

Abstract
We previously investigated novel therapies for pediatric ependymoma and found 5‐fluorouracil (5‐FU) i.v. bolus increased survival in a representative mouse model. However, without a quantitative framework to derive clinical dosing recommendations, we devised a translational pharmacokinetic‐pharmacodynamic (PK‐PD) modeling and simulation approach. Results from our preclinical PK‐PD model suggested tumor concentrations exceeded the 1‐hour target exposure (in vitro IC90), leading to tumor growth delay and increased survival. Using an adult population PK model, we scaled our preclinical PK‐PD model to children. To select a 5‐FU dosage for our clinical trial in children with ependymoma, we simulated various 5‐FU dosages for tumor exposures and tumor growth inhibition, as well as considering tolerability to bolus 5‐FU administration. We developed a pediatric population PK model of bolus 5‐FU and simulated tumor exposures for our patients. Simulations for tumor concentrations indicated that all patients would be above the 1‐hour target exposure for antitumor effect.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC? ☑ Although 5‐FU is often administered as a continuous infusion, our preclinical studies in pediatric ependymoma showed bolus administration improved survival. However, no bolus pediatric PK data existed to provide dosing recommendations. • WHAT QUESTION DID THIS STUDY ADDRESS? ☑ We utilized a modeling and simulation approach using a preclinical PK‐PD model and an adult PK model to recommend initial dosages for our phase I trial and determined the disposition of bolus 5‐FU in children with recurrent ependymoma. • WHAT THIS STUDY ADDS TO OUR KNOWLEDGE ☑ The recommended phase II dosage from our phase I trial derived from our translational PK‐PD M&S approach was tolerable and the simulated tECF concentrations were above the targeted exposure (in vitro IC90). • HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS ☑ Because ependymomas lack effective chemotherapy, we utilized PK‐PD M&S to translate bolus 5‐FU to the clinic. This approach is applicable to other settings for translating new or optimizing existing therapies for other diseases to improve drug development.
Pediatric ependymomas are predominantly localized to the posterior fossa with a median age at diagnosis of 6 years old and a 7‐year event‐free survival nearing 70%.1 Primary treatment for nonmetastatic disease consists of both surgery and local field radiation. In children <3 years old, chemotherapy has been used to delay radiation‐related side effects, including neurocognitive and endocrine deficits.2 Approximately 33% to 50% of patients relapse because of local failure, distant disease, or a combination, leading to fatal outcomes in 90% of these patients.3 The median 5‐year progression‐free survival and overall survival from a recent retrospective study in relapsed patients with ependymoma were 17% and 27.6%, respectively.3
Because of the rarity of this disease, clinical evaluation of novel therapies remains challenging. We previously reported on a process for investigating novel therapies for pediatric ependymomas that showed i.v. bolus 5‐fluorouracil (5‐FU) was efficacious in a mouse model of pediatric ependymoma.4 A purine antimetabolite, 5‐FU, is used to treat several adult malignancies, most notably colorectal cancer. It rapidly enters cells where it is converted to several active metabolites that cause downstream DNA and RNA damage.5 In pediatric malignancies, 5‐FU is usually administered as a continuous infusion for the treatment of adolescent colorectal cancer and nasopharyngeal carcinoma, whereas in smaller clinical trials, i.v. bolus 5‐FU has been used to treat recurrent solid tumors.6, 7, 8, 9, 10 Little is known regarding 5‐FU pharmacokinetics (PKs) in pediatrics; one study in 31 children with refractory solid tumors reported the mean 5‐FU clearance after a 12‐hour infusion to be similar to adult estimates.11
The clinical use of 5‐FU dates back decades, but due to the paucity of pediatric 5‐FU PK data, we lacked a rigorous quantitative framework for scaling preclinical findings for use in the clinic. Therefore, we devised a translational modeling and simulation (M&S) approach to determine an initial dosage for bolus 5‐FU for a phase I clinical trial in children with recurrent ependymoma.12 The objectives of this report are to describe our preclinical PK‐pharmacodynamic (PD) studies and M&S techniques used to determine the initial bolus 5‐FU dosing regimen for a phase I study in pediatric patients with recurrent ependymoma. Additionally, we describe the first population PK analysis for bolus 5‐FU in children, which when linked with a preclinical tumor PK model was used to simulate target exposures.
METHODS AND MATERIALS
Preclinical PK and efficacy studies
The preclinical plasma PK, cerebral microdialysis, and efficacy studies used here for preclinical PK‐PD model development were published in our previous manuscript4 or are described in the Supplementary Material. Briefly, plasma PK studies of 5‐FU were performed in two groups of nontumor bearing mice receiving 5‐FU 75 mg/kg i.v. bolus via tail vein injection or subcutaneous continuous infusion over 3 days. Cerebral microdialysis studies were performed in mice bearing pediatric ependymoma receiving 5‐FU 75 mg/kg i.v. bolus or 13 mg/kg/hr over 24 hours via subcutaneous infusion. Plasma samples and dialysate fractions were collected at predetermined timepoints after drug administration and analyzed using a validated high‐performance liquid chromatography method.13 For preclinical efficacy studies, mice were randomized to control or treatment groups 7 days after tumor implantation based on their luciferase‐mediated bioluminescence signal, a measure of tumor growth. Mice were dosed according to the following schedules: (1) 5‐FU 75 mg/kg i.v. bolus weekly for 4 weeks; (2) 5‐FU 75 mg/kg subcutaneous infusion over 3 or 5 days every 3 weeks; and (3) 5‐FU 37.5 or 75 mg/kg i.v. bolus weekly for 4 weeks.
PK‐PD modeling of preclinical data
PK‐PD data were analyzed by nonlinear mixed effects modeling using NONMEM 7.3 (ICON Development Solutions, Ellicott City, MD). The interindividual and residual variability were parameterized exponentially and proportionally in the model, respectively. The Akaike information criterion value and inspection of diagnostic plots were used to select between competing models. A visual predictive check was performed by simulating 1,000 concentration time or tumor growth profiles at each dosage. The standard errors of PK model parameter estimates were determined by running covariance steps, whereas confidence intervals of the PD parameter estimates were determined by performing bootstrap analysis consisting of 500 runs.
First, a one or two‐compartment model with linear or Michaelis–Menten elimination was tested on all 5‐FU plasma concentration‐time data. Parameters from the final plasma PK model were fixed and used as inputs to the tumor PK model, which included the 5‐FU tumor extracellular fluid concentration (tECF). Several different tumor PK models were tested, including one or two‐compartment models with linear or Michaelis–Menten kinetics. Based on the assumption that only free drug crosses the blood‐brain barrier, the 5‐FU amount in the central compartment (A(1)) was multiplied by the fraction unbound in the differential equations.14 Because the microdialysate was collected in fractions (30 min/fraction for bolus and 4 h/fraction for infusion), each fraction was modeled as a separate compartment using the MTIME option in NONMEM to code when a new fraction was collected. The microdialysate flow rate, tubing lag time, and the total volume of each fraction were fixed to experimental values. The area under the concentration‐time curve profiles were estimated by integrating the predicted concentration‐time profiles to a specific time after dosing (t). The tumor to plasma partition coefficient for unbound 5‐FU (Kpt,uu) was estimated as a ratio of unbound tumor to plasma area under concentration values.
Bioluminescence data for each mouse were normalized to the value at day 7 after tumor cell implantation. The tumor growth model, based on a previously described model, contains exponential and linear tumor growth phases, and three transit compartments representing drug‐induced cell death.15 The differential equations representing tumor growth and drug induced cell death are as follows:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
L 1(t) represents proliferating cells within whole tumor; L 2(t), L 3(t), and L 4(t) are tumor growth of cells in transit compartments after drug treatment. W(t) is the fold increase in tumor bioluminescence at any time (t) after 0. Kexp,m and K lin,m are the exponential and linear growth rates, respectively; Ψm is the constant that related to switching tumor growth from exponential to linear; K m is the drug inhibitory effect on tumor growth; K del,m is a first order rate constant for the transit compartments. Because the tumor size was normalized to baseline bioluminescence, W(0) was fixed to 1. The inhibitory effect of the drug on tumor growth (Km) was modeled with a sigmoidal Emax equation15:
| (6) |
where kmax,m is the maximal effect of the drug; EC50,m is the concentration producing half the maximal effect; CtECF is the model predicted 5‐FU concentration in the tECF; and Hm is the Hill coefficient.
Scaling of adult PK model and qualification for pediatric patients
To translate our preclinical PK‐PD model to the clinic for use in children with ependymoma, an adult 5‐FU bolus population PK model was used because of the limited pediatric 5‐FU PK information available.16 Terret et al.16 published a two‐compartment plasma PK model with Michaelis–Menten elimination and body surface area (BSA) as a covariate on Vmax. To account for the wider range of BSA in children, we BSA‐normalized the volume of the central compartment (V1) (Supplementary Table S1).16 We simulated 1,000 pediatric 5‐FU concentration time profiles after a 12‐hour 5‐FU infusion using the scaled‐adult model and compared the simulated concentrations to published pediatric data.11
Simulation of tumor exposure and tumor growth inhibition
The scaled‐adult plasma PK model was linked to the preclinical PK‐PD model by fixing the tumor growth and drug effect parameters and changing the plasma fraction unbound (fu) value from mouse to human. Tumor exposure and tumor growth inhibition were simulated for various 5‐FU bolus dosages based on those used in a previous pediatric trial.9 Dosage levels were assessed for percentage of simulated profiles reaching the targeted exposure (1‐hour in vitro IC90) and for tumor growth inhibition. The simulated results were used along with a knowledge of the tolerability to i.v. bolus 5‐FU to determine a recommended phase I dosage for use in a clinical trial to treat children with recurrent ependymoma.
Pediatric population PK study
Patient population and PK sampling strategy
Details regarding eligibility, study design, and PK sampling strategy are described in a previous publication.17
Population PK modeling
In total, 570 concentration‐timepoints were utilized for the development of the population PK model. Various compartmental structural models were tested to describe 5‐FU plasma concentrations including one, two, and three‐compartment models with linear and/or saturable elimination. Interindividual (between subject) and interoccasion (within‐subject) variability were explored assuming a log‐normal distribution of the parameters. Treatment occasions were defined as a group of concentration‐time data after a single 5‐FU bolus administration. A combined additive and proportional error model was chosen to explain the residual variability in observed concentrations.
BSA was incorporated in the model parameterization a priori and the following covariates were tested and evaluated graphically to explain the interindividual variability of the population PK model: baseline age, race, actual body weight, sex, albumin, alanine aminotransferase, aspartate aminotransferase, estimated creatinine clearance,18, 19 total protein, and total bilirubin. A covariate was considered significant in this analysis if the addition to the model reduced the −2 log‐likelihood by 3.84 units or greater (P < 0.05) based on the χ2 goodness‐of‐fit test and a difference of one degree of freedom.
Approaches for analyzing PK data below the limit of quantitation (BLQ) were explored and parameter estimation was performed using the LAPLACIAN and Monte Carlo Importance Sampling estimation method with the covariance step in NONMEM 7.3.20 The predictive performance of the model was evaluated by visual predictive check simulating 1,000 versions of the original data using the final model parameters.
Pediatric tECF simulations
We linked the pediatric 5‐FU population plasma PK model to the preclinical tECF PK model and simulated individual tECF concentration‐time profiles using individual post hoc parameter estimates to estimate tECF exposures in our patient population. We estimated the percentage of patients reaching the targeted exposure (1‐hour in vitro IC90) to evaluate the success of our translational approach.
RESULTS
Plasma protein binding
The 5‐FU protein binding in mouse plasma was determined using equilibrium dialysis at concentrations of 10 and 100 μM. As shown in Supplementary Figure S1, equilibrium between the donor and receptor chamber was reached within 8 hours. Mean 5‐FU fraction unbound in mouse plasma was 0.37 ± 0.05. No significant difference was observed in fraction unbound at equilibrium between the two 5‐FU concentrations, suggesting concentration‐independent binding of 5‐FU in mouse plasma within this range of concentrations.
Development of preclinical PK‐PD model
We first developed a preclinical plasma PK model representing the 5‐FU total plasma concentration‐time data. Among the models tested, a two‐compartment model with Michaelis–Menten elimination from the central compartment (Figure 1 , Step 1a) best represented the 5‐FU plasma profiles (Figure 2 a–c). The 5‐FU showed nonlinear, saturable elimination in mouse plasma with a mean ± SD maximum plasma concentration (Cmax) of 536 ± 162 μM after a 75 mg/kg i.v. bolus dose, ∼4 times higher than the estimated mean ± SD Km,plasma, 125 ± 22 μM. The mean ± SD 5‐FU plasma steady‐state concentration observed after a 1‐day or 3‐day continuous infusion was 7.11 ± 2.64 μM and 0.43 ± 0.18 μM, ∼17 and 300 times lower than the estimated Km,plasma, respectively. This nonlinear PK produced higher plasma exposures (area under the curve) at the same dosages after i.v. bolus administration compared to the 3‐day or 5‐day infusion (Supplementary Figure S2).
Figure 1.
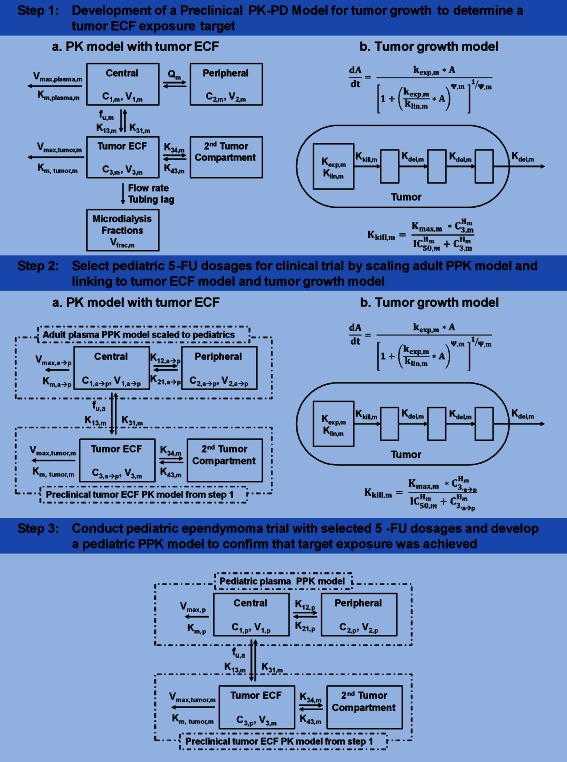
A workflow diagram detailing the preclinical and clinical studies discussed in this article. PPK, population pharmacokinetics; C defines concentration in the corresponding compartment; subscript m defines mouse; subscript “a” defines adult; subscript “p” defines pediatric; subscript “a → p” defines scaled‐adult parameter; the remainder of the parameters are defined in Table 1.
Figure 2.
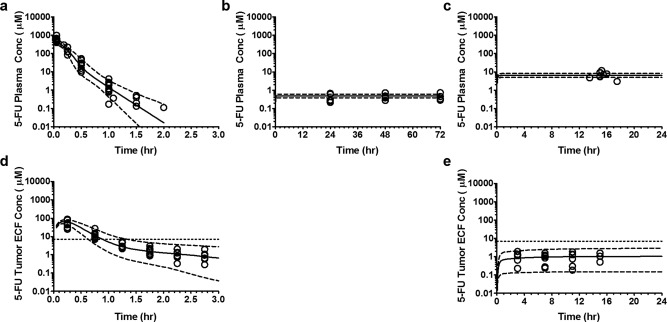
Visual predictive check of preclinical pharmacokinetic model representing plasma and tumor extracellular fluid concentration (tECF) disposition of 5‐fluorouracil (5‐FU) in ependymoma bearing mice. (a, b, and c) Plasma concentration‐time profile of 5‐FU at dosage of 75 mg/kg given as i.v. bolus injection, 75 mg/kg given as 3‐day infusion, and 13 mg/kg/hr infusion over 24 hours, respectively, plotted on semi‐log scale. (d and e) The tECF concentration‐time profile of 5‐FU at dosage of 75 mg/kg given as i.v. bolus injection and 13 mg/kg/hr infusion over 24 hours, respectively, plotted on semi‐log scale. (Open circle represents observed plasma or tECF concentration; solid line represents the median (50th percentile) of model predicted individual concentrations; dashed line represents the 5th and 95th percentile of model predicted individual plasma concentrations.) Supplementary Figure 4 shows this figure in the linear scale.
Next, we developed a PK model representing 5‐FU unbound tECF concentration‐time data, conditioned on the given plasma PK model. To avoid over‐parameterization, plasma PK parameters were fixed to previously estimated values. The volume of tECF compartment (V3) was fixed to 0.001 L/kg based on published data and assuming tumor weight to be 15% of the mouse brain.21, 22 A two‐compartment model with Michaelis–Menten elimination from the tECF compartment best described the tECF concentration‐time data (Figure 2 d,e). The tECF compartment was linked to the plasma compartment using rate constants (K13 and K31). The final plasma and tECF PK parameter estimates are listed in Table 1. Based on prior in vitro assays with ependymoma cell lines, 7.1 μM was the extracellular IC90 for 5‐FU induced cell death after a 1‐hour incubation.4 Therefore, 7.1 μM was used as our target exposure for tECF. The 5‐FU i.v. bolus tECF concentrations were above this target exposure for ∼1 hour, and tECF concentrations after a continuous infusion were at least sevenfold lower.4
Table 1.
Final parameter estimates for preclinical PK‐PD model fitted to mouse 5‐FU plasma and tECF disposition and efficacy studies
| Parameters | Unit | Estimate ± SE | IIV (% CV) |
|---|---|---|---|
| Plasma PK study | |||
| Maximum plasma 5‐FU elimination rate (Vmax,plasma,m) | μmol/hr/kg | 2040 ± 239 | 12.2 |
| Michaelis–Menten constant for plasma 5‐FU elimination (Km,plasma,m) | μM | 125 ± 22 | NE |
| Volume of central compartment (V1,m) | L/kg | 0.962 ± 0.691 | 22.1 |
| Intercompartmental clearance (Qm) | L/hr/kg | 1.67 ± 1.03 | 31.1 |
| Volume of peripheral compartment (V2,m) | L/kg | 0.332 ± 0.50 | NE |
| Residual variability for 5‐FU plasma concentrations (σplasma, prop) | % CV | 30.7 | |
| Tumor microdialysis study | |||
| Rate constant for 5‐FU movement from plasma to tECF (K13,m) | 1/hr | 0.0043 ± 0.001 | 7.8 |
| Rate constant for 5‐FU movement from tECF to plasma (K31,m) | 1/hr | 3.24 ± 0.58 | 26.1 |
| 5‐FU fraction unbound in mouse plasma (fu,m) | 0.37 FIX | NE | |
| Volume of tECF compartment (V3,m) | L/hr | 0.001 FIX | NE |
| Rate parameter for intratumoral 5‐FU disposition (K34,m) | 1/hr | 1.82 ± 0.30 | NE |
| Rate parameter for intratumoral 5‐FU disposition (K43,m) | 1/hr | 0.334 ± 0.077 | NE |
| Maximum tumor 5‐FU elimination rate (Vmax,tumor,m) | μmol/hr/kg | 0.0063 ± 0.004 | NE |
| Michaelis–Menten constant for tumor 5‐FU elimination (Km,tumor,m) | μM | 0.012 ± 0.023 | NE |
| Residual variability for 5‐FU plasma concentrations (σplasma, prop) | % CV | 35.9 | |
| Residual variability for 5‐FU tumor concentrations (σtECF, prop) | % CV | 27.9 | |
| Efficacy study | |||
| Exponential tumor growth parameter ( ) for experiment one | 1/hr | 0.158 (0.079–0.337)a | 40.6 |
| Linear covariate effect on for experiments two and three ( ) | 1/hr | 0.102 (0.031–0.287)a | |
| Linear tumor growth parameter (Klin,m) | 1/hr | 214,000 (66,700–1,480,000)a | NE |
| Constant related to switching tumor growth from exponential to linear (Ψm) | 0.0936 (0.085–0.104)a | NE | |
| Maximum 5‐FU tumor inhibitory effect (Kmax,m) | 1/hr | 0.658 (0.316–1.134)a | NE |
| 5‐FU tECF concentration producing half the maximum tumor inhibitory effect (IC50,m) | μM | 2.12 (0.28–5.84)a | 30.4 |
| Hill coefficient (Hm) | 1.22 (0.64–1.89)a | NE | |
| Rate constant transit tumor compartment (Kdel,m) | 1/hr | 0.946 (0.124–1.950)a | NE |
| Residual variability for tumor growth (σefficacy, prop) | % CV | 37.0 | |
5‐FU, 5‐fluorouracil; CV, coefficient of variation; FIX, value fixed during estimation; IIV, interindividual variability; NE, not estimated; PK‐PD, pharmacokinetic‐pharmacodynamic; tECF, tumor extracellular fluid concentration.
aValues represent 2.5th and 97.5th percentile of bootstrap‐derived parameter estimates.
Last, we developed a PD model representing ependymoma tumor growth in the control and 5‐FU‐treated CD1 nude mice, conditioned on the plasma‐tECF PK model. The 5‐FU efficacy studies were performed in three different experimental cohorts with treatment(s) and control groups. During experiment one, mice received 5‐FU 75 mg/kg i.v. bolus every week for 4 weeks or no treatment. In experiment two, mice received 5‐FU 75 mg/kg subcutaneous infusion over 3 or 5 days every 3 weeks or no treatment. Experiment three compared mice receiving 5‐FU 37.5 or 75 mg/kg i.v. bolus every week for 4 weeks. As shown in Figure 3 a–c, tumor growth data in the control groups (top panels) were best described by exponential and linear growth phases, whereas tumor inhibition by 5‐FU was best described by sigmoidal Emax model (middle and bottom panels). Because the tumor growth rate in control animals from experiment one was different compared with experiments two and three, the exponential tumor growth term (Kexp) was allowed to vary for experiment one and a linear covariate effect was applied to Kexp1 for experiments two and three using the following structure:
| (7) |
| (8) |
| (9) |
Figure 3.
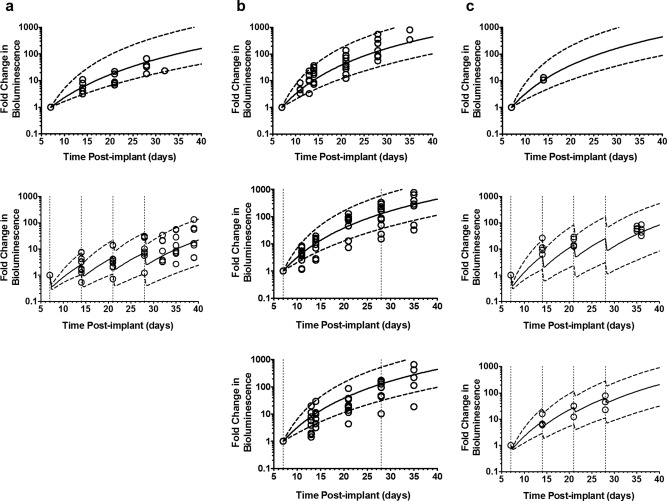
Visual predictive check of preclinical pharmacodynamics model representing tumor growth in control and treated groups in ependymoma bearing mice. (a) Experiment one in which mice were either not treated (top) or treated with 75 mg/kg i.v. bolus 5‐fluorouracil (5‐FU) once every week (bottom); (b) represents experiment two in which mice were either not treated (top) or treated with 75 mg/kg 5‐FU infusion over 3 (middle) or 5 days (bottom) once every 21 days; (c) represents experiment three in which mice were either not treated (top) or treated with 75 (middle) or 37.5 (bottom) mg/kg i.v. bolus 5‐FU once every week. (Open circle represents observed fold increase in bioluminescence; solid line represents the median (50th percentile) of model predicted individual fold increase in bioluminescence; dashed line represents the 5th and 95th percentile of model predicted individual fold increase in bioluminescence; vertical dotted lines indicate 5‐FU dosing.)
The final PD model parameters are provided in Table 1. The in vivo tECF concentration producing half maximum tumor inhibitory effect (IC50, 2.12 μM) estimated using our PK‐PD model was within twofold of our 1‐hour in vitro IC50 (3.5 μM).4 The 5‐FU i.v. bolus resulted in significant tumor growth suppression and subsequent increase in overall survival, whereas 5‐FU continuous infusion failed to exhibit tumor suppression, resulting in no difference in survival compared to the control mice.4 This is consistent with our plasma‐tumor PK model that showed 5‐FU i.v. bolus achieved tECF concentrations above the target exposure (Figure 2 d), whereas 5‐FU continuous infusion did not (Figure 2 e). This supports using 5‐FU i.v. bolus in children with ependymoma. However, without published pediatric PK data to support dosing recommendations, we decided to utilize our preclinical PK‐PD model with available adult 5‐FU PK data to establish dosages for our clinical trial.
Model‐based determination of efficacious clinical dosage
Scaling of adult 5‐FU plasma PK model for pediatrics
To apply our preclinical PK‐PD model to pediatric patients, we first scaled an adult plasma two‐compartment population PK model with Michaelis–Menten elimination.16 The scaled‐adult model was qualified by performing Monte Carlo simulations of 1,000 pediatric patients and comparing the simulated steady‐state concentrations to PK data from a pediatric 5‐FU continuous infusion (80 mg/m2/h infusion over 12 hours).11 The simulated profiles had a mean steady‐state concentration of 4.1 μM, within twofold of the pediatric mean steady‐state concentration (6.7 μM), supporting further use of the adult scaled‐model for pediatric dosage simulations.
Application of scaled‐adult model to determine pediatric dosage
By linking the scaled‐adult plasma PK model with the preclinical PK‐PD model, we compared predicted tECF concentrations to the 1‐hour IC90 (7.1 μM) and investigated tumor growth inhibition to determine a potentially efficacious 5‐FU bolus dosage in children with brain tumors. To accurately estimate unbound 5‐FU tumor concentrations, we used published data showing the 5‐FU plasma fraction unbound was 0.9.23 The tECF simulations showed 68%, 90%, and 99% of subjects would have tECF concentrations above the target exposure (7.1 μM) for at least 1‐hour at 400, 500, and 650 mg/m2, respectively (Figure 4 a–c). Combined with our simulations for tumor growth inhibition (Figure 4 d–f), 500 and 650 mg/m2 dosages were suggested to be highly efficacious. Based on the reported tolerability of 5‐FU bolus in pediatrics, we recommended 500 mg/m2 as the starting dosage with 400 mg/m2 as dosage level zero (dose deescalation) and 650 mg/m2 as dose level two (dose escalation) for our clinical trial of single‐agent 5‐FU in children with recurrent/refractory ependymoma.
Figure 4.
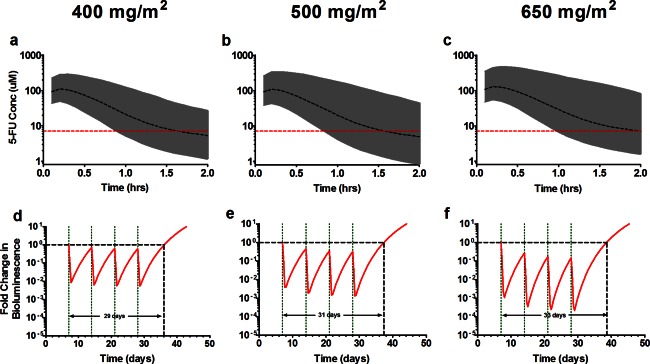
Simulation of the combined scaled‐adult plasma pharmacokinetic (PK) model and preclinical PK‐pharmacodynamic (PD) model for pediatric dosing regimens. The top row represents predicted tumor extracellular fluid concentration (tECF) concentration‐time profiles and the bottom predicted tumor growth profiles. Top: Shown in the black dashed line is the median (50th) percentile, the gray shaded region represents the 95% confidence interval of the Monte Carlo model simulations, and the red horizontal dashed line is the 1‐hour in vitro IC90 (7.1μM). Bottom: The red line represents fold change in bioluminescence in 5‐fluorouracil (5‐FU) treated tumors. The black horizontal dashed line represents tumor condition at baseline and serves as a reference to calculate tumor growth delay. The green vertical dotted lines represent 5‐FU dosing.
Population PK modeling of pediatric data and simulation of tumor exposures
Study population
The pediatric phase I study consisted of 26 children and young adults with recurrent/refractory ependymoma. The patient cohort had a median age of 7.1 years old with 18 girls (69.2%) and 8 boys (30.8%), and the majority were white (73.1%). The PK data were available for patients receiving 400, 500, and 650 mg/m2 dosage of 5‐FU. The baseline demographics are shown in Supplementary Table S2.
Model development
We explored a one or two‐compartment model structure with linear or saturable elimination and various model parameterizations.24 Approximately 40% of samples assayed were found to be BLQ, 15 μM, and approaches for modeling PK data BLQ were explored.20
A two‐compartment structural model best fit the 5‐FU plasma concentration‐time data, which followed a bi‐exponential decay. The final model had first‐order elimination from the central compartment using a BSA normalized dosage. The M3 method was used for BLQ data, leading to improved accuracy in parameter estimation (lower relative standard error %), a lower Akaike information criterion, and improved model diagnostics (visual predictive checks). The model was parameterized in terms of total systemic clearance (CLt), volume of the central compartment (Vc), inter‐compartmental clearance (Q), and volume of the peripheral compartment (Vp). The individual predicted vs. observed concentrations were equally distributed above and below the line of unity and individual weighted residuals vs. predicted concentrations were symmetrically distributed about zero, suggesting an adequate structural model (Figure 5). The parameter estimates for the final structural model are presented in Table 2. Moderate interindividual variability (28.8% for CLt and 26.6% for Vc) and low interoccasion variability (13.0% for CLt) were estimated for 5‐FU parameters. A visual predictive check (Figure 5 and Supplementary Figure S5) supports the structural model is appropriate for 5‐FU bolus PK data from our pediatric population.
Figure 5.

Goodness‐of‐fit plots for the population pharmacokinetic model of bolus 5‐fluorouracil (5‐FU) in pediatric patients. The 5‐FU observed vs. population (a) and individual (b) predicted concentrations. Individually weighted residual vs. population predicted concentrations (c). (Open circle represents individual values; red dashed line represents the line of identity or a horizontal line at y = 0; purple dashed line represents local regression line (LOWESS)). Visual predictive check for pediatric patients receiving 400 (d), 500 (e), or 650 (f) mg/m2 5‐FU i.v. bolus. (Open circle represents observed individual plasma concentrations; solid line represents the median (50th percentile) of model predicted individual concentrations; dashed lines represent the 5th and 95th percentile of model predicted individual concentrations; the red horizontal dashed line is the lower limit of quantitation.)
Table 2.
Final pediatric population pharmacokinetic parameter estimates
| Parameters | Unit | Estimate ± SE | IIV (% CV) | IOV |
|---|---|---|---|---|
| Systemic clearance (CLp) | L/hr/m2 | 16.6 ± 1.2 | 28.8 | 13.0 |
| Volume of central compartment (V1,p) | L/m2 | 4.2 ± 0.3 | 26.6 | NE |
| Intercompartmental clearance (Qp) | L/hr/m2 | 1.6 ± 0.7 | 80.1 | NE |
| Volume of peripheral compartment (V2,p) | L/m2 | 20.1 ± 8.6 | 93.9 | NE |
| Residual variability | ||||
| Proportional | 0.51 ± 0.04 | |||
| Additive | 1.98 ± 1.5 |
CV, coefficient of variation; IIV, interindividual variability; IOV, interoccasion variability; NE, not estimated.
The effects of baseline patient demographics and laboratory values on each parameter were assessed graphically. Given the model parameterization of BSA as a covariate a priori, no other definitive relationship was noted between 5‐FU PK parameters and other covariates explored.
Simulation of tECF concentrations using the pediatric population PK model
To estimate tECF concentrations in our patients, we linked the pediatric 5‐FU population PK model to the preclinical tECF PK model and used individual post hoc parameter estimates to simulate individual tECF concentration‐time profiles. All 26 patients on each occasion achieved tECF concentrations at or above the target exposure at 1 hour with a median (range) concentration at 1 hour of 182.5 μM (51.8–754.1 μM).
DISCUSSION
Ependymomas are one of the most challenging malignancies to treat in pediatric oncology because of extensive biological heterogeneity and lack of effective chemotherapeutic agents to date. We previously reported on the preclinical efficacy of 5‐FU in a subtype of ependymoma.4 Herein, we discussed the M&S approach used to translate the results of our 5‐FU in vitro and in vivo studies in ependymoma to patients. Using our preclinical PK‐PD model with available data from the literature, we defined a dosage regimen for bolus 5‐FU in children with ependymoma. Last, we reported the first population PK model for bolus 5‐FU in children and simulated the expected target exposures in our patients.
For the preclinical 5‐FU PK‐PD model of pediatric ependymoma, we performed PK studies in plasma and tECF, and PD studies measuring tumor growth by luciferase mediated bioluminescence. The 5‐FU plasma and tumor concentrations exhibited a bi‐exponential elimination and dose‐dependent clearance, best described using a two‐compartment model with Michaelis–Menten elimination (Figure 1 , Step 1). The delay in tumor growth between 5‐FU treated and control mice was estimated using the PK‐PD model (Supplementary Figure S3). Mice receiving 75 mg/kg 5‐FU i.v. bolus had tumor growth delayed by 16–19 days compared with controls, similar to the 14‐day increase in overall survival observed previously (Supplementary Figure S3a,c).4 We acknowledge that bioluminescence has limitations for monitoring tumor growth and may not be applicable across all tumor models25, 26, 27, 28; however, we found good correlation between survival and tumor growth delay measured using bioluminescence.
To scale the preclinical PK‐PD model for use in children with ependymomas, we chose a two‐compartment Michaelis–Menten saturable elimination model commonly used to describe the plasma PKs of bolus 5‐FU in adults.16, 24, 29, 30 The adult model was previously validated on external datasets with different dosages and schedules of 5‐FU bolus and continuous infusion. The model was qualified on available pediatric data and linked to the tECF PK model and the tumor growth inhibition PD model. After simulating bolus 5‐FU dosages for tECF exposure (Figure 4 a–c), tumor growth inhibition (Figure 4 d–f), and incorporating knowledge of 5‐FU bolus tolerability, we selected 500 mg/m2 as the phase I dosage for our clinical trial in ependymoma.9 Using PK data from our phase I trial, we first described 5‐FU disposition with a saturable elimination model. However, the two‐compartment linear model had comparable model diagnostics and a lower Akaike information criterion (2,439 vs. 2,729).
The 5‐FU clearance for this trial was linear across dosages and lower (16.6 L/h/m2) than previously reported values. Di Paolo et al.31 evaluated the PK of i.v. bolus 5‐FU (370 mg/m2) in patients with colorectal cancer and reported an overall clearance of 51.5 L/h/m2. Bocci et al.32 investigated the clinical PK of 5‐FU and its metabolite in 20 patients with colorectal cancer receiving two different bolus doses (250 and 370 mg/m2). They found a lower clearance, 25.4 L/h/m2, in patients receiving 370 mg/m2 and a higher clearance, 54.6 L/h/m2, in the same patients receiving a “test” dose of 250 mg/m2 with the difference attributed to the saturable elimination of 5‐FU. Other 5‐FU bolus PK models include saturable elimination processes with an approximate Vmax and Km of 1,450 mg/h (11,150 μmol/h) and 5 mg/L (40 μM), respectively.16, 29, 30 The maximum 5‐FU concentrations observed in our phase I study are ≥1,000 μM, 50‐fold greater than the adult Km values, which suggests that 5‐FU is likely in the concentration range where elimination processes are saturated, resulting in a lower overall clearance (16.6 L/h/m2). Thus, the high maximum 5‐FU plasma concentrations observed relative to the assay BLQ, and the fact that the study was not designed to assess the nonlinearity of 5‐FU in children, prevented us from accurately estimating Vmax and Km for the Michaelis–Menten model.
In our 5‐FU population PK analysis, BSA was a covariate a priori on clearance and volume, and no other relationships significantly explained the variability in the PK parameters. The use of adult parameter estimates as priors in the model did not improve model performance. A two‐compartment, first order elimination from the central compartment evaluated using the M3 method was an appropriate model for the data.
Approximately 40% of the 5‐FU data were BLQ with the majority of these data located in the terminal elimination phase. However, censoring these data would increase the likelihood for bias in parameter estimates; thus, we investigated methods to handle BLQ data, particularly the M3 method.20, 33 Studies over the past decade describe the M3 method as a reliable approach for data BLQ20, 33, 34, 35 and, recently, Keizer et al.36 indicated the least bias may be present by including all BLQ data without incorporating any likelihood‐based approaches. The M3 approach resulted in an increased precision in parameter estimation and improvement in model diagnostics.
Given the high rate of drug failures, it is important to perform preclinical experiments in representative tumor models and incorporate clinically relevant dosages/schedules to increase the translation of preclinical research. The translational PK‐PD M&S described above is an approach that should serve as a template for drug development in all therapeutic areas, especially rare diseases where the need for promising therapies is high and few patients are available for clinical trials. The results of our preclinical PK‐PD M&S confirm our dosage selection for bolus 5‐FU and indicate our translational approach achieved the required target exposure for antitumor effect in this phase I trial of recurrent/refractory ependymoma.
Grants and Financial Support
This work was supported by the Collaborative Ependymoma Research Network (CERN), United States National Institutes of Health Cancer Center Support (CORE) Grant P30 CA21765, and by the American Lebanese Syrian Associated Charities (ALSAC).
Disclosure
The authors have no conflicts of interest to report.
Author Contributions
V.M.D., Y.T.P., M.T., D.C.T., A.M.C., J.M.A., A.G., R.J.G., K.D.W., and C.F.S. wrote the manuscript. M.T., A.M.C., J.M.A., A.G., R.J.G., K.D.W., and C.F.S. designed the research. V.M.D., Y.T.P., M.T., D.C.T., A.M.C., J.M.A., A.G., R.J.G., K.D.W., and C.F.S. performed the research. V.M.D., Y.T.P., M.T., D.C.T., A.M.C., J.M.A., A.G., R.J.G., K.D.W., and C.F.S. analyzed the data. V.M.D. and Y.T.P contributed equally to this work. K.D.W and C.F.S. are co‐senior authors of this work.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
We are grateful to the staff of Akaike information criterion and ARC at St. Jude Children's Research Hospital for their technical assistance in our work. We are also grateful to Kristen Haddock and K. Elaine Harstead for the bioanalysis of clinical 5‐FU samples.
References
- 1. Taylor, M.D. et al Radial glia cells are candidate stem cells of ependymoma. Cancer Cell 8, 323–335 (2005). [DOI] [PubMed] [Google Scholar]
- 2. Grundy, R.G. et al Primary postoperative chemotherapy without radiotherapy for intracranial ependymoma in children: the UKCCSG/SIOP prospective study. Lancet Oncol. 8, 696–705 (2007). [DOI] [PubMed] [Google Scholar]
- 3. Zacharoulis, S. , Ashley, S. , Moreno, L. , Gentet, J.C. , Massimino, M. & Frappaz, D. Treatment and outcome of children with relapsed ependymoma: a multi‐institutional retrospective analysis. Childs Nerv. Syst. 26, 905–911 (2010). [DOI] [PubMed] [Google Scholar]
- 4. Atkinson, J.M. et al An integrated in vitro and in vivo high‐throughput screen identifies treatment leads for ependymoma. Cancer Cell 20, 384–399 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Longley, D.B. , Harkin, D.P. & Johnston, P.G. 5‐fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer 3, 330–338 (2003). [DOI] [PubMed] [Google Scholar]
- 6. Hill, D.A. et al Colorectal carcinoma in childhood and adolescence: a clinicopathologic review. J. Clin. Oncol. 25, 5808–5814 (2007). [DOI] [PubMed] [Google Scholar]
- 7. Katzenstein, H.M. et al Hepatocellular carcinoma in children and adolescents: results from the Pediatric Oncology Group and the Children's Cancer Group intergroup study. J. Clin. Oncol. 20, 2789–2797 (2002). [DOI] [PubMed] [Google Scholar]
- 8. Ortega, J.A. et al Randomized comparison of cisplatin/vincristine/fluorouracil and cisplatin/continuous infusion doxorubicin for treatment of pediatric hepatoblastoma: a report from the Children's Cancer Group and the Pediatric Oncology Group. J. Clin. Oncol. 18, 2665–2675 (2000). [DOI] [PubMed] [Google Scholar]
- 9. Patel, R. et al Pharmacology and phase I trial of high‐dose oral leucovorin plus 5‐fluorouracil in children with refractory cancer: a report from the Children's Cancer Study Group. Cancer Res. 51, 4871–4875 (1991). [PubMed] [Google Scholar]
- 10. Pratt, C.B. et al Phase II study of 5‐fluorouracil/leucovorin for pediatric patients with malignant solid tumors. Cancer 74, 2593–2598 (1994). [DOI] [PubMed] [Google Scholar]
- 11. Balis, F.M. et al Phase II trial of sequential methotrexate and 5‐fluorouracil with leucovorin in children with sarcomas. Invest. New Drugs 8, 181–182 (1990). [DOI] [PubMed] [Google Scholar]
- 12. Stroh, M. , Duda, D.G. , Takimoto, C.H. , Yamazaki, S. & Vicini, P. Translation of anticancer efficacy from nonclinical models to the clinic. CPT Pharmacometrics Syst. Pharmacol. 3, e128 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alsarra, I.A. & Alarifi, M.N. Validated liquid chromatographic determination of 5‐fluorouracil in human plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 804, 435–439 (2004). [DOI] [PubMed] [Google Scholar]
- 14. Di, L. , Rong, H. & Feng, B. Demystifying brain penetration in central nervous system drug discovery. Miniperspective. J. Med. Chem. 56, 2–12 (2013). [DOI] [PubMed] [Google Scholar]
- 15. Simeoni, M. et al Predictive pharmacokinetic‐pharmacodynamic modeling of tumor growth kinetics in xenograft models after administration of anticancer agents. Cancer Res. 64, 1094–1101 (2004). [DOI] [PubMed] [Google Scholar]
- 16. Terret, C. et al Dose and time dependencies of 5‐fluorouracil pharmacokinetics. Clin. Pharmacol. Ther. 68, 270–279 (2000). [DOI] [PubMed] [Google Scholar]
- 17. Wright, K.D. et al Phase I study of 5‐fluorouracil in children and young adults with recurrent ependymoma. Neuro. Oncol. 17, 1620–1627 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brandt, J.R. , Wong, C.S. , Hanrahan, J.D. , Qualls, C. , McAfee, N. & Watkins, S.L. Estimating absolute glomerular filtration rate in children. Pediatr. Nephrol. 21, 1865–1872 (2006). [DOI] [PubMed] [Google Scholar]
- 19. Schwartz, G.J. et al New equations to estimate GFR in children with CKD. J. Am. Soc. Nephrol. 20, 629–637 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beal, S.L. Ways to fit a PK model with some data below the quantification limit. J. Pharmacokinet. Pharmacodyn. 28, 481–504 (2001). [DOI] [PubMed] [Google Scholar]
- 21. Brown, R.P. , Delp, M.D. , Lindstedt, S.L. , Rhomberg, L.R. & Beliles, R.P. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol. Ind. Health 13, 407–484 (1997). [DOI] [PubMed] [Google Scholar]
- 22. Hofstein, R. , Hesse, G. & Shashoua, V.E. Proteins of the extracellular fluid of mouse brain: extraction and partial characterization. J. Neurochem. 40, 1448–1455 (1983). [DOI] [PubMed] [Google Scholar]
- 23. Czejka, M. & Schüller, J. The binding of 5‐fluorouracil to serum protein fractions, erythrocytes and ghosts under in vitro conditions [in German]. Arch. Pharm. (Weinheim) 325, 69–71 (1992). [DOI] [PubMed] [Google Scholar]
- 24. van Kuilenburg, A.B. & Maring, J.G. Evaluation of 5‐fluorouracil pharmacokinetic models and therapeutic drug monitoring in cancer patients. Pharmacogenomics 14, 799–811 (2013). [DOI] [PubMed] [Google Scholar]
- 25. Jost, S.C. , Collins, L. , Travers, S. , Piwnica–Worms, D. & Garbow, J.R. Measuring brain tumor growth: combined bioluminescence imaging‐magnetic resonance imaging strategy. Mol. Imaging 8, 245–253 (2009). [PMC free article] [PubMed] [Google Scholar]
- 26. Czupryna, J. & Tsourkas, A. Firefly luciferase and RLuc8 exhibit differential sensitivity to oxidative stress in apoptotic cells. PLoS One 6, e20073 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fan, F. & Wood, K.V. Bioluminescent assays for high‐throughput screening. Assay Drug Dev. Technol. 5, 127–136 (2007). [DOI] [PubMed] [Google Scholar]
- 28. Zhang, Y. et al ABCG2/BCRP expression modulates D‐Luciferin based bioluminescence imaging. Cancer Res. 67, 9389–9397 (2007). [DOI] [PubMed] [Google Scholar]
- 29. Maring, J.G. , Piersma, H. , van Dalen, A. , Groen, H.J. , Uges, D.R. & De Vries, E.G. Extensive hepatic replacement due to liver metastases has no effect on 5‐fluorouracil pharmacokinetics. Cancer Chemother. Pharmacol. 51, 167–173 (2003). [DOI] [PubMed] [Google Scholar]
- 30. van Kuilenburg, A.B. et al Evaluation of 5‐fluorouracil pharmacokinetics in cancer patients with a c.1905 + 1G>A mutation in DPYD by means of a Bayesian limited sampling strategy. Clin. Pharmacokinet. 51, 163–174 (2012). [DOI] [PubMed] [Google Scholar]
- 31. Di Paolo, A. et al 5‐fluorouracil pharmacokinetics predicts disease‐free survival in patients administered adjuvant chemotherapy for colorectal cancer. Clin. Cancer Res. 14, 2749–2755 (2008). [DOI] [PubMed] [Google Scholar]
- 32. Bocci, G. et al Comparative pharmacokinetic analysis of 5‐fluorouracil and its major metabolite 5‐fluoro‐5,6‐dihydrouracil after conventional and reduced test dose in cancer patients. Clin. Cancer Res. 6, 3032–3037 (2000). [PubMed] [Google Scholar]
- 33. Bergstrand, M. & Karlsson, M.O. Handling data below the limit of quantification in mixed effect models. AAPS J. 11, 371–380 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ahn, J.E. , Karlsson, M.O. , Dunne, A. & Ludden, T.M. Likelihood based approaches to handling data below the quantification limit using NONMEM VI. J. Pharmacokinet. Pharmacodyn. 35, 401–421 (2008). [DOI] [PubMed] [Google Scholar]
- 35. Yang, S. & Roger, J. Evaluations of Bayesian and maximum likelihood methods in PK models with below‐quantification‐limit data. Pharm. Stat. 9, 313–330 (2010). [DOI] [PubMed] [Google Scholar]
- 36. Keizer, R.J. et al Incorporation of concentration data below the limit of quantification in population pharmacokinetic analyses. Pharmacol. Res. Perspect. 3, e00131 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information