

Abstract
Objective:
The objective was to provide a brief history of J wave syndromes and to summarize our current understanding of their molecular, ionic, cellular mechanisms, and clinical features. We will also discuss the existing debates and further direction in basic and clinical research for J wave syndromes.
Data Sources:
The publications on key words of “J wave syndromes”, “early repolarization syndrome (ERS)”, “Brugada syndrome (BrS)” and “ST-segment elevation myocardial infarction (STEMI)” were comprehensively reviewed through search of the PubMed literatures without restriction on the publication date.
Study Selection:
Original articles, reviews and other literatures concerning J wave syndromes, ERS, BrS and STEMI were selected.
Results:
J wave syndromes were firstly defined by Yan et al. in a Chinese journal a decade ago, which represent a spectrum of variable phenotypes characterized by appearance of prominent electrocardiographic J wave including ERS, BrS and ventricular fibrillation (VF) associated with hypothermia and acute STEMI. J wave syndromes can be inherited or acquired and are mechanistically linked to amplification of the transient outward current (Ito)-mediated J waves that can lead to phase 2 reentry capable of initiating VF.
Conclusions:
J wave syndromes are a group of newly highlighted clinical entities that share similar molecular, ionic and cellular mechanism and marked by amplified J wave on the electrocardiogram and a risk of VF. The clinical challenge ahead is to identify the patients with J wave syndromes who are at risk for sudden cardiac death and determine the alternative therapeutic strategies to reduce mortality.
Keywords: Brugada Syndrome, Early Repolarization Syndrome, J Wave Syndromes, ST-Segment Elevation Myocardial Infarction, Transient Outward Potassium Current
INTRODUCTION
J wave syndromes were first defined by Yan et al. in a Chinese journal in 2004[1] and has gained worldwide recognition in the past decade.[2,3,4] J wave syndromes are a spectrum of variable phenotypes characterized by the appearance of prominent electrocardiographic J waves (or Osborn waves) with a risk of ventricular fibrillation (VF), including the inherited Brugada syndrome (BrS), traditional early repolarization syndrome (ERS), idiopathic ventricular fibrillation (IVF) with J wave in inferior leads as well as acquired arrhythmias linked to the acute ST-segment elevation myocardial infarction (MI) and hypothermia. Although they may bear differences with regard to the electrocardiogram (ECG) lead location, amplitude, and underlying causes of J wave, these disease entities share a similar ionic and cellular basis, risk factors, and similar clinical outcomes. In this review, we aim to provide a brief history of J wave syndromes, summarize a decade of their research progress in molecular, ionic, cellular mechanisms, and clinical features. We will also discuss the existing debates pertaining to J wave syndromes.
J WAVE AND TRANSIENT OUTWARD POTASSIUM CURRENT (ITO)
J wave is a positive deflection immediately following the QRS complex of the surface ECG or be in part buried inside of the QRS as notching or slurring.[3,5] J wave may be accompanied by an ST segment elevation, an electrocardiographic feature that is, traditionally referred to as an early repolarization (ER) pattern.[6,7,8]
J wave or elevated J point was demonstrated as early as in the last century. J wave (QRS slurring or notching) was first reported in an experimental model of hypercalcemia,[9] followed by hypothermia-induced J waves in an accidentally frozen man by Tomaszewski, who described the wave as a very slowly inscribed deflection between the QRS complex and the ST segment of the ECG.[10] Shipley and Hallaran described J wave in healthy young individuals shortly afterward.[11] J wave was later named as Osborn wave after being highlighted by a landmark study in which Osborn described hypothermia-induced J wave in hypothermic dogs and its accentuation prior to VF.[12] Over the past decades, J waves have been increasingly recognized in subjects with central nervous system disorders,[13] clinical hypercalcemia,[14] BrS,[15,16] IVF,[17,18] and myocardial ischemia.[19,20] Especially, J wave has gained a great deal of attention after determining it as a sign of a substrate capable of generating fatal ventricular arrhythmias.
Underlying ionic and cellular basis of Ito-mediated J wave was elucidated in the days when the arterially perfused ventricular wedge preparation was first developed in 1996.[16] The action potential (AP) of human ventricular myocytes can be divided into 5 distinct phases (phases 0–4). Activation of inward Na+ current triggers a rapid depolarization (phase 0).[21] Phase 1 proceeds rapidly, lasts a few milliseconds and is followed by phase 2. During phase 3 of the AP, the L-type Ca2+ channels close, while the slow delayed rectifier K+ channels are still open, allowing more K+ channels to open, causing the cell to repolarize. The third phase of repolarization terminates the AP and returns the membrane potential to phase 4.[21,22] Ito is the main current contributing to the repolarizing phase 1 of the AP.[23] It is a result of the movement of K+ ions from the intracellular to the extracellular space.[23] Ito is rapidly activated and deactivated.[24] It is activated after the fast increase of the membrane potential following the phase 0 of the AP.[23] Once activated, the outward flow of (K+) ions from inside the cells constitutes Ito and causes the transmembrane potential to decrease. This decrease of the transmembrane potential is known as repolarization. Ito is then quickly deactivated, stopping the repolarization and ending the phase 1 of the AP.[23,24] A distinct AP notch mediated by Ito in epicardium, rather than endocardium, produces a transmural voltage gradient during early ventricular repolarization that is, contributory to registration of J waves on the ECG. Several lines of evidence determined the higher density of Ito in the epicardium compared to the midmyocardial (M) region and significantly greater than the endocardial region of canine ventricle. Similar results were obtained in subepicardial and subendocardial myocytes from human ventricles.[25,26] Factors that affect the gating properties of Ito or ventricular activation sequence can modify the appearance of the J waves [Figure 1]. For example, because of its slow recovery from inactivation, Ito is reduced following faster heart rate, resulting in a decrease in the amplitude of the J waves.
Figure 1.
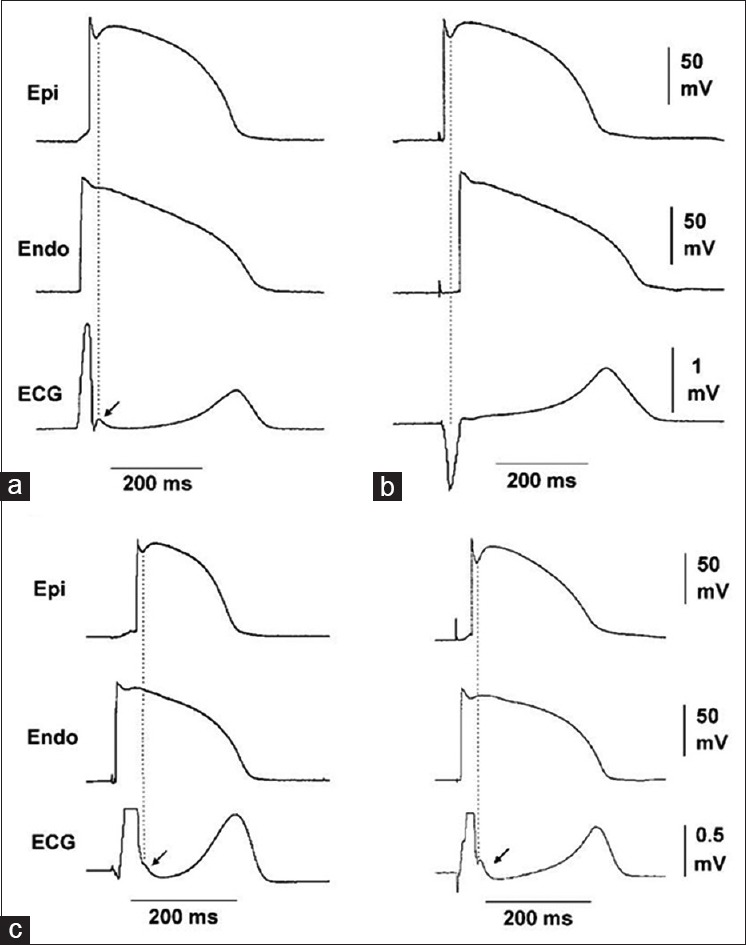
The sequence of ventricular activation impacts the appearance of J wave in a pseudo-electrocardiogram recorded from coronary-perfused canine right ventricular wedge preparation. (a) Stimulation of the endocardial (Endo) surface causes the epicardial (Epi) surface to be activated last; (b) stimulation of the epicardial surface activates it before the endocardial surface; (c) endocardial activation at different locations can cause the J wave to occur at the end of the QRS, manifesting as slurred (left panel) or notched (right panel) QRS.
CLINICAL AND ELECTROCARDIOGRAM FEATURES OF J WAVE SYNDROMES
Historically, traditional ERS (i.e., concave ST segment elevation with or without J wave in lateral precordial leads), BrS, some cases of IVF that are linked to J wave in inferior leads were considered as different ECG or disease entities. Increasing basic and clinical evidences in the past decade supports our initial proposal a decade ago that it is appropriate to group these syndromes and entities under the heading of J wave syndromes because of their similarity in ECG characteristics, clinical outcomes, risk factors, a common arrhythmic platform associated with amplification of Ito-mediated J waves [Table 1]. For example, the clinical and ECG characteristics of these syndromes can co-exist in an individual or among members of the same family.[27,28,29] Qi et al. reported a young Chinese man with recurrent VF who had distinct J waves and ST segment elevation in almost all ECG leads, showing the ECG attributes of all of these syndromes [Figure 2].[27,28] Furthermore, in a family observed by Matsuo et al., one individual with recurrent syncope and inducible VF showed J wave in the inferior leads; whereas his brother displayed a typical Brugada-like ECG features with ST-segment elevation in the right precordial leads.[29] In addition, some genetic overlap among these syndromes is also existing. For example, the mutations in SCN5A are linked to ST-segment elevation in the inferior leads as well as in BrS.[5,30,31,32]
Table 1.
J wave syndromes
| Characteristics | Inherited | Acquired | ||||
|---|---|---|---|---|---|---|
| ERS type 1 | ERS type 2 | ERS type 3 | BrS | Ischemia- mediated VT/VF | Hypothermia mediated VT/VF | |
| Average age of first event | 35 years | 30–40 years | ||||
| Anatomic location | Anterolateral left ventricle | Inferior left ventricle | Left and right ventricles | Right ventricle | Left and right ventricles | Left and right ventricles |
| Leads displaying J point/J wave | I, V4-6 | II, III, aVF | Global | V1-3 | Any of 12 leads | Any of 12 leads |
| Response of J wave/ST elevation to | ||||||
| Bradycardia or pause | ↑ | ↑ | ↑ | ↑ | NA | NA |
| Na-channel blockers | ↓ → | ↓ → | ↓ → | ↑ | NA | NA |
| Male predominance | 75% | 80% | ||||
| Sex dominance | Male | Male | Male | Male | Male | Either |
| VT/VF | Rare common in healthy athletes | Yes | Yes, electrical storms | Yes | Yes | Yes |
| Response to quinidine | Limited data | |||||
| J wave/ST elevation | ↓ | ↓ | ↓ | ↓ | ||
| VT/VF | ↓ | ↓ | ↓ | ↓ | ↓ | |
| Response to isoproterenol | Limited data | NA | NA | |||
| J wave/ST elevation | ↓ | ↓ | ↓ | |||
| VT/VF | ↓ | ↓ | ↓ | |||
| Gene mutations | CACNA1C, CACNB2B | KCNJ8, CACNA1C, CACNB2B | CACNA1C | SCN5A, CACNA1C, CACNB2B, GPD1L, SCN1B, KCNE3, SCN3B, KCNJ8, CACNA2D1, KCND3, MOG1, ABCC9, HCN4, KCNH2, KCNE5 | SCN5A | NA |
ERS: Early repolarization syndrome; BrS: Brugada syndrome; VT: Ventricular tachycardia; VF: Ventricular fibrillation; NA: Not available.
Figure 2.
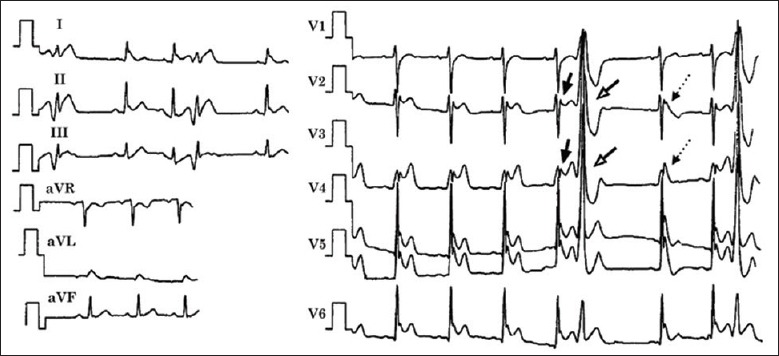
Electrocardiogram obtained from a 34-year-old Chinese man who survived cardiac arrest displaying characteristics of Brugada syndrome, early repolarization syndrome and idiopathic ventricular fibrillation.
Since BrS was first described by Brugada et al. in 1992,[15] it has aroused tremendous interest worldwide. The BrS is a clinical entity related to a high risk of VF in young adults with structurally normal hearts and affects as many as 1 in 2500 individuals.[15,33] BrS is characterized electrocardiographically by an accentuated J wave resembling incomplete or complete right branch bundle block (commonly called coved-type ST segment elevation) in the right precordial leads (V1–V3).[15,34] The ST segment elevation in BrS individuals is affected by heart rate and autonomic tone,[29,34] and its coved ST morphology is associated with a higher incidence of sudden cardiac death. BrS is an autosomal congenital disorder with variable transmission and is related to mutations in seven genes.[35,36,37,38,39,40,41,42] These genetic defects lead to dysfunction of cardiac channels like Ito, L-type calcium current, and INa. Around 60%–70% of BrS probands show genotype-negative.[3] Individuals with BrS are typically males at the age of around mid-30 s with an increased risk of syncope and sudden death that is, often triggered by increased vagal tone. Similar patients have been reported in China.[43] The male predominance in BrS is likely the consequence of a higher density of Ito in men probably due to a higher level of testosterone.[44,45] BrS is thought to be contributory to around 5% of all sudden deaths and up to 20% of sudden deaths in individuals with obvious structurally normal heart diseases. However, current therapeutic strategies for BrS are not adequate. Although quinidine, an Ito inhibitor, and implantable cardioverter defibrillator (ICD) are effective in treating patients with BrS, the side effects of quinidine and ICD in younger patients over a lifetime are the concerns.[46,47] Existing evidences from case reports point out that the catheter ablation may be an emerging therapeutic option for BrS by normalizing the Brugada ECG pattern and preventing ventricular tachycardia (VT)/VF, however, further population-based studies are needed to be in support of this option.
The ERS was first described as early take-off of the ST segment with up to 4 mm elevation and symmetrical high-amplitude T wave in 1961.[48] The ER pattern was traditionally regarded as a “benign” ECG phenomenon, which is commonly found in young healthy men and athletes. However, some authors have shifted the definition of ERS from the traditional description of ST segment elevation to J wave (or J point elevation) only since 2008.[18] This change in ERS definition has shifted away from a benign view of ER to the potential of fatal arrhythmias.[18] An ERS pattern in the arterially-perfused wedge preparations is associated with phase 2 reentry capable of leading to polymorphic VT/VF.[49] Clinical evidence in support of the basic research observation is the development of VF in individuals with distinct J wave and ST segment elevation in the inferior leads. The ERS in the inferior leads with a VF risk was first postulated to represent a variant of the BrS in 2000.[17,50] A relationship between VF and J wave with ER pattern was demonstrated in relatively large numbers of patients by Haissaguerre et al.[18] and Nam et al.[8]
In J wave syndromes, there are three inherited subtypes in terms of ER pattern plus BrS.[3] Subtype 1 is related to ER pattern predominantly in the lateral precordial leads and this type, which is equivalent to traditional ERS, is very prevalent in healthy male athletes and rarely found in VF survivors. Subtype 2 shows an ER pattern mainly in the inferior or inferolateral leads, which is similar to previously called IVF in the setting of J wave in the inferior leads, and is related to a higher level of VF risk.[17,18] This subtype can be observed among healthy young males. Subtype 3 shows an ER pattern globally in the inferior, lateral and right precordial leads and is associated with the highest level of risk for the development of VF storms.[8] In subtype 3, the Brugada waves may be seen together with giant J waves in other ECG leads. Although the Brugada waves are not called ER, their underlying mechanism is identical to that of the ER patterns.
CURRENT CONTROVERSIES IN J WAVE SYNDROMES
One of the hot debates pertaining to J wave syndromes is whether J wave syndromes are repolarization or depolarization disorders.[6,51] Basic and clinical evidence indicates that J waves are repolarization rather than depolarization/conduction abnormalities. J waves as repolarization components are commonly seen in young males with structurally normal hearts. However, depolarization/conduction abnormalities are commonly found in aged individuals with obvious structural heart diseases. J waves display a frequency dependency property [Figure 3], that is, that J waves demonstrate a characteristic pause-dependent accentuation.[52,53,54] In addition, isoproterenol diminishes J waves and can prevent VF in subjects with J wave syndromes,[52,55,56] which does not favor J wave syndromes as depolarization/conduction disorders. This is because that depolarization/conduction defects should become more prominent with tachycardia in the presence of isoproterenol and improve during bradycardia. The magnitude of J waves decreases during tachycardia because the Ito-mediated AP notch decreases from inadequate time for the Ito to full recover.[57] More solid evidence in support of J wave syndromes as repolarization abnormalities is the inhibitory effect of quinidine on both INa and Ito. Quinidine diminishes J wave amplitude and the concealed phase 2 reentry by inhibiting Ito and suppressing so-called “late ventricular potentials” and VF in BrS and other J wave syndromes.[56,58,59]
Figure 3.
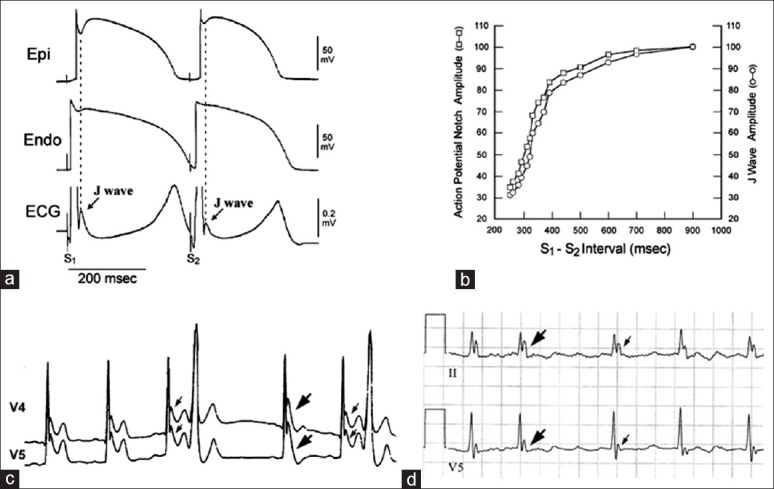
Frequency-dependent changes in epicardial notch and J wave amplitude. (a) Simultaneously recorded transmural electrocardiogram (ECG) and transmembrane action potentials (APs) from a canine right ventricular wedge; (b) plot of epicardial AP notch and J wave amplitude across a range of S1-S2 intervals. Epicardial AP notch and J wave amplitudes changed in parallel to changes in S1-S2 intervals; (c) ECG lead V4-V5 recorded from a man with J wave syndromes; (d) a J-wave-like deflection at the terminal portion of the QRS in a patient with intraventricular conduction delay.
PHASE 2 REENTRY, AN INITIATOR FOR VENTRICULAR FIBRILLATION
If the Ito-mediated epicardial AP notch is deep enough, complete loss of epicardial AP dome may occur. During transition to complete loss of epicardial AP dome, a few of interesting electrical alterations occur: (1) The dome is markedly delayed immediately prior to its complete loss, resulting in paradoxical AP prolongation and so-called “downslope ST segment elevation,” which in fact is a giant J wave, followed by a negative T wave on the ECG [Figure 4]; (2) once the epicardial AP dome is completely lost, AP duration shortens by around 40%,[58] causing a marked increase in transmural dispersion of repolarization; (3) complete loss of the dome is often heterogeneous across the epicardium: That is, complete loss of the dome with significantly AP shortening occurs in some areas, but the delayed AP dome remains in others.[58,60] Due to a marked difference in AP duration and the property of the delayed dome similar to early afterdepolarization,[61] the dome may produce a new AP in the areas where complete loss of epicardial AP is present, leading to formation of short-coupled ectopic beats, which can be capable of originating polymorphic VT or VF. Because it is the propagation of the dome at AP phase 2, it is termed as phase 2 reentry, which has also been demonstrated in humans.[62] Phase 2 reentry is the initiator for VF in all of the J wave syndromes regardless of the locations of J wave on the surface ECG [Figure 5].
Figure 4.
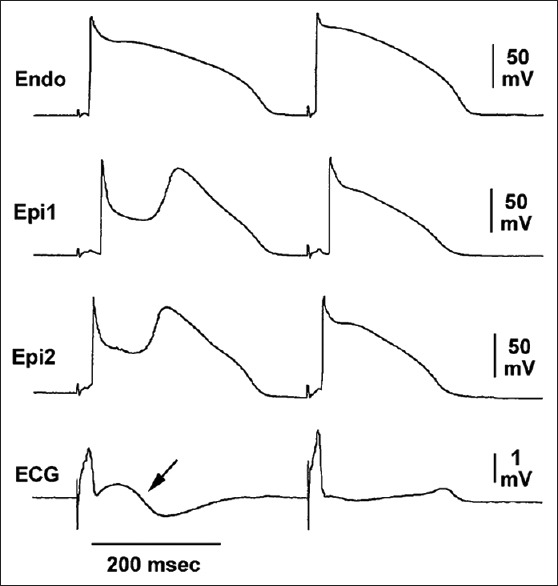
Cellular basis for “downslope ST segment elevation.” Transmembrane action potential (AP) recording in a canine ventricular wedge showing markedly deep and prolonged epicardial (Epi1 and Epi2) but not endocardial (Endo) AP notch reflected on a transmural electrocardiogram as a “downslope ST segment elevation” (arrow) followed by T inversion. The “downslope ST segment elevation” is, in fact, a giant J wave.
Figure 5.
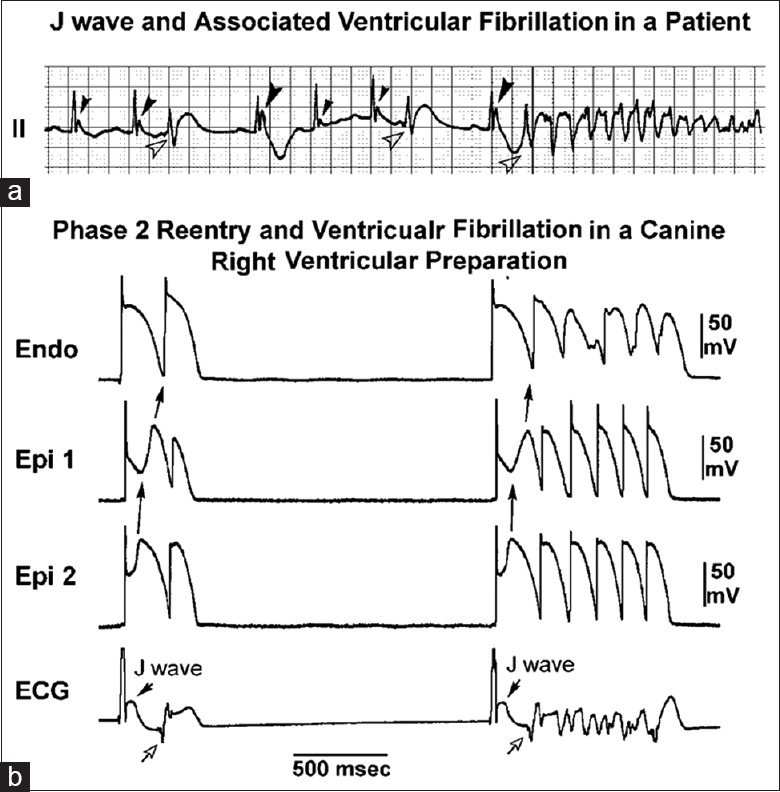
J wave and ventricular fibrillation (VF) via phase 2 reentry. (a) VF in a patient with J wave in lead II, note the larger amplitude of J wave in the beat preceding VF, following a longer R-R interval; (b) phase 2 reentry predisposing to VF in a canine right ventricular wedge preparation in the presence of the K+ channel opener pinacidil. Loss of action potential dome in Epi1 but not Epi2 caused propagation of the dome at Epi2 to Epi1, that is, phase 2 reentry (solid arrows), which manifested a short-coupled R-on-T beats (open arrows) capable of triggering VF.
ACQUIRED J WAVE SYNDROMES
J wave syndromes can be acquired, which share the similar properties with those of inherited J wave syndromes, including ECG features and the underlying mechanism for VF.[3,63]
Hypothermia-induce J wave is well-known, and the study that showed J wave accentuation prior to VF can be dated back to 1953.[12] Hypothermia can produce distinct J waves, resulting in phase 2 reentry and VF.[64] Note, hypothermia-induced J waves can be confined to some selected leads or manifest globally in all leads. Figure 6 shows the hypothermia-induced J wave. Under normal conditions, much of the J wave is buried inside the QRS complex. With hypothermia, the epicardial AP notch is evidently accentuated, and transmural conduction is slowed bringing about to a prominent J wave.[6] It seems that there is no prominent gender-related discrepancy in manifestation of hypothermia-induced VF. This may be due to the powerful potential of hypothermia to significantly amplify the magnitude of J waves, which can then abate the basically gender-related diversity of J wave.
Figure 6.
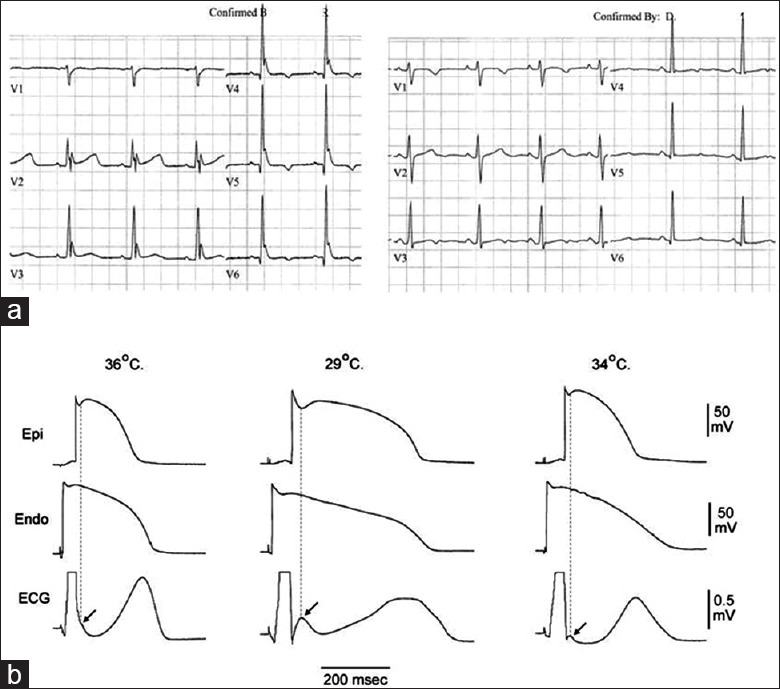
(a) Hypothermia-induced J wave in humans. Appearance of prominent J (Osborn) wave on electrocardiogram of a 32-year-old female at 32°C (left panel) and abolishment of J wave by rewarming at 36.3°C (right panel); (b) hypothermia-induced J wave in animal experiments.
Another more common type of acquired J wave syndromes is ischemia-induced J wave syndrome.[19,63,65,66] During early phase of acute MI in canine experiments, phase 2 reentry causes R-on-T ectopic beats capable of initiating VF.[19] Intrinsically, much higher density of Ito in right compared to left ventricular epicardium may be responsible for an increased incidence of ischemia-induced VF. This is further supported by clinical observation of higher incidence of primary VF in individuals with acute inferior MI who have right ventricular involvement (8.4%) than those without (2.7%), or with an anterior MI (5.0%).[67]
FURTHER DIRECTION
Although a decade of progress in research has greatly enhanced our understanding of J wave syndromes, confusion in terminology and debates on the mechanisms and early diagnosis are still present. First, the terminology of Ito-dependent ER should be standardized. Second, the mechanisms underlying ischemia-induced J wave should be better understood because sudden arrhythmic death is a major cause for mortality associated with acute coronary syndrome. Finally, established role of Ito in IVF may set us thinking about its potential in other causes of VF. An example is commotion cordis where patients develop VF after a sport-related mechanical impact on the chest, and some individuals seem to be more prone to it than others. Since most patients affected with this disorder are males and that the right ventricle-which lies anteriorly in the chest wall-is more directly affected by the impact, therefore, the effort to determine the potential of Ito is needed.
Footnotes
Edited by: Yuan-Yuan Ji
Source of Support: Sharpe-Strumia Research Foundation, and National Natural Science Foundation of China (No. 81400258, 81370289, 81270236).
Conflict of Interest: None declared.
REFERENCES
- 1.Yan GX, Yao QH, Wang DQ, Cui CC. J wave and J wave syndromes (in Chinese) Chin J Card Arrhythm. 2004;8:360–5. [Google Scholar]
- 2.Shu J, Zhu T, Yang L, Cui C, Yan GX. ST-segment elevation in the early repolarization syndrome, idiopathic ventricular fibrillation, and the Brugada syndrome: Cellular and clinical linkage. J Electrocardiol. 2005;38(4 Suppl):26–32. doi: 10.1016/j.jelectrocard.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Antzelevitch C, Yan GX. J wave syndromes. Heart Rhythm. 2010;7:549–58. doi: 10.1016/j.hrthm.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Badri M, Patel A, Yan G. Cellular and ionic basis of J-wave syndromes. Trends Cardiovasc Med. 2015;25:12–21. doi: 10.1016/j.tcm.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Antzelevitch C. Genetic, molecular and cellular mechanisms underlying the J wave syndromes. Circ J. 2012;76:1054–65. doi: 10.1253/circj.cj-12-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antzelevitch C, Yan GX, Viskin S. Rationale for the use of the terms J-wave syndromes and early repolarization. J Am Coll Cardiol. 2011;57:1587–90. doi: 10.1016/j.jacc.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koncz I, Gurabi Z, Patocskai B, Panama BK, Szél T, Hu D, et al. Mechanisms underlying the development of the electrocardiographic and arrhythmic manifestations of early repolarization syndrome. J Mol Cell Cardiol. 2014;68:20–8. doi: 10.1016/j.yjmcc.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nam GB, Kim YH, Antzelevitch C. Augmentation of J waves and electrical storms in patients with early repolarization. N Engl J Med. 2008;358:2078–9. doi: 10.1056/NEJMc0708182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kraus F. Concerning the effect of calcium on the circulation (in German) Dtsch Med Wochenschr. 1920;46:201–3. [Google Scholar]
- 10.Tomaszewski W. Electrocardiographic changes observed in a man frozen to death (in French) Arch Mal Coeur. 1938;31:525–8. [Google Scholar]
- 11.Shipley RA, Hallaran WR. The four lead electrocardiogram in two hundred normal men and women. Am Heart J. 1936;11:325–45. [Google Scholar]
- 12.Osborn JJ. Experimental hypothermia; respiratory and blood pH changes in relation to cardiac function. Am J Physiol. 1953;175:389–98. doi: 10.1152/ajplegacy.1953.175.3.389. [DOI] [PubMed] [Google Scholar]
- 13.Hersch C. Electrocardiographic changes in head injuries. Circulation. 1961;23:853–60. doi: 10.1161/01.cir.23.6.853. [DOI] [PubMed] [Google Scholar]
- 14.Sridharan MR, Horan LG. Electrocardiographic J wave of hypercalcemia. Am J Cardiol. 1984;54:672–3. doi: 10.1016/0002-9149(84)90273-x. [DOI] [PubMed] [Google Scholar]
- 15.Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20:1391–6. doi: 10.1016/0735-1097(92)90253-j. [DOI] [PubMed] [Google Scholar]
- 16.Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic J wave. Circulation. 1996;93:372–9. doi: 10.1161/01.cir.93.2.372. [DOI] [PubMed] [Google Scholar]
- 17.Kalla H, Yan GX, Marinchak R. Ventricular fibrillation in a patient with prominent J (Osborn) waves and ST segment elevation in the inferior electrocardiographic leads: A Brugada syndrome variant? J Cardiovasc Electrophysiol. 2000;11:95–8. doi: 10.1111/j.1540-8167.2000.tb00743.x. [DOI] [PubMed] [Google Scholar]
- 18.Haïssaguerre M, Derval N, Sacher F, Jesel L, Deisenhofer I, de Roy L, et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med. 2008;358:2016–23. doi: 10.1056/NEJMoa071968. [DOI] [PubMed] [Google Scholar]
- 19.Yan GX, Joshi A, Guo D, Hlaing T, Martin J, Xu X, et al. Phase 2 reentry as a trigger to initiate ventricular fibrillation during early acute myocardial ischemia. Circulation. 2004;110:1036–41. doi: 10.1161/01.CIR.0000140258.09964.19. [DOI] [PubMed] [Google Scholar]
- 20.Jastrzebski M, Kukla P. Ischemic J wave: Novel risk marker for ventricular fibrillation? Heart Rhythm. 2009;6:829–35. doi: 10.1016/j.hrthm.2009.02.036. [DOI] [PubMed] [Google Scholar]
- 21.Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–9. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- 22.Li G, Cheng G, Wu J, Zhou X, Liu P, Sun C. Drug-induced long QT syndrome in women. Adv Ther. 2013;30:793–802. doi: 10.1007/s12325-013-0056-x. [DOI] [PubMed] [Google Scholar]
- 23.Niwa N, Nerbonne JM. Molecular determinants of cardiac transient outward potassium current (I (to)) expression and regulation. J Mol Cell Cardiol. 2010;48:12–25. doi: 10.1016/j.yjmcc.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wettwer E, Amos G, Gath J, Zerkowski HR, Reidemeister JC, Ravens U. Transient outward current in human and rat ventricular myocytes. Cardiovasc Res. 1993;27:1662–9. doi: 10.1093/cvr/27.9.1662. [DOI] [PubMed] [Google Scholar]
- 25.Wettwer E, Amos GJ, Posival H, Ravens U. Transient outward current in human ventricular myocytes of subepicardial and subendocardial origin. Circ Res. 1994;75:473–82. doi: 10.1161/01.res.75.3.473. [DOI] [PubMed] [Google Scholar]
- 26.Näbauer M, Beuckelmann DJ, Uberfuhr P, Steinbeck G. Regional differences in current density and rate-dependent properties of the transient outward current in subepicardial and subendocardial myocytes of human left ventricle. Circulation. 1996;93:168–77. doi: 10.1161/01.cir.93.1.168. [DOI] [PubMed] [Google Scholar]
- 27.Qi X, Sun F, Xiao A, Yang J. A case of Brugada syndrome with ST segment elevation through entire precordial leads (in Chinese) Chin J Cardiol. 2004;32:272–3. [Google Scholar]
- 28.Cui C, Yan GX. Which is appropriate, Brugada syndrome, Brugda wave or J wave syndrome (in Chinese)? Chin J Cardiol. 2004;32:960. [Google Scholar]
- 29.Matsuo K, Shimizu W, Kurita T, Suyama K, Aihara N, Kamakura S, et al. Increased dispersion of repolarization time determined by monophasic action potentials in two patients with familial idiopathic ventricular fibrillation. J Cardiovasc Electrophysiol. 1998;9:74–83. doi: 10.1111/j.1540-8167.1998.tb00869.x. [DOI] [PubMed] [Google Scholar]
- 30.Potet F, Mabo P, Le Coq G, Probst V, Schott JJ, Airaud F, et al. Novel brugada SCN5A mutation leading to ST segment elevation in the inferior or the right precordial leads. J Cardiovasc Electrophysiol. 2003;14:200–3. doi: 10.1046/j.1540-8167.2003.02382.x. [DOI] [PubMed] [Google Scholar]
- 31.Takehara N, Makita N, Kawabe J, Sato N, Kawamura Y, Kitabatake A, et al. A cardiac sodium channel mutation identified in Brugada syndrome associated with atrial standstill. J Intern Med. 2004;255:137–42. doi: 10.1046/j.0954-6820.2003.01247.x. [DOI] [PubMed] [Google Scholar]
- 32.Watanabe H, Nogami A, Ohkubo K, Kawata H, Hayashi Y, Ishikawa T, et al. Electrocardiographic characteristics and SCN5A mutations in idiopathic ventricular fibrillation associated with early repolarization. Circ Arrhythm Electrophysiol. 2011;4:874–81. doi: 10.1161/CIRCEP.111.963983. [DOI] [PubMed] [Google Scholar]
- 33.Antzelevitch C, Brugada P, Brugada J, Brugada R, Towbin JA, Nademanee K. Brugada syndrome: 1992-2002: A historical perspective. J Am Coll Cardiol. 2003;41:1665–71. doi: 10.1016/s0735-1097(03)00310-3. [DOI] [PubMed] [Google Scholar]
- 34.Miyazaki T, Mitamura H, Miyoshi S, Soejima K, Aizawa Y, Ogawa S. Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome. J Am Coll Cardiol. 1996;27:1061–70. doi: 10.1016/0735-1097(95)00613-3. [DOI] [PubMed] [Google Scholar]
- 35.Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–6. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 36.Schulze-Bahr E, Eckardt L, Breithardt G, Seidl K, Wichter T, Wolpert C, et al. Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: Different incidences in familial and sporadic disease. Hum Mutat. 2003;21:651–2. doi: 10.1002/humu.9144. [DOI] [PubMed] [Google Scholar]
- 37.London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na current and causes inherited arrhythmias. Circulation. 2007;116:2260–8. doi: 10.1161/CIRCULATIONAHA.107.703330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–9. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watanabe H, Koopmann TT, Le Scouarnec S, Yang T, Ingram CR, Schott JJ, et al. Sodium channel ß1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest. 2008;118:2260–8. doi: 10.1172/JCI33891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delpón E, Cordeiro JM, Núñez L, Thomsen PE, Guerchicoff A, Pollevick GD, et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol. 2008;1:209–18. doi: 10.1161/CIRCEP.107.748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu D, Barajas-Martinez H, Burashnikov E, Springer M, Wu Y, Varro A, et al. A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ Cardiovasc Genet. 2009;2:270–8. doi: 10.1161/CIRCGENETICS.108.829192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7:33–46. doi: 10.1016/j.hrthm.2009.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang Z, Patel C, Li W, Xie Q, Wu R, Zhang L, et al. Role of signal-averaged electrocardiograms in arrhythmic risk stratification of patients with Brugada syndrome: A prospective study. Heart Rhythm. 2009;6:1156–62. doi: 10.1016/j.hrthm.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 44.Di Diego JM, Cordeiro JM, Goodrow RJ, Fish JM, Zygmunt AC, Pérez GJ, et al. Ionic and cellular basis for the predominance of the Brugada syndrome phenotype in males. Circulation. 2002;106:2004–11. doi: 10.1161/01.cir.0000032002.22105.7a. [DOI] [PubMed] [Google Scholar]
- 45.Shimizu W, Matsuo K, Kokubo Y, Satomi K, Kurita T, Noda T, et al. Sex hormone and gender difference - role of testosterone on male predominance in Brugada syndrome. J Cardiovasc Electrophysiol. 2007;18:415–21. doi: 10.1111/j.1540-8167.2006.00743.x. [DOI] [PubMed] [Google Scholar]
- 46.Belhassen B, Glick A, Viskin S. Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation. 2004;110:1731–7. doi: 10.1161/01.CIR.0000143159.30585.90. [DOI] [PubMed] [Google Scholar]
- 47.Nademanee K, Veerakul G, Mower M, Likittanasombat K, Krittayapong R, Bhuripanyo K, et al. Defibrillator Versus beta-Blockers for Unexplained Death in Thailand (DEBUT): A randomized clinical trial. Circulation. 2003;107:2221–6. doi: 10.1161/01.CIR.0000066319.56234.C8. [DOI] [PubMed] [Google Scholar]
- 48.Wasserburger RH, Alt WJ. The normal RS-T segment elevation variant. Am J Cardiol. 1961;8:184–92. doi: 10.1016/0002-9149(61)90204-1. [DOI] [PubMed] [Google Scholar]
- 49.Gussak I, Antzelevitch C. Early repolarization syndrome: Clinical characteristics and possible cellular and ionic mechanisms. J Electrocardiol. 2000;33:299–309. doi: 10.1054/jelc.2000.18106. [DOI] [PubMed] [Google Scholar]
- 50.Takagi M, Aihara N, Takaki H, Taguchi A, Shimizu W, Kurita T, et al. Clinical characteristics of patients with spontaneous or inducible ventricular fibrillation without apparent heart disease presenting with J wave and ST segment elevation in inferior leads. J Cardiovasc Electrophysiol. 2000;11:844–8. doi: 10.1111/j.1540-8167.2000.tb00062.x. [DOI] [PubMed] [Google Scholar]
- 51.Borggrefe M, Schimpf R. J-wave syndromes caused by repolarization or depolarization mechanisms a debated issue among experimental and clinical electrophysiologists. J Am Coll Cardiol. 2010;55:798–800. doi: 10.1016/j.jacc.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 52.Aizawa Y, Sato A, Watanabe H, Chinushi M, Furushima H, Horie M, et al. Dynamicity of the J-wave in idiopathic ventricular fibrillation with a special reference to pause-dependent augmentation of the J-wave. J Am Coll Cardiol. 2012;59:1948–53. doi: 10.1016/j.jacc.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 53.Mizumaki K, Fujiki A, Tsuneda T, Sakabe M, Nishida K, Sugao M, et al. Vagal activity modulates spontaneous augmentation of ST elevation in the daily life of patients with Brugada syndrome. J Cardiovasc Electrophysiol. 2004;15:667–73. doi: 10.1046/j.1540-8167.2004.03601.x. [DOI] [PubMed] [Google Scholar]
- 54.Kaneko K. Treatment for nocturnal enuresis: The current state in Japan. Pediatr Int. 2012;54:8–13. doi: 10.1111/j.1442-200X.2011.03554.x. [DOI] [PubMed] [Google Scholar]
- 55.Tanaka H, Kinoshita O, Uchikawa S, Kasai H, Nakamura M, Izawa A, et al. Successful prevention of recurrent ventricular fibrillation by intravenous isoproterenol in a patient with Brugada syndrome. Pacing Clin Electrophysiol. 2001;24:1293–4. doi: 10.1046/j.1460-9592.2001.01293.x. [DOI] [PubMed] [Google Scholar]
- 56.Haïssaguerre M, Sacher F, Nogami A, Komiya N, Bernard A, Probst V, et al. Characteristics of recurrent ventricular fibrillation associated with inferolateral early repolarization role of drug therapy. J Am Coll Cardiol. 2009;53:612–9. doi: 10.1016/j.jacc.2008.10.044. [DOI] [PubMed] [Google Scholar]
- 57.Antzelevitch C. J wave syndromes: Molecular and cellular mechanisms. J Electrocardiol. 2013;46:510–8. doi: 10.1016/j.jelectrocard.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100:1660–6. doi: 10.1161/01.cir.100.15.1660. [DOI] [PubMed] [Google Scholar]
- 59.Szél T, Antzelevitch C. Abnormal repolarization as the basis for late potentials and fractionated electrograms recorded from epicardium in experimental models of Brugada syndrome. J Am Coll Cardiol. 2014;63:2037–45. doi: 10.1016/j.jacc.2014.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yan GX, Lankipalli RS, Burke JF, Musco S, Kowey PR. Ventricular repolarization components on the electrocardiogram: Cellular basis and clinical significance. J Am Coll Cardiol. 2003;42:401–9. doi: 10.1016/s0735-1097(03)00713-7. [DOI] [PubMed] [Google Scholar]
- 61.Guo DL, Zhao XJ, Wu Y, Liu TX, Kowey PR, Yan GX. L-type calcium current reactivation contributes to arrhythmogenesis associated with action potential triangulation. J Cardiovasc Electrophysiol. 2007;18:196–203. doi: 10.1111/j.1540-8167.2006.00698.x. [DOI] [PubMed] [Google Scholar]
- 62.Bloch Thomsen PE, Joergensen RM, Kanters JK, Jensen TJ, Haarbo J, Hagemann A, et al. Phase 2 reentry in man. Heart Rhythm. 2005;2:797–803. doi: 10.1016/j.hrthm.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 63.Cui C, Yan G. Update of the J wave syndrome and acute ischemia J wave (in Chinese) Chin J Cardiol. 2010;38:299–300. [PubMed] [Google Scholar]
- 64.Gurabi Z, Koncz I, Patocskai B, Nesterenko VV, Antzelevitch C. Cellular mechanism underlying hypothermia-induced ventricular tachycardia/ventricular fibrillation in the setting of early repolarization and the protective effect of quinidine, cilostazol, and milrinone. Circ Arrhythm Electrophysiol. 2014;7:134–42. doi: 10.1161/CIRCEP.113.000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang DQ, Su XM, Li HB, Ma Y, Yang HT, Yu ZX, et al. The clinical characteristics of J wave syndrome during early ischemia in acute myocardial infaction (in Chinese) Chin J Card Pacing Electrophysiol. 2008;22:31–3. [Google Scholar]
- 66.Li H, Wang D, Yang H, Gong C, Cui C, Yan G. Clinical characteristics of ST elevation acute myocardial infarction with J wave syndrome (in Chinese) Chin J Card Arrhythm. 2009;13:357–60. [Google Scholar]
- 67.Mehta SR, Eikelboom JW, Natarajan MK, Diaz R, Yi C, Gibbons RJ, et al. Impact of right ventricular involvement on mortality and morbidity in patients with inferior myocardial infarction. J Am Coll Cardiol. 2001;37:37–43. doi: 10.1016/s0735-1097(00)01089-5. [DOI] [PubMed] [Google Scholar]