

Abstract
Here we describe a robust, microfluidic technique to generate and analyze 3D tumor spheroids, which resembles tumor microenvironment and can be used as a more effective preclinical drug testing and screening model. Monodisperse cell-laden alginate droplets were generated in Polydimethylsiloxane (PDMS) microfluidic devices that combine T-junction droplet generation and external gelation for spheroid formation. The proposed approach has the capability to incorporate multiple cell types. For the purposes of our study, we generated spheroids with breast cancer cell lines (MCF-7 drug sensitive and resistant) and co-culture spheroids of MCF-7 together with a fibroblast cell line (HS-5). The device has the capability to house 1000 spheroids on chip for drug screening and other functional analysis. Cellular viability of spheroids in the array part of the device was maintained for two weeks by continuous perfusion of complete media into the device. The functional performance of our 3D tumor models and a dose dependent response of standard chemotherapeutic drug, Doxorubicin (Dox) and standard drug combination Dox and Paclitaxel (PCT) was analyzed on our chip-based platform. Altogether, our work provides a simple and novel, in vitro platform to generate, image and analyze uniform, 3D monodisperse Alginate hydrogel tumors for various Omic studies and therapeutic efficiency screening, an important translational step before in vivo studies.
Graphical Abstract
Introduction
Our current understanding of cellular functions is primarily derived from two dimensional (2D) cell-based assays, which constrain cells to a rigid substrate thereby altering cell morphology, polarity, signalling, extracellular matrix (ECM) production and gene expression 1, 2. Technological advances in engineering three dimensional (3D) microenvironments have resulted in a paradigm shift in in vitro cell culture, where 3D models provide improved approximation of cell-cell and cell-matrix interactions, nutrient and oxygen gradients, and overall cellular architecture compared to 2D monolayer cultures. Multi-cellular aggregates, with or without exogenously added ECM, have been widely utilized for characterization of stem cell and cancer biology. These aggregates also referred to as tumor spheroids, simulate avascular tumors and small metastases and can serve as an intermediate model between 2D cultures and in vivo animal studies 3. The screening of anti-cancer drugs and evaluation of new therapeutic targets have yielded significantly different outcomes in 3D cell spheroids compared to 2D molonolayer cultures 4,5. Tumor spheroids exhibit stronger resistance to anti-cancer drugs or radiation when cultured in spheroids compared to 2D monolayers since mass transport is mediated primarily by diffusion in 3D aggregates as opposed to convection in 2D systems 6. This survival advantage has been observed even in small spheroids comprised of 25–50 cells. The complexity of the tumor spheroids can be increased further by addition of ECM such as collagen and matrigel as well as heterotypic cell populations such as cancer-associated fibroblasts and macrophages, thus providing a more physiologically relevant platform to investigate the dynamics of tumorigenic processes 7– 9.
Multicellular spheroids are generally formed in hanging drops, spinner flasks, liquid overlay cultures and bioreactors10–12. However, there are a number of drawbacks including the limited spatial organization, non-uniform size distribution, static cell culture and shear-related cell damage. The size of tumor spheroids can have a profound effect in regulating response to anticancer treatments, as demonstrated by the size-dependent resistance of colon cancer cells to photodynamic therapy 13. Moreover, imaging and biochemical analyses of the spheroids cannot be performed in situ under the above-mentioned culture conditions, requiring transfer of the spheroids to a different platform. To circumvent some of the issues mentioned above, Kim et al. recently developed a well-based pump less perfusion culture system 14. However none of these systems permit high throughput screening (HTS), which has been made possible using microfluidic methods for generation and processing of multicellular spheroids. A number of microfluidic systems have been employed for 3D cell culture and drug screening over prolonged periods, including patterned hydrogels, microwells, microbubbles and droplets 15–18. Microfluidic droplets are suitable as a platform for 3D spheroid formation as highly monodisperse droplets can be generated at rates greater than 1000 droplets/sec using flow-focusing strategies 18. This allows greater control over spheroid sizes, massive parallel processing of individual spheroids and on-chip integration of live imaging and downstream analysis. The composition of hydrogels can be adjusted to vary matrix stiffness and permeability so as to characterize mass transport and mechanical effect of microenvironment on cell-cell and cell-ECM interaction. Furthermore, miniaturization of cell culture in the micro-scale hydrogel spheroids permits rapid and efficient transport of oxygen, nutrients and drugs to all cells in the spheroids, eliminating heterogeneity in external signaling cues. Microfluidic droplet based methods have been used to develop cell spheroids embedded in single polymers such as agarose, gelatin, alginate, poly (ethylene glycol) as well as in mixed hydrogels such as alginate-Matrigel 18,19. Alginate hydrogels have been used in the past as scaffolds for culture of tumor cells, hepatocytes and pancreatic cells and as carriers for drug and protein delivery with controlled release characteristics 20–23. It has also been employed in microfluidic devices to encapsulate yeast, mammalian cells and antibodies 24, 25. Using the droplet generation strategy, bacteria-alginate microspheres were developed in a single device by flowing cell-alginate precursor solution in a 3D micro channel with variable height, followed by calcium dissolved in organic phase to promote gelation 26. Double emulsion (water-in-oil-in-water) droplets were used to encapsulate human mesenchymal stem cells in alginate and alginate-arginine-glycine-aspartic acid (RGD) at a size of 30–80μm 27. Tumor spheroids were also generated by encapsulating HeLa cells in alginate-matrigel mixed solutions 28. Inspite of the extensive research, there is still a need for development of a simple yet robust microfluidic device capable of high throughput generation of hydrogel- based multicellular tumor spheroids for preclinical drug and therapeutic testing applications.
In this study we report the development of an integrated high-throughput microfluidic droplet array capable of generating and entrapping up to 1000 individual multicellular spheroids. The array allows the generation, maintenance, stimulation and analysis of the multicellular tumor spheroids sequentially in the same device. We encapsulated two types of MCF-7 breast cancer cells, sensitive or resistant to doxorubicin (Dox) treatment, in alginate using this platform. The multicellular tumor spheroids were maintained up to 14 days on-chip, with minimal adverse effect on cell viability. Co-encapsulation of tumor and stromal fibroblasts in the spheroids were also achieved using this approach, thus increasing the tunability of the local tumor microenvironment. Next, the tumor spheroids were exposed to varying doses of Dox, and a combination of Dox and Paclitaxel (PCT), followed by cytotoxicity analysis, to determine the efficacy of this platform in anticancer drug screening. The high-capacity droplet array not only permits large-scale parallel processing of the spheroids, but also resolution of spatiotemporal dynamics within each spheroid due to secure entrapment of the spheroids in known locations within the array. This microfluidic 3D spheroid generation methodology can be employed for automated, high-throughput screening of drugs, antibodies and experimental therapeutics along with proteomic and metabolomics detection for tissue engineering applications.
Materials and Methods
Microfluidic device fabrication
Microfluidic devices were prepared using soft lithography as discussed previously 29. Silicon wafers were patterned by depositing negative photo resist SU-8 2100 (MicroChem, Newton, MA) to a thickness of 150 μm, and exposed to UV light through a transparent photomask (CAD/Art Services, Bandon, OR). Poly(dimethylsiloxane) (PDMS) (Sylgard 184, Dow Corning, Midland, MI) was mixed with the crosslinker at a of ratio 10:1, poured onto the template wafers, degassed for 2 hours and cured for 12 hours at 75°C. The PDMS devices were detached from the wafers and bonded to glass slides after oxygen-plasma treatment of both surfaces. The devices were first treated with Aquapel (PPG Industries, Pittsburg, PA) for 20 minutes to make the surfaces hydrophobic and allow smooth droplet generation. Subsequently Aquapel was expelled by flushing with air. All solutions were added to the devices through Tygon Micro Bore PVC Tubing of the following dimension: 100f, 0.010" ID, 0.030" OD, 0.010" Wall (Small Parts Inc, FL, USA). 1ml syringes were used to load cell suspensions, alginate solution and oil into the devices, which were regulated via syringe pumps (Harvard Apparatus, USA) to control flow rates. The oil phase consisted of mineral oil and 3% (w/v) of surfactant Span80 (Sigma-Aldrich, St. Louis, MO).
Cell culture and treatment
MCF-7 human breast adenocarcinoma cells were originally purchased from American Type Culture Collection (ATCC, Manassas, VA). Two types of MCF-7 cell were used in these experiments: wild type (sensitive to doxorubicin treatment) and resistant to doxorubicin, referred to as MCF-7S and MCF-7R respectively. The resistant MCF-7 phenotype has been previously characterized as overexpressing the MDR-1 gene 30. Both cell lines were maintained in DMEM medium with 4.5g/L glucose and L-glutamine (Corning Cellgro, Manassas, VA) supplemented with 10% Fetal Bovine Serum (Corning) and 1% antibiotic-antimycotic (Corning). All cells were grown at 37°C under 5% CO2 in a humidified atmosphere. Cells were routinely passaged every three days and harvested at a density of 1×106 viable cells/mL.
For co-culture experiments, bone marrow stromal fibroblast line HS-5 was purchased from ATCC and maintained as described above in DMEM complete growth media.
Multicellular alginate spheroid formation in microfluidic device
Sodium alginate and calcium chloride was purchased from Sigma. 2% (w/v) alginate solutions were prepared by mixing the appropriate amount of alginate with complete growth medium, and allowed to dissolve completely at 50°C for 2hrs. The alginate solutions were either used immediately or stored under sterile conditions at 4°C for later use. Calcium chloride stock solutions (4% w/v) were also prepared in complete growth media, filtered using a 0.22μm syringe filter and stored at 4°C. Prior to use, all solutions were warmed to 37°C to dissolve any precipitations.
The process of spheroid formation consisted of two steps: (a) generation of alginate droplets containing cells in suspension and (b) gelation of droplets in situ in droplet incubation array. Following trypsinization, single cell suspensions were prepared in 2% alginate solution in media and added to one inlet of the microfluidic devices at a flow rate of 80 μl/hr. The cell concentration for MCF-7 cells was optimized to 10×106/mL to achieve a reasonable cell density in the droplets. Droplets were formed by flowing mineral oil with 3% w/v surfactant at a flow rate of 300 μl/hr through the second inlet. After droplet generation and stabilization in the incubation array, the third inlet was opened and calcium chloride flown in at a constant flow rate of 2 μl/hr. Gelation was allowed to occur over a period of 3–4hrs. Once the multicellular alginate spheroids were formed, complete growth media was perfused continuously at a steady rate of 20 μl/hr to maintain cell viability throughout the duration of the experiment.
For co-culture experiments, the above protocol was modified slightly to add each cell type (MCF-7 and HS-5) at a concentration of 7.5×106 cells/mL in alginate solutions through individual inlets. Droplet generation and gelation were performed as described above. To distinguish between the cancer and fibroblast cells, MCF-7S cells were labeled with Qtracker 705 cell labeling kit (Life Technologies, Carlsbad, CA) as per the manufacturer’s protocol. Briefly, 10nM labeling solution was prepared by mixing the nanocrystals and carrier solutions and incubating at room temperature for 5min. The labeling solutions was added to the cells in suspension and incubated for 45–60min at 37°C. Excess labeling solution was removed by aspiration and the cells visualized using a Cy5 filter set (Qtracker 705 ex/em: 405–665/705nm). Alternatively, MCF-7S Cell nuclei was stained using Hoechst 33342 dye and visualized using a DAPI filter (ex/em: 361/497nm).
Doxorubicin (Dox) treatment
Doxorubicin hydrochloride (Dox) was purchased from LC labs D-4000 (LC lab, Woburn, MA). Stock solutions of Dox were prepared at a concentration of 7mM in Sterile water and stored at 4°C. Dox solutions were diluted to the indicated concentrations (0.8 μM, 1.6 μM, 3.2 μM and 12.8 μM) in complete growth media immediately prior to use. Following multicellular tumor (or tumor/stroma) spheroid formation in the microfluidic array, the cells were allowed to stabilize for 24hrs. The drug-free perfusion media syringe was then replaced with Dox-containing media at various concentrations and perfused for a period of 48hrs at the flow rate of 20 μl/hr. The microfluidic device containing spheroids as well as the syringe pumps were maintained in a humidified incubator at 37°C and 5% CO2 for the duration of the experiment.
Paclitaxel (PCT) Treatment
Paclitaxel was purchased from LC labs (LC lab, Woburn, MA) and a stock concentration of 3.2mM in DMSO was prepared and stored at −20°C. The stock was diluted to working concentration (12.8 μM) in complete growth media before use. For combination chemotherapeutic experiments, Paclitaxel is administered for 24hrs sequentially after a 24hr administration of Dox in the same manner as described in the previous section.
2D monolayer experiment
As control for 3D multicellular spheroids, control experiments were performed by seeding 5,000 cells/well of MCF-7S and MCF-7R cells in a 96-well plate. The cells were allowed to adhere to the surface for 24 hours and treated with the indicated Dox concentrations for 48 hrs. Each condition was tested in triplicate wells for statistical analysis. Subsequently, Dox-containing media was aspirated out and cell viability assessed as described below.
Cytotoxicity Assays
The cell viability was determined at the end of the 48-hour drug perfusion period using Live/Dead Viability/Cytotoxicity Kit (#L3224, Life Technologies, Grand Island, NY) as per the manufacturer’s instructions. Briefly, a stock solution was prepared containing 4μM ethidium homodimer-1 (EthD-1) and 2.5μM calcein-AM and perfused into the chip for 30–45 minutes. The cell spheroids were imaged immediately using standard FITC and DsRed filters (Calcein fluorescence ex/em: 495/515nm; EthD-1 fluorescence ex/em: 495/635nm). Live cells were identified by the green fluorescence of calcein, produced by the conversion of non-fluorescent calcein-AM by the intracellular esterase activity. Dead cells were identified by the nuclear red fluorescence of EthD-1 intercalating with DNA. The exposure time required for the detection of EthD-1 is low and fluorescence intensity is high when compared to auto-fluorescence observed in the presence of Dox. Thus nuclear EthD-1 staining can be clearly observed.
Characterization of alginate spheroids by SEM
Microspheres were fixed in 2.5% glutaraldehyde in sodium cacodylate (0.1 M) for 1 hr at room temperature, followed by dehydration with a graded ethanol series (30, 50, 70, 85, 95, and 100%) for 10 minutes in each solution. Microspheres were maintained in 100% ethanol until critically point dried (Samdri-PVT-3B Critical Point Dryer, Tousimis Research Corporation, Rockville, Maryland). After sectioning the microspheres with a blade, they were sputter-coated with Platinum (5nm) using Cressington 208HR High Resolution Sputter Coater and observed using a Hitachi S-4800 Field Emission Scanning Electron Microscope (FESEM) at an accelerating voltage of 1-3 kV.
Image Analysis
All images were obtained using Zeiss Axio Observer.Z1 Microscope (Zeiss, Germany) equipped with Hamamatsu digital camera C10600 Orca-R2. For tumor and tumor/stromal viability studies, a minimum of 30 spheroids was analyzed for every drug concentration. The percent cell viability was calculated for each spheroid by obtaining the ratio of the number of live cells to the total number of cells in that spheroid. Data analysis for 2D monolayer was done in the same fashion by analysing the cell viability obtained from 3 wells for each condition. Statistical analysis was done by comparing the data sets using the student’s T-test, and p < 0.05 was considered statistically significant.
Results and discussion
Device design
The microfluidic platform described in this study allows robust yet simple 3D cell-hydrogel construct development within one integrated device. The microarray consists of 1000 droplet docking sites, thereby allowing simultaneous drug toxicity screening in a large number of tumor spheroids. The microfluidic device is equipped with multiple inlets that facilitate incorporation of varying cell types, thus allowing robust control over the local microenvironment of the tumor spheroids. Additionally, media and drugs could be perfused continuously through the same inlets so as to promote delivery of nutrients to all the spheroids throughout the experimental duration. Continuous perfusion accurately mimics in vivo nutrient and drug delivery to the tumors as opposed to static delivery methods using conventional systems. Thus the system not only permits high throughput tumor spheroid generation based on the principles of droplet microfluidics but also microscopic functional analysis in the same device due to the integrated spheroid-trapping microarray.
A variety of microfluidic device designs have been utilized for long-term tumor spheroid culture and drug efficacy evaluation. The SpheroChip system designed by Kwapiszewska et al. had compact, micro-well based single microfluidic chip for spheroid generation and functional analysis 16. Though the authors reported precise control over spheroid size and microscopic observation capabilities, the generated spheroids did not incorporate exogenous extracellular matrix like alginate, collagen and chitosan. There was no continuous medium perfusion to the spheroids to maintain the metabolite/nutrient balance in the extracellular environment. Yu et al. 31 employed the microfluidic droplet approach to generate cell-loaded alginate droplets that were crosslinked off-chip and subsequently introduced to a second microfluidic chip for culture and drug-based cytotoxicity studies. Matrigel was also used as an extracellular matrix in spheroids for therapeutic efficacy screening, as reported by Shin et al. 32. Their design lacked individual docking sites for individual spheroids and the throughput of the device was very low. More recently, McMillan et al. described a droplet based spheroid generation system utilizing the self-aggregation of cells in liquid droplets. While a high-throughput method, the spheroids lacked the incorporation of exogenous ECM and could be described as an aggregated cell cluster. Additionally, spheroids were enclosed in a liquid droplet, which made continuous renewal of media and delivery of therapeutic reagents difficult without disruption of the droplet. This limits the lifetime of the spheroid to 3–4 days and hence, practical applicability of the system 33. Here, we describe an integrated approach allowing the generation, culture and toxicity screening of multicellular tumor and tumor-stromal spheroids in a single microfluidic system.
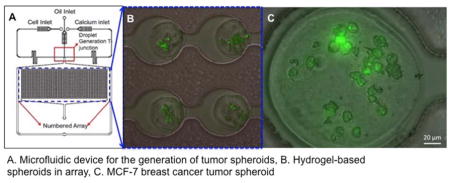
In our device, droplet generation occurred at a T-junction, where the aqueous inlet channel perpendicularly intersected the continuous phase channel. Droplets were formed by shearing force of the continuous phase (Fig 1.C) 34. The droplet docking array consisted of circular holding sites, 200 μm in diameter, separated by constricted regions of 100 μm width and 150 μm length (Fig 1.D, E) . This aspect of the array separates individual alginate droplets, thus preventing fusion of the droplets prior to or during the gelation process and resulting in stable alginate spheroids. By adjusting the fluid flow rates, an optimum droplet size of 170±20 μm was obtained. The device further consisted of two outlets: one connected to the droplet docking array and the second for bulk waste removal from the chip to prevent flow-blocking plaque formations after gelation. (Fig 1. A, B). The serpentine region in the inlet channels enabled lateral cell alignment to promote near - uniform cellular distribution in the droplets.
Fig 1.
Spheroid generation and docking array in microfluidic platform. Schematic of microfluidic device for spheroid generation for single cell type encapsulation (A) and co-encapsulation of two cell types (B). (C) T-junction for droplet formation. Scale bar: 200μm. (D) Droplets entering the docking array before gelation. Scale bar: 200μm. (E) Gelled cell-laden alginate spheroids in docking array. Scale bar: 100μm. (F) Cells in spheroids stained with Calcein-AM as a live cell indicator. Scale bar: 100μm
Hydrogels have been used extensively for in vitro bio-mimetic applications due to its properties such as high water retention mimicking in vivo extracellular matrix, biocompatibility, inertness and prevention of shear stress-mediated cell damage during culture media perfusion 35. In addition to these properties, alginate exhibits mechanical strength to maintain the structural integrity of the spheroids, does not interfere with innate cellular function and permits cellular encapsulation and processing at physiological pH and temperature and thus is extremely useful for tissue engineering applications 23, 36, 37.
The concentration of cations required for gelation of alginate has to be precisely controlled to maintain appropriate porous conditions of the hydrogel, which is pertinent for cellular viability. The ratio of calcium ions (Ca++) to alginate used for crosslinking has to be regulated since structural resistance to ion and nutrient flow in the spheroids could have an undesirable effect on cellular viability 38. It has been previously reported that calcium chloride concentrations of 100–500 mM have little or no detrimental effect on cell health if the period of calcium ion exposure is controlled 38, 39. We thus used an intermediate concentration of calcium (350 mM) for the gelation of the alginate spheroids in situ. Also, cell survival and proliferation are best achieved at alginate concentration of 2–4% w/v 40, 41. However, the increased viscosity of solutions with high alginate concentration impeded smooth droplet generation in microfluidic platforms in our study. Therefore, we utilized an optimal alginate concentration of 2%w/v in complete media for spheroid generation.
Previously reported methods of alginate droplet polymerization include both internal and external gelation methods. For external gelation, the alginate droplets were crosslinked in a divalent cation solution 31, 42. Internal gelation was achieved by incorporating initiators or crosslinking agents in the alginate solution such as soluble calcium carbonate as reported previously 43. Another study utilized photopatterning of alginate due to UV light-mediated release of calcium caged within alginate gels 44. Though these methods have their advantages, they are less than ideal for maintaining cell viability owing to the use of low pH or UV exposure for cross-linking 45, 46. We thus incorporated the use of external gelation technique for polymerization of alginate to avoid using any harmful gelation strategies for maintaining highest possible cellular viability.
Generation and incubation of multicellular spheroids
Herein we used MCF-7 wild type (MCF-7S) and MCF-7 doxorubicin resistant (MCF-7R) cell line for establishment of tumor spheroids. The MCF-7 wild type cells are hormone responsive breast cancer cell line, positive for estrogen and progesterone receptors 47. The drug resistant cell line has been previously characterized for overexpression of MDR-1 gene, which causes an overexpression of p-glycoprotein (P-gp) on cell surface. P-gp uses ATP to transport drugs and other xenobiotics from the intracellular to the extracellular compartment and has been deemed as one of the primary reasons for chemotherapy failure in different MDR cancer types 48, 49. Furthermore, stromal cells such as fibroblasts play an important role in altering anti-tumor activity of chemotherapeutic agents by cytokine and adhesion mediated mechanisms 60. However, conventional anti- cancer drug screening rarely employs accessory cell types during evaluation of drug cytotoxicity. To address this drawback of conventional platforms, we incorporated a fibroblast component (HS-5 cell line) for generation of composite tumor spheroids 50, 51.
The cells were suspended in 2% w/v alginate solution and incubated in microfluidic channels to generate monodisperse droplets of 170 μm in diameter. (Fig 1). Once the cell-laden droplets were stabilized within the incubation array, calcium chloride solution (4% w/v 350mM) was flown in at a slow controlled rate (2 μL/hr) to promote gelation of the droplets. This external gelation method allowed crosslinking of alginate droplets in situ, thus maintaining the spheroidal structure of the cell-hydrogel construct as well as retaining them on-chip for characterization of cellular functions downstream. The sol-gel transition of the alginate by calcium is further facilitated by the rapid mixing kinetics observed within droplets as a result of large surface area with respect to volume 29. Furthermore, the small sizes of constructs (170 μm with a 10% coefficient of variation) allow fast diffusion-based mass exchange, thereby providing cells with nutrition during long-term culture, removal of wastes and drug transport for toxicity studies. The spheroids were constantly perfused with complete media via syringe pumps at a steady flow rate of 20 μL/hr (equivalent to 230 μm/s) throughout the experimental duration (96 hours). This flow rate mimics the blood velocity in the tumor, which has been reported to vary over a range of 100–800 μm/s, depending on the tumor size in vivo 52.
Structural characterization of the cellular alginate spheroids
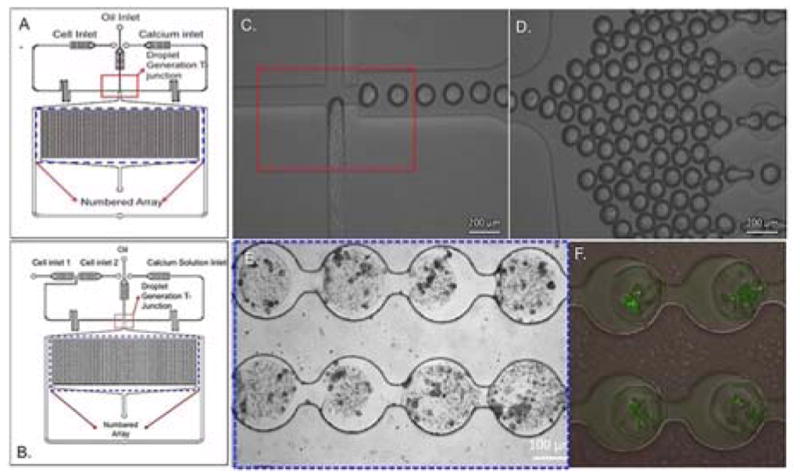
A critical difference between tumor spheroids formed with or without hydrogels is the survival advantage conferred by the supporting hydrogel matrix. Hydrogels are inherently porous, and these pores can be occupied by either the culture media for nutrient, gas and metabolite exchange promoting survival of encapsulated cells, or space to facilitate cell physiological activities, such as metabolism, proliferation, migration, etc. 23,53. The pore size of hydrogels is directly proportional to higher diffusivity of nutrient-laden media and oxygen throughout the hydrogel, which corresponds to higher nutrient gradient for enhanced cellular survival and functional activity 54. The SEM images of the alginate-only (Fig 2.A) and multicellular alginate spheroids (Fig 2.B–D) revealed an ‘egg-box’ morphology, which is a typical characteristic of alginate hydrogels obtained by ionic cross-linking with pore sizes ranging from 5 nm to 100 nm. This broad range of pore sizes allows for water, oxygen, amino acid, protein, nutrient and small molecule exchange but restricts the movement of larger substances such as genetic material (DNA; >100nm) and whole cells 23.
Fig. 2.
Internal structure of cell-laden spheroids using Scanning Electron Microscopy. (A) Internal structure of plain alginate spheroid. Scale bar: 1μm (B), (C), (D) Internal structure of MCF-7S cells encapsulated in alginate spheroid Scale bar (B): 40μm, Scale bar (C), (D): 10μm
Cytotoxicity screening with breast cancer spheroid models
To validate the generated 3D spheroids for drug-based cytotoxicity screening, standard chemotherapeutic drugs, Doxorubicin (Dox) and Paclitaxel (PCT) were utilized as model agents. Doxorubicin acts by intercalating with the DNA of the cell to inhibit the action of the enzyme Topoisomerase II required for supercoil relaxation during transcription. Cytotoxicity results from inhibition of replication and free radical formation 55. Paclitaxel on the other hand intercalates with tubulin in cytoskeleton to stabilize the spindle microtubules, which causes cell cycle arrest and subsequent cell death 56.
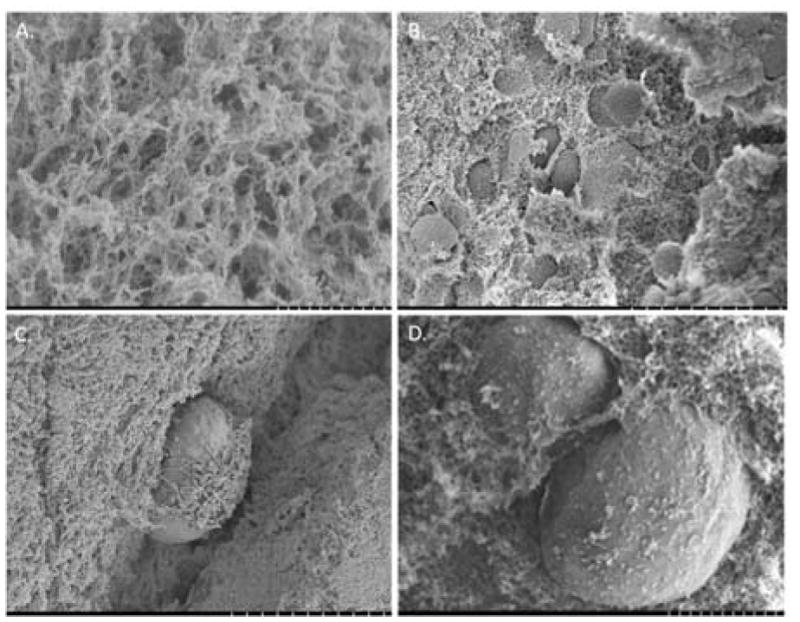
The aim of our study was to compare and contrast the difference in cytotoxicity profiles of free drug in 2D monolayer and 3D microfluidic spheroid model. The addition of hydrogel matrix and human bone marrow fibroblasts allowed us to mimic a complex tumor model, taking into account the tumor-tumor, tumor-stroma, and tumor/stroma-matrix interactions in the same model. The following timeline was employed for drug screening analysis: spheroid formation (day 1); drug treatment (day 2, day 3); live/dead viability staining and readout (day 4). Fluorescent microscopic images of intact spheroids (Fig 3.A–F) subjected to increasing concentration of doxorubicin were utilized for end-point cell viability assessment using live/dead assay. Two fluorescent dyes are employed to distinguish between live and dead cells; live cells emitting a bright green fluorescence and nuclei of dead cells emitting a red fluorescence.
Fig 3.
Spheroids under various treatment conditions. Live/Dead assay images of various spheroid models in the docking array with/without treatment of Doxorubicin (Dox) after 4 days. (A) MCF-7R cell spheroids, no Dox treatment. (B) MCF-7R cell spheroids, 12.8 μM Dox treatment. (C) MCF-7S cell spheroids, no Dox treatment. (B) MCF-7S cell spheroids, 12.8 μM Dox treatment. (E) MCF-7S and HS-5 co-culture spheroids, no Dox treatment. MCF-7S cells are labeled with hoechst 33342 nuclei stain (blue fluorescent) to distinguish between two cell types. (F) MCF-7S and HS-5 co-culture spheroids, 12.8 μM Dox treatment. MCF-7S cells are labeled with Qtracker 705 tracker (far red fluorescent). Scale bar: 20 μm
The results indicated dose-dependent decrease in cell viability as the cells are treated with increasing concentrations of doxorubicin, in 2D as well as 3D format (Fig 4. A–C). As expected, the cytotoxicity observed in 3D spheroids was markedly less compared to the 2D monolayers. MCF-7R cells treated with higher concentration of Dox (12.8 μM) demonstrated a statistically significant difference in cell viability (p<0.05) between 2D and 3D cultures (Fig 4.A). This suggests that the MCF-7R cells were more susceptible to drug treatment in a 2D environment compared to 3D. The cell viability in spheroids is as high as 90% for the highest concentration tested as compared to 60% in 2D monolayers (Fig 4.B). In contrast, MCF-7S spheroids depicted statistically significant differences in cell viability compared to 2D monolayer cultures over a range of Dox doses (1.6–12.8 μM) (Fig 4B). This suggests that MCF-7S cells acquired higher drug resistivity in 3D architecture as compared to their monolayer counterparts. When the microfluidic spheroid models of MCF-7S and the MCF-7R were compared, both cell types exhibited high and comparable survival rates in spheroids (Fig 4.C). These results clearly indicate that cell survival is regulated differentially in 2D vs. 3D structures, as also noted previously 57,58. The tissue architecture and ECM organization in vivo differs significantly from the simulated tumor microenvironment described here, thereby resulting in altered diffusion and transport mechanisms. However, the alginate hydrogels are able to modify the drug uptake phenomena and subsequent cytotoxicity compared to 2D monolayer cultures. Our microfluidic tumor spheroids established that ECM contributes to drug-induced cell responses, even at micro-scale.
Fig 4.

Comparing dose-dependent cytotoxicity observed with Doxorubicin in 2D versus 3D models. (A) Effect of increasing concentration of Dox (0.8 μM, 1.6 μM, 3.2 μM and 12.8 μM) on cell viability of MCF-7R cells in 2D monolayer and 3D spheroids after 48 hr of drug treatment. (B) Effect of increasing concentration of Dox (0.8 μM, 1.6 μM, 3.2 μM and 12.8 μM) on cell viability of MCF-7S cells in 2D monolayer and 3D spheroids after 48 hr of drug treatment. (C) Comparing the effect of increasing Dox concentrations between MCF-7R and MCF-7s cell spheroids. (p < 0.05)
Co-culture spheroids for cytotoxicity screening
Stromal fibroblasts are known to play key roles in the tumor microenvironment. Fibroblasts are activated at the site of tissue injury or inflammation, which present a similar pathophysiological condition as seen in tumors. These activated fibroblasts, referred as Cancer-Associated Fibroblasts (CAFs), promote survival, progression and metastases of tumors by cytokine and chemokine secretion (CXCL12, CCL2, TGF-β, IL-6, MMPs) and downregulation of suppressor genes (p53, p21, PTEN) 59, 60. Also, Non-Cancer Associated Fibroblasts (NAFs) have been shown to transform into CAFs in tumor microenvironment in vivo 61. Thus, monitoring the interaction of fibroblasts with tumor cells in a 3D microenvironment will allow us to optimize drug therapy protocols by assessing the contribution of these cells to cancer cell survival. We investigated the effect of various chemotherapeutic regimens on MCF-7S and HS-5 co-culture spheroids in our microfluidic platform (Fig 5.A–C). The overall cell survival rate in spheroids was statistically higher in co-culture spheroids compared to MCF-7S spheroids for lower and higher concentration of free doxorubicin treatment. (Fig 5.A.) For lower concentration of drug (3.2 μM) the total cell viability was 98% in co-culture spheroids compared to 79% for MCF-7S spheroids. Similarly for higher concentration (12.8 μM), co-culture viability was 20% higher in contrast to MCF-7S spheroid. We postulate that the HS-5 confers greater survival to the breast cancer cells in the spheroids, at least within the experimental duration (48 hours). This could be due to biophysical effects, cytoprotective paracrine signaling, or a combination of both. Our next step will be to characterize these paracrine-signaling events in intact spheroids.
Fig 5.

Analysing drug and dose dependent cytotoxicity in MCF-7S and HS-5 co-culture spheroids. (A) Comparing cell viability between MCF-7S spheroids and MCF-7S and HS-5 spheroids for higher concentrations of Doxorubicin (3.2 μM and 12.8 μM) after 48 hours of treatment. (B) Comparing difference in cell viability in MCF-7S and HS-5 spheroids treated with either 12.8 μM Dox for 48 hrs or 12.8 μM Dox for 24 hrs followed by 12.8 μM of PCT for 24 hrs. (C) Comparing difference in viability between MCF-7S cells and HS-5 cells in co-culture spheroids upon treatment with either 12.8 μm Dox for 48 hrs or sequential treatment of 12.8 μM Dox and12.8 μM PCT for 24 hrs each. (p < 0.05)
To further test the functionality of our system to screen combination chemotherapeutic regimens effectively, we assessed a well-established and clinically relevant drug combination consisting of Dox and PCT, administered in a sequential manner for a total time period of 48 hours for the highest concentration of Dox and PCT in co-culture spheroids (Fig 5.B.). There was a 12% decrease in total cell viability with combination treatment as compared to single drug regimen. This sequential treatment has been shown to be effective in inhibiting breast cancer growth 62, 63. Another important observation was that the drug based cytotoxicity of Dox or the combination regimen affected the fibroblasts and the tumor cells similarly (Fig 5.C.), which reiterates the well-established fact that chemotherapeutic effect of free drug is non-targeted 64.
Thus this platform is not only capable of generating and docking individual multicellular spheroids, it also has the capability of screening drug and dose dependent cellular responses in intact spheroids. The spheroids are held in specific locations throughout the experiment, which enables tracking cell and ECM modifications in the same spheroid over prolonged periods. Therapeutic screening in this platform has additional advantages such as improved simulation of cell-cell and cell-matrix behavior and well defined spherical geometry that allows the correlation of structure with function, in contrast with traditional 3D spheroids. Our future goal is to develop larger spheroids to monitor establishment of nutrient gradients to accurately predict drug action in in vivo-mimetic avascular tumors in a high throughput manner.
Conclusions
In the present work we have described a robust microfluidic device for in situ production of multicellular 3D spheroids in alginate hydrogel matrix. The device has the capability to generate and incubate up to 1000 spheroids of 170μM in size, in a precisely controlled and reproducible manner. We generated three spheroid models: drug resistant breast cancer spheroid, drug sensitive breast cancer spheroid, and co-culture spheroid consisting drug sensitive breast cancer cells and fibroblast cells. To prove the functional performance of spheroids on-chip we conducted cytotoxicity experiments using standard chemotherapeutic drugs utilized in breast cancer treatment (Doxorubicin and Paclitaxel) and compared the results to 2D monolayer cultures. Our future studies will focus on further characterization of cell-cell interactions by quantifying the released cytokines and chemokines to track their effect on cell viability in the presence of anticancer drugs in spheroids of larger dimensions, which are known to develop chemical gradients similar to in vivo avascular tumors. The flexibility of the microfluidic design can be further exploited to include various types of stromal components capable of remodeling the tumor microenvironment to characterize the effect of drug therapies in a comprehensive in vitro tumor spheroid model.
Our proposed system is the next step towards development and semi-automation of more sophisticated spheroid culture approaches, which have the potential to replace 2D drug testing platforms. Efficacy evaluations of various therapeutic agents (i.e. small molecules, antibodies, nucleic acid polymers) could benefit significantly from this sensitive and rapid detection methodology.
Supplementary Material
Acknowledgments
The authors would like to acknowledge NIH/NCI as their funding source (R21 awarded to T.K- RM11-014). The authors would also like to acknowledge Abhinav Gupta and Sneha Varghese at Northeastern University for assistance in cell culture and fabrication of microfluidic devices and William Fowle and Wentao Liang from Northeastern University electron Microscopy core facilities for their assistance in generating SEM images.
Footnotes
Electronic Supplementary Information (ESI) available: [submitted]. See DOI: 10.1039/x0xx00000x
References
- 1.Aggarwal BB, Danda D, Gupta S, Ghelot P. Biochem Pharmacol. 2009;78(9):1083–1094. doi: 10.1016/j.bcp.2009.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van der Worp HB, Howells DW, Sena ES, Porritt MJ, Rewell S, O’Collin V, Macleod MR. PLoS Med. 2010;7(3) doi: 10.1371/journal.pmed.1000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Regehr KJ, Domenech M, Koepsel JT, Carver KC, Ellison-Zelski SJ, Murphy WL, Schuler LA, Alarid ET, Beebe DJ. Lab Chip. 2009;9:2132–2139. doi: 10.1039/b903043c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel NR, Aryasomayajula B, Abouzeid AH, Torchilin VP. Ther Deliv. 2015;6(4):509–520. doi: 10.4155/tde.15.1. [DOI] [PubMed] [Google Scholar]
- 5.Mehta G, Hsiao AY, Ingram M, Luker GD, Takayama S. J Control Release. 2012;164(2):192–204. doi: 10.1016/j.jconrel.2012.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu H, Meyvantsson I, Shkel IA, Beebe DJ. Lab Chip. 2005;5:1089–1095. doi: 10.1039/b504403k. [DOI] [PubMed] [Google Scholar]
- 7.Fischbach1 C, Chen R, Matsumoto T, Schmelzle T, Brugge JS, Polverini PJD, Mooney J. Nat Methods. 2007;4(10):855–860. doi: 10.1038/nmeth1085. [DOI] [PubMed] [Google Scholar]
- 8.Dvir T, Benishti N, Shachar M, Cohen S. Tissue Eng. 2006;12(10):2843–2852. doi: 10.1089/ten.2006.12.2843. [DOI] [PubMed] [Google Scholar]
- 9.Radisic M, Park H, Shing H, Consi T, Schoen FJ, Langer R, Freed LE, Vunjak-Novakovic G. Proc Natl Acad Sci USA. 2004;101(52):18129–18134. doi: 10.1073/pnas.0407817101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Timmins NEI, Nielsen LK. Methods Mol Med. 2007;140:141–151. doi: 10.1007/978-1-59745-443-8_8. [DOI] [PubMed] [Google Scholar]
- 11.Kelm JM, Timmins NE, Brown CJ, Fussenegger M, Nielsen LK. Biotechnol Bioeng. 2003;83(2):173–180. doi: 10.1002/bit.10655. [DOI] [PubMed] [Google Scholar]
- 12.Mehta G, Hsiao AY, Ingram M, Luker GD, Takayama S. J Control Release. 2012;164(2):192–204. doi: 10.1016/j.jconrel.2012.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.West CML, West DC, Kumar S, Moore JV. Int J Radiat Biol. 1990;58(1):145–156. doi: 10.1080/09553009014551501. [DOI] [PubMed] [Google Scholar]
- 14.Kim T, Doh I, Cho YH. Biomicrofluidics. 2012;6(3):034107. doi: 10.1063/1.4739460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeon SE, No DY, Lee SH, Nam SW, Oh IH, Lee J, Kuh HJ. PLoS ONE. 2013;8(9) doi: 10.1371/journal.pone.0073345. eCollection 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwapiszewska K, Michalczuk A, Rybka M, Kwapiszewski R, Brzózka Z. Lab Chip. 2014;14(12):2096–2104. doi: 10.1039/c4lc00291a. [DOI] [PubMed] [Google Scholar]
- 17.Agastin S, Giang UT, Geng Y, DeLouise LA, King MR. Biomicrofluidics. 2011;5(2):24110. doi: 10.1063/1.3596530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alessandri K, Sarangi BR, Gurchenkov VV, Sinha B, Kieβlingg TR, Fetlera,b,c L, Ricoh F, Scheuringh S, Lamazea,e C, Simona,e A, Geraldoa,e S, Vignjevića,e D, Doméjeanc,i,j H, Rollandc,i,j L, Funfakc,i,j A, Bibettec,i,j J, Bremondc,i,j N, Nassoy P. Proc Natl Acad Sci USA. 2013;110(37):14843–14848. doi: 10.1073/pnas.1309482110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Jinyi W. Analyst. 2014;139(10):2449–2458. doi: 10.1039/c4an00015c. [DOI] [PubMed] [Google Scholar]
- 20.Xu XX, Liu C, Liu Y, Li N, Guo X, Wang SJ, Sun GW, Wang W, Ma XJ. Exp Cell Res. 2013;319(14):2135–2144. doi: 10.1016/j.yexcr.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 21.Tonnesen HH, Karlsen J. Drug Dev Ind Pharm. 2002;28(6):621– 630. doi: 10.1081/ddc-120003853. [DOI] [PubMed] [Google Scholar]
- 22.Leonard M, De Boisseson AR, Hubert P, Dalencon F, Dellacherie E. J Control Release. 2004;98(3):395–405. doi: 10.1016/j.jconrel.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 23.Lee KY, Mooney DJ. Prog Polym Sci. 2012;1:106–126. doi: 10.1016/j.progpolymsci.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen W, Kim JH, Zhang D, Lee KH, Cangelosi GA, Soelberg SD, Furlong CE, Chung JH, Shen AQ. J R Soc Interface. 2013;10(88) doi: 10.1098/rsif.2013.0566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akbari S, Pirbodaghi T. Lab Chip. 2014;14(17):3275–3280. doi: 10.1039/c4lc00082j. [DOI] [PubMed] [Google Scholar]
- 26.Lian M, Patrick Collier C, Doktycz MJ, Retterer ST. Biomicrofluidics. 2012;6(4) doi: 10.1063/1.4765337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan HF, Zhang Y, Ho YP, Chiu YL, Jung Y, Leong KW. Sci Rep. 2013;10(3):3462. doi: 10.1038/srep03462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Wang J. Analyst. 2014;139(10):2449–2458. doi: 10.1039/c4an00015c. [DOI] [PubMed] [Google Scholar]
- 29.Konry T, Golberg A, Yarmush M. Sci Rep. 2013;11(3):3179. doi: 10.1038/srep03179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saad M, Garbuzenko OB, Minko T. Nanomedicine (Lond) 2008;3(6):761–776. doi: 10.2217/17435889.3.6.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu L, MC, Chen W, Cheung KC. Lab Chip. 2010;10(18):2424–2432. doi: 10.1039/c004590j. [DOI] [PubMed] [Google Scholar]
- 32.Shin CS, Kwak B, Han B, Park K. Mol Pharmaceutics. 2013;10(6):2167–2175. doi: 10.1021/mp300595a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McMillan KS, McCluskey AG, Sorensen A, Boyd M, Zagnoni M. Analyst. 2016 doi: 10.1039/C5AN01382H. [DOI] [PubMed] [Google Scholar]
- 34.Tran TM, Lan F, Thompson CS, Abate AR. J Phys D Appl Phys. 2013;46(11):114004. [Google Scholar]
- 35.Verhulsel M, Vignes M, Descroix S, Malaquin L, Vignjevic DM, Viovy JL. Biomaterials. 2014;35(6):1816–1832. doi: 10.1016/j.biomaterials.2013.11.021. [DOI] [PubMed] [Google Scholar]
- 36.de Vos P, Faas MM, Strand B, Calafiore R. Biomaterials. 2006;27(32):5603–5617. doi: 10.1016/j.biomaterials.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 37.Tibbitt MW, Anseth KS. Biotechnol Bioeng. 2009;103(4):655–663. doi: 10.1002/bit.22361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blandino A, Macias M, Cantero D. J Biosci Bioeng. 1999;88(6):686–869. doi: 10.1016/s1389-1723(00)87103-0. [DOI] [PubMed] [Google Scholar]
- 39.Sugiura S, Oda T, Izumida Y, Aoyagi Y, Satake M, Ochiai A, Ohkohchi N, Nakajima M. Biomaterials. 2005;26(16):3327–3331. doi: 10.1016/j.biomaterials.2004.08.029. [DOI] [PubMed] [Google Scholar]
- 40.Cao N, Chen XB, Schreyer DJ. ISRN Chemical Engineering. 2012:516461. doi: 10.5402/2012/516461. [DOI] [Google Scholar]
- 41.Choi CH, Jung JH, Rhee YW, Kim DP, Shim SE, Lee CS. Biomed Microdevices. 2007;9(6):855–862. doi: 10.1007/s10544-007-9098-7. [DOI] [PubMed] [Google Scholar]
- 42.Huang KS, Lai TH, Lin YC. Lab Chip. 2006;13(6):954–957. doi: 10.1039/b606424h. [DOI] [PubMed] [Google Scholar]
- 43.Tan WH, Takeuchi S. Adv Mater. 2007;19:2696. [Google Scholar]
- 44.Chueh B, Zheng Y, Torisawa Y, Hsiao AY, Ge C, Hsiong S, Huebsch N, Franceschi R, Mooney DJ, Takayama S. Biomed Microdevices. 2010;12(1):145–151. doi: 10.1007/s10544-009-9369-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Workman VL, Dunnett SB, Kille P, Palmer DD. Biomicrofluidics. 2007;1(1):14105. doi: 10.1063/1.2431860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang H, Tumarkin E, May R, Sullan A, Walker GC, Kumacheva E. Macromol Rapid Commun. 2007;28:527–538. [Google Scholar]
- 47.Levenson AS, Jordan VC. Cancer Res. 1997;57(15):3071–3078. [PubMed] [Google Scholar]
- 48.AbuHammad S, Zihlif M. Genomics. 2013;101(4):213–220. doi: 10.1016/j.ygeno.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 49.Shen F, Chu S, Bence AK, Bailey B, Xue X, Erickson PA, Montrose MH, Beck WT, Erickson LC. J Pharmacol Exp Ther. 2008;324(95):95–102. doi: 10.1124/jpet.107.127704. [DOI] [PubMed] [Google Scholar]
- 50.McMillin DW, Delmore J, Weisberg E, Negri JM, Geer DC, Klippel S, Mitsiades N, Schlossman RL, Munshi NC, Kung AL, Griffin JD, Richardson PG, Anderson KC, Mitsiades CS. Nat Med. 2010;16(4):483–489. doi: 10.1038/nm.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McMillin DW, Negri JM, Mitsiades CS. Nat Rev Drug Discov. 2013;12(3):217–228. doi: 10.1038/nrd3870. [DOI] [PubMed] [Google Scholar]
- 52.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK. Cancer Res. 1995;55(17):3752–3756. [PubMed] [Google Scholar]
- 53.Huang X, Zhang X, Wang X, Wang C, Tang B. J Biosci Bioeng. 2012;114(1):1–8. doi: 10.1016/j.jbiosc.2012.02.024. [DOI] [PubMed] [Google Scholar]
- 54.Yao R, 1,2,3,5, Zhang1 R, Luan J, Lin F. Biofabrication. 2012;4(2):025007. doi: 10.1088/1758-5082/4/2/025007. [DOI] [PubMed] [Google Scholar]
- 55.Gewirtz DA. Biochem Pharmacol. 1999;57(7):727–741. doi: 10.1016/s0006-2952(98)00307-4. [DOI] [PubMed] [Google Scholar]
- 56.Jordan MA. Curr Med Chem Anticancer Agents. 2002;2(1):1–17. doi: 10.2174/1568011023354290. [DOI] [PubMed] [Google Scholar]
- 57.Xu F, Burg KJ. Cytotechnology. 2007;54(3):135–143. doi: 10.1007/s10616-007-9065-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang X, Wang W, Yu W, Xie Y, Zhang X, Zhang Y, Ma X. Biotechnol Prog. 2005;21(4):1289–1296. doi: 10.1021/bp050003l. [DOI] [PubMed] [Google Scholar]
- 59.Mao Y, Kelle ET, Garfield DH, Shen K, Wang J. Cancer Metastasis Rev. 2013;32(1–2):303–315. doi: 10.1007/s10555-012-9415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang W, Huang P. Cancer Biol Ther. 2011;11(2):150–156. doi: 10.4161/cbt.11.2.14623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bremnes RM, Dønnem T, Al-Saad S, Al-Shibli K, Andersen S, Sirera R, Camps C, Marinez I, Busund LT. J Thorac Oncol. 2011;6(1):209–217. doi: 10.1097/JTO.0b013e3181f8a1bd. [DOI] [PubMed] [Google Scholar]
- 62.Holmes FA, Madden T, Newman RA, Valero V, Theriault RL, Fraschini G, Walters RS, Booser DJ, Buzdar AU, Willey J, Hortobagyi GN. J Clin Oncol. 1996;14(10):2713–2721. doi: 10.1200/JCO.1996.14.10.2713. [DOI] [PubMed] [Google Scholar]
- 63.Paridaens R, Biganzoli L, Bruning P, Klijn JG, Gamucci T, Houston S, Coleman R, Schachter J, Van Vreckem A, Sylvester R, Awada A, Wildiers J, Piccart M. J Clin Oncol. 2000;18(4):724–733. doi: 10.1200/JCO.2000.18.4.724. [DOI] [PubMed] [Google Scholar]
- 64.Torchilin VP. Nat Rev Drug Discov. 2014;13(11):813–827. doi: 10.1038/nrd4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.