

Summary
A complete understanding of host-parasite interactions must necessarily include the identification and characterization of gene products expressed by both parties during the infectious process. We have developed a new screen to identify bacterial genes that are transcriptionally induced during infection of a host animal. The method is based on pre-selection of strains carrying tnpR operon fusions (encoding resolvase, a site-specific DNA recombinase) which are not expressed in vitro, followed by screening for a subset of these strains that subsequently express resolvase within the host environment. The latter subset was recognized as recombinants that had deleted a resolvase-specific reporter construct. Thirteen transcription units of Vibrio cholerae were identified that were induced during infection in an infant mouse model of cholera. Five of these were predicted to encode polypeptides with diverse functions in metabolism, biosynthesis and motility; one encoded a secreted lipase; two appear to be antisense to genes involved in motility; and five are predicted to encode polypeptides of unknown function. Three of the transcripts were shown to be required for full virulence in infant mice, as assessed by competition experiments.
Introduction
Vibrio cholerae is a Gram-negative bacterium responsible for severe epidemic and endemic diarrhoea in many parts of the world (Wachsmuth et al., 1994). The profuse watery diarrhoea, which serves to spread V. cholerae into the environment and eventually to other human hosts, is caused, in large part, by the action of cholera toxin (Ctx) on the small intestinal epithelium (Gill, 1977). Several other virulence factors of this pathogen, such as the major colonizing factor TCP pili have been identified in the laboratory by virtue of their co-ordinate regulation with Ctx (Peterson and Mekalanos, 1988; Taylor et al., 1987). However, given the complexity of the host gastrointestinal tract, it is likely that there are additional virulence factors which are not co-regulated with Ctx in vitro. It is further likely that a multitude of other bacterial factors which facilitate survival, growth and subsequent spread from the host have escaped our detection because the in vivo conditions required to induce their expression have not been duplicated in the laboratory.
Several experimental approaches have recently been reported that have allowed identification of genes that are expressed by pathogens within host animals or cells. Mahan et al. (1993) recently reported the development of in vivo expression technology (IVET), a method that allows the direct selection of bacterial gene fusions expressed in the host compartment via complementation of an attenuating auxotrophic mutation. Subsequent in vitro screening of a co-expressed lacZY operon fusion allowed the identification of gene fusions that were not expressed during growth on laboratory media. Gene fusions to an antibiotic resistance and other selectable genes have also recently been used to select directly for gene fusions expressed in host animals (Mahan et al., 1995) and tissue culture (Berger and Isberg, 1993; Klarsfeld et al., 1994). Finally, subtractive hybridization has been used to identify in vivo-induced transcripts (Plum and Clark-Curtiss, 1994). However, the short half life of bacterial mRNA and the difficulty in obtaining sufficient numbers of bacterial cells from infected tissues has so far limited this last approach to tissue culture models. A limitation of all these approaches is that they inherently select more strongly for those genes that are continuously and highly expressed within the host. In vivo-expressed genes that display transient bursts of expression would not be expected to be revealed by any of these approaches. Accordingly, we have designed a genetic ‘screen’ to identify bacterial genes that are transcriptionally silent during growth in the laboratory but which are then transiently or constitutively induced within the host to either low or high levels of transcription. Our approach is based on the recently developed method of assaying gene expression via transcriptional fusions to the site-specific recombinase resolvase (Camilli et al., 1994). In this report, we describe the application of this approach to identify genes of V. cholerae which are transcriptionally induced in an infant mouse model of cholera (Wachsmuth et al., 1994).
Results
Construction of V. cholerae transcriptional fusion libraries
Plasmid pIVET5 was constructed to facilitate the generation of random V. cholerae transcriptional fusions to a promoterless synthetic operon consisting of tnpR and lacZY reporter genes (Fig. 1). tnpR codes for the site-specific recombinase resolvase from Tnγδ (Reed, 1981). Random fragments of genomic DNA from both V. cholerae classical and El Tor biotypes were cloned immediately in front of the synthetic operon and the resulting recombinant plasmid libraries were transferred into classical and El Tor reporter strains, respectively; whereupon the non-replicating plasmids integrated into the genomes by homologous recombination. As illustrated in Fig. 1, each resulting integration strain contained a duplication of the cloned putative promoter-containing V. cholerae sequence such that each strain remained wild type with respect to expression of the downstream gene or operon. The classical and El Tor reporter strains used in these library constructions contained the resolvase substrate sequences res1–tet–res1 and res–tet–res on their respective chromosomes within the lacZ locus (see the Experimental procedures), which resulted in LacZ− and tetracyline-resistant (TcR) backgrounds. Note that the res1–tet–res1 and res–tet–res sequences are unlinked to the integrated tnpR fusions in each strain, in contrast to the system previously described by Camilli et al. (1994). The mutant res1 sequences contained a T to C transition at their crossover sites resulting in approximately 10-fold decreased recombination efficiency (Newman and Grindley, 1984). This allowed us to examine two levels of gene expression in the different V. cholerae biotypes, whereby the El Tor biotype reporter strain was more sensitive to low levels of tnpR expression than the classical biotype reporter strain. Expression of resolvase in each strain background catalysed excision of tet out of the bacterial chromosome (Camilli et al., 1994). The excised tet gene is present on a non-replicating DNA circle which is segregated by cell division leading to a TcS phenotype in descendent bacteria (Camilli et al., 1994).
Fig. 1.
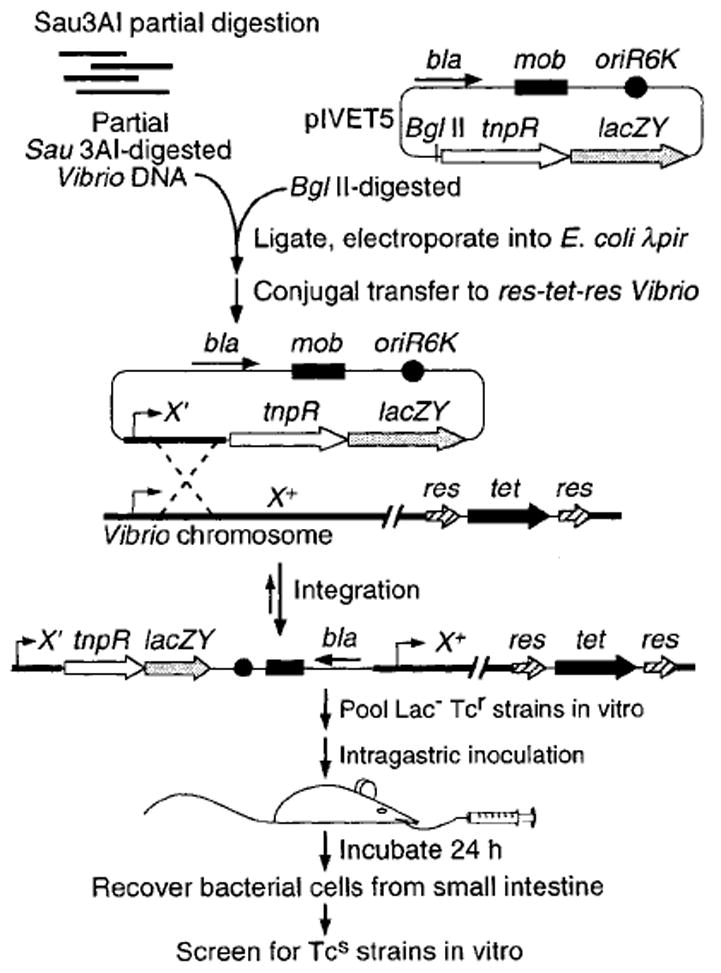
Schematic diagram of the screening protocol used to identify V. cholerae transcripts that are induced in the host. See the Results for details.
Screening for V. cholerae transcripts induced in vivo
When each transcriptional fusion library was plated onto a rich agar media containing Xgal, approximately 40% of the resulting colonies were phenotypically LacZ− and, therefore, were predicted to have transcriptionally inactive fusions to the synthetic operon. Pools of 200 LacZ− TcR strains were introduced individually into single CD-1 suckling mice. After 24 h, bacteria were recovered from homogenates of upper intestinal tissue by plating on Luria–Bertani (LB) agar containing streptomycin. Replica plating was then used to identify those in vivo recovered clones that were TcS (Fig. 1). Of approximately 13000 LacZ− and TcR strains screened, 56 TcS strains were recovered from the infant mice small intestines after 24h. Therefore, approximately 0.1–0.2% of strains from the original library contained in vivo-induced transcriptional fusions to the tnpR–lacZY synthetic operon. In control experiments in which the parental strain AC-V66 (TnpR−) was used to infect infant mice, no TcS bacteria were recovered. Therefore, the res–tet–res reporter substrate for TnpR was completely stable during infection in the absence of tnpR fusions.
Confirmation of in vivo induction of V. cholerae transcriptional fusions
From each of the 56 TcS strains isolated above, the integrated plasmids containing the synthetic operon fusions were recovered into Escherichia coli (see the Experimental procedures) for subsequent analysis and to allow the reconstruction of each fusion in a fresh reporter strain background. Restriction enzyme analysis of each of the 56 plasmids enabled us to eliminate those having identical V. cholerae DNA inserts (data not shown), resulting in 37 plasmids that were unique. In all cases of identical plasmid inserts, their parent strains were found to have originated from the same pool and were, therefore, considered to have been siblings.
Each of the 37 unique plasmids was integrated into the chromosome of their respective parental strain to regenerate each transcriptional fusion in a fresh res–tet–res (El Tor) or res1–tet–res1 (classical) background. This allowed us to independently test each strain for its resolution levels when grown in vitro and during infection of infant mice. Of the 37 reconstructed fusion strains examined, 13 reproducibly resolved to a greater extent during infection of infant mice than when grown in vitro (Table 1). Of the remaining 24 strains, most had very low resolution levels when grown either in vitro or in infant mice (data not shown) and, therefore, apparently arose as a result of low level constitutive resolution events that occurred during the original mouse infections. These 24 strains were considered ‘background’ inherent in our screening protocol and were not analysed further.
Table 1.
Resolution levels in vitro and in vivo.
| Strain | TcS (% cfu after growth)a
|
Nb | P(<)c | |
|---|---|---|---|---|
| in vitro | in vivo | |||
| AC-V226 | 4 | 21 | 4 | 0.07 |
| AC-V227 | 9 | 43 | 4 | 0.07 |
| AC-V228 | 0 | 40 | 5 | 0.04 |
| AC-V229 | 51 | 88 | 6 | 0.03 |
| AC-V230 | 0 | 31 | 7 | 0.04 |
| AC-V231 | 0 | 87 | 4 | 0.07 |
| AC-V232 | 0 | 69 | 4 | 0.07 |
| AC-V233 | 0 | 13 | 6 | 0.03 |
| AC-V234 | 1 | 11 | 6 | 0.04 |
| AC-V235 | 6 | 36 | 5 | 0.04 |
| AC-V236 | 26 | 66 | 5 | 0.04 |
| AC-V237 | 2 | 14 | 8 | 0.05 |
| AC-V238 | 0 | 11 | 6 | 0.06 |
Value given is the mean from 4–8 independent experiments.
N equals the number of independent experiments, with each experiment consisting of two animals.
P is the probability that each strain resolved to a greater extent (%TcS) in infant mice than when grown in vitro, using the Wilcoxon paired-sample test (Zar, 1974).
None of the tnpR–lacZY fusions to putative in vivo-induced genes were inducible by growth of the strains in a low-iron medium, under microaerophilic conditions in a rich medium or when grown under in vitro conditions (Iwanga et al., 1986; Miller and Mekalanos, 1988), which maximally induce expression of the ToxR regulon in V. cholerae classical and El Tor biotypes (data not shown). Given that the ToxR regulon includes many of the known virulence determinants of V. cholerae, these data suggested that the TnpR fusion approach was apparently identifying new classes of potential virulence genes.
A DNA sequencing primer designed to read out from the N-terminal coding end of tnpR allowed us to determine the sequence of approximately 300 bases of V. cholerae DNA fused to the synthetic operon in the 13 strains shown in Table 1. In 11 of the strains, the synthetic operon was found to be transcriptional fused within a predicted open reading frame (ORF) based on preferred codon usage (data not shown) and, in the majority of cases, on extensive similarity of the predicted amino acid sequences to known bacterial proteins (see below). The remaining two strains contained fusions of the synthetic operon to putative anti-sense transcripts.
In vivo-induced loci involved in amino acid biosynthesis and general metabolism
Strain AC-V226 contained a synthetic operon fusion within the N-terminal end of a predicted ORF, designated iviI (in vivo-induced transcript I) (Fig. 2). The deduced amino acid sequence of ivil displayed a high degree of similarity to the Salmonella typhimurium Cysl protein (J. Ostrowski, J. Y. Wu, D. C. Rueger, B. E. Miller, L. M. Siegel, and N. M. Kredich, unpublished data), which catalyses an intermediate step in the biosythesis of l-cysteine from inorganic sulphate (Siegel et al., 1973). Evidence of an upstream ORF, orfU, encoding a homologue of the S. typhimurium CysJ protein was also obtained (data not shown). Therefore, V. cholerae appears to have maintained the order of at least the first two genes of the cysJIH operon as found in S. typhimurium (Jones-Mortimer, 1973). Beta-galactosidase activity was assayed for strain AC-V226 (iviI∷tnpR–lacZY) and was found to be induced 21-fold in a minimal medium lacking l-cysteine (667 versus 32 units of activity during growth in media without or with l-cysteine, respectively). An iviI (cysl) mutant strain of V. cholerae, AC-V172 (see below), was accordingly found to be auxotrophic for l-cysteine.
Fig. 2.

A. Similarities of deduced V. cholerae polypeptide partial sequences. Vertical lines indicate identical residues, highly conserved substitutions are represented by colons and conserved substitutions are represented by dots. Gaps were introduced into the sequences to maximize the similarities (see the Experimental procedures). For each V. cholerae polypeptide sequence shown, the C-terminal amino acid delineates the corresponding DNA site of fusion to tnpR–lacZY.
B. Schematic diagram (not to scale) of predicted V. cholerae genes containing fusions to tnpR–lacZY (designated by triangles). Transcriptional orientations are shown by the arrows.
Strain AC-V228 contained a fusion within the middle of iviIII (Fig. 2) which was predicted to encode a polypeptide that had high similarity to the Escherichia coli ArgA protein. ArgA catalyses the first step in the biosynthesis of l-arginine (Brown et al., 1987). Beta-galactosidase activity was assayed for strain AC-V228 (iviIII∷tnpR–lacZY) and was found to be induced 3.5-fold in a minimal medium lacking l-arginine (98 versus 28 units of activity during growth in media without or with l-arginine, respectively). An iviIII (argA) mutant strain of V. cholerae, AC-V176 (see below), was accordingly found to be auxotrophic for l-arginine.
Strain AC-V227 contained a fusion within the middle of iviII (Fig. 2) which was predicted to encode a polypeptide that had high similarity to the E. coil SucA protein (Darlison et al., 1984). SucA is part of a multisubunit enzyme that catalyses a step in the tricarboxylic acid (TCA) cycle whereby α-ketoglutarate is oxidatively decarboxylated to succinyl CoA and carbon dioxide. We were unable to modulate β-galactosidase activity from this strain by growth in conditions known to induce transcription of sucA in E. coli (Nimmo, 1987) (data not shown).
Strain AC-V230 contained a fusion within the C-terminal end of iviV (Fig. 2) which was predicted to encode a polypeptide that had high similarity to the Pseudomonas stutzeri NirT protein, nirT codes for a tetrahaem protein of unknown function; the gene is located within a nitrite reduction operon (nirSTBM) and its product is required for respiratory nitrate reduction (Zumft et al., 1988). Expression of nirT in P. stutzeri was hypothesized to be induced by anaerobic growth in the presence of nitrate and/or nitrite (Juengst et al., 1991; Liu et al., 1983). However, we were unable to induce expression of the iviV∷tnpR–lacZY fusion in V. cholerae under similar growth conditions, as measured by β-galactosidase activity (data not shown).
In vivo-induced loci involved in motility and chemotaxis
Strain AC-V229 contained a fusion within the middle of iviIV (Fig. 3) which was predicted to encode a polypeptide that had similarity to members of the bacterial methyl-accepting chemoreceptor protein family. A moderate level of iviIV transcription during in vitro growth was suggested by the resolution data in Table 1.
Fig. 3.
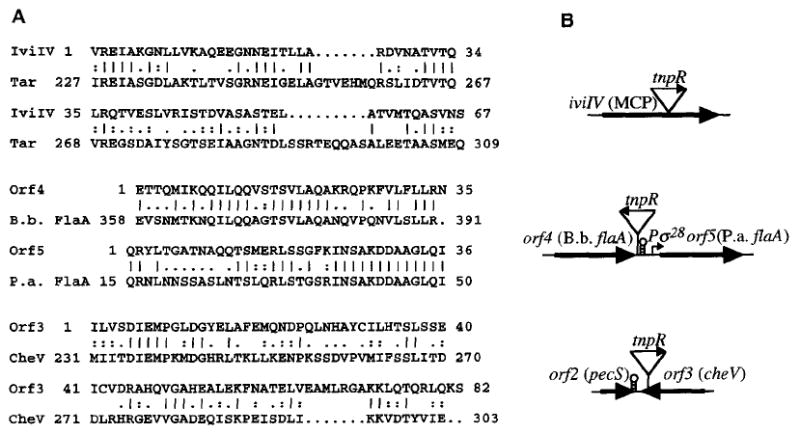
Similarities of deduced V. cholerae polypeptide partial sequences (A) and predicted genes (B). Details are as in the caption to Fig. 2, except that only for IvilV does the C-terminal amino acid delineate the corresponding DNA site of fusion to tnpR–lacZY. The DNA site of fusion to tnpR–lacZY in AC-V233 and AC-V234 occurred 51 bp 3′ of the orf4 stop codon and within the codon for amino acid 45, respectively. Putative transcriptional terminators are designated by short stem–loop symbols. A putative σ28-dependent promoter is shown immediately upstream of orf5. B. b. and P. a. refer to Bordetella pertussis and Pseudomonas aeruginosa, respectively.
Strain AC-V233 contained a fusion within the non-coding 3′ end of an RNA transcript containing orf4; this was predicted to encode a polypeptide that was a flagellin subunit protein (Fig. 3). However, the fusion junction occurred in the opposite transcriptional orientation to and between the predicted stop codon and transcriptional terminator for orf4. This suggested that the tnpR fusion occurred within an RNA transcript induced during infection which was anti-sense to at least the non-coding end of the orf4 RNA transcript. Interestingly, a downstream ORF, orf5, also was predicted to encode a polypeptide that had high similarity to other flagellin subunit proteins. A putative σ28-dependent promoter was located immediately upstream of orf5 (data not shown). Therefore, V. cholerae may express multiple flagellin subunit proteins, as do many other motile Gram-negative bacterial species (Nuijten et al., 1990; Pleier and Schmitt, 1989).
Strain AC-V234 contained a fusion within the C-terminal end of orf3 (Fig. 3) which was predicted to encode a polypeptide that had similarity to the Bacillus subtilis CheV protein (Fredrick and Helmann, 1994). The B. subtilis CheV protein is a novel ‘chimaeric’ protein which has CheY and CheW homologous domains and was shown to be required for normal chemotaxis (Fredrick and Helmann, 1994). However, the tnpR fusion in AC-V234 occurred in the opposite transcriptional orientation to that predicted for orf3. This suggested that the synthetic operon fusion occurred within an RNA transcript induced during infection which was anti-sense to the C-terminal coding end of orf3. This putative RNA transcript may have originated from an upstream ORF, orf2, which is in the proper transcriptional orientation to induce the synthetic operon (Fig. 3). However, orf2 had a predicted transcriptional terminator at its 3′ end which may prevent transcription into the downstream orf3 region, orf2 was predicted to encode a polypeptide that had similarity to the Erwinia chrysanthemi PecS regulatory protein (data not shown) which regulates expression of several proteins including the secreted pectinase and cellulase enzymes (Reverchon et al., 1994).
In vivo-induced locus encoding a secreted lipase
Strain AC-V237 contained a fusion within the middle of iviXII which was identical in DNA sequence to a partially sequenced ORF designated the hlyC gene of V. cholerae (Casanova and Peterson, 1995; Manning et al., 1984). hlyC is an uncharacterized gene which was predicted to encode a secreted triacylglyceride-specific lipase, based on similarity to other bacterial lipases. Our analysis confirmed this observation by finding similarity of the predicted HlyC (IviXII) polypeptide to the Pseudomonas aeruginosa LipA protein (Fig. 4), including the heptapeptide consensus motif (IGHSHGG) thought to lie at the active site of this family of lipases (Wohlfarth et al., 1992) (data not shown). A hlyC (iviXII)-containing mutant strain of V. cholerae that we constructed, AC-V195 (see below), lost the ability to produce a clearing zone around colonies grown for 48 h on agar media containing emulsified tributyrin (Jorgensen et al., 1991) (Fig. 5), thus demonstrating that the htyC (iviXII) locus is required for this secreted lipase activity.
Fig. 4.
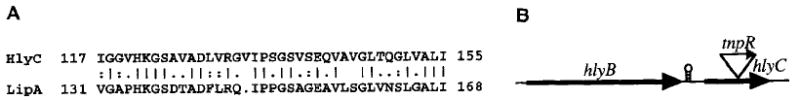
Similarities of deduced V choterae polypeptide partial sequences (A) and predicted genes (B). Details as in the legend to Fig. 2.
Fig. 5.
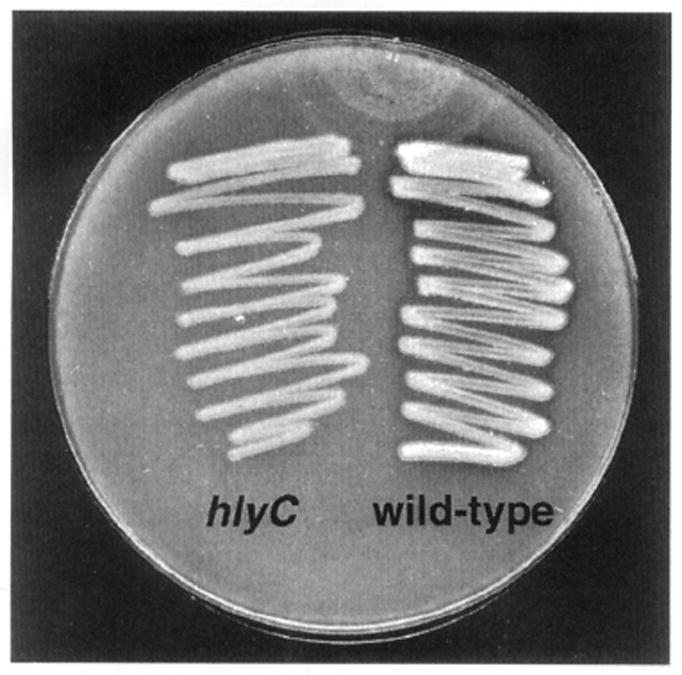
Secreted lipase activity from wild-type V. cholerae C6709-1 and the hlyC mutant AC-V195. The plate contained LB agar plus emulsified tributyrin and was incubated at 37°C for 48h.
In vivo-induced loci encoding polypeptides of unknown function
Strain AC-V231, which showed the highest level of in vivo-induction (Table 1), contained a fusion within the middle of iviVI (Fig. 6) which was predicted to encode a polypeptide that had high similarity to members of the ATP-binding cassette family of transporters. The sequenced portion of iviVI included an N-terminal ATP-binding Walker A box motif (GPS/TGXGKS/T), which is highly conserved in members of this transporter family (Ames et al., 1990). The best similarity obtained was to the Synechococcus PCC 7942 CysA protein, which is part of a three-component transporter for sulphate (Green et al., 1989). Accordingly, β-galactosidase activities and resolution were assayed for strain AC-V231 (iviVI∷tnpR–lacZY) after growth in a minimal medium lacking l-cysteine and containing sulphate as the sole source of sulphur. However, the AC-V231 gene fusion was not induced in this medium. In addition, an iviVI-containing mutant strain of V. cholerae, AC-V184 (see below), was not auxotrophic for l-cysteine in a minimal medium containing sulphate as the sole source of sulphur. Therefore, iviVI appeared not to encode the sole V. cholerae transporter of sulphate.
Fig. 6.

Similarities of deduced V. cholerae polypeptide partial sequences (A) and predicted genes (B). Details are as in the caption to Fig. 2 except that the N-terminal amino acid for IviVI delineates the corresponding DNA site of fusion to tnpR–lacZY.
Strain AC-V232 contained a fusion within the N-terminal end of iviVII (Fig. 6B) whose predicted polypeptide sequence had no significant similarity to any sequences in the searched databases. Evidence of a downstream ORF, orf1, was also obtained, which was predicted to encode a polypeptide that had high similarity to the Vibrio alginolyticus collagenase protein (Takeuchi et al., 1992). It is not known at this time whether iviVII and orf1 are co-regulated and/or are functionally related in any way.
Strain AC-V236 contained a fusion within the middle of iviXI (Fig. 6) which was predicted to encode a polypeptide with high similarity to a hypothetical 21.7 kDa E. coli polypeptide of unknown function. AC-V236 exhibited a moderate level of resolution during in vitro growth but resolved to a higher level in infant mice (Table 1).
Strains AC-V235 and AC-V238 contained fusions within iviX and the C-terminal end of iviXIII, respectively, whose predicted polypeptide sequences had no significant similarities to any sequences in the searched databases.
Competition assays of V. cholerae mutant strains in infant mice
To test whether any of the in vivo-induced and associated loci discussed above contributed to colonization and growth in infant mice, we examined strains mutated in each locus in an infant mouse competition assay. Each gene was mutated by single integration of the non-replicating plasmid pGP704, with the exception of iviII for which we repeatedly failed to obtain an integration, suggesting that insertional inactivation of the V. cholerae sucA homologue was lethal for growth on standard laboratory media. Each mutant strain was competed against its isogenic, virulent parent strain, which was phenotypically LacZ− as a result of integration of pGP704 into the endogenous V. cholerae lacZ gene. The majority of mutant strains had competitive indices of approximately 1 (Table 2) and, therefore, exhibited no colonization defects in this competition assay. However, mutant strains AC-V176 (iviIII (argA)), AC-V186 (iviVII) and AC-V196 (iviXIII) reproducibly exhibited small colonization defects in two independent experiments and their numbers were reduced by five-, two- and threefold, respectively, compared with the wild-type strain. Interestingly, the mutant strain AC-V189 (orf3 (CheV similarity)) colonized at least twofold better than the wild type in two independent experiments. This mutant exhibited a hyperswarming phenotype when grown in semi-solid agar, consistent with a possible role for orf3 in the chemotaxis phenotype of V. cholerae.
Table 2.
Competition assays of V. cholerae mutant strains.
| Test strain | Gene mutated | Competitive indexa |
|---|---|---|
| AC-V172 | iviI | 1 |
| AC-V176 | iviIII | 0.2b |
| AC-V179 | iviIV | 1 |
| AC-V180 | iviV | 1 |
| AC-V184 | iviVI | 1 |
| AC-V186 | iviVII | 0.5b |
| AC-V187 | orf1 | 0.9 |
| AC-V188 | orf2 | 1 |
| AC-V189 | orf3 | 2 |
| AC-V191 | iviX | 0.9 |
| AC-V193 | iviXI | 0.9 |
| AC-V195 | iviXII | 0.7 |
| AC-V196 | iviXIII | 0.3b |
Competitive indices from one representative experiment are shown for each strain. Each competitive index was calculated by dividing the ratio of test strain to wild-type bacteria from 6–8 infant mice outputs by that from the in vitro competition output, after correcting the ratios for deviations of the inoculum ratio from a value of 1. All 13 mutant strains had in vitro competitive ratios of approximately 1 and, therefore, did not have any general growth defects caused by the mutations (data not shown).
P<0.0078 that the competitive indices were equal to or greater than a value of 1 using the sign test (Zar, 1974).
Discussion
The data presented here demonstrate the utility of using a gene encoding a site-specific DNA recombinase as a reporter to identify those RNA transcripts of a pathogenic bacterium which are induced during infection of a host animal. Thirteen transcription units of V. cholerae were identified which were induced after intragastric inoculation of infant mice. Five of these are predicted to encode polypeptides that could be linked with diverse functions in metabolism, biosynthesis and motility; one was similar to a secreted lipase; five are predicted to encode polypeptides of unknown function in that they either failed to exhibit any significant similarity to protein sequences in the databases or had similarity to proteins of unknown function; and, two may be antisense RNA transcripts, both of which lie within or near genes important for bacterial motility. Three of the thirteen in vivo-induced transcripts identified in this study contributed to the virulence of V. cholerae in the infant mouse model of cholera. This fulfilled our expectation that the identification of genes specifically induced during infection will yield an enrichment for genes which contribute to colonization, survival and/or growth within the host.
In vivo-induced transcripts involved in biosynthesis and metabolism
Two in vivo-induced amino acid biosynthetic genes were identified, including one for l-cysteine and for l-arginine. A mutant strain for the latter gene was attenuated fivefold in competition assays, suggesting that l-arginine was limiting for bacterial growth in the infant mouse small intestine. Klarsfeld et al. (1994) recently reported that the Listeria monocytogenes arpJ gene, encoding a subunit of an l-arginine transporter, was transcriptionally induced during infection of a tissue-culture cell line. Perhaps free l-arginine, which is an essential amino acid supplied only through the diet, is in low concentration in host tissues, or, alternatively, bacteria have an increased demand for arginine in vivo. The arginine biosynthetic pathway feeds into the pathway for polyamines (Glansdorff, 1987). It is possible that increased iviII (argA) transcription occurs in response to decreased intracellular arginine pools that result from utilization of arginine for polyamine biosynthesis. Perhaps bacterial polyamines play undefined roles in the virulence of a variety of bacterial pathogens.
A mutant strain for the l-cysteine biosynthetic gene iviI (cysl) appeared fully virulent in infant mouse competition assays. Adult rats have been found to maintain high concentrations of cysteine in their small intestinal mucosa and lumen regardless of diet or short-term starvation (Dahm and Jones, 1994). However, cysteine concentrations in the infant mouse small intestine are not known. Therefore, it is possible that the concentrations of available l-cysteine in the infant mouse small intestine may have been limiting enough to induce transcription of the V. cholerae iviI (cysl) gene, yet not limiting for bacterial growth. Alternatively, if available l-cysteine was limited, it is possible that the iviI (cysl) mutation, which would block reduction of sulphite to sulphide in the l-cysteine biosynthetic pathway, may have been bypassed by available sulphide and/or thiosulphate within the infant mouse small intestine.
Interestingly, a second gene predicted to be involved in sulphate metabolism was identified in this study, iviVI is predicted to code for an ATP-binding cassette transporter with similarity to the sulphate uptake transporter CysA of Synechococcus PCC 7942 (Green et al., 1989). However, transcription of iviVI was not inducible by growth of V. cholerae AC-V231 under conditions of l-cysteine limitation with sulphate present as the sole source of sulphur. Furthermore, an iviVI mutant strain of V. cholerae was competent for growth in a minimal medium containing sulphate as the sole source of sulphur. Therefore, it is unlikely that iviVI codes for a sulphate transporter, iviVI was the most highly in vivo-induced gene found in this study yet was not inducible during growth in the laboratory under microaerophilic conditions, in a minimal medium, a low-iron medium or in a medium known to maximally induce expression of the ToxR virulence gene regulon. Although an iviVI mutant strain of V. cholerae was not attenuated in infant mouse competition experiments, it would be of interest to understand the regulation of this gene, which appears to respond specifically to signals which occur in the host.
The in vivo-induced metabolic genes identified included one encoding a TCA cycle enzyme subunit and one encoding a tetrahaem protein presumably involved in nitrite reduction. In E. coli, the gene for the former, sucA, is induced by aerobic growth on acetate and repressed by growth anaerobically or in the presence of high glucose concentrations (Nimmo, 1987). We were unable to alter expression of the V. cholerae iviII (sucA) gene under similar growth conditions, which suggested differing regulation to that in E. coli. A V. cholerae mutant strain for iviII was not obtained, presumably because of lethality, at least under standard conditions of laboratory growth.
The P. stutzeri nirTgene codes for a tetrahaem polypeptide that is required for anaerobic respiratory nitrate reduction and whose expression is presumably induced by anaerobic conditions (Juengst et al., 1991; Liu et al., 1983). A mutant strain for the V. cholerae nirT homologue (iviV) appeared fully virulent in the infant mouse competition assay. Therefore, either V. cholerae has a redundant mechanism for reducing nitrite or, alternatively, reduction of nitrite is not required for survival and/or growth in the infant mouse assay. Oxygen tension in the adult rat (both germ-free and conventional) intestinal lumen and at the mucosal surface is low (Bornside et al., 1976). Growth of V. cholerae in the infant mouse intestinal lumen is believed to reduce the presumed low oxygen tension in this compartment further (Freter et al., 1981). We could not, however, show that the V. cholerae nirT homologue (iviV) was induced by anaerobic growth conditions in vitro. It is, therefore, possible that iviV may represent one member of a regulon that is induced by another host signal(s) that acts as a surrogate signal for the low oxygen tension in the intestinal lumen.
In vivo-induced transcripts involved in motility and chemotaxis
Three of the in vivo-induced transcription units identified in this study relate to bacterial motility and chemotaxis. There is evidence that suggests that motility and chemotaxis are required for full virulence of V. cholerae in various adult animal models of cholera (Freter et al., 1981; Richardson, 1991). In contrast, in the infant mouse model of cholera, chemotactic motility is actually a detriment in that non-chemotactic mutant strains survive in greater numbers than wild-type vibrios and produce a more rapid and severe disease (Freter and O’Brien, 1981). One of the in vivo-induced transcripts we found, iviIV, was predicted to encode a methyl-accepting chemoreceptor and was shown to be transcribed at significant levels both in vitro and in vivo, as measured by resolution levels. Expression of MIV appeared to be independent of ToxR regulation, in contrast to the recently described acfB and tcpl genes which encode putative methyl-accepting chemoreceptor proteins and which are positively regulated by ToxR (Everiss et al., 1994; Harkey et al., 1994). Strains mutated in acfB and tcpl exhibited altered chemotaxis in vitro (Everiss et al., 1994; Harkey et al., 1994) but their contribution to colonizing the small intestine, if any, is not known at present. A mutant strain for iviIV exhibited normal chemotaxis in vitro and appeared fully virulent in the infant mouse competition assay. Therefore, chemotaxis mediated by this protein was neither required for nor a detriment to colonization of the infant mouse.
Another in vivo-induced fusion (iviIX) was found to be anti-sense to orf3, which encoded a protein with similarity to CheV of B. subtilis. An orf3 mutant strain of V. cholerae exhibited a hyperswarming phenotype in vitro and appeared to outcompete the wild-type strain in infant mice. Therefore, chemotaxis mediated by Orf3 was a detriment to wild-type vibrios; this is in agreement with the observations of Freter and O’Brien (1981). A second in vivo-induced fusion (iviVIII) was also found to be antisense, in this case to the non-coding sequence 3′ of orf4 which is predicted to encode a flagellin protein. Any biological significance for these anti-sense transcripts is not known at present. However, it is tempting to speculate that they are either directly involved in down-regulating orf3 and orf4 expression or are artifactual in nature but reflect reduced levels of orf3 and orf4 transcription at some stage of infection. Recent evidence indicates that motility and ToxR-activated gene expression are reciprocally regulated, suggesting that at a stage in the infection cycle where cholera toxin and TCP pili are optimally expressed, motility gene expression may be repressed (Gardel and Mekalanos, 1994; Harkey et al., 1994). Our identification of various motility and chemotaxis genes and associated RNA transcripts should aid further study to assess the role(s) of these bacterial properties in the virulence and pathogenicity of V. cholerae.
In vivo-induced transcript coding for a secreted lipase
One in vivo-induced gene found in this study was identical to the previously sequenced hlyC gene of V. cholerae, which was predicted (Casanova and Peterson, 1995; Manning et al., 1984) and demonstrated here to encode a secreted lipase, hlyC lies downstream from the hlyAB genes, which encode a secreted haemolysin and a methyl-accepting chemoreceptor homologue, respectively (Aim and Manning, 1990; Jeffery and Koshland, 1993; Manning et al., 1984). Although by examination of their nucleotide sequence, the hlyA, hlyB and hlyC genes do not appear to comprise a classical operon, it is possible they are co-regulated at least in part by HlyU, a recently described transcriptional activator of hlyA (Williams et al., 1993). A strain containing the hlyU mutation was recently shown to be attenuated for virulence in the infant mouse model and furthermore, the observed attenuation was not fully explained by loss of HlyA expression (Williams et al., 1993). Therefore, HlyU may regulate additional virulence factors and, if it is responsible for expression of hlyC, it follows that HlyU may be a regulator that responds to in vivo environmental signals. Stoebner and Payne (1988) have reported an increase in haemolysin (hlyA) expression under low iron conditions. However, we did not observe induction of our hlyC∷lacZ fusion by growth in media containing low iron levels. Therefore, at one level at least (i.e. low iron induction), hlyA and hlyC appear to be regulated independently.
A hlyC mutant strain appeared fully virulent in the infant mouse competition assay. However, it is possible that the co-inoculated wild-type strain was able to complement a lipase defect of the mutant strain in trans. Roles for the haemolysin and lipase in virulence and/or pathogenesis are not known, but it is possible that they act within the host small intestine, serving perhaps a defensive and/or nutritional role, for example, by damaging host immune cells by hydrolysing membrane lipids and thus releasing fatty acids; V. cholerae could potentially metabolize the released fatty acids. Mahan et al. (1995) have recently described an in vivo-induced S. typhimurium gene, fadB, which codes for a subunit of a multienzyme complex capable of degrading long-chain fatty acids. Therefore, metabolism of host lipids may be a common activity of bacterial pathogens.
In vivo-induced transcripts are not inducible in vitro under various growth conditions
None of the in vivo-induced V. cholerae transcription units identified in this study was regulated in vitro by conditions in which iron was limiting for growth, under microaerophilic growth conditions or by growth conditions which modulate members of the ToxR virulence gene regulon. The last result was not unexpected because, in preliminary experiments, we found that ToxR-activated genes (tag) exhibited some transcriptional activity even when grown under non-permissive conditions in the laboratory and, therefore, resolvase fusions to these tag genes were induced enough in vitro to result in resolution of the reporter in such newly constructed strains. This hypothesis was confirmed by the successful construction of unresolved tag∷tnpR fusion strains (e.g. acfB∷tnpR and ctx∷tnpR) in a toxR-null genetic background (A. Camilli and J. J. Mekalanos, unpublished data).
As with most genetic screens or selections, only a subset of possible genes will be identifiable by any one method. In our case, we chose only those gene fusions that were initially transcriptionally silent or expressed at a low level during growth on a rich laboratory medium under aerobic conditions but were subsequently induced during infection. Our choice of a rich medium for the in vitro pre-screen was done to broaden the set of obtainable genes to include those that might be involved in general metabolism and nutrient scavenging in the host. Our protocol could easily be altered, with respect to both the pre-screening conditions as well as the infection model used, in order to identify other classes of genes. In addition, further modifications of our system can be envisaged that would broaden the set of identifiable genes to include those having higher basal levels of transcription. For example, the generation of gene fusions to a tnpR allele in which the ribosome-binding site has been mutated would reduce subsequent levels of resolvase expression. In addition, the incorporation of other selectable markers into the resolvase-excisible cassette (e.g. that allow positive selection of cassette-deleted progeny) should facilitate the recovery of rare resolved clones within the animal passaged pool. Because resolvase requires only a negatively supercoiled DNA substrate and Mg2+ as a cofactor (Reed, 1981), modified versions of our method should be broadly applicable for study of other bacterial pathogens, both Gram negative and Gram positive, and even, perhaps, unicellular eukaryotic pathogens.
The method described here has allowed us to identify 13 transcription units of V. cholerae that are specifically induced in vivo during infection. Our results demonstrate that only a subset of such transcripts will contribute measurably to virulence in animal infection models. However, we believe that a full understanding of host–parasite interactions must include the study of the complete set of pathogen genes expressed in response to host environments. The identification of pathogen gene products which are expressed during infection may facilitate the identification of new targets for antimicrobial drug development. The gene products expressed in vivo by pathogens may represent new, unrecognized targets for immune therapy or prophylaxis. Finally, the in vivo-induced promoters identified through these studies have applications in the expression of heterologous antigens in live, attenuated vaccines (J. R. Butterton, D. T. Beattie, C. L. Gardel, P. A. Carrol, T. Hyman, K. P. Killeen, J. J. Mekalanos and S. B. Calderwood, submitted).
Experimental procedures
Plasmid constructions
Plasmids are listed in Table 3. Plasmids pAC20 and pAC21, which are derivatives of pACYC184 (Chang and Cohen, 1978; Rose, 1988), were used to place the res–tet–res and res1–tet–res1 sequences into the V. cholerae chromosome by allelic replacement of the endogenous lacZ gene. A 5.6 kb AccI–EcoRV fragment of the V. cholerae chromosome containing lacZ and surrounding sequences was inserted into the 2.55kb HincII–ClaI fragment of pACYC184 to generate pAC18. The 3.6kb PacI–DrdI fragment containing res–tet–res was isolated from pRR51(Reed, 1981), treated with T4 DNA polymerase to blunt the DNA ends and had SacI and KpnI linkers (New England Biolabs) added sequentially. The resulting fragment was inserted into the unique KpnI site of pAC18, which lay within the lacZ gene, to generate pAC20. The 3.6 kb Kpnl fragment containing res1–tet–res1 was isolated from pSKCAT2tnpR (Camilli et al., 1994) and inserted into the unique Kpnl site of pAC18 to generate pAC21.
Table 3.
Bacterial strains and plasmids.
| Strain/Plasmid | Relevant genotype and phenotype | Source/Reference |
|---|---|---|
| Strain | ||
| SM10λpir | thi recA thr leu tonA lacY supE RP4-2-Tc∷Mu λ∷pir | Laboratory strain |
| DH5αλpir | F− Δ(lacZYA–argF)U169 recA1 endA1 hsdR17 supE44 thi-1 gyrA96 relA1 λ∷pir | Hanahan (1983); Kolter et al. (1978) |
| AC-V66 | C6709-1 El Tor biotype, lacZ∷ res–tet–res, SmR | This work |
| AC-V87 | O395 classical biotype, lacZ∷ res1–tet–res1, SmR | This work |
| AC-V226 | AC-V66 iviI∷pIVET5 | This work |
| AC-V227 | AC-V87 iviII∷pIVET5 | This work |
| AC-V228 | AC-V87 iviIII∷pIVET5 | This work |
| AC-V229 | AC-V66 iviIV∷pIVET5 | This work |
| AC-V230 | AC-V87 iviV∷pIVET5 | This work |
| AC-V231 | AC-V66 iviVI∷pIVET5 | This work |
| AC-V232 | AC-V66 iviVII∷pIVET5 | This work |
| AC-V233 | AC-V66 iviVIII∷pIVET5 | This work |
| AC-V234 | AC-V66 iviIX∷pIVET5 | This work |
| AC-V235 | AC-V66 iviX∷pIVET5 | This work |
| AC-V236 | AC-V66 iviXI∷pIVET5 | This work |
| AC-V237 | AC-V66 iviXII∷pIVET5 | This work |
| AC-V238 | AC-V87 iviXIII∷pIVET5 | This work |
| AC-V167 | O395 lacZ∷pGP704 | This work |
| AC-V168 | C6709-1 lacZ∷pGP704 | This work |
| AC-V172 | C6709-1 iviI∷pGP704 | This work |
| AC-V176 | O395 iviIII∷pGP704 | This work |
| AC-V179 | C6709-1 iviIV∷pGP704 | This work |
| AC-V180 | O395 iviV∷pGP704 | This work |
| AC-V184 | C6709-1 iviVI∷pGP704 | This work |
| AC-V186 | C6709-1 iviVII∷pGP704 | This work |
| AC-V187 | C6709-1 orf1∷pGP704 | This work |
| AC-V188 | C6709-1 orf2∷pGP704 | This work |
| AC-V189 | C6709-1 orf3∷pGP704 | This work |
| AC-V191 | C6709-1 iviX∷pGP704 | This work |
| AC-V193 | C6709-1 iviXI∷pGP704 | This work |
| AC-V195 | C6709-1 iviXII∷pGP704 | This work |
| AC-V196 | O395 iviXIII∷pGP704 | This work |
| Plasmid | ||
| pGP704 | oriR6K mobRP4, ApR | Miller and Mekalanos (1988) |
| pAC20 | pACYC184∷lacZ∷res–tet–res | This work |
| pAC21 | pACYC184∷lacZ∷res1–tet–res1 | This work |
| pIVET5 | pSKCAT2tnpR∷lacZYA’ trpAt | This work |
Plasmid pIVET5 is a derivative of pSKCAT2tnpR (Camilli et al., 1994). Plasmid pSKCAT2tnpR was digested with Sacl, treated with T4 DNA polymerase to blunt the DNA ends and the 28bp trpA transcriptional terminator trpAt (Pharmacia) was inserted in the proper orientation to terminate RNA transcripts originating from the tnpR gene; this generated pSKCAT2tnpRt. The 6.2 kb BamHI–NruI fragment of pMC903 (Casadaban et al., 1980) containing the E. coli lacZYA′ sequences was treated with T4 DNA polymerase to blunt the DNA ends; KpnI linkers (New England Biolabs) were added and the resulting fragment was inserted into the unique KpnI site of pSKCAT2tnpRt. A recombinant plasmid, designated pIVET5, was isolated in which the transcriptional orientation of the inserted lacZYA′ genes was the same as that of the tnpR gene.
Construction of bacterial strains and libraries
Bacterial strains are listed in Table 3. Allelic exchange of the chromosomal lacZ gene in the virulent streptomycin (Sm)-resistant parental V. cholerae strains O395 (classical biotype) and C6709-1 (El Tor biotype) was done using pAC21 and pAC20, respectively. Each plasmid was electroporated into V. cholerae and transformants in which the plasmids had integrated into the chromosomal lacZ gene were selected on LB agar (Sambrook et al., 1989) containing 50 μg Sm, 2 μg Tc, and 3μg chloramphenicol (Cm). Several transformants were pooled and then passaged for approximately 20 generations in LB broth at 37°C. Beta-galactosidase-deficient allelic exchange mutant strains were screened for on LB agar containing 50 μg Sm, 1 μg Tc and 60 μg Xgal. Loss of plasmid-encoded chloramphenicol resistance (CmR) was confirmed for each of the allelic exchange mutant strains, which were designated AC-V66 (El Tor) and AC-V87 (classical).
The transcriptional gene fusion libraries in V. cholerae were constructed by first generating Sau3AI partial digests of total V. cholerae O395 and C6709-1 DNA. The partial digests were electrophoretically separated on a 1% low-melting-temperature agarose gel and the 1 to 3.5 kb range of fragments were isolated and then inserted into pIVET5 that had been cut with BglII and treated with calf intestinal alkaline phosphatase (New England Biolabs). Each pool of recombinant plasmids was electroporated into SM10λpir. The resulting O395 and C6709-1-derived plasmid libraries were mobilized into AC-V87 and AC-V66, respectively, by biparental mating (Ditta et al., 1980). Exconjugates in which the plasmids had integrated into the V. cholerae chromosome resulting in transcriptionally silent fusions to the tnpR–lacZYA′ synthetic operon (Lac−) were selected and then screened for on LB agar containing 100 μg Sm, 60 μg ampicillin (Ap) and 60 μg Xgal. Pools of 200 Lac− colonies were collected by transferring an approximately equivalent portion of each colony to LB agar containing 2 μg Tc. Each agar plate was incubated at 37 °C for 2 h and the bacteria were collected in 1 ml of LB broth and stored at −70°C in 50% glycerol.
Each in vivo-induced gene fusion strain was reconstructed in a fresh genetic background. The integrated pIVET5 derivatives from each of the TcS strains recovered from the infant mice were able to excise out of the chromosome at the same low frequency at which they had originally integrated. These excised but non-replicating plasmids were recovered by plasmid preparation from 100 ml of overnight LB broth cultures using Qiagen midi-prep columns (Qiagen) and using 1 μg tRNA as carrier for the isopropanol-mediated precipitation reaction. Each plasmid preparation was electroporated into SM10λpir and transformants were selected for on LB agar containing 100 μg Ap. A single transformant from each electroporation was isolated and the plasmids were purified and their structures confirmed by restriction enzyme analysis (data not shown). Each plasmid was mobilized into either AC-V66 or AC-V87 by biparental mating as above, thus regenerating each gene fusion in the TcR V. cholerae strain backgrounds.
V. cholerae strains mutated in individual genes were constructed by inserting an internal coding fragment of DNA from each gene into pGP704 (Miller and Mekalanos, 1988). The pGP704 recombinant plasmids were electroporated into SM10λpir and then subsequently mobilized into either O395 or C6709-1 by biparental mating. Exconjugates in which the plasmid had integrated into the chromosome were selected for on LB agar containing 100μg Sm and 60μg Ap. Correct integration into the chromosomal gene of interest was confirmed by Southern blot analysis (data not shown).
Initial screen for V. cholerae strains having in vivo-induced gene fusions
Each V. cholerae Lac− gene fusion pool was grown to stationary phase at 30°C with aeration in LB broth containing 50 μg Sm, 30 μg Ap and 1 μg Tc. One 5-day-old infant CD-1 mouse (Charles River Breeding Laboratories) was intragastrically inoculated with 105 colony-forming units (cfu) per pool as previously described (Camilli et al., 1994). The bacteria were recovered from the small intestines after 24 h as described (Camilli et al., 1994) and approximately 1000 cfu were grown on LB agar containing 100 μg Sm. TcS strains were screened for by replica-plating the colonies onto LB agar containing 2 μg Tc and incubating the replica plates at 37°C for 8 h.
Quantification of resolution of V. cholerae gene fusion strains
Each of the reconstructed V. cholerae gene fusion strains was grown as above and used to inoculate 2 ml of 37°C LB broth with 4 × 104 cfu and to intragastrically inoculate infant mice as above. The LB broth cultures were grown 24 h at 37°C with aeration after which approximately 300cfu were grown on LB agar containing 100 μg Sm. Bacteria were recovered from the infant mouse small intestines after 24 h, as above, and approximately 300 cfu were grown on LB agar containing 100 μg Sm. The percentages of the bacterial populations recovered from the in vitro cultures and from the infant mice that were TcS were determined by replica-plating as above.
Competition assays
Each of the V. cholerae mutant strains to be tested and the fully virulent Lac− wild-type strains AC-V167 and AC-V168 were grown to stationary phase at 30°C with aeration in LB broth containing 50 μg Sm and 30 μg Ap. A portion of each test strain culture was mixed with an equal volume of the appropriate wild-type strain culture and the resulting mixed culture was washed, diluted and approximately 105 cfu in total were used to intragastrically inoculate eight infant mice, as previously described (Camilli et al., 1994). In vitro competitions were done by using each of the prepared inoculi above to inoculate 2 ml of 37°C LB broth with 4 × 104 cfu; after which the cultures were grown for 24 h at 37°C with aeration. The ratio of test strain to wild-type strain in each inoculum, as well as in the resulting bacterial populations recovered from the in vitro and from the in vivo competitions after 24 h, was determined by growing approximately 300cfu on LB agar containing 50 μg Sm, 30 μg Ap and 60 μg Xgal at 37°C and scoring the number of Lac+ and Lac− colonies after 24 h. A competitive index for each mutant strain tested was calculated as described in the legend to Table 2.
Sequencing of tnpR fusion junctions and flanking DNA
At least 300 bases of V. cholerae DNA flanking the 5′ end of tnpR was sequenced from each pIVET5 derivative recovered from the gene fusion strains, using primer 5′-GTTGATACCCGTGCGTAACC-3′, which hybridized to a sequence near the 5′ end of tnpR (Reed et al., 1982). Sequencing was done using an ABI 373A automated sequencer according to the manufacturer’s directions (Applied Biosystems Incorporated). In certain cases, V. cholerae DNA sequences 3′ of the fusion junction were obtained by inverse polymerase chain reaction (PCR) (Ochman et al., 1988) from O395 or C6709-1 chromosomal DNA and sequenced as above using primers which hybridized to flanking V. cholerae sequences (data not shown).
Predicted amino acid sequences from all six reading frames of DNA sequence obtained above were subjected to similarity searches of the PIR-protein database (Release 43.0) using the FASTA program (Genetics Computer Group (GCG) version 7.3.1). Homologous polypeptide segments (see Figs 2, 3, 4 and 6) were compared using the BESTFIT program (GCG version 7.3.1) with a gap weight equal to 3 and a length weight equal to 0.1. V. cholerae DNA sequences which coded for the polypeptides segments shown in part A of Figs 2, 3, 4 and 6 were deposited into the GenBank database as iviI (database accession number U25709), iviII (U25710), iviIII (U25807), ivIV (U25711), iviIV (U25726), orf4 (U25727), orf5 (U25820), orf3 (U25728), iviVI (U25729), orf1 (U25730) and iviXI (U25731). The known bacterial proteins used for these comparisons were Cysl (M23007), SucA (A30256), ArgA (A30372), NirT (S13937), Tar (A29053), B. bronchiseptica FlaA (A40594), P. aeruginosa FlaA (A37853), CheV (Z29584), CysA (A30301), HlyC (X16945), LipA (S25768), collagenase (S19658) and hypothetical 21.7kDa protein (P32892).
Beta-galactosidase activity assays
Beta-galactosidase assays were performed using a modification of the method of Miller (Miller, 1972): the method of Slauch and Silhavy (Slauch and Silhavy, 1991). The reaction was monitored by reading A420 on a microplate reader. Activities were calculated as (UA600−1 ml−1 cell suspension) × 103, where U = μmol min−1 ONP formed. Assays were carried out on 37°C log-phase cultures of V. cholerae strain AC-V226 (cysl∷tnpR–lacZY) grown in M9 minimal media (Sambrook et al., 1989) supplemented with either 0.2% d-glucose, 0.2 mM MgCl2, 1 mM glutathione and 1 mM O-acetyl-l-serine (inducing conditions) or 0.2% d-glucose, 0.2 mM MgCI2 and 0.2 mM l-cysteine (repressing conditions). Assays were carried out on 37°C log-phase cultures of V. cholerae strain AC-V228 (argA∷tnpR–lacZY) grown in M9 minimal media supplemented with either 0.2% d-glucose and 0.2 mM MgCl2 (inducing conditions) or 0.2% d-glucose, 0.2 mM MgCl2 and 0.2 mM l-arginine (repressing conditions).
Lipase activity assay
Secreted lipase activity was detected visually by formation of a clearing zone surrounding a heavily grown streak of V. cholerae after 48 h at 37 °C on LB agar supplemented with emulsified tributyrin. The media was prepared by adding 1 g Gum arabic (Sigma Chemical Co.) and 10 ml tributyrin (Sigma Chemical Co.) per litre of LB agar and emulsifying with a homogenizer prior to pouring the media.
Acknowledgments
We thank M. K. Waldor for critically reading the manuscript, and N. Grindley and G. Hatfull for plasmid constructs. This research was supported by Cancer Research Fund of the Damon Runyon–Walter Winchell Foundation Fellowship DRG-1201 (to A.C.) and National Institutes of Health Grant AI-26289 (to J.J.M.).
References
- Aim RA, Manning PA. Characterization of the hlyB gene and its role in the production of the El Tor haemolysin of Vibrio cholerae. Mol Microbiol. 1990;4:413–425. doi: 10.1111/j.1365-2958.1990.tb00608.x. [DOI] [PubMed] [Google Scholar]
- Ames GF, Mimura CS, Shyamala V. Bacterial periplasmic permeases belong to a family of transport proteins operating from Escherichia coil to human: traffic ATPases. FEMS Microbiol Rev. 1990;6:429–46. doi: 10.1111/j.1574-6968.1990.tb04110.x. [DOI] [PubMed] [Google Scholar]
- Berger KH, Isberg RR. Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol Microbiol. 1993;7:7–19. doi: 10.1111/j.1365-2958.1993.tb01092.x. [DOI] [PubMed] [Google Scholar]
- Bornside GH, Donovan WE, Myers MB. Intracolonic tensions of oxygen and carbon dioxide in germfree, conventional, and gnotobiotic rats. Proc Soc Exp Biol Med. 1976;151:437–441. doi: 10.3181/00379727-151-39229. [DOI] [PubMed] [Google Scholar]
- Brown K, Finch PW, Hickson ID, Emmerson PT. Complete nucleotide sequence of the Escherichia coil argA gene. Nucl Acids Res. 1987;15:10586. doi: 10.1093/nar/15.24.10586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilli A, Beattie D, Mekalanos J. Use of genetic recombination as a reporter of gene expression. Proc Natl Acad Sci USA. 1994;91:2634–2638. doi: 10.1073/pnas.91.7.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadaban M, Chou J, Cohen SN. In vitro gene fusions that join an enzymatically active β-galactosidase segment to amino-terminal fragments of exogenous proteins: Escherichia coil plasmid vectors for the detection and cloning of translational initiation signals. J Bacteriol. 1980;143:971–980. doi: 10.1128/jb.143.2.971-980.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova TB, Peterson KM. The Vibrio cholerae hlyC gene encodes a protein that is related to lipases of Pseudomonas species. DNA Sequence. 1995;5:181 –184. doi: 10.3109/10425179509029360. [DOI] [PubMed] [Google Scholar]
- Chang ACY, Cohen SN. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol. 1978;134:1141–1156. doi: 10.1128/jb.134.3.1141-1156.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahm LJ, Jones DP. Secretion of cysteine and glutathione from mucosa to lumen in rat small intestine. Am J Physiol. 1994;267:G292–G300. doi: 10.1152/ajpgi.1994.267.2.G292. [DOI] [PubMed] [Google Scholar]
- Darlison MG, Spencer ME, Guest JR. Nucleotide sequence of the sucA gene encoding the 2-oxoglutarate dehydrogenase of Escherichia coil K-12. Eur J Biochem. 1984;141:351–359. doi: 10.1111/j.1432-1033.1984.tb08199.x. [DOI] [PubMed] [Google Scholar]
- Ditta G, Stanfield S, Corbin D, Helinski DR. Broad host range DNA cloning system for Gram-negative bacteria: construction of a gene bank of Rhizobium meliloti. Proc Natl Acad Sci USA. 1980;77:7347–7351. doi: 10.1073/pnas.77.12.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everiss KD, Hughes KJ, Kovach ME, Peterson KM. The Vibrio cholerae acfB colonization determinant encodes an inner membrane protein that is related to a family of signal-transducing proteins. Infect Immun. 1994;62:3289–3298. doi: 10.1128/iai.62.8.3289-3298.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredrick KL, Helmann JD. Dual chemotaxis signaling pathways in Bacillus subtilis: a sigma D-dependent gene encodes a novel protein with both CheW and CheY homologous domains. J Bacteriol. 1994;176:2727–2735. doi: 10.1128/jb.176.9.2727-2735.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freter R, O’Brien PCM. Role of chemotaxis in the association of motile bacteria with intestinal mucosa: fitness and virulence of nonchemotactic Vibrio cholerae mutants in infant mice. Infect Immun. 1981;34:222–233. doi: 10.1128/iai.34.1.222-233.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freter R, O’Brien PCM, Macsai MMS. Role of chemotaxis in the association of motile bacteria with intestinal mucose in vivo studies. Infect Immun. 1981;34:234–240. doi: 10.1128/iai.34.1.234-240.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardel CL, Mekalanos JJ. Modus operandi of Vibrio cholerae: swim to arrive stop to kill. The relationship among chemotaxis, motility and virulence. J Cell Biochem. 1994;18A:65. [Google Scholar]
- Gill DM. Mechanism of action of cholera toxin. Adv Cyclic Nucleotide Res. 1977;8:85–118. [PubMed] [Google Scholar]
- Glansdorff N. Biosynthesis of arginine and polyamines. In: Neidhardt FC, Ingraham JL, Low KB, Magasanik B, Schaechter M, Umbarger HE, editors. Escherichia coil and Salmonella typhimurium, Cellular and Molecular Biology. Washington, D.C: American Society for Microbiology; 1987. pp. 321–344. [Google Scholar]
- Green LS, Laudenbach DE, Grossman AR. A region of a cyanobacterial genome required for sulphate transport. Proc Natl Acad Sci USA. 1989;86:1949–1953. doi: 10.1073/pnas.86.6.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. Studies on transformation of Escherichia coil with plasmids. J Mol Biol. 1983;166:557–580. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- Harkey CW, Everiss KD, Peterson KM. The Vibrio cholerae toxin-co-regulated-pilus gene tcpl encodes a homolog of methyl-accepting chemotaxis proteins. Infect Immun. 1994;62:2669–2678. doi: 10.1128/iai.62.7.2669-2678.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwanga M, Yamamoto K, Higa N, Ichinose Y, Nakasone N, Tanabe M. Culture conditions for stimulating cholera toxin production by Vibrio cholerae O1 El Tor. Microbiol Immunol. 1986;30:1075–1083. doi: 10.1111/j.1348-0421.1986.tb03037.x. [DOI] [PubMed] [Google Scholar]
- Jeffery CJ, Koshiand DE., Jr Vibrio cholerae hlyB is a member of the chemotaxis receptor gene family. Prot Sci. 1993;2:1532–1535. doi: 10.1002/pro.5560020918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones-Mortimer MC. Mapping of structural genes for the enzymes of cysteine biosynthesis in Escherichia coil K-12 and Salmonella typhimurium LT2. Heredity. 1973;31:213–221. doi: 10.1038/hdy.1973.76. [DOI] [PubMed] [Google Scholar]
- Jorgensen S, Skov KW, Diderichsen B. Cloning, sequence, and expression of a lipase gene from Pseudomonas cepacia: lipase production in heterologous hosts requires two Pseudomonas genes. J Bacteriol. 1991;173:559–567. doi: 10.1128/jb.173.2.559-567.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juengst A, Wakabayashi S, Matsubara H, Zumft WG. The nirSTBM region coding for cytochrome cd(1)-dependent nitrite respiration of Pseudomonas stutzeri consists of a cluster of mono-, di-, and tetrahaem proteins. FEBS Lett. 1991;279:205–209. doi: 10.1016/0014-5793(91)80150-2. [DOI] [PubMed] [Google Scholar]
- Klarsfeld AD, Goossens PL, Cossart P. Five Listeria monocytogenes genes preferentially expressed in infected mammalian cells: picA, purH, purD, pyrE and an arginine ABC transporter gene, arpJ. Mol Microbiol. 1994;13:585–597. doi: 10.1111/j.1365-2958.1994.tb00453.x. [DOI] [PubMed] [Google Scholar]
- Kolter R, Inuzuka M, Helinski DR. Trans-complementation-dependent replication of a low molecular weight origin fragment from plasmid R6K. Cell. 1978;15:1199–1208. doi: 10.1016/0092-8674(78)90046-6. [DOI] [PubMed] [Google Scholar]
- Liu MC, Payne WJ, Peck HD, Jr, LeGall J. Comparison of cytochromes from anaerobically and aerobically grown cells of Pseudomonas perfectomarinus. J Bacteriol. 1983;154:278–286. doi: 10.1128/jb.154.1.278-286.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahan MJ, Slauch JM, Mekalanos JJ. Selection of bacterial virulence genes that are specifically induced in host tissues. Science. 1993;259:686–688. doi: 10.1126/science.8430319. [DOI] [PubMed] [Google Scholar]
- Mahan MJ, Tobias JW, Slauch JM, Hanna PC, Collier RJ, Mekalanos JJ. Antibiotic-based IVET selection for bacterial virulence genes that are specifically induced during infection of a host. Proc Natl Acad Sci USA. 1995;92:669–673. doi: 10.1073/pnas.92.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning PA, Brown MH, Heuzenroeder MW. Cloning of the structural gene (hly) for the haemolysin of Vibrio cholerae El Tor strain 017. Gene. 1984;31:225–231. doi: 10.1016/0378-1119(84)90213-0. [DOI] [PubMed] [Google Scholar]
- Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1972. [Google Scholar]
- Miller VL, Mekalanos JJ. A novel suicide vector and its use in construction of insertion mutations: osmo-regulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol. 1988;170:2575–2583. doi: 10.1128/jb.170.6.2575-2583.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman BJ, Grindley ND. Mutants of the gamma delta resolvase: a genetic analysis of the recombination function. Cell. 1984;38:463–469. doi: 10.1016/0092-8674(84)90501-4. [DOI] [PubMed] [Google Scholar]
- Nimmo HG. The tricarboxylic acid cycle and anaplerotic reactions. In: Neidhardt FC, Ingraham JL, Low KB, Magasanik B, Schaechter M, Umbarger HE, editors. Escherichia coli and Salmonella typhimurium, Cellular and Molecular Biology. Washington, D.C: American Society for Microbiology; 1987. pp. 156–169. [Google Scholar]
- Nuijten PJ, van Asten FJ, Gaastra W, van der Zeijst BA. Structural and functional analysis of two Campylobacter jejuni flagellin genes. J Biol Chem. 1990;265:17798–17804. [PubMed] [Google Scholar]
- Ochman H, Gerber AS, Hartl DL. Genetic applications of an inverse polymerase chain reaction. Genetics. 1988;120:621–623. doi: 10.1093/genetics/120.3.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson KM, Mekalanos JJ. Characterization of the Vibrio cholerae ToxR regulon: identification of novel genes involved in intestinal colonization. Infect Immun. 1988;56:2822–2829. doi: 10.1128/iai.56.11.2822-2829.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleier E, Schmitt R. Identification and sequence analysis of two related flagellin genes in Rhizobium meliloti. J Bacteriol. 1989;171:1467–1475. doi: 10.1128/jb.171.3.1467-1475.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plum G, Clark-Curtiss JE. Induction of Mycobacterium avium gene expression following phagocytosis by human macrophages. Infect Immun. 1994;62:476–483. doi: 10.1128/iai.62.2.476-483.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed RR. Transposon-mediated site-specific recombination: a defined in vitro system. Cell. 1981;25:713–719. doi: 10.1016/0092-8674(81)90178-1. [DOI] [PubMed] [Google Scholar]
- Reed RR, Shibuya GI, Steitz JA. Nucleotide sequence of gamma delta resolvase gene and demonstration that its gene product acts as a repressor of transcription. Nature. 1982;300:381–383. doi: 10.1038/300381a0. [DOI] [PubMed] [Google Scholar]
- Reverchon S, Nasser W, Robert-Baudouy J. pecS: a locus controlling pectinase, cellulase and blue pigment production in Erwinia chrysanthemi. Mol Microbiol. 1994;11:1127–1139. doi: 10.1111/j.1365-2958.1994.tb00389.x. [DOI] [PubMed] [Google Scholar]
- Richardson K. Roles of motility and flagellar structure in pathogenicity of Vibrio cholerae: analysis of motility mutants in three animal models. Infect Immun. 1991;59:2727–2736. doi: 10.1128/iai.59.8.2727-2736.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose RE. The nucleotide sequence of pACYC184. Nucl Acids Res. 1988;16:355. doi: 10.1093/nar/16.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Siegel LM, Murphy MJ, Kamin H. Reduced nicotinamide adenine dinucleotide phosphate-sulphite reductase of Enterobacteria. I. The Escherichia coli hemoflavoprotein: molecular parameters and prosthetic groups. J Biol Chem. 1973;248:251–261. [PubMed] [Google Scholar]
- Slauch JM, Silhavy TJ. cis-acting ompF mutations that result in OmpR-dependent constitutive expression. J Bacteriol. 1991;173:4039–4048. doi: 10.1128/jb.173.13.4039-4048.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoebner JA, Payne SM. Iron-regulated hemolysin production and utilization of heme and hemoglobin by Vibrio cholerae. Infect Immun. 1988;56:2891–2895. doi: 10.1128/iai.56.11.2891-2895.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Shibano Y, Morihara K, Fukushima J, Inami S, Keil B, Gilles AM, Kawamoto S, Okuda K. Structural gene and complete amino acid sequence of Vibrio alginolyticus collagenase. Biochem J. 1992;281:703–708. doi: 10.1042/bj2810703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc Natl Acad Sci USA. 1987;84:2833–2837. doi: 10.1073/pnas.84.9.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachsmuth IK, Blake PA, Olsvik O. Vibrio cholerae and cholera: molecular to global perspectives. Washington, DC: American Society for Microbiology; 1994. [Google Scholar]
- Williams SG, Attridge SR, Manning PA. The transcriptional activator HlyU of Vibrio cholerae: nucleotide sequence and role in virulence gene expression. Mol Microbiol. 1993;9:751–760. doi: 10.1111/j.1365-2958.1993.tb01735.x. [DOI] [PubMed] [Google Scholar]
- Wohlfarth S, Hoesche C, Strunk C, Winkler UK. Molecular genetics of the extracellular lipase of Pseudomonas aeruginosa PAOI. J Gen Microbiol. 1992;138:1325–1335. doi: 10.1099/00221287-138-7-1325. [DOI] [PubMed] [Google Scholar]
- Zar JH. Biostatistical Analysis. Englewood, N J: Prentice-Hall; 1974. p. 124. [Google Scholar]
- Zumft WG, Dohler K, Korner H, Lochelt S, Viebrock A, Frunzke K. Defects in cytochrome cdl-dependent nitrite respiration of transposon Tn5-induced mutants from Pseudomonas stutzeri. Arch Microbiol. 1988;149:492–498. doi: 10.1007/BF00446750. [DOI] [PubMed] [Google Scholar]