

Abstract
Discrete Molecular Dynamics (DMD) is a physics-based simulation method using discrete energetic potentials rather than traditional continuous potentials, allowing microsecond time scale simulations of biomolecular systems to be performed on personal computers rather than supercomputers or specialized hardware. With the ongoing explosion in processing power even in personal computers, applications of DMD have similarly multiplied. In the past two years, researchers have used DMD to model structures of disease-implicated protein folding intermediates, study assembly of protein complexes, predict protein-protein binding conformations, engineer rescue mutations in disease-causative protein mutants, design a protein conformational switch to control cell signaling, and describe the behavior of polymeric dispersants for environmental cleanup of oil spills, among other innovative applications.
Introduction
The Discrete Molecular Dynamics (DMD) simulation method utilizes the physics of ballistic motion to generate trajectories of particles in space over time according to discrete, distance-based energetic potentials [1–5]. Originally developed for hard-sphere fluid systems [6], in the past 20 years DMD has emerged as a key tool in the study of biomacromolecules and their assemblies, due primarily to the vastly increased time and length scales allowed by advanced event scheduling and search algorithms used to progress the simulation. In place of integrating continuous energetic potentials at set time steps to determine forces that will impact new velocities and position, DMD assumes ballistic motion and assigns time step as the time until the next occurring interaction (“event”), saving time and computational resources. Upon interaction, energy is assessed with a distance-based step function, and velocity and position change instantaneously upon collision according to the conservation of momentum [7]. The use of energetic step potentials also readily allows for incorporation of distance constraints derived from experimentally-derived proximity and solvent exposure information [8–10]. Here, we review several applications in biology and medicine for which DMD has made a key impact in advancing understanding and accelerating technological innovation (Figure 1).
Figure 1. Length and time scales of molecular phenomena studied with DMD.
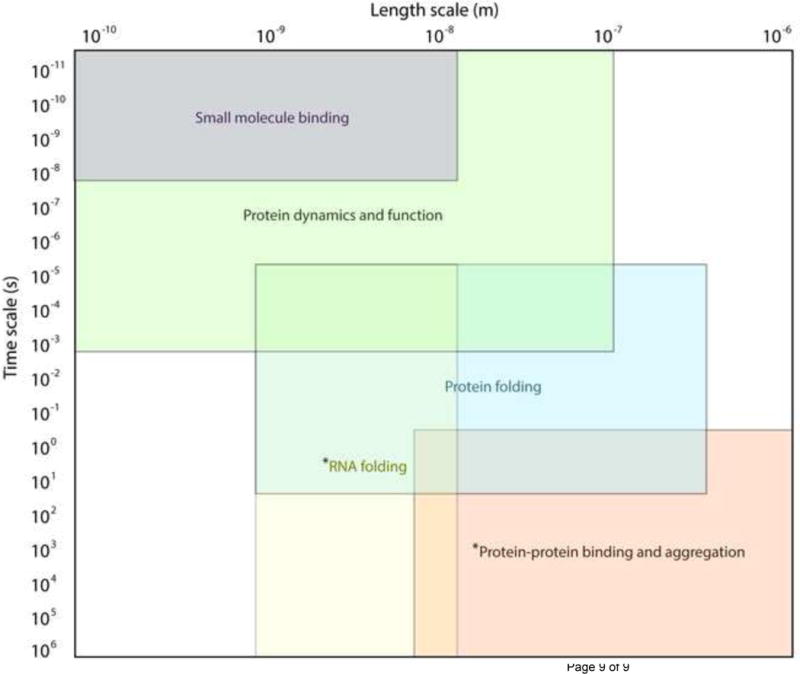
Asterisk (*) denotes that RNA folding and Protein aggregation can extend to time scales of seconds. Various molecular models (coarse-grained vs. all-atom) are appropriate for reaching different length and time scales, with larger scales being better represented by coarse-grained models. The incorporation of experimental information can accelerate simulations by several orders of magnitude.
Protein folding and aggregation
The protein folding problem, determining the three-dimensional folded structure of a protein given its amino acid sequence, has been a challenge in the field of physics and computer science since Anfinsen’s landmark paper in 1973 [11]. Because it allows for increased sampling of the folding landscape while retaining physically relevant dynamics [3,5], DMD is a tool well-suited for the study of aberrant folding intermediates relevant to protein misfolding diseases such as Alzheimer’s disease (AD) and Amyotrophic Lateral Sclerosis (ALS). Recently, Williams et al. [12] utilized DMD simulations to identify a misfolded intermediate of the protein ApoE4, an isoform of the ApoE protein associated with dramatically increased risk of AD. Ding et al. [10] combined a coarse-grained protein model with experimentally-derived structural information to determine that different ALS-associated SOD1 mutants display distinct patterns of misfolding, causing them to form differently-shaped aggregates of 8 SOD1 monomers (153 residues per monomer). Distinct aggregate morphology suggests an explanation for differences in disease progression for patients with different SOD1 mutations. Meral and Urbanc [13] investigated oligomeric formation in four different peptides of amyloid-β, a protein that forms brain plaques in the vast majority of AD patients, finding that those peptides known to be more toxic feature more flexible and solvent-exposed N-termini. Kimura et al. [14] performed DMD simulations to observe the folding of HIV-1 protease monomers and their assembly into active dimers, finding that the precursor to the mature protein can form non-native dimers of natively folded monomers.
Molecular modeling
Despite recent advances in experimental methods and technology, the number of high-resolution protein structures available is dwarfed by the number of known proteins and complexes. Computational molecular modeling is often faster and cheaper than experimental methods, and can also circumvent the technical issues encountered in solving structures of large, insoluble, and/or meta-stable proteins and aggregates. As discussed in the previous sections, the rapid sampling of the DMD simulation method, as well as the incorporation of experimental information to reduce the size of conformational space that must be explored, allows for accurate modeling of proteins and their assemblies for which experimentally-obtained structures do not exist. For example, Konrad et al. [15] utilized DMD simulations to construct three-dimensional structures of deoxyribonucleoside kinases between arthropods and vertebrates from their reconstructed ancestral sequence information, in order to determine evolutionary differentiation in substrate specificity. Szöllősi et al. [16] performed simulations of intrinsically disordered proteins and found evidence of transient secondary structural elements called helical prestructured motifs, which can play an important role in the binding of these proteins. Emperador et al. [17] designed a method for the efficient modeling of protein-protein binding using DMD simulations to address the outstanding problem of conformational change and protein flexibility upon docking. Similarly, Freeman et al. [18] used DMD simulations to identify and model novel protein-protein binding interactions.
Protein dynamics and function
Molecular function is often reliant on dynamic behavior. Proteins are highly flexible structures whose function is enabled by a change in shape. This conformational change may be a subtle rearrangement of a binding pocket to accommodate a drug or binding partner, or a dramatic allosteric global shift in shape. Because in many cases we cannot know a priori whether conformational changes will be mostly local or necessitate global rearrangement, use of an unconstrained, physics-based atomistic force field is ideal. DMD allows for increased sampling with fewer computational resources, which is imperative in dynamics studies where multiple replicates of long trajectories are necessary for statistically distinguishing thermodynamic fluctuations from meaningful rearrangements in structure. For example, Sfriso et al. [19] implemented coarse-grained DMD simulations to map protein conformational transitions. By taking advantage of the increased sampling of conformational space made possible by DMD event search algorithms [3–5], the authors create a tool that can trace complex, non-linear transitions in agreement with experiment without distortions in protein structure. Kota et al. [20] performed DMD simulations to elucidate the structural and energetic basis for substrate recognition in the proteolytic regulation of the epithelial sodium channel (ENaC), a key player in salt balance, blood pressure, and organ function. Proctor et al. [21] studied the dynamics of disease-causative mutants of the cystic fibrosis transmembrane conductance regulator (CFTR) using DMD. They discovered networks of correlated motion allowing transfer of aberrant dynamics from the mutation site to distal regions associated with ion channel function and stability. Stiffening of a “hotspot” residue located along the determined path of propagation by mutation to proline abolished the aberrant correlated dynamics and rescued CFTR function. The increased time and length scales of DMD are especially evident in atomistic simulations uncovering mechanistic details. Using multiple replicate simulations of the integral membrane protein cytochrome b6f, part of the electron transfer chain that generates energy from light in plants and is analogous to the mitochondrial protein cytochrome bc1 in animals, Hasan et al. [22] determined that the presence of chlorophyll in the plant protein increased residence time of quinone redox reaction product in the binding pocket, explaining the order of magnitude greater production of superoxide in the plant protein as compared to the animal protein.
Protein engineering
An understanding of protein structure and dynamics allows for rational perturbation in order to inhibit, enhance, or alter protein function. DMD simulations are useful in the design of proteins as both a method of determining the structural effects of the designed perturbations and, conversely, to determine the best shape or energetic properties of a desired binding partner. As an example of these uses of DMD, Dagliyan et al. [23] built upon previous work [24] to design a single-chain protein conformational switch controlled by binding of the drug rapamycin. Using DMD simulations, the authors built a structural model of the desired kinase-switch-rapamycin complex to test and refine conformational switching ability. Inserted into conserved kinase allosteric sites, the stabilization of the protein conformational switch upon binding of rapamycin activates the kinase, offering a powerful tool for the study of signaling pathways in vivo.
RNA tertiary structure
The recent discovery of the importance of non-coding RNA in catalysis, development, and gene regulation has led to an increased interest in the three-dimensional structure and dynamics of RNA molecules. However, the additional flexibility of RNA in comparison to proteins, with six backbone dihedrals as compared to two, exponentially increases the complexity of ab initio structural determination. As discussed in the above sections, DMD is an exceptionally appropriate tool for structural determination and description of dynamics in systems with high degrees of freedom, and sampling can be even further accelerated by use of a coarse-grained model. Ding et al. [25] addressed this problem by introducing a three-bead model (one bead each for the sugar, base, and phosphate) of RNA for use with DMD, which was used by Tsao et al. [26] to study differences in the dynamics of mRNA transcripts for variants of the protein catechol-O-methyltransferase (COMT), which plays a major role in the perception of pain. The authors determined that a silent mutation to the gene for COMT results in identical protein sequence but significantly influences the structural stability and dynamics of mRNA structure near the 5′ end, affecting translation efficiency and hence protein levels of COMT, influencing pain sensitivity. As in proteins, discussed above, the incorporation of experimental information as constraints can significantly decrease the size of conformational space necessary to be explored in simulation, and therefore increase the size of the system that can be evaluated. Ding et al. [27] incorporated base-pairing and solvent accessibility information at each nucleotide, derived from hydroxyl radical probing (HRP), as constraints in DMD simulations for the structural refinement of RNAs from 80 to 230 nucleotides in length. After rough tertiary structure determination using coarse-grained DMD simulations with base-pairing information, the authors applied a bias potential derived from the HRP data and constructed ensembles of low-energy, high-experimental agreement structures. Cole et al. [28] used this method of incorporating experimental data into DMD to model minimal telomerase RNA complex, revealing conformational changes that occur during telomerase assembly. Prior to this study, no high-resolution structural data on telomerase complexes had been available, due to the large size and high flexibility of the RNA molecules.
Surface chemistry
Surface chemistry of materials can play an important role in binding properties and outcomes. For example, the role of nanoparticles as potentially revolutionary drug delivery vehicles has attracted attention in recent years, but their binding properties must be carefully controlled in order to be safe and effective. The accelerated sampling of DMD is well-suited characterization of binding properties in simulations, which requires study of many interactions. Radic et al. [29] utilized DMD simulations to illustrate the effects of surface chemistry on the binding of nanoparticles to their target proteins. They demonstrated that highly hydroxylated fullerene nanoparticles form hydrogen bonds with protein surfaces, stabilizing them without inducing structural changes, while less hydroxylated fullerene particles are hydrophobic and can cause proteins to misfold. Geitner et al. [30] conducted DMD simulations of poly(amidoamine) dendrimers (PMAMs), a class of highly branched polymers, to determine differences in oil dispersion interactions between cationic, anionic, and neutral charged PMAMs with various hydrocarbons. Their results can be applied to inform design of polymers used in environmental cleanup, water purification, and drug delivery, among other industrial and biomedical applications.
Therapeutic development
Computational drug screening methods have long been employed to decrease the cost of drug development, but oversimplification of the docking process can result in high numbers of false negatives and false positives. Most docking methods match rigid bodies, while some generate ensembles of static conformations to imitate dynamic movement. Neither of these scenarios matches reality, where factors such as the entropy of binding, protein allostery, and synergistic dynamics between ligand and target can all affect drug binding. Proctor et al. [31] utilized many short replicate DMD simulations to obtain statistics of the residence time of a drug in the binding pocket in many different generated poses. The authors were able to identify the correct binding pose of the drug in “difficult” targets: those for which traditional computational drug screening methods could not do so. In a similar study, Dagliyan et al. [32] used DMD simulations to correctly identify native binding sites of peptides on their protein targets, as well as recapitulating native-like poses of the protein-peptide complex. The authors stressed the importance of protein target flexibility and dynamics as well as electrostatic interactions in identification of binding site and pose, highlighting the crucial use of ultrafast DMD simulation for this application.
Conclusions
As advanced technology has allowed us to make discoveries on larger scales and in finer detail, both experimentally and computationally, the nearly overwhelming complexity of biological processes and human diseases has become increasingly evident. Despite the explosion of revolutionary, “textbook-rewriting” discoveries in recent years, we are left with more questions, not fewer, regarding interactions between signaling pathways, drug resistance, the influence of the immune system on diverse tissues, and other phenomena beyond the molecular scale that nonetheless influence the environment in which molecules interact. While emerging fields such as systems biology and systems pharmacology are beginning to untangle signaling pathways on the cellular and tissue levels, the atomistic details of molecular interactions are critical to the elucidation of mechanism and the engineering of effective therapeutic strategies. The computational power of DMD is capable of providing these details on relevant time and length scales to real biological processes. The implicit solvent used with DMD provides an opportunity for parameterization to take into account the crowded cellular milieu, and the event-driven nature of the algorithms that advance the simulations are more appropriate to large, crowded systems than are continuous potentials with necessary integration over increasingly complex energetic potentials. In the near future, the combination of parallelization [7,33] and increasing processing power will likely make DMD the method of choice for the first atomistic cellular simulations and beyond.
Highlights.
Discrete Molecular Dynamics (DMD) accelerates simulation of large biomolecules
Increased sampling over MD allows microsecond simulations on a personal computer
DMD simulations are used in mechanistic, aggregation, and molecular design studies
As processing power increases, DMD applications can expand to the cellular level
Acknowledgments
N.V.D is supported by a grant from the National Institutes of Health (R01GM080742).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dokholyan NV, Buldyrev SV, Stanley HE, Shakhnovich EI. Discrete molecular dynamics studies of the folding of a protein-like model. Fold Des. 1998;3:577–87. doi: 10.1016/S1359-0278(98)00072-8. [DOI] [PubMed] [Google Scholar]
- 2.Zhou Y, Karplus M. Folding thermodynamics of a model three-helix-bundle protein. Proc Natl Acad Sci U A. 1997;94:14429–32. doi: 10.1073/pnas.94.26.14429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Proctor EA, Ding F, Dokholyan NV. Discrete molecular dynamics. Wiley Interdiscip Rev Comput Mol Sci. 2011;1:80–92. [Google Scholar]
- 4.Ding F, Dokholyan NV. Discrete Molecular Dynamics Simulation of Biomolecules. Computational Modeling of Biological Systems: From Molecules to Pathways. 2012:55–73. [Google Scholar]
- 5.Sfriso P, Emperador A, Gelpí JL, Orozco M. Computational Approaches to Protein Dynamics. CRC Press; 2015. Discrete Molecular Dynamics: Foundations and Biomolecular Applications; pp. 339–362. [Google Scholar]
- 6.Alder BJ, Wainwright TE. Studies in Molecular Dynamics.1. General Method. J Chem Phys. 1959;31:459–466. [Google Scholar]
- 7.Shirvanyants D, Ding F, Tsao D, Ramachandran S, Dokholyan NV. Discrete Molecular Dynamics: An Efficient And Versatile Simulation Method For Fine Protein Characterization. J Phys Chem B. 2012 doi: 10.1021/jp2114576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Ding F, Dokholyan NV. Fidelity of the protein structure reconstruction from inter-residue proximity constraints. J Phys Chem B. 2007;111:7432–8. doi: 10.1021/jp068963t. [DOI] [PubMed] [Google Scholar]
- 9.Gherghe CM, Leonard CW, Ding F, Dokholyan NV, Weeks KM. Native-like RNA tertiary structures using a sequence-encoded cleavage agent and refinement by discrete molecular dynamics. J Am Chem Soc. 2009;131:2541–6. doi: 10.1021/ja805460e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding F, Furukawa Y, Nukina N, Dokholyan NV. Local unfolding of Cu, Zn superoxide dismutase monomer determines the morphology of fibrillar aggregates. J Mol Biol. 2012;421:548–560. doi: 10.1016/j.jmb.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 12.Williams BI, Convertino M, Das J, Dokholyan NV. ApoE4-specific misfolded intermediate identified by molecular dynamics simulations. PLoS Comput Biol. doi: 10.1371/journal.pcbi.1004359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meral D, Urbanc B. Discrete molecular dynamics study of oligomer formation by N-terminally truncated amyloid β-protein. J Mol Biol. 2013;425:2260–2275. doi: 10.1016/j.jmb.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14•.Kimura S, Caldarini M, Broglia RA, Dokholyan NV, Tiana G. The maturation of HIV-1 protease precursor studied by discrete molecular dynamics. Proteins. 2014;82:633–639. doi: 10.1002/prot.24440. The authors observed the folding of HIV-1 protease monomers and their assembly into active dimers, finding that the precursor to the mature protein can form non-native dimers of natively folded monomers. Using DMD, this study achieves extensive sampling of a large system in order to observe the assembly of large protein chains, which would require global computing resources using traditional simulation methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konrad A, Lai J, Mutahir Z, Piškur J, Liberles DA. The phylogenetic distribution and evolution of enzymes within the thymidine kinase 2-like gene family in metazoa. J Mol Evol. 2014;78:202–216. doi: 10.1007/s00239-014-9611-6. [DOI] [PubMed] [Google Scholar]
- 16.Szöllősi D, Horváth T, Han K-H, Dokholyan NV, Tompa P, Kalmár L, Hegedűs T. Discrete molecular dynamics can predict helical prestructured motifs in disordered proteins. PloS One. 2014;9:e95795. doi: 10.1371/journal.pone.0095795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emperador A, Solernou A, Sfriso P, Pons C, Gelpi JL, Fernandez-Recio J, Orozco M. Efficient Relaxation of Protein–Protein Interfaces by Discrete Molecular Dynamics Simulations. J Chem Theory Comput. 2013;9:1222–1229. doi: 10.1021/ct301039e. [DOI] [PubMed] [Google Scholar]
- 18.Freeman TC, Black JL, Bray HG, Dagliyan O, Wu YI, Tripathy A, Dokholyan NV, Leisner TM, Parise LV. Identification of novel integrin binding partners for calcium and integrin binding protein 1 (CIB1): structural and thermodynamic basis of CIB1 promiscuity. Biochemistry. 2013;52:7082–7090. doi: 10.1021/bi400678y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19••.Sfriso P, Hospital A, Emperador A, Orozco M. Exploration of conformational transition pathways from coarse-grained simulations. Bioinforma Oxf Engl. 2013;29:1980–1986. doi: 10.1093/bioinformatics/btt324. The authors implemented coarse-grained DMD simulations to create a tool that can trace complex, non-linear conformational transitions in agreement with experiment without distortions in protein structure. The mapping of mechanistically accurate conformational changes involves transitions through meta-stable state, requiring extensive sampling, a strength of the DMD method, especially paired with a coarse-grained model, as in this study. [DOI] [PubMed] [Google Scholar]
- 20.Kota P, García-Caballero A, Dang H, Gentzsch M, Stutts MJ, Dokholyan NV. Energetic and structural basis for activation of the epithelial sodium channel by matriptase. Biochemistry. 2012;51:3460–3469. doi: 10.1021/bi2014773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21•.Proctor EA, Kota P, Aleksandrov AA, He L, Riordan JR, Dokholyan NV. Rational Coupled Dynamics Network Manipulation Rescues Disease-Relevant Mutant Cystic Fibrosis Transmembrane Conductance Regulator. Chem Sci R Soc Chem 2010. 2015;6:1237–1246. doi: 10.1039/c4sc01320d. The authors studied the dynamics of disease-causative mutants of the cystic fibrosis transmembrane conductance regulator (CFTR) using DMD. They discovered networks of correlated motion allowing transfer of aberrant dynamics from the mutation site to distal regions associated with ion channel function and stability. Stiffening of a “hotspot” residue located along the determined path of propagation by mutation to proline abolished the aberrant correlated dynamics and rescued CFTR function. This study established a method using DMD and network analysis for rational determination of the optimal sites in a protein to perturb (e.g., by mutation or drug binding) in order to restore native behavior. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hasan SS, Proctor EA, Yamashita E, Dokholyan NV, Cramer WA. Traffic within the cytochrome b6f lipoprotein complex: gating of the quinone portal. Biophys J. 2014;107:1620–1628. doi: 10.1016/j.bpj.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dagliyan O, Shirvanyants D, Karginov AV, Ding F, Fee L, Chandrasekaran SN, Freisinger CM, Smolen GA, Huttenlocher A, Hahn KM, et al. Rational design of a ligand-controlled protein conformational switch. Proc Natl Acad Sci U S A. 2013;110:6800–6804. doi: 10.1073/pnas.1218319110. The authors designed a single-chain protein conformational switch controlled by binding of the drug rapamycin. Using DMD simulations, the authors built a structural model of the desired kinase-switch-rapamycin complex to test and refine conformational switching ability. Inserted into conserved kinase allosteric sites, the stabilization of the protein conformational switch upon binding of rapamycin activates the kinase, offering a powerful tool for the study of signaling pathways in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karginov AV, Ding F, Kota P, Dokholyan NV, Hahn KM. Engineered allosteric activation of kinases in living cells. Nat Biotechnol. 2011;28:743–7. doi: 10.1038/nbt.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding F, Sharma S, Chalasani P, Demidov VV, Broude NE, Dokholyan NV. Ab initio RNA folding by discrete molecular dynamics: from structure prediction to folding mechanisms. RNA. 2008;14:1164–73. doi: 10.1261/rna.894608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsao D, Shabalina SA, Gauthier J, Dokholyan NV, Diatchenko L. Disruptive mRNA folding increases translational efficiency of catechol-O-methyltransferase variant. Nucleic Acids Res. 2011;39:6201–6212. doi: 10.1093/nar/gkr165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding F, Lavender CA, Weeks KM, Dokholyan NV. Three-dimensional RNA structure refinement by hydroxyl radical probing. Nat Methods. 2012;9:603–8. doi: 10.1038/nmeth.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cole DI, Legassie JD, Bonifacio LN, Sekaran VG, Ding F, Dokholyan NV, Jarstfer MB. New models of Tetrahymena telomerase RNA from experimentally derived constraints and modeling. J Am Chem Soc. 2012;134:20070–20080. doi: 10.1021/ja305636u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29•.Radic S, Nedumpully-Govindan P, Chen R, Salonen E, Brown JM, Ke PC, Ding F. Effect of fullerenol surface chemistry on nanoparticle binding-induced protein misfolding. Nanoscale. 2014;6:8340–8349. doi: 10.1039/c4nr01544d. The authors utilized DMD simulations to illustrate the effects of surface chemistry on the binding of nanoparticles to their target proteins, demonstrating that highly hydroxylated fullerene nanoparticles form hydrogen bonds with protein surfaces, stabilizing them without inducing structural changes, while less hydroxylated fullerene particles are hydrophobic and can cause proteins to misfold. This study investigates binding properties of nanoparticles crucial to the safety of their potential role as revolutionary drug delivery vehicles, highlighting the potential of DMD for investigating novel behavior in multi-molecular interactions. [DOI] [PubMed] [Google Scholar]
- 30.Geitner NK, Wang B, Andorfer RE, Ladner DA, Ke PC, Ding F. Structure-function relationship of PAMAM dendrimers as robust oil dispersants. Environ Sci Technol. 2014;48:12868–12875. doi: 10.1021/es5038194. [DOI] [PubMed] [Google Scholar]
- 31.Proctor EA, Yin S, Tropsha A, Dokholyan NV. Discrete molecular dynamics distinguishes nativelike binding poses from decoys in difficult targets. Biophys J. 2012;102:144–51. doi: 10.1016/j.bpj.2011.11.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dagliyan O, Proctor EA, D’Auria KM, Ding F, Dokholyan NV. Structural and dynamic determinants of protein-peptide recognition. Structure. 2011;19:1837–45. doi: 10.1016/j.str.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khan MA, Herbordt MC. Parallel Discrete Molecular Dynamics Simulation With Speculation and In-Order Commitment. J Comput Phys. 2011;230:6563–6582. doi: 10.1016/j.jcp.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]