mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Phox−/−/iNOS−/− mice and fibrin(ogen)−/− mice were generously provided by Li Chen (Xinan Hospital, Chongqing). Animals were housed in a specific pathogen-free facility and cared for according to the General Hospital of Chinese People's Liberation Army and Yuhuangding Hospital Animal Care and Use Committee guidelines. Five to ten mice were used for each group in our study.
Infections
Toxoplasma gondii strain ME49 was originally provided by Li Chen (Xinan Hospital, Chongqing) and maintained by serial passage in C57BL/6 mice. For experimental infections, mice were infected perorally with indicated numbers of cysts in 0.1 ml diluted brain suspension obtained from infected animals, as previously described.[21] Sham-infected mice received similarly diluted brain suspensions from uninfected animals. At the indicated times, samples from each mouse were harvested, processed, and analyzed individually. Where indicated, mice were treated with polyclonal anti-IP-10 (100 μg, i.p. every other day) antibody.
Blood parameters
Blood samples from anticoagulated mice (500 U heparin, intravenously) were obtained by cardiac puncture immediately following euthanasia by carbon dioxide narcosis. For the determination of hematocrits and total number of circulating RBCs, blood was diluted 20-fold in 5 mm EDTA and analyzed using a Coulter Counter (Beckman Coulter, USA). Fecal occult blood was identified using a Hemoccult kit (Beckman Coulter). IFNγ protein levels in sera were measured by sandwich ELISA (BD Biosciences, USA).
To quantify numbers of circulating reticulocytes, 5 μl of blood was stained with 500 μl thiazole orange solution (Retic-Count, BD Biosciences) for 1 h at room temperature, centrifuged at 800×g for 5 min, and resuspended in 1% formaldehyde.[22] The percentage of thiazole orange-stained RBC (i.e., reticulocytes) was determined by flow cytometry, using forward and side scatter to gate on RBC. The total number of reticulocytes was then calculated by multiplying the percentage of reticulocytes by the total number of RBC as determined by Coulter Counter.
Red blood cell survival was measured by biotinylating all circulating cells in vivo, and then monitoring the loss of biotinylated RBC over time.[23] In vivo biotinylation was performed the day prior to infection by retro-orbital injection of 1.2 mg biotin, dissolved in 10% dimethyl sulfoxide, given in a split dose 2 h apart. Mice were then euthanized on the indicated days, and 5 μl blood was stained with streptavidin conjugated with phycoerythrin (BD Pharmingen) and TER119 mAb conjugated with fluorescein isothiocyanate. The percentage of streptavidin-positive TER119-positive cells in each sample was identified by flow cytometry and multiplied by the total number of RBC determined by Coulter Counter to obtain total numbers of biotinylated RBC.
Real-time polymerase chain reaction
Levels of TNF-α, iNOS, and IP-10 mRNA in liver tissue were measured by real-time polymerase chain reaction (PCR) (PerkinElmer). Real-time PCR primer and probe sequences for TNF-α and iNOS were described by Smiley laboratory.[21] Primer combinations for IP-10 are as follows; IP-10-forward CTGCCGTCATTTTCTGCCTC, IP-10-reverse CACTGGCCCGTCATCGATAT, IP-10-probe CGCAAGGACGGTCCGCTGC.
Statistics
The data about hematocrit drop, RBC production and RBC loss were analyzed by one-way analysis of variance (ANOVA). Student's t-test was used for other data analysis. Both Student's t-test and one-way ANOVA were performed using the computer program Prism 5.0 (GraphPad Software Inc. San Diego, USA).
RESULTS
Production of red blood cell is impaired during infection but does not fully account for Toxoplasma gondii-induced anemia
Infection of C57BL/6 mice with T. gondii induced robust IFNγ production [Figure 1a] that correlated kinetically with decreased hematocrits [Figure 1b] and numbers of circulating RBC [Figure 1c]. To further delineate the mechanisms underlying this anemia, we quantified RBC production throughout the course of T. gondii infection. After their release from bone marrow as erythroblasts, RBC retain RNA for 24–48 h. During this period, RBC was referred to as reticulocytes and can be enumerated using flow cytometric assays employing RNA-binding dyes. Using such an assay, we found that numbers of reticulocytes in infected mice were comparable to those of sham-infected mice up through day 6. At day 8, post infection reticulocyte numbers decreased significantly, but rebounded by day 10 and then remained elevated until the anemia resolved [Figure 1d]. These results indicate that RBC production only briefly decreases during T. gondii infection, and fully recovers prior to the peak of anemia at day 10.
Figure 1.
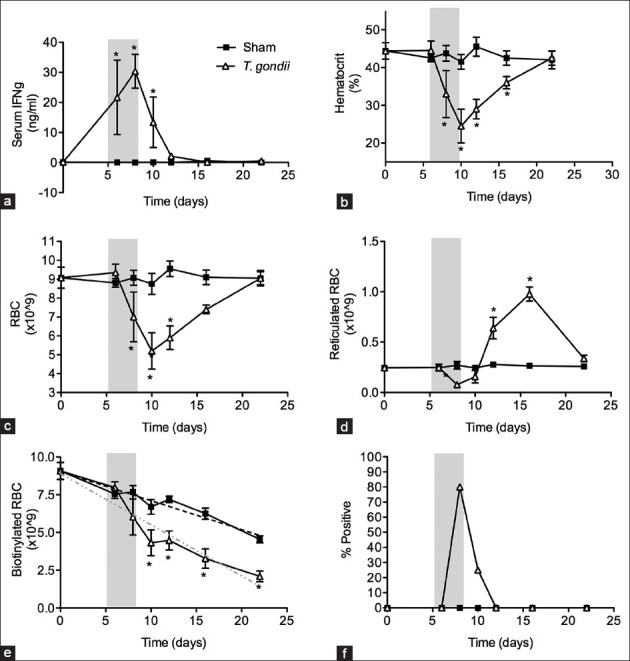
Interferon gamma (IFN-γ) dependent hematocrit drop during Toxoplasma gondii infection is a result of decreased red blood cell (RBC) production and increased RBC loss. Wild type C57BL/6 mice were infected with 10 ME49 cysts and injected with biotin as described in materials and methods. (a) Serum IFNγ levels; (b) Hematocrit; (c) RBC numbers; (d) Total number of reticulocytes; (e) Total number of biotinylated RBC and (f) Occult blood were evaluated on days indicated. Significant differences between sham-infected controls and T. gondii-infected mice are depicted (*P < 0.02). All data are represented as mean ± SD of four mice per group.
Increased red blood cell loss and hemorrhage accompany Toxoplasma gondii infection
Based on data in Figure 1d, decreased production of RBC can only account for at most a deficit of 0.8 × 109 RBC. Since T. gondii-infected mice have approximately 3.55 × 109 fewer circulating RBC than sham control mice on day 10 of infection [Figure 1c], an alternative mechanism to decreased erythropoiesis must account for the 2.75 × 109 decrease in total RBC in infected mice. Therefore, we investigated the possibility that anemia results also from RBC loss. To estimate the rate of RBC loss, we pulse-labeled RBC with biotin in vivo[23] and measured the number of biotin-labeled RBC remaining at various time points throughout T. gondii infection. Figure 1e indicates that sham-infected mice lost circulating biotinylated RBC at an average rate of 2.1% per day. In contrast, T. gondii-infected mice lost circulating biotinylated RBC at an average of 11.4% per day between days 6 and 10 of infection accounting for a total of 2.4 × 109 RBC. After that time, the rate of loss returned to that observed in sham-infected controls. Thus, T. gondii-infected mice display a significant increase in RBC loss during the same period at which they display anemia.
To evaluate whether the loss of RBC resulted from T. gondii-induced hemorrhage, we obtained fecal samples from infected animals at various days of infection and analyzed for the presence of occult blood. We detected fecal occult blood on days 8 and 10 post infection [Figure 1f] indicative of an intestinal hemorrhage in infected animals. Together, our findings of brief decline in erythropoiesis, increased RBC loss and the presence of fecal occult blood in infected mice strongly suggest that hemorrhage accounts for a majority of the RBC lost during T. gondii-infection.
Fibrin-deficiency exacerbates Toxoplasma gondii induced hemorrhage without impacting red blood cell production
Fibrin is a product of the coagulation cascade that aids clotting thereby suppressing blood loss. To determine whether the exacerbated hematocrit drop in fibrin(ogen)−/− mice results from decreased RBC production or increased RBC loss, both parameters were measured in fibrin(ogen)−/− and littermate control mice on day 8 post infection [Figure 2]. As previously reported, the hematocrits of fibrin(ogen)−/− mice infected with T. gondii were significantly decreased as compared with infected littermate controls [Figure 2a]. The number of reticulocytes in fibrin(ogen)−/− animals did not differ significantly from controls [Figure 2b], however; the loss of circulating biotinylated RBC was significantly greater in fibrin(ogen)−/− mice compared with control mice [Figure 2c]. Taken together, these data indicate an important role for RBC loss in the T. gondii-induced hematocrit reduction in fibrin(ogen)−/− mice, supporting our conclusion that decreases in hematocrit primarily reflect hemorrhage caused by infection.
Figure 2.
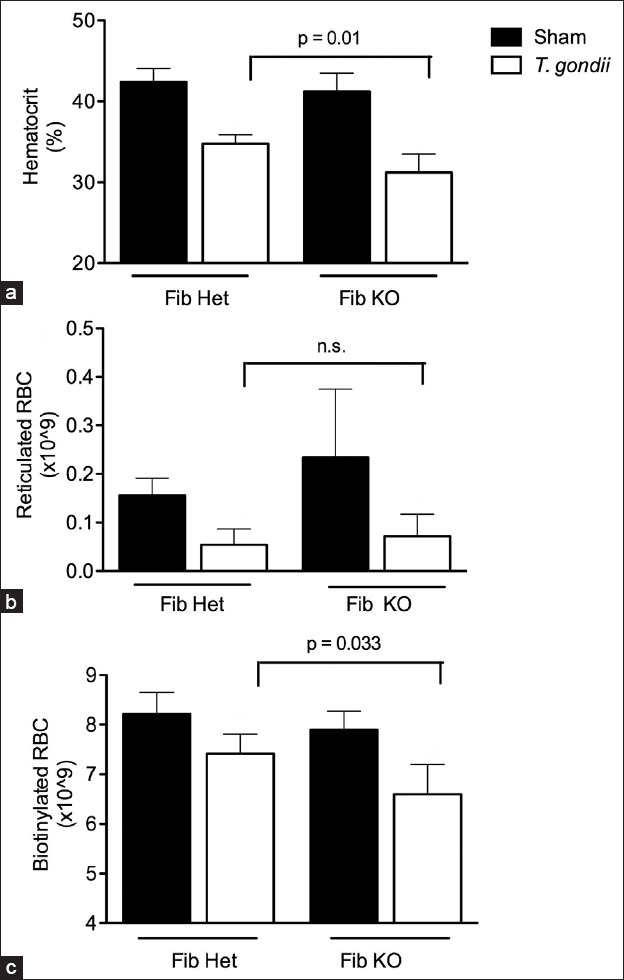
Fibrin is protective against interferon gamma-induced hemorrhage during Toxoplasma gondii infection. Fibrin (ogen) heterozygous and knockout mice were biotinylated and infected with T. gondii on day 0. Blood samples were collected on day 8 of infection. (a) Hematocrit levels; (b) Total number of reticulocytes and (c) Total number of biotinylated red blood cell are shown for fibrin (ogen)−/− mice and heterozygous littermates.
Anemia that accompanies Toxoplasma gondii infection is dependent upon interferon gamma and interferon gamma receptor, but is tumor necrosis factor, inducible nitric oxide synthase, phagocyte NADPH oxidase and inducible protein 10 independent
Mice lacking the capacity to produce IFNγ fail to exhibit the T. gondii-induced hematocrit reduction [Figure 3a]. Consistent with those findings, mice unable to respond to IFNγ due to a genetic deficiency of IFNγ-R also do not demonstrate the T. gondii-induced hematocrit reduction [Figure 3a]. While infection of WT mice caused a significant decrease in reticulocyte numbers, infection of IFNγ−/− or IFNγ-R−/− mice did not alter reticulocyte numbers as compared with sham controls [Figure 3b]. Furthermore, there was no significant evidence of hemorrhage in infected IFNγ−/− or IFNγ-R−/− mice as measured through loss of biotinylated RBC or fecal occult blood [Figure 3c and d]. These data demonstrate that both the decreased erythropoiesis and the increased RBC loss components of T. gondii-induced anemia are dependent upon the production of IFNγ and subsequent signaling through the IFNγ-R.
Figure 3.
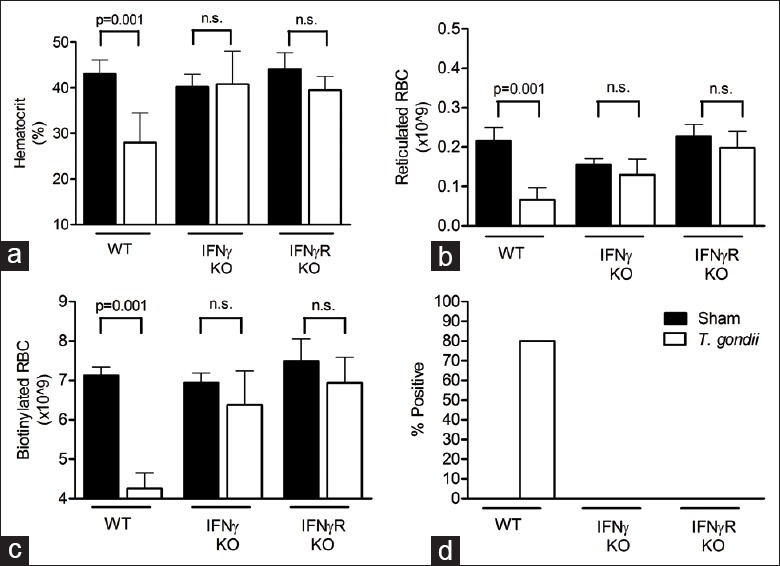
Interferon gamma (IFNγ) and IFNγ receptor are required for Toxoplasma gondii induce anemia. Indicated knockout mice were infected with 10 ME49 cysts and blood and fecal samples collected on day 8. (a) Hematocrit levels; (b) Total number of reticulocytes; (c) Total number of biotinylated red blood cell and (d) Percent positive hemoccults are shown for both infected and uninfected animals (ND: None detected). Significant differences are shown (*P < 0.05). All data are represented as mean ± SD of at least three mice per group.
Interferon gamma has been implicated in the induction of the inflammatory cytokine TNF-α, resulting in pathology during malaria infection.[24] Also, previous literature has suggested a synergistic role for IFNγ and TNF-α in eliciting an immune response to T. gondii[25,26,27] and in the suppression of hematopoietic progenitor cell growth in vitro.[28] Following confirmation of TNF-α mRNA induction during infection with T. gondii [Figure 4a], we asked whether TNF-α plays a role in T. gondii-induced anemia. Hematocrit levels and numbers of biotinylated RBC were significantly decreased in TNF-α−/− mice infected with T. gondii as compared with sham controls [Figure 4b], indicating that T. gondii-induced anemia is TNF-α-independent.
Figure 4.
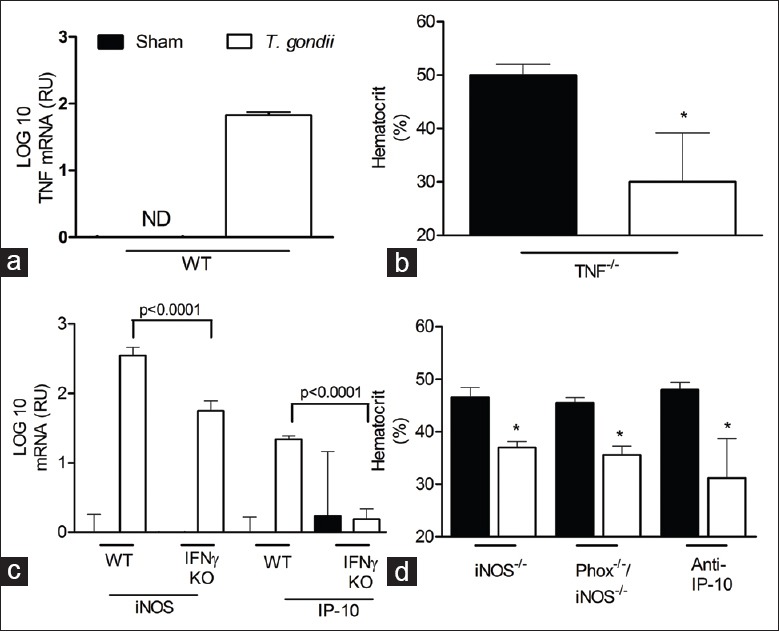
Toxoplasma gondii induces hemorrhage in a tumor necrosis factor (TNF), phagocyte NADPH oxidase, inducible nitric oxide synthase (iNOS), and inducible protein 10 (IP-10) independent manner. Indicated knockout mice were infected with 10 ME49 cysts and blood and fecal samples collected on day 8. (a) Levels of TNF mRNA in C57BL/6 mice and (b) hematocrit levels in TNF knockout mice, are shown for both infected and uninfected animals. Similarly, induction of iNOS and IP-10 mRNA (c) and decreased hematocrit for indicated KO mice (d) are shown for both infected and uninfected animals. Significant differences are shown (*P < 0.02) (ND: None detected). All data are represented as mean ± SD of at least three mice per group.
Having ruled out a role for TNF-α in the reduction of hematocrit levels during T. gondii infection, we focused on IFNγ-dependent effector molecules upregulated during T. gondii infection. Both iNOS and Phox are IFNγ inducible genes involved in the regulation of the immune response and intestinal pathology during infection with Francisella tularensis.[29] The products of these genes generate reactive nitrogen and oxygen species, which could be responsible for tissue and endothelial damage leading to vascular leakage and subsequent hemorrhage. Induction of iNOS mRNA on D8 of infection was significantly abrogated in IFNγ−/− mice compared with C57BL/6 infected mice [Figure 4c], indicating that iNOS is inducible by IFNγ during T. gondii infection. To determine whether hemorrhage during T. gondii infection was a consequence of host cell damage due to reactive oxygen, we evaluated hematocrit levels in both iNOS−/− and Phox/iNOS double knock-out animals. Hematocrit levels decreased significantly in both iNOS−/− and Phox−/−/iNOS−/− mice on day 8 postinfection compared with sham-infected controls [Figure 4d], and the extent of this decrease was comparable to that seen in WT mice. In addition to iNOS and Phox, IFNγ stimulates production also of IP-10, which has been implicated in downstream effector functions resulting in immunopathology during Crohn's disease.[30] Similarly to iNOS mRNA, induction of IP-10 mRNA during T. gondii infection is reduced in IFNγ−/− mice compared with WT controls [Figure 4c]. Nevertheless, wild type B6 mice treated with anti-IP-10 displayed a significant hematocrit decrease on day 8 of infection [Figure 4d]. Thus, while the T. gondii-induced anemia is IFNγ- and IFNγ-R-dependent [Figure 3], these data demonstrate that anemia is independent of TNF-α, iNOS, Phox and IP-10 gene products [Figure 4].
Toxoplasma gondii-induced, interferon gamma-dependent hemorrhage is signal transducer and activator of transcription 1 independent
Identification of the signaling cascades involved in IFNγ-mediated anemia during T. gondii infection may provide critical insight into the downstream effector molecules regulating infection-associated hemorrhage. IFNγ induces gene expression primarily through STAT1-dependent pathways.[31,32] To evaluate the role of STAT1 in T. gondii-stimulated anemia, we infected STAT1−/− mice and measured blood parameters on day 8 postinfection. Unexpectedly, hematocrit levels in STAT1−/− mice were significantly lower than those of sham controls [Figure 5a], and comparable to genetically matched 129S6 mice infected with T. gondii. Reticulocyte production did not decrease as a result of infection in STAT1−/− mice or their 1296 controls [Figure 5b], but there was a significant loss of biotinylated RBC in both strains compared with sham-infected mice [Figure 5c]. Based on these results, we conclude that the IFNγ-induced hemorrhage in T. gondii infected animals is mediated through a STAT1-independent mechanism.
Figure 5.
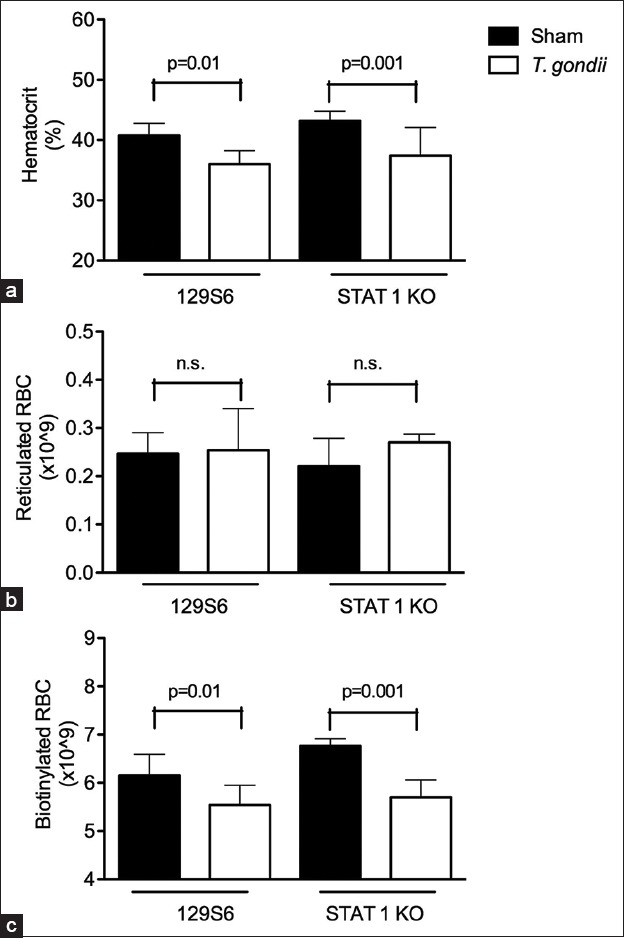
Interferon gamma mediated hemorrhage is STAT1-independent. STAT1 KO mice and genetically matched 129S6 controls were injected with biotin and infected with 10 ME49 cysts of Toxoplasma gondii. On day 8 of infection, blood and fecal samples were collected and analyzed. (a) Hematocrit levels in 129S6 and STAT1−/− mice were significantly decreased as compared with sham controls; (b) Reticulocyte numbers were not significantly different between Toxoplasma gondii infected and sham control mice; (c) Both 129S6 and STAT1−/− mice lost a significant number of biotinylated red blood cell as a result of T. gondii infection. Significant differences are shown (*P < 0.05). All data are represented as mean ± SD of at least four mice per group.
DISCUSSION
Anemia accompanies a variety of infectious diseases including malaria,[24] trypanosomiasis,[33] mononucleosis[34] and secondary infections during HIV.[2,35] Clinical studies have implicated important roles from inflammatory cytokines in the anemia's associated with infectious disease. In this study, an in vivo model was used to evaluate roles for the inflammatory cytokine, IFNγ, in the onset of anemia due to infectious disease. Specifically, this study identifies a STAT1-independent IFNγ-induced pathology during infection with the protozoan parasite T. gondii.
Infection-induced anemia could potentially result from decreased RBC production, RBC loss due to hemorrhage or destruction, as described in disseminated intravascular coagulation (DIC), or pathogen-associated hemolysis. In the present study, we show that T. gondii-induced anemia results from both decreased erythropoiesis and increased loss of circulating RBC [Figure 1]. We believe hypochromic microcytic anemia is the only morphological features for hemorrhagic anemia, and red cell loss caused by infection is the only reason during this process. Results of mean corpuscular volume mean corpuscular hemoglobin support our idea (data not shown). Anemia resulting from decreased erythropoiesis has been described in murine malaria, in part resulting from effects of increased TNF-α levels on RBC precursor cells in the bone marrow of infected mice.[36] Additionally, decrease numbers of erythroid progenitor cells have been previously observed in CBA mice infected with the virulent RH strain of T. gondii.[37] Together, these reports support our conclusion that a decrease in erythropoiesis during T. gondii infection is in part responsible for the induced anemia. Nevertheless, the decrease in RBC production accounts for only a small portion of the total number of RBC lost during T. gondii infection, indicating that an additional mechanism must contribute to this anemia. The possibility that anemia during T. gondii infection is not solely due to a reduction of erythropoiesis is supported by similar data during malaria infections.[38]
Another candidate for the cause of infection-induced anemia includes the destruction of RBC in circulation.[39] During T. gondii infection, RBC destruction does not appear to be a mechanism for anemia, as there are no detectable spherocytes or schistocytes, hallmarks of such destruction,[39] in peripheral blood smears from infected mice (data not shown). Alternatively, hemophagocytosis accompanies certain infections and can result in decreased erythrocyte numbers due to their phagocytosis by activated macrophages.[34] However, it is unclear why loss of fibrin would predispose to increased hemophagocytosis or lysis, whereas it is obvious that it predisposes to hemorrhage [Figure 2]. Indeed, hemorrhage, as measured through loss of biotinylated RBC and positive hemoccult, accounts for the majority of RBC loss during T. gondii-induced anemia [Figure 1].
Hemorrhagic pathology, including bleeding from the respiratory and gastrointestinal organs, has been described during both bacterial and viral infections. During dengue hemorrhagic fever, hemorrhage is a result of vasculopathy and coagulopathy due to an imbalance between coagulation and fibrinolysis.[40] Activation of the coagulation system can result in increased fibrin deposition in the vasculature causing DIC and a subsequent depletion of RBC due to thrombosis and bleeding as described in viral hemorrhagic fevers.[41] Dengue-virus induced hemorrhage is correlated to platelet abnormalities and DIC.[40] Though we have not formally ruled out DIC as a possible cause of T. gondii induced anemia, exacerbation of anemia in fibrin (ogen)−/− animals suggests that excessive coagulation is a not a primary cause of anemia. Nevertheless, our data indicated that RBC loss and hemorrhage are primary causes of IFNγ-dependent anemia in T. gondii-infected mice.
Previous research has implicated several IFNγ-IPs in pathology associated with inflammation. Treating mice with nitric oxide inhibitors prevented intestinal necrosis during T. gondii infection.[25] Additionally, IP-10 has been implicated in the induction of liver, kidney, and intestinal pathology during Crohn's disease.[30] However, we were unable to identify a role for nitric oxide, or IP-10 in the induction of anemia during T. gondii infection. Additionally, no role for TNF-α was identified in T. gondii induced anemia. Though anemia during Trypanosoma cruzi is not accompanied by a decrease in reticulocyte numbers, similar to our findings, chemical inhibition of nitric oxide or treatment with anti-TNF did not rescue mice from anemia even though these treatments resulted in increased numbers of reticulocytes and suppressed decreases in bone marrow erythroblast numbers during infection.[33] Our results suggest that divergent mechanisms may exist for IFNγ-induced protection compared with IFNγ-induced hemorrhagic immunopathology during T. gondii infection.
Identification of the signaling cascades involved in IFNγ-mediated anemia during T. gondii infection may provide insight into the downstream effector molecules regulating hemorrhage. Recent studies established that IFNγ production is STAT1 independent[14,42] during T. gondii infection. This led us to assess roles for STAT1 as a downstream signaling molecule involved in IFNγ-induced hemorrhage during T. gondii infection. Based on previous literature describing the induction of pathological IFNγ effector mechanisms through STAT1, we anticipated that this signaling mechanism also would be required in induction of IFNγ-dependent pathology during T. gondii infection. Surprisingly, in this setting, T. gondii infection stimulated anemia in a STAT1-independent fashion. These data suggest that both the induction of IFNγ and the subsequent activation of an IFNγ-induced effector mechanism resulting in hemorrhage is a STAT1-independent pathway. Recent in vitro studies with macrophages have identified MCP-1, MIP-1α and β, as STAT1-independent, IFNγ-inducible genes.,[43,44] However, infections of CCL2, CCR2 or CCR5 deficient mice did not indicate a role for these chemokines in T. gondii-induced anemia (data not shown). Future in vivo studies will continue to investigate potential IFNγ-dependent, STAT1-independent effector molecules that are responsible for T. gondii induced hemorrhagic anemia. Understanding the pathways that result in IFNγ-mediated protection versus IFNγ-mediated pathology will provide valuable information to prevent immune pathology while, not compromising the protective effects of the immune response.
ACKNOWLEDGMENTS
We are indebted to the employees of The Genernal Hospital of People's Liberation Army and Yuhuangding Hospital Animal Breeding and Maintenance Facilities for dedicated care of the mice used in these studies.
Footnotes
Edited by: Li-Shao Guo
Source of Support: This work was supported by a grant from Yuhuangding Hospital.
Conflict of Interest: None declared.
REFERENCES
- 1.Means RT, Jr, Krantz SB. Progress in understanding the pathogenesis of the anemia of chronic disease. Blood. 1992;80:1639–47. [PubMed] [Google Scholar]
- 2.Gu L, Chen X, Li H, Qu J, Miao M, Zhou F, et al. A case of lethal hemolytic anemia associated with severe pneumonia caused by Mycoplasma pneumoniae. Chin Med J (Engl) 2014;127:3839. [PubMed] [Google Scholar]
- 3.Papadaki HA, Kritikos HD, Valatas V, Boumpas DT, Eliopoulos GD. Anemia of chronic disease in rheumatoid arthritis is associated with increased apoptosis of bone marrow erythroid cells: Improvement following anti-tumor necrosis factor-alpha antibody therapy. Blood. 2002;100:474–82. doi: 10.1182/blood-2002-01-0136. [DOI] [PubMed] [Google Scholar]
- 4.Means RT., Jr The anaemia of infection. Baillieres Best Pract Res Clin Haematol. 2000;13:151–62. doi: 10.1053/beha.1999.0065. [DOI] [PubMed] [Google Scholar]
- 5.MacMicking JD, Taylor GA, McKinney JD. Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science. 2003;302:654–9. doi: 10.1126/science.1088063. [DOI] [PubMed] [Google Scholar]
- 6.Ostler T, Davidson W, Ehl S. Virus clearance and immunopathology by CD8(+) T cells during infection with respiratory syncytial virus are mediated by IFN-gamma. Eur J Immunol. 2002;32:2117–23. doi: 10.1002/1521-4141(200208)32:8<2117::AID-IMMU2117>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 7.Denkers EY, Gazzinelli RT. Regulation and function of T-cell-mediated immunity during Toxoplasma gondii infection. Clin Microbiol Rev. 1998;11:569–88. doi: 10.1128/cmr.11.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: An overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–89. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 9.Rutitzky LI, Ozkaynak E, Rottman JB, Stadecker MJ. Disruption of the ICOS-B7RP-1 costimulatory pathway leads to enhanced hepatic immunopathology and increased gamma interferon production by CD4 T cells in murine schistosomiasis. Infect Immun. 2003;71:4040–4. doi: 10.1128/IAI.71.7.4040-4044.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blass SL, Puré E, Hunter CA. A role for CD44 in the production of IFN-gamma and immunopathology during infection with Toxoplasma gondii. J Immunol. 2001;166:5726–32. doi: 10.4049/jimmunol.166.9.5726. [DOI] [PubMed] [Google Scholar]
- 11.Liesenfeld O, Kosek J, Remington JS, Suzuki Y. Association of CD4+ T cell-dependent, interferon-gamma-mediated necrosis of the small intestine with genetic susceptibility of mice to peroral infection with Toxoplasma gondii. J Exp Med. 1996;184:597–607. doi: 10.1084/jem.184.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raefsky EL, Platanias LC, Zoumbos NC, Young NS. Studies of interferon as a regulator of hematopoietic cell proliferation. J Immunol. 1985;135:2507–12. [PubMed] [Google Scholar]
- 13.Zoumbos NC, Djeu JY, Young NS. Interferon is the suppressor of hematopoiesis generated by stimulated lymphocytes in vitro. J Immunol. 1984;133:769–74. [PubMed] [Google Scholar]
- 14.Gavrilescu LC, Butcher BA, Del Rio L, Taylor GA, Denkers EY. STAT1 is essential for antimicrobial effector function but dispensable for gamma interferon production during Toxoplasma gondii infection. Infect Immun. 2004;72:1257–64. doi: 10.1128/IAI.72.3.1257-1264.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumatori A, Yang D, Suzuki S, Nakamura M. Cooperation of STAT-1 and IRF-1 in interferon-gamma-induced transcription of the gp91(phox) gene. J Biol Chem. 2002;277:9103–11. doi: 10.1074/jbc.M109803200. [DOI] [PubMed] [Google Scholar]
- 16.Delgado M. Inhibition of interferon (IFN) gamma-induced Jak-STAT1 activation in microglia by vasoactive intestinal peptide: Inhibitory effect on CD40, IFN-induced protein-10, and inducible nitric-oxide synthase expression. J Biol Chem. 2003;278:27620–9. doi: 10.1074/jbc.M303199200. [DOI] [PubMed] [Google Scholar]
- 17.Majumder S, Zhou LZ, Chaturvedi P, Babcock G, Aras S, Ransohoff RM. p48/STAT-1alpha-containing complexes play a predominant role in induction of IFN-gamma-inducible protein, 10 kDa (IP-10) by IFN-gamma alone or in synergy with TNF-alpha. J Immunol. 1998;161:4736–44. [PubMed] [Google Scholar]
- 18.Klein RS, Izikson L, Means T, Gibson HD, Lin E, Sobel RA, et al. IFN-inducible protein 10/CXC chemokine ligand 10-independent induction of experimental autoimmune encephalomyelitis. J Immunol. 2004;172:550–9. doi: 10.4049/jimmunol.172.1.550. [DOI] [PubMed] [Google Scholar]
- 19.Gao XP, Standiford TJ, Rahman A, Newstead M, Holland SM, Dinauer MC, et al. Role of NADPH oxidase in the mechanism of lung neutrophil sequestration and microvessel injury induced by Gram-negative sepsis: Studies in p47phox-/- and gp91phox-/- mice. J Immunol. 2002;168:3974–82. doi: 10.4049/jimmunol.168.8.3974. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki Y, Orellana MA, Schreiber RD, Remington JS. Interferon-gamma: The major mediator of resistance against Toxoplasma gondii. Science. 1988;240:516–8. doi: 10.1126/science.3128869. [DOI] [PubMed] [Google Scholar]
- 21.Johnson LL, Berggren KN, Szaba FM, Chen W, Smiley ST. Fibrin-mediated protection against infection-stimulated immunopathology. J Exp Med. 2003;197:801–6. doi: 10.1084/jem.20021493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Hove L, Goossens W, Van Duppen V, Verwilghen RL. Reticulocyte count using thiazole orange. A flow cytometry method. Clin Lab Haematol. 1990;12:287–99. doi: 10.1111/j.1365-2257.1990.tb00039.x. [DOI] [PubMed] [Google Scholar]
- 23.Hoffmann-Fezer G, Maschke H, Zeitler HJ, Gais P, Heger W, Ellwart J, et al. Direct in vivo biotinylation of erythrocytes as an assay for red cell survival studies. Ann Hematol. 1991;63:214–7. doi: 10.1007/BF01703446. [DOI] [PubMed] [Google Scholar]
- 24.Artavanis-Tsakonas K, Tongren JE, Riley EM. The war between the malaria parasite and the immune system: Immunity, immunoregulation and immunopathology. Clin Exp Immunol. 2003;133:145–52. doi: 10.1046/j.1365-2249.2003.02174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liesenfeld O, Kang H, Park D, Nguyen TA, Parkhe CV, Watanabe H, et al. TNF-alpha, nitric oxide and IFN-gamma are all critical for development of necrosis in the small intestine and early mortality in genetically susceptible mice infected perorally with Toxoplasma gondii. Parasite Immunol. 1999;21:365–76. doi: 10.1046/j.1365-3024.1999.00237.x. [DOI] [PubMed] [Google Scholar]
- 26.Chang HR, Grau GE, Pechère JC. Role of TNF and IL-1 in infections with Toxoplasma gondii. Immunology. 1990;69:33–7. [PMC free article] [PubMed] [Google Scholar]
- 27.Däubener W, Remscheid C, Nockemann S, Pilz K, Seghrouchni S, Mackenzie C, et al. Anti-parasitic effector mechanisms in human brain tumor cells: Role of interferon-gamma and tumor necrosis factor-alpha. Eur J Immunol. 1996;26:487–92. doi: 10.1002/eji.1830260231. [DOI] [PubMed] [Google Scholar]
- 28.Broxmeyer HE, Williams DE, Lu L, Cooper S, Anderson SL, Beyer GS, et al. The suppressive influences of human tumor necrosis factors on bone marrow hematopoietic progenitor cells from normal donors and patients with leukemia: Synergism of tumor necrosis factor and interferon-gamma. J Immunol. 1986;136:4487–95. [PubMed] [Google Scholar]
- 29.Lindgren H, Stenmark S, Chen W, Tärnvik A, Sjöstedt A. Distinct roles of reactive nitrogen and oxygen species to control infection with the facultative intracellular bacterium Francisella tularensis. Infect Immun. 2004;72:7172–82. doi: 10.1128/IAI.72.12.7172-7182.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh UP, Singh S, Iqbal N, Weaver CT, McGhee JR, Lillard JW., Jr IFN-gamma-inducible chemokines enhance adaptive immunity and colitis. J Interferon Cytokine Res. 2003;23:591–600. doi: 10.1089/107999003322485099. [DOI] [PubMed] [Google Scholar]
- 31.Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–21. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 32.Platanias LC, Fish EN. Signaling pathways activated by interferons. Exp Hematol. 1999;27:1583–92. doi: 10.1016/s0301-472x(99)00109-5. [DOI] [PubMed] [Google Scholar]
- 33.Malvezi AD, Cecchini R, de Souza F, Tadokoro CE, Rizzo LV, Pinge-Filho P. Involvement of nitric oxide (NO) and TNF-alpha in the oxidative stress associated with anemia in experimental Trypanosoma cruzi infection. FEMS Immunol Med Microbiol. 2004;41:69–77. doi: 10.1016/j.femsim.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 34.Fisman DN. Hemophagocytic syndromes and infection. Emerg Infect Dis. 2000;6:601–8. doi: 10.3201/eid0606.000608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sullivan P. Associations of anemia, treatments for anemia, and survival in patients with human immunodeficiency virus infection. J Infect Dis. 2002;185(Suppl 2):S138–42. doi: 10.1086/340203. [DOI] [PubMed] [Google Scholar]
- 36.Miller KL, Silverman PH, Kullgren B, Mahlmann LJ. Tumor necrosis factor alpha and the anemia associated with murine malaria. Infect Immun. 1989;57:1542–6. doi: 10.1128/iai.57.5.1542-1546.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petakov M, Stojanovic N, Jovcic G, Bugarski D, Todorovic V, Djurkovic-Djakovic O. Hematopoiesis during acute Toxoplasma gondii infection in mice. Haematologia (Budap) 2002;32:439–55. [PubMed] [Google Scholar]
- 38.Jakeman GN, Saul A, Hogarth WL, Collins WE. Anaemia of acute malaria infections in non-immune patients primarily results from destruction of uninfected erythrocytes. Parasitology. 1999;119(Pt 2):127–33. doi: 10.1017/s0031182099004564. [DOI] [PubMed] [Google Scholar]
- 39.Mabiala Babela JR, Ollandzobo LC, Nika ER, Moyen G. Post-malaria immune hemolytic anemia: 11 cases. Med Mal Infect. 2014;44:441–3. doi: 10.1016/j.medmal.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 40.Lei HY, Yeh TM, Liu HS, Lin YS, Chen SH, Liu CC. Immunopathogenesis of dengue virus infection. J Biomed Sci. 2001;8:377–88. doi: 10.1007/BF02255946. [DOI] [PubMed] [Google Scholar]
- 41.Wiersinga WJ, Leopold SJ, Cranendonk DR, van der Poll T. Host innate immune responses to sepsis. Virulence. 2014;5:36–44. doi: 10.4161/viru.25436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lieberman LA, Banica M, Reiner SL, Hunter CA. STAT1 plays a critical role in the regulation of antimicrobial effector mechanisms, but not in the development of Th1-type responses during toxoplasmosis. J Immunol. 2004;172:457–63. doi: 10.4049/jimmunol.172.1.457. [DOI] [PubMed] [Google Scholar]
- 43.Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1-dependent and-independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002;23:96–101. doi: 10.1016/s1471-4906(01)02118-4. [DOI] [PubMed] [Google Scholar]
- 44.Lin W, Lin Y. Interferon-γ inhibits central nervous system myelination through both STAT1-dependent and STAT1-independent pathways. J Neurosci Res. 2010;88:2569–77. doi: 10.1002/jnr.22425. [DOI] [PMC free article] [PubMed] [Google Scholar]