

Summary
AMP-activated protein kinase (AMPK) plays a critical role both in sensing and regulating cellular energy state. In experimental animals, its activation has been shown to reduce the risk of obesity and diabetes-related co-morbidities such as insulin resistance, the metabolic syndrome and atherosclerotic cardiovascular disease. However, in humans, AMPK activation alone often does not completely resolve these conditions. Thus, an improved understanding of AMPK action and regulation in metabolic and other diseases is needed. Herein, we provide a brief description of the enzymatic regulation of AMPK and review its role in maintaining energy homeostasis. We then discuss tissue-specific actions of AMPK that become distorted during such conditions as obesity, type 2 diabetes and certain cancers. Finally, we explore recent findings regarding the interactions of AMPK with mTORC1 and the lysosome and discuss how changes in these relationships during overnutrition may lead to AMPK dysfunction. A more thorough understanding of AMPK's molecular interactions during diseases of overnutrition may provide key insights for the development of AMPK-based combinatorial treatments for metabolic disease.
Keywords: AMPK, type 2 diabetes, insulin resistance, lysosome, mTORC1
1. Introduction
Escalating rates of obesity and its associated co-morbidities highlight the critical need for an improved understanding of energy metabolism during disease.[1] AMP-activated protein kinase (AMPK) is an enzyme that responds to decreases in cellular energy state by stimulating processes that generate ATP such as fatty acid oxidation and glucose transport, and inhibiting others that consume ATP such as fatty acid and protein synthesis.[2] AMPK also counteracts cellular abnormalities that commonly occur during obesity and diabetes such as endoplasmic reticulum (ER) stress, oxidative stress and inflammation. Evidence from experimental animals with obesity and/or diabetes and obese humans indicates that AMPK may become inhibited or dysfunctional, yet the mechanisms responsible for this are poorly understood.[3] What is clear is that the consequences of dysfunctional AMPK include an increased risk of insulin resistance, hypertension and cardiovascular disease (CVD) and possibly a predilection to certain cancers.[4-6] Finally, several endogenous factors have been identified that could contribute to such AMPK dysregulation. They include high concentrations of glucose, fatty acids such as palmitate, and glucocorticoids and a decreased concentration of adiponectin; the same factors that predispose individuals to metabolic diseases.[3, 7] However, the mechanisms by which AMPK is dysregulated are not clear and may vary among these factors and between different types of cells.
Conversely, AMPK is activated in multiple tissues by physical exercise, which has long been known to increase insulin sensitivity and decrease adiposity, hypertension, atherosclerotic CVD and certain cancers.[8, 9] In addition, highly-prescribed medications such as metformin and statins that have shown some efficacy in treating these conditions are, among their other actions, AMPK activators at least in certain tissues. Intriguingly these drugs do not completely ameliorate disease risk in all patients.[10] We propose that this could be attributed to metabolic disease induced-modifications of the pathways these drugs normally activate that make them less efficacious, as may be the case with AMPK activators.
We will briefly discuss how AMPK is regulated enzymatically and during exercise and review its regulation in different tissues during obesity, type 2 diabetes and cancer, metabolic conditions associated with chronic overnutrition. We will then explore recent findings describing its molecular interactions with mTORC1 and the lysosome and discuss how alterations of these relationships during overnutrition may contribute to AMPK dysregulation.
2. AMPK: Structure and Regulation
AMPK is a heterotrimer consisting of a catalytic subunit (α) and two regulatory subunits (β and γ) (Fig. 1). The existence of two α, two β and three γ subunit isoforms creates a diversity of trimeric complexes which likely contributes to the similar, but not always identical, actions of AMPK in different tissues of a single organism. Recent structural analyses have confirmed decades of experimental findings indicating that AMP, ADP and ATP regulate AMPK, as extensively reviewed by Hardie.[2] The γ subunit contains 4 CBS domains, 1 of which binds AMP constitutively, and two others on which AMP can displace ATP. When the AMP/ATP ratio is high (e.g. during glucose deprivation, hypoxia or exercise), AMP displaces ATP at these two sites (Fig. 1). In addition to this allosteric activation which moderately (2-10 fold) activates the enzyme, binding of AMP increases the susceptibility of the α subunit to phosphorylation at Thr172 by upstream kinases and decreases its accessibility to phosphatases. ADP can also activate AMPK by decreasing the accessibility of phosphorylated Thr172 to phosphatases.[2] Phosphorylation of Thr172 is not required for activity, but increases it nearly 100-fold, making it a more potent modulator of activity than allosteric activation alone. This has led to the use of p-AMPKThr172 as a surrogate indicator for AMPK activity. Depending upon the context, there are at least three protein kinases that may phosphorylate Thr172 and activate AMPK (Fig. 1). There are also many pharmaceutical and naturally-occurring activators of AMPK that operate through a variety of mechanisms, as previously reviewed.[2] Conversely, AMPK activity may be inhibited by several factors such as glycogen synthase kinase 3β (GSK3β), Akt[2] and possibly other molecules including protein kinase C (PKC) (Coughlan et al, unpublished) (Fig. 1).
Figure 1. Regulation of AMPK Activity.
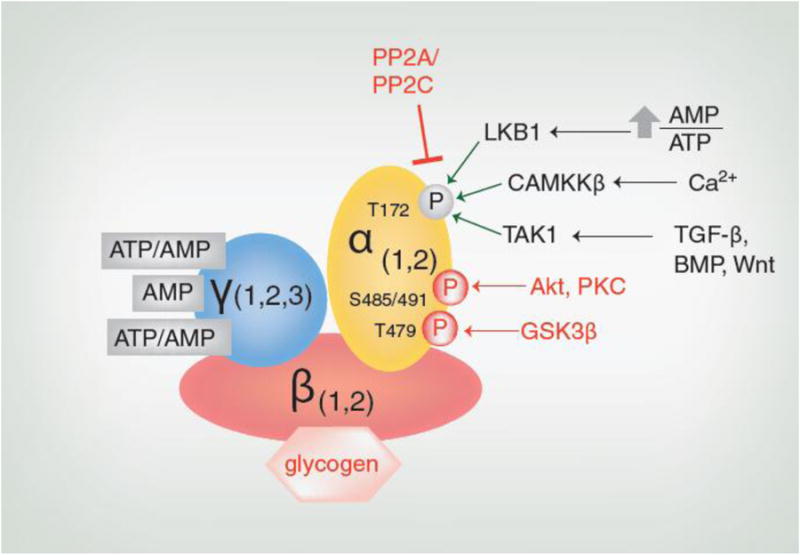
When the AMP/ATP ratio increases, tumor suppressor liver kinase B1 (LKB1) phosphorylates Thr172. Increases in intracellular calcium levels activate calcium calmodulin-dependent protein kinase kinase β (CAMKKβ), which also phosphorylates Thr172. Transforming growth factor-β activated kinase 1 (TAK1) phosphorylates AMPK at Thr172 in response to signaling from transforming growth factor-β (TGF-β), bone morphogenic protein (BMP) or Wnt. AMPK activity also depends on the activity of protein phosphatases such as PP2A and PP2C that target Thr172. In addition, AMPK activity may be inhibited by the binding of its β subunit to glycogen or the phosphorylation of its α subunit at Thr479 by glycogen synthase kinase 3β (GSK3β) or Ser485 (α1)/Ser491(α2) by Akt and possibly other molecules including protein kinase C (PKC). Factors which inhibit AMPK activity are shown in red.
3. AMPK in Exercise
It has long been established that regular exercise is beneficial for maintaining optimal health. Many studies have shown that it reduces the risk of developing type 2 diabetes, CVD and certain cancers.[8, 9] Studies in both humans and experimental animals suggest that such benefits are related to its ability to decrease insulin resistance, obesity and ectopic lipid deposition, and increase muscle capillary density, as previously reviewed.[3, 11, 12] Despite this, the cellular mechanisms responsible for these effects are incompletely understood.
One important insight into the cellular mechanisms by which exercise acts was the discovery in rats that both treadmill running and electrical stimulation of the hindlimb skeletal muscle activate AMPK.[13] Activation of AMPK and its upstream kinase LKB1 in skeletal muscle during exercise has been extensively confirmed in experimental animals and in humans.[11, 14] In this context, it has been hypothesized that activation of AMPK increases muscle glucose uptake and mitochondrial biogenesis, and decreases protein synthesis.[11] Whether all of these processes directly rely on AMPK remains to be determined. Data from muscle-specific AMPK knock-out mice currently suggest that AMPK is necessary for exercise-induced glucose uptake and efficient mitochondrial biogenesis,[15] but the involvement of factors that may be independent of AMPK (e.g. ERK1/2, PKC) in fatty acid oxidation in muscle (as is suggested by some animal and human studies[8]) remains to be clearly established.[16]
Given the important role of skeletal muscle in glucose disposal during exercise, dysfunction of skeletal muscle AMPK could impact the development of diabetes, other metabolic syndrome-associated disorders and insulin resistance. There is also a more comprehensive physiological benefit of exercise that involves a systemic response of AMPK. Experimental models have shown that exercise activates AMPK in adipose tissue, heart, liver, pancreas and aortic endothelium, as well as muscle and that this may contribute to its many benefits.[17-21] Interestingly, exercise-induced activation of AMPK is attenuated in patients with obesity or type 2 diabetes compared to healthy adults, suggesting that chronic metabolic disease may suppress AMPK activity and/or result in AMPK dysfunction.[22] In this context, whether the decrease in AMPK activity is a marker of the metabolic disease or a causative factor remains to be determined.
4. AMPK Dysfunction in Specific Tissues and in Cancer
4.1 Tissue-Specific Mechanisms of AMPK Action
4.1.1 Skeletal Muscle
During exercise, AMPK in skeletal muscle is activated which in turn leads to the phosphorylation and activation of Akt substrate of 160 kDa (AS160), TBC1 domain family member 1 (TBC1D1) and possibly nitric oxide synthase (NOS), all enzymes that are necessary for efficient GLUT4 translocation and glucose entry into the cell. AMPK also plays a significant role in enhancing insulin sensitivity following exercise. It is currently hypothesized that post-exercise, AMPK is necessary for TBC1D1 priming (by phosphorylation) that would allow for efficient secondary phosphorylation by Akt when the cells are stimulated by insulin.[15, 23] There is also speculation that AMPK-mediated stimulation of autophagy (a process for protein/organelle degradation) may stimulate glucose uptake in concert with sestrin2, a member of the sestrin family of stress-inducible proteins, although the underlying mechanism is unknown.[24] Fatty acid oxidation, which is necessary for constant ATP production during exercise, is stimulated by AMPK's phosphorylation of acetyl CoA carboxylase (ACC) which leads to decreases in malonyl CoA, and secondarily, increases in fatty acid oxidation.[12, 25] AMPK also stimulates mitochondrial biogenesis via phosphorylation and activation of PGC-1α.[26] For a more thorough analysis of the actions of AMPK in skeletal muscle metabolism, see ref [12].
Conversely, exposure of muscle to high concentrations of glucose or the branched chain amino acid leucine, such as occurs during diabetes or insulin resistance, causes AMPK activity to decline. Recent studies suggest that this may be mediated by multiple factors including AMPK phosphorylation at Ser485 by Akt and PKC and decreased activity of SIRT1, an indirect AMPK activator.[27, 28]
4.1.2 Liver
AMPK plays a complex role in regulating hepatic metabolism during fasting and re-feeding, as previously reviewed.[29] During starvation, AMPK activation decreases glycerolipid synthesis via diminishing glycerol-3-phosphate acyltransferase activity, and it inhibits the transcription factor SREBP-1c which diminishes the expression of lipogenic genes.[2, 29]
In systemic states characterized by insulin resistance, hepatic glucose output is typically increased. Conversely, one key mechanism by which metformin decreases insulin resistance is by inhibiting hepatic gluconeogenesis. Whether this is mediated specifically by activation of AMPK (which is suppressed during insulin resistance) is not clear.[30] In addition to metformin, salicylate, the active component of aspirin, has been shown to reduce hepatic lipid production and insulin resistance perhaps through its activation of AMPK in livers from healthy patients and those with dysglycemia.[31] Likewise, anti-diabetic thiazolidinediones such as rosiglitazone, appear to decrease hepatic glucose production at least in part by increasing adipose tissue production of adiponectin, which activates AMPK in liver as it does elsewhere.[32]
4.1.3 Endothelium
In endothelial cells, activation of AMPK inhibits ACC and fatty acid synthesis, but these cells are not major sites for the synthesis or storage of lipids. On the other hand, in the endothelium AMPK plays an important role in modulating oxidative stress and inflammation, processes directly related to the production of nitric oxide which governs vasodilation, one of its major functions. Mice expressing a constitutively active form of AMPK in the endothelium are protected against diabetes-induced impairment of vessel relaxation and carotid injury.[33] Conversely, homozygous deletion of the upstream kinase LKB1 in the endothelium reduces nitric oxide production and endothelial function, changes that are attenuated by overexpression of AMPK.[4]
During insulin resistance or type 2 diabetes, endothelial cells are continuously exposed to high concentrations of glucose and fatty acids, both of which have been shown to induce inflammatory, oxidative, and ER stresses. Such changes are associated with decreased activity of AMPK and many of them are attenuated when AMPK is activated by AICAR or exercise.[20, 34] In human aortic endothelial cells (HAECs), the same concentrations of glucose and palmitate that suppress AMPK activity and increase inflammation and apoptosis also impair basal (constitutive) autophagy. Autophagy is the process by which proteins or organelles are engulfed by a double-membraned autophagosome and subsequently degraded by lysosomal proteases. Although AMPK stimulates autophagy under low-nutrient conditions, activation of AMPK alone does not restore this process when cells are passaged in high concentrations of glucose and then acutely exposed to high concentrations of free fatty acids as might occur in the setting of diabetes. [7] Intriguingly, these data suggest that epigenetic changes are another potential mechanism that could lead to AMPK inhibition, impairment of basal autophagy and distortion of AMPK-mediated signaling in metabolic disease.
4.1.4 Adipose Tissue
In adipose tissue, fasting and exercise stimulate AMPK activity as do such hormones as leptin and adiponectin. Similar to skeletal muscle, AMPK activation in adipose tissue inhibits ACC and activates malonyl-CoA decarboxylase to decrease fatty acid synthesis and increase fatty acid oxidation. It is not clear how AMPK activation affects lipolysis in the adipocyte because data from AMPKα2 knockout mice suggest that AMPK inhibits lipolysis, whereas expression of dominant negative AMPKα1 in adipocytes has no effect, and exercise-induced AMPK activation in rats is associated with increased lipolysis.[17, 35, 36]
In humans who are insulin-resistant, AMPK activity is decreased and oxidative stress increased in subcutaneous, epiploic and omental adipose tissue. Compared to tissues from insulin-sensitive humans, expression of pro-inflammatory genes is higher and the AMPK activator SIRT1 is lower in some of these depots.[37] Although the cause of this AMPK inhibition is uncertain, levels of the AMPK activator adiponectin have been shown to decrease in patients with obesity or type 2 diabetes.[38]
Further evidence for an important role of dysregulation of adipose tissue AMPK in the pathology of obesity and diabetes is found in recent studies of bariatric surgery patients. Bariatric surgery, in particular the Roux-en-Y gastric bypass, greatly improves or resolves type 2 diabetes and reduces the risk for myocardial infarction and certain cancers. The mechanism underlying these benefits, independent of its effects on weight loss, is not clear. Recent evidence from subcutaneous adipose tissue of bariatric surgery patients suggests that AMPK may be involved in this response. Xu et al[39] found that three months post-surgery, reduction of BMI, increased insulin sensitivity and decreased plasma insulin levels were associated with several fold increases in AMPK activity and reduced oxidative stress in subcutaneous adipose tissue. Other post-operative events that could contribute to this improvement include increased adiponectin and decreases in both free fatty acid release from adipocyte lipid droplets and type 1 macrophage activation in the stromal vascular fraction.[3]
These snapshots are not the only examples of AMPK dysregulation during metabolic disease; however they are meant to illustrate how chronic overnutrition exerts systemic effects. In fact, AMPK dysfunction may play a role in the development of type 2 diabetes also through its actions in the pancreas and brain, as reviewed extensively by others.[2, 40] Although the correlations of AMPK activity with disease in each of these tissues does not distinguish whether AMPK dysfunction is a cause or result of pathology, a more thorough understanding of how AMPK regulation is altered during disease may help clarify its role.
4.2 Cancer
Regulation of AMPK in cancer is rather complicated, due to its role in multiple signaling pathways, some of which promote, and others which inhibit cancer cell development, as recently reviewed in [6]. For example, AMPK can inhibit hypoxia-inducible factor-a to antagonize the glycolytic metabolism of cancer cells.[41] On the other hand, it can also activate Unc-51-like kinase 1 (ULK1) to induce autophagy, a process that promotes cancer cell survival.[42] Thus, the effects of AMPK activation on cancer pathology may vary depending upon the stage and the type of cancer. Although experimental models have shown that AMPK activators such as metformin may be promising anti-cancer treatments, there is no consensus among epidemiological studies about the efficacy of metformin.[6] For this reason, many ongoing clinical trials are studying the effects of metformin either alone or in combination with other chemotherapeutic agents on a variety of cancers.
5. Deciphering AMPK's Role in Nutrient Sensing at the Molecular Level
Thus far, we have shown that both exercise-induced activation and disease-induced inhibition of AMPK can have important ramifications. For instance, diminished AMPK activity could lead to insulin resistance in numerous tissues, whereas increased AMPK activity could enhance insulin sensitivity. These tissue-specific actions likely contribute to AMPK's involvement in systemic regulation of metabolism, as recently reviewed.[43] Although there are several contributing factors to metabolic disease involving AMPK such as inflammation, oxidative stress and dysregulation of its interaction with SIRT1,[3] the remainder of this review will focus on what is known about a relatively more nascent topic: how specific stimuli involved in overnutrition (e.g. excess glucose or fatty acids) alter AMPK function. We will discuss the actions of AMPK in response to changes in nutrient status at the metabolic level and how it may be compromised by the excess nutrient environment of obesity and type 2 diabetes. A more thorough understanding of AMPK's role in nutrient sensing during overnutrition is crucial to the development of improved AMPK-based therapeutics for metabolic disease.
5.1 AMPK and mTORC1 in the Molecular Response to Starvation
Studies in many different cell lines and tissues indicate that adaptations to changes in nutrient availability are largely coordinated by AMPK and mammalian target of rapamycin complex 1 (mTORC1). Whereas AMPK activation during low-nutrient conditions stimulates catabolic and inhibits anabolic processes, mTORC1 activation under nutrient-replete conditions initiates anabolic processes such as protein synthesis and proliferation (Fig. 2).[44]
Figure 2. AMPK Signaling During Fed and Fasted States.

(A) In the fed state, insulin receptor signaling activates Akt which phosphorylates and inhibits the tuberous sclerosis complex (TSC1/TSC2). Inhibition of TSC1/TSC2 converts Ras homolog enriched in brain (Rheb) into its GTP-bound state, thus activating mTORC1. Akt can also directly phosphorylate and activate the mTORC1 complex and directly phosphorylate and inhibit AMPK (Fig. 1). Active mTORC1 inhibits autophagy by phosphorylating ULK1 at Ser757. (B) During starvation, increased levels of AMP activate AMPK which phosphorylates and activates TSC2. This promotes the formation of Rheb-GDP, thus decreasing mTORC1 activation and releasing its inhibition of ULK1. AMPK can directly inhibit mTORC1 activity by phosphorylating one of its components, Raptor. This, in conjunction with AMPK-induced phosphorylation (e.g. Ser555) of ULK1 activates ULK1 and stimulates autophagy.
Metabolism in the fed state is largely controlled by insulin activation of Akt, one of the major upstream regulators of mTORC1 (Fig. 2A). An active mTORC1 complex promotes cell growth and proliferation via 4EBP1, SREBP 1/2 and PPAR-γ.[44] In addition, it decreases energy-recycling processes such as autophagy, both by phosphorylating and inactivating ULK1 and transcription factor EB (TFEB) and by phosphorylating the death associated protein-1 complex.[45, 46] Akt has also been shown to inhibit AMPK activity by phosphorylating it at Ser485 (Fig. 1).[2] When Akt is active, hyperactivation of GSK3β can inhibit AMPK activity by phosphorylating it at another inhibitory site, Thr479 (Fig. 1).[47]
Under starvation conditions, an increased ratio of AMP/ATP activates AMPK which can subsequently inhibit mTORC1 activity via several of its upstream modulators (Fig. 2B).[48, 49] This releases mTORC1's inhibitory phosphorylation on ULK1 and allows autophagy to proceed.[46] Meanwhile, active AMPK can induce autophagy by directly phosphorylating and activating the ULK1 complex (Fig. 2B).[42, 50] Interestingly, active ULK1 has been shown to inactivate AMPK in vitro by phosphorylating all three of its subunits at a total of 9 phosphorylation sites.[51] Although this has not been confirmed in vivo, it suggests that inhibition of mTORC1 might indirectly inhibit AMPK via ULK1-mediated feedback.
Emerging evidence from mouse embryonic fibroblasts (MEFs), podocytes and HEK293T cells has identified a common location, the lysosome, as the site of activation for both mTORC1 and AMPK during the switch between starvation and nutrient repletion. The presence of certain amino acids and fatty acids induces mTORC1 localization to the lysosomal surface where it is activated by the v-ATPase/Ragulator complex.[52, 53] Active mTORC1 on the lysosomal membrane can then coordinate autophagy suppression by phosphorylating and retaining TFEB in the cytoplasm (Fig. 3A). Upon starvation or lysosomal stress, mTORC1 activity declines, allowing TFEB to migrate to the nucleus where it activates autophagic and lysosomal genes.[45]
Figure 3. mTORC1 and AMPK Activation on the Lysosome.
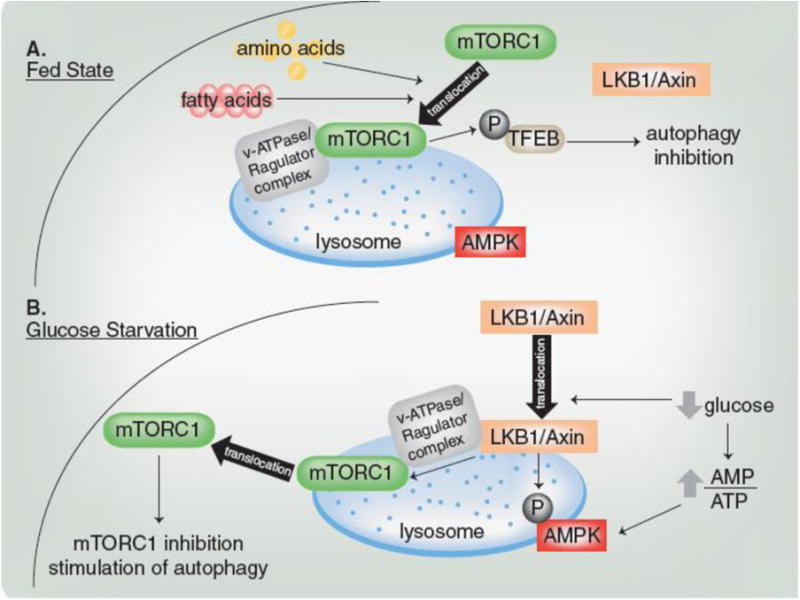
(A) In the fed state, mTORC1 is recruited to the v-ATPase/Ragulator complex on the lysosome and activated. Active mTORC1 phosphorylates TFEB to inhibit autophagy. (B) During glucose starvation, LKB1/Axin is recruited to the v-ATPase/Ragulator complex on the lysosome and both of these complexes activate AMPK. Lysosomal LKB1/Axin also causes mTORC1 to dissociate from the lysosome, which decreases its activity.
Under nutrient replete conditions, when mTORC1 is active at the lysosome, the kinase upstream of AMPK, LKB1, remains in the cytosol with its scaffolding protein Axin (Fig. 3A). A recent study in MEFs[54] demonstrated that glucose starvation relocalizes the LKB1/Axin complex to the v-ATPase/Ragulator complex on the lysosome, where a population of AMPK resides (Fig. 3B). Depletion of glucose increases AMPK's affinity for the LKB1/Axin complex, which phosphorylates AMPK (Fig. 3B). This phosphorylation, in conjunction with the binding of AMP and Ragulator induced-increased sensitivity of AMPK to AMP, results in AMPK activation (Fig. 3B). Binding of LKB1/Axin to v-ATPase/Ragulator also inhibits GEF-activity of the Ragulator complex, which causes mTORC1 to dissociate from the lysosome and decrease activity (Fig. 3B).[54] Thus, Ragulator activity is required for mTORC1 activation in the fed state[52, 53] and AMPK activation during glucose starvation.[54]
5.2 AMPK and mTORC1 in the Molecular Response to Overnutrition
In conditions associated with the metabolic syndrome such as insulin resistance, AMPK activity declines in a number of tissues. Changes in mTORC1 activity under the same circumstances are more complex owing to its intrinsic pathway of negative feedback, as reviewed by Laplante et al.[44] A robust literature of cell culture data regarding the regulation of AMPK and mTORC1 when nutrients are supplied to a starved system suggests that activation of these two kinases is reciprocal. However, the limited quantity of data describing the AMPK-mTORC1 relationship under conditions of chronic overnutrition, when excess nutrients are added to an already nutrient-replete system, suggests that the activity of these kinases is not always opposing. For example, in HAECs, nutrient excess conditions decrease AMPK activity but do not increase mTORC1 activity.[7] Impairment of lysosomal cathepsin activity in HAECs by excess nutrients,[7] coupled with evidence of mTORC1 and AMPK regulation on the lysosomal membrane in other contexts[54] suggests that the lysosome may be a key factor regulating mTORC1 and AMPK in HAECs.
5.3 Lysosomal Changes During Overnutrition: Consequences for mTORC1 and AMPK
In contrast to the catabolic-anabolic transition, changes in the mTORC1-AMPK relationship during chronic overnutrition are less defined. This may be due to a still primitive understanding of how diseases of overnutrition affect the lysosome and regulate mTORC1 and AMPK activity in this setting. Zhang et al[54] have shown that the lysosome is a crucial component of AMPK activation in the absence of glucose; however, the role of the lysosome in AMPK activation in the presence of excess nutrients is less clear. Nevertheless, lysosomal stress could potentially alter mTORC1 and AMPK localization on the lysosome, or indirectly alter their activity through accumulation of oxidative stress, inflammation or cholesterol.
Obesity, insulin resistance, type 2 diabetes, and its related complications are often associated with increased levels of oxidative stress and inflammatory mediators such as TNF-α that can cause an accumulation of cholesterol inside the lysosome. As previously reviewed,[55] excessive levels of lysosomal cholesterol in the macrophage can inhibit autophagy, activate the inflammasome, induce leakage of cathepsins and increase lysosomal membrane permeability, events that increase risk for lysosomal storage disorders and metabolic diseases such as atherosclerosis and non-alcoholic steatohepatitis. Accumulation of cholesterol or reactive oxygen species (ROS) has been shown to induce lysosomal permeabilization in a number of cell types,[56] but how such permeabilization affects recruitment and activation of mTORC1 and AMPK at the lysosome is not known. Also unknown is how diet-induced changes in lysosomal membrane composition affect these kinases. An analysis of mouse fibroblasts and livers found that consumption of diets rich in fat and cholesterol increases cholesterol and ceramide content in the lysosome membrane, which decreases chaperone-mediated autophagy.[57] Much of the current literature linking lysosomal stress with chronic disease has been carried out in macrophages, β cells, liver and endothelial cells and so it remains to be determined if similar findings occur in the adipose and muscle.
Consistent with the link between oxidative/inflammatory stress and lysosomal permeabilization, exposure to excess free fatty acids and glucose can destabilize lysosomes and increase lysosomal pH in HAECs and pancreatic β cells.[7, 58] Studies in HeLa cells, HEK293 cells and rat hepatocytes have also found that lysosomal pH is associated with mTORC1 activity, likely via TFEB.[45, 59, 60] In addition to genes directly involved in lysosome function, genomic analysis of HeLa cells indicated that TFEB regulates the expression of PRKAG2, the gene that encodes for AMPKγ2.[61] Thus, lysosomal stress may regulate AMPK via TFEB. In support of such a theory, stress induced by chloroquine (increases lysosomal pH) has been found to reduce AMPK activity in several cancer cell lines.[62] Similarly, treatment of C2C12 cells with the autophagy inhibitor 3-methyladenine suppressed phosphorylation of AMPK at Thr172.[24] However, lysosomal stress induced by bafilomycin (inhibits autophagosome-lysosome fusion, increases lysosomal pH) decreased mTORC1 activity but not AMPK activity.[59] Thus, AMPK may respond to slightly different types of lysosomal stress or lysomotropic drugs than mTORC1. Clearly, more research is needed to further our emerging understanding of how AMPK is affected by lysosomal stress. Whether AMPK can affect lysosomal health is also not known, but reduction of lysosomal pH with metformin suggests that the relationship between AMPK activation and lysosomal stress deserves further inquiry.[63]
Excess nutrient-induced increases in lysosomal pH can cause inactivation of lysosomal enzymes.[7, 58] Due to their roles in sphingolipid/ceramide metabolism, this inactivation can induce lysosomal ceramide accumulation. Inactivation of acid ceramidase (which converts ceramide to sphingosine in the lysosome) in pancreatic β cells and cardiomyocytes results in ceramide accumulation,[64] whereas its overexpression in C2C12 myotubes reduces free fatty acid-induced insulin resistance.[65] This suggests that accumulation of lysosomal ceramides may contribute to insulin resistance much like accumulation of total intracellular ceramides has been linked to insulin resistance.[66] Increased levels of total ceramides have been shown to inactivate AMPK through activation of protein phosphatase 2A[67] and possibly GSK3β[47, 68] in a variety of cell lines but whether the same applies to lysosomal ceramides is not clear. Interestingly, we found that overexpression of acid ceramidase in HAECs prevents excess nutrient-induced decreases in ULK1 phosphorylation at Ser555, an AMPK-targeted site.[7] This suggests that accumulation of lysosomal ceramides may play a role in deactivating AMPK.
Additional secondary effects of lysosomal storage diseases include macrophage activation and cytokine release, and increased oxidative stress; processes inversely related to AMPK activity.[69] Pro-inflammatory stimulation of macrophages decreases phosphorylation of Thr172 on AMPK. AMPKα1 knockout increases LPS-stimulated release of pro-inflammatory cytokines, whereas expression of constitutively active AMPK decreases the release of proinflammatory cytokines and increases the release of the anti-inflammatory cytokine IL-10.[70] AMPK is also a redox sensor that can reduce formation of ROS through its regulation of NADPH oxidase and NOS.[71] These data support the notion that one of the consequences of lysosomal stress-induced macrophage activation and oxidative stress may be suppression of AMPK. With the recent discovery of AMPK's regulation on the lysosome, the available literature has thus far shown correlations between AMPK activity and various aspects of lysosome function, from which we suggest potential molecular mechanisms. Importantly, whether the relationship between AMPK and lysosome function is causal remains to be shown.
6. Translational Potential of AMPK Therapies and Future Directions
Pharmaceuticals that target either a single or multiple mechanisms of AMPK activation[12] are invaluable tools in dissecting its regulation during health and disease. Recently, the combination of metformin with salicylate, two drugs which activate AMPK by distinct mechanisms, has been shown to cause greater AMPK activation and amelioration of insulin resistance than either drug alone[31], demonstrating the enormous potential of combination therapies. However, further optimization of AMPK-targeted treatments for metabolic disease in the clinic demands consideration of the molecular factors with which AMPK interacts. As discussed in this review, perhaps therapies that target AMPK and mTORC1 or lysosomes could be such a novel combination therapy.
Thus far, most therapies involving AMPK and mTORC1 in basic and clinical research target cancer. For example, in a prostate cancer mouse model, the combination of the mTORC1 inhibitor rapamycin and metformin was more effective than either agent alone in reducing prostate cancer progression and inflammation.[72] In keeping with this, an upcoming clinical trial will evaluate the efficacy of combining everolimus, another mTORC1 inhibitor, with metformin for the treatment of neuroendocrine pancreatic tumors.[73]
The use of AMPK-based combination therapies for the treatment of type 2 diabetes has not been as widely studied as that of cancer, but there is one ongoing trial, “A Pharmacodynamic Study of Sirolimus and Metformin in Patients With Advanced Solid Tumors,” in patients with solid tumors that will evaluate the efficacy of combining sirolimus (an mTORC1 inhibitor) with metformin. Several of the secondary outcomes include mTORC1, AMPK and insulin signaling in tumors, as well as insulin, glucose and triglyceride levels in blood.[74] These results are not yet available, but will provide some insight into how this drug combination may affect outcomes relevant to diabetes. Experimental models are also beginning to explore the effects of combining metformin with other compounds. For instance, in db/db mice, the combination of metformin with oleanolic acid, a glycogen phosphorylase inhibitor, improved insulin sensitivity and liver pathology, increased AMPK activity and decreased mTORC1 activity more than either treatment alone.[75]
Overnutrition and insulin resistance, which often lead to metabolic diseases such as type 2 diabetes, are associated with suppressed AMPK in many tissues. This exerts an array of effects that ultimately may contribute to systemic disease. Common among these tissues is the way in which individual cells respond to the stress of excess nutrients. Although much of this process remains to be clarified, a key component is the interaction between AMPK and mTORC1, coordinated by the lysosome. In this context, targeting AMPK along with mTORC1 and/or mediators of lysosomal stress may be an attractive approach for clinical reduction of insulin resistance and cellular stress (Fig. 4). While combining mTORC1 inhibitors with lysomotropic agents such as choloroquine may be beneficial for cancer treatment, the effects of such a combination (or one including an AMPK activator) on insulin resistance and its related pathologies remains to be determined.[76] Such a systemic treatment would be a powerful force to combat overnutrition and the metabolic diseases that follow, much like the proven benefits of exercise on overall health.
Figure 4. Therapeutic Potential for AMPK-Combination Therapies in Overnutrition.
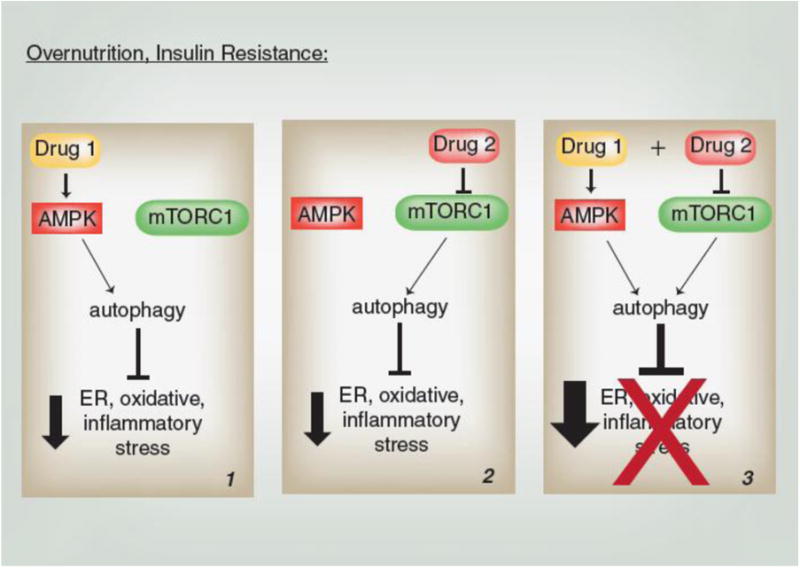
Autophagy is inversely associated with ER, oxidative and inflammatory stresses which increase risk for metabolic disease. Panels 1-3 use autophagy as an example to illustrate the potentially improved suppression of these stresses by targeting both AMPK and mTORC1.
Crucial to the development of AMPK-mTORC1-lysosome combination therapies is a more refined understanding of their relationship during disease (Fig. 5). Whether AMPK can be activated at the lysosome in the presence of excess nutrients, and if so, how the v-ATPase/Ragulator complex and mTORC1 may be involved is not known. Furthermore, the effect of lysosomal stress on AMPK is an under-explored topic, especially in tissues other than macrophages, but one that could provide key new insights into how oxidative stress and inflammation dysregulate AMPK. A clearer contextual understanding of AMPK in the stages that precede frank type 2 diabetes may also provide clues as to its regulation during other chronic diseases such as cancer and neurodegeneration.
Figure 5. Molecular Regulation of AMPK During Overnutrition.
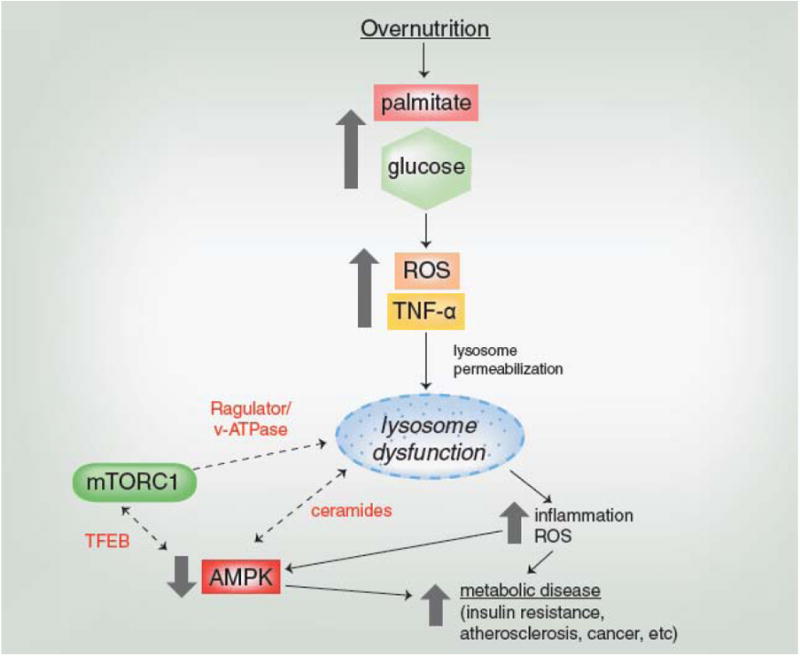
During overnutrition, excess glucose and palmitate induce oxidative and inflammatory stresses that permeabilize and damage the lysosome. It remains to be determined how the damaged lysosome and AMPK regulate one another, and how their interaction influences mTORC1 activity. Dotted arrows and red text indicate those relationships and factors that need further study and which may help clarify if and how lysosomal stress and suppression of AMPK lead to metabolic disease.
In summary, the health-promoting effects of AMPK activation can be observed during exercise, while impairment of AMPK activity is observed during metabolic diseases such as insulin resistance (Fig. 6). Investigation into how metabolic disease damages the lysosome and its interactions with AMPK and mTORC1 may not only improve our understanding of disease-associated AMPK dysregulation, but may provide insight for the development of new treatment combinations (Fig. 6).
Figure 6. Linking AMPK with exercise and metabolic disease.
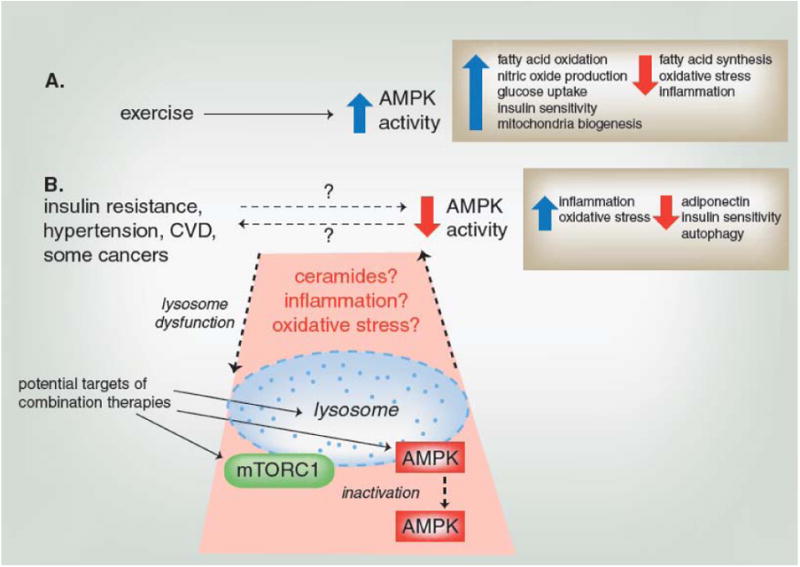
(A) Exercise has been shown to increase AMPK activity in several tissues, which can alter a number of processes (examples shown in box) to promote systemic health. (B) Metabolic diseases have been associated with decreased AMPK activity and other cellular stress indicators such as inflammation and oxidative stress (examples shown in box). While it is not clear whether decreased AMPK activity is a cause or result of metabolic disease, evaluation of disease-induced changes in the lysosome, a site of AMPK activation, may help clarify the role of AMPK. Lysosome dysfunction, such as that resulting from a change in membrane permeability or pH, may impair AMPK activation or disrupt its relationship with mTORC1 through accumulation of ceramides, inflammation or oxidative stress. The shaded area indicates those relationships that are not well-established and require further inquiry, but that may lead to new AMPK-based combination therapies for metabolic diseases.
Highlights.
AMPK is activated by nutrient depletion, anti-diabetic drugs and exercise.
AMPK becomes dysregulated in obesity, type 2 diabetes and certain cancers.
Lysosomal stress may contribute to AMPK inhibition during overnutrition.
Acknowledgments
We would like to acknowledge the Boston University Medical Campus Educational Media Center for their assistance in preparing the figures.
Funding: This work was supported by American Diabetes Association 7-11-MN-43 (NBR), NIH RO1 DK067509-06 (NBR), NIH R01 DK019514-34 (NBR), and Boston University (JMC). KAW was supported by NIH T32 Multidisciplinary Training in Cardiovascular Research HL007224 (VB).
Abbreviations
- ACC
acetyl CoA carboxylase
- AMPK
AMP-activated protein kinase
- AS160
Akt substrate of 160 kDa
- ER
endoplasmic reticulum
- GSK3β
glycogen synthase kinase 3β
- HAECs
human aortic endothelial cells
- LKB1
liver kinase B1
- MEF
mouse embryonic fibroblast
- NOS
nitric oxide synthase
- mTORC1
mammalian target of rapamycin complex 1
- PGC-1α
peroxisome proliferator-activated receptor gamma coactivator-1α
- PKC
protein kinase C
- Rheb
Ras homolog enriched in brain
- ROS
reactive oxygen species
- SREBP-1c
sterol regulatory element binding protein-1c
- TBC1D1
TBC1 domain family member 1
- TFEB
transcription factor EB
- TSC
tuberous sclerosis complex
- ULK1
Unc-51-like kinase 1
Footnotes
Conflict of interest: none
Contributions of Authors: KAW and JMC wrote the manuscript with editorial assistance from NBR.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.An R. Prevalence and Trends of Adult Obesity in the US, 1999-2012. ISRN Obes. 2014;2014:185132. doi: 10.1155/2014/185132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hardie DG. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol. 2015;33:1–7. doi: 10.1016/j.ceb.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. The Journal of clinical investigation. 2013;123:2764–72. doi: 10.1172/JCI67227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang W, Wang Q, Wu Y, Moriasi C, Liu Z, Dai X, et al. Endothelial cell-specific liver kinase B1 deletion causes endothelial dysfunction and hypertension in mice in vivo. Circulation. 2014;129:1428–39. doi: 10.1161/CIRCULATIONAHA.113.004146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinberg GR, O'Neill HM, Dzamko NL, Galic S, Naim T, Koopman R, et al. Whole body deletion of AMP-activated protein kinase {beta}2 reduces muscle AMPK activity and exercise capacity. J Biol Chem. 2010;285:37198–209. doi: 10.1074/jbc.M110.102434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zadra G, Batista JL, Loda M. Dissecting the Dual Role of AMPK in Cancer: From Experimental to Human Studies. Mol Cancer Res. 2015 doi: 10.1158/1541-7786.MCR-15-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weikel KA, Cacicedo JM, Ruderman NB, Ido Y. Glucose and palmitate uncouple AMPK from autophagy in human aortic endothelial cells. American journal of physiology Cell physiology. 2015;308:C249–63. doi: 10.1152/ajpcell.00265.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson AM, Church TS, Janssen I, Katzmarzyk PT, Earnest CP, Blair SN. Cardiorespiratory fitness as a predictor of cancer mortality among men with pre-diabetes and diabetes. Diabetes care. 2008;31:764–9. doi: 10.2337/dc07-1648. [DOI] [PubMed] [Google Scholar]
- 10.Fruchart JC, Sacks F, Hermans MP, Assmann G, Brown WV, Ceska R, et al. The Residual Risk Reduction Initiative: a call to action to reduce residual vascular risk in patients with dyslipidemia. The American journal of cardiology. 2008;102:1k–34k. doi: 10.1016/S0002-9149(08)01833-X. [DOI] [PubMed] [Google Scholar]
- 11.Richter EA, Ruderman NB. AMPK and the biochemistry of exercise: implications for human health and disease. Biochem J. 2009;418:261–75. doi: 10.1042/BJ20082055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinberg GR. Role of the AMP-activated protein kinase in regulating fatty acid metabolism during exercise. Appl Physiol Nutr Metab. 2009;34:315–22. doi: 10.1139/H09-009. [DOI] [PubMed] [Google Scholar]
- 13.Vavvas D, Apazidis A, Saha AK, Gamble J, Patel A, Kemp BE, et al. Contraction-induced changes in acetyl-CoA carboxylase and 5′-AMP-activated kinase in skeletal muscle. J Biol Chem. 1997;272:13255–61. doi: 10.1074/jbc.272.20.13255. [DOI] [PubMed] [Google Scholar]
- 14.Fujii N, Hayashi T, Hirshman MF, Smith JT, Habinowski SA, Kaijser L, et al. Exercise induces isoform-specific increase in 5′AMP-activated protein kinase activity in human skeletal muscle. Biochemical and biophysical research communications. 2000;273:1150–5. doi: 10.1006/bbrc.2000.3073. [DOI] [PubMed] [Google Scholar]
- 15.O'Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jorgensen SB, Schertzer JD, et al. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci U S A. 2011;108:16092–7. doi: 10.1073/pnas.1105062108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rockl KS, Witczak CA, Goodyear LJ. Signaling mechanisms in skeletal muscle: acute responses and chronic adaptations to exercise. IUBMB Life. 2008;60:145–53. doi: 10.1002/iub.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koh HJ, Hirshman MF, He H, Li Y, Manabe Y, Balschi JA, et al. Adrenaline is a critical mediator of acute exercise-induced AMP-activated protein kinase activation in adipocytes. Biochem J. 2007;403:473–81. doi: 10.1042/BJ20061479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coven DL, Hu X, Cong L, Bergeron R, Shulman GI, Hardie DG, et al. Physiological role of AMP-activated protein kinase in the heart: graded activation during exercise. American journal of physiology Endocrinology and metabolism. 2003;285:E629–36. doi: 10.1152/ajpendo.00171.2003. [DOI] [PubMed] [Google Scholar]
- 19.Park H, Kaushik VK, Constant S, Prentki M, Przybytkowski E, Ruderman NB, et al. Coordinate regulation of malonyl-CoA decarboxylase, sn-glycerol-3-phosphate acyltransferase, and acetyl-CoA carboxylase by AMP-activated protein kinase in rat tissues in response to exercise. J Biol Chem. 2002;277:32571–7. doi: 10.1074/jbc.M201692200. [DOI] [PubMed] [Google Scholar]
- 20.Cacicedo JM, Gauthier MS, Lebrasseur NK, Jasuja R, Ruderman NB, Ido Y. Acute exercise activates AMPK and eNOS in the mouse aorta. American journal of physiology Heart and circulatory physiology. 2011;301:H1255–65. doi: 10.1152/ajpheart.01279.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calegari VC, Zoppi CC, Rezende LF, Silveira LR, Carneiro EM, Boschero AC. Endurance training activates AMP-activated protein kinase, increases expression of uncoupling protein 2 and reduces insulin secretion from rat pancreatic islets. J Endocrinol. 2011;208:257–64. doi: 10.1530/JOE-10-0450. [DOI] [PubMed] [Google Scholar]
- 22.Sriwijitkamol A, Coletta DK, Wajcberg E, Balbontin GB, Reyna SM, Barrientes J, et al. Effect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: a time-course and dose-response study. Diabetes. 2007;56:836–48. doi: 10.2337/db06-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vichaiwong K, Purohit S, An D, Toyoda T, Jessen N, Hirshman MF, et al. Contraction regulates site-specific phosphorylation of TBC1D1 in skeletal muscle. Biochem J. 2010;431:311–20. doi: 10.1042/BJ20101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu X, Niu Y, Yuan H, Huang J, Fu L. AMPK binds to Sestrins and mediates the effect of exercise to increase insulin-sensitivity through autophagy. Metabolism. 2015;64:658–65. doi: 10.1016/j.metabol.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 25.Long YC, Zierath JR. Influence of AMP-activated protein kinase and calcineurin on metabolic networks in skeletal muscle. American journal of physiology Endocrinology and metabolism. 2008;295:E545–52. doi: 10.1152/ajpendo.90259.2008. [DOI] [PubMed] [Google Scholar]
- 26.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–22. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saha AK, Xu XJ, Lawson E, Deoliveira R, Brandon AE, Kraegen EW, et al. Downregulation of AMPK accompanies leucine- and glucose-induced increases in protein synthesis and insulin resistance in rat skeletal muscle. Diabetes. 2010;59:2426–34. doi: 10.2337/db09-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coughlan KA, Balon TW, Valentine RJ, Petrocelli R, Schultz V, Brandon A, et al. Nutrient Excess and AMPK Downregulation in Incubated Skeletal Muscle and Muscle of Glucose Infused Rats. PloS one. 2015;10:e0127388. doi: 10.1371/journal.pone.0127388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hasenour CM, Berglund ED, Wasserman DH. Emerging role of AMP-activated protein kinase in endocrine control of metabolism in the liver. Molecular and cellular endocrinology. 2013;366:152–62. doi: 10.1016/j.mce.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sliwinska A, Drzewoski J. Molecular action of metformin in hepatocytes: an updated insight. Curr Diabetes Rev. 2015;11:175–81. doi: 10.2174/1573399811666150325233108. [DOI] [PubMed] [Google Scholar]
- 31.Ford RJ, Fullerton MD, Pinkosky SL, Day EA, Scott JW, Oakhill JS, et al. Metformin and salicylate synergistically activate liver AMPK, inhibit lipogenesis and improve insulin sensitivity. Biochem J. 2015;468:125–32. doi: 10.1042/BJ20150125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nawrocki AR, Rajala MW, Tomas E, Pajvani UB, Saha AK, Trumbauer ME, et al. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J Biol Chem. 2006;281:2654–60. doi: 10.1074/jbc.M505311200. [DOI] [PubMed] [Google Scholar]
- 33.Li FY, Lam KS, Tse HF, Chen C, Wang Y, Vanhoutte PM, et al. Endothelium-selective activation of AMP-activated protein kinase prevents diabetes mellitus-induced impairment in vascular function and reendothelialization via induction of heme oxygenase-1 in mice. Circulation. 2012;126:1267–77. doi: 10.1161/CIRCULATIONAHA.112.108159. [DOI] [PubMed] [Google Scholar]
- 34.Cacicedo JM, Yagihashi N, Keaney JF, Jr, Ruderman NB, Ido Y. AMPK inhibits fatty acid-induced increases in NF-kappaB transactivation in cultured human umbilical vein endothelial cells. Biochemical and biophysical research communications. 2004;324:1204–9. doi: 10.1016/j.bbrc.2004.09.177. [DOI] [PubMed] [Google Scholar]
- 35.Villena JA, Viollet B, Andreelli F, Kahn A, Vaulont S, Sul HS. Induced adiposity and adipocyte hypertrophy in mice lacking the AMP-activated protein kinase-alpha2 subunit. Diabetes. 2004;53:2242–9. doi: 10.2337/diabetes.53.9.2242. [DOI] [PubMed] [Google Scholar]
- 36.Chakrabarti P, English T, Karki S, Qiang L, Tao R, Kim J, et al. SIRT1 controls lipolysis in adipocytes via FOXO1-mediated expression of ATGL. J Lipid Res. 2011;52:1693–701. doi: 10.1194/jlr.M014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu XJ, Gauthier MS, Hess DT, Apovian CM, Cacicedo JM, Gokce N, et al. Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot-specific changes in gene expression in adipose tissue. J Lipid Res. 2012;53:792–801. doi: 10.1194/jlr.P022905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arteriosclerosis, thrombosis, and vascular biology. 2000;20:1595–9. doi: 10.1161/01.atv.20.6.1595. [DOI] [PubMed] [Google Scholar]
- 39.Xu XJ, Apovian C, Hess D, Carmine B, Ruderman N. Improved insulin sensitivity 3 months after RYGB surgery is associated with increased subcutaneous adipose tissue AMPK activity and decreased oxidative stress. Diabetes. 2015 doi: 10.2337/db14-1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fu A, Eberhard CE, Screaton RA. Role of AMPK in pancreatic beta cell function. Molecular and cellular endocrinology. 2013;366:127–34. doi: 10.1016/j.mce.2012.06.020. [DOI] [PubMed] [Google Scholar]
- 41.Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell metabolism. 2013;17:113–24. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–61. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Huynh T, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. Embo J. 2012;31:1095–108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–91. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suzuki T, Bridges D, Nakada D, Skiniotis G, Morrison SJ, Lin JD, et al. Inhibition of AMPK catabolic action by GSK3. Mol Cell. 2013;50:407–19. doi: 10.1016/j.molcel.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 49.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Loffler AS, Alers S, Dieterle AM, Keppeler H, Franz-Wachtel M, Kundu M, et al. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy. 2011;7:696–706. doi: 10.4161/auto.7.7.15451. [DOI] [PubMed] [Google Scholar]
- 52.Averous J, Lambert-Langlais S, Carraro V, Gourbeyre O, Parry L, B'Chir W, et al. Requirement for lysosomal localization of mTOR for its activation differs between leucine and other amino acids. Cellular signalling. 2014;26:1918–27. doi: 10.1016/j.cellsig.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 53.Yasuda M, Tanaka Y, Kume S, Morita Y, Chin-Kanasaki M, Araki H, et al. Fatty acids are novel nutrient factors to regulate mTORC1 lysosomal localization and apoptosis in podocytes. Biochim Biophys Acta. 2014;1842:1097–108. doi: 10.1016/j.bbadis.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 54.Zhang CS, Jiang B, Li M, Zhu M, Peng Y, Zhang YL, et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell metabolism. 2014;20:526–40. doi: 10.1016/j.cmet.2014.06.014. [DOI] [PubMed] [Google Scholar]
- 55.Hendrikx T, Walenbergh SM, Hofker MH, Shiri-Sverdlov R. Lysosomal cholesterol accumulation: driver on the road to inflammation during atherosclerosis and non-alcoholic steatohepatitis. Obesity reviews : an official journal of the International Association for the Study of Obesity. 2014;15:424–33. doi: 10.1111/obr.12159. [DOI] [PubMed] [Google Scholar]
- 56.Aits S, Jaattela M. Lysosomal cell death at a glance. Journal of cell science. 2013;126:1905–12. doi: 10.1242/jcs.091181. [DOI] [PubMed] [Google Scholar]
- 57.Rodriguez-Navarro JA, Kaushik S, Koga H, Dall'Armi C, Shui G, Wenk MR, et al. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc Natl Acad Sci U S A. 2012;109:E705–14. doi: 10.1073/pnas.1113036109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Las G, Serada SB, Wikstrom JD, Twig G, Shirihai OS. Fatty acids suppress autophagic turnover in beta-cells. J Biol Chem. 2011;286:42534–44. doi: 10.1074/jbc.M111.242412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu Y, Parmar A, Roux E, Balbis A, Dumas V, Chevalier S, et al. Epidermal growth factor-induced vacuolar (H+)-atpase assembly: a role in signaling via mTORC1 activation. J Biol Chem. 2012;287:26409–22. doi: 10.1074/jbc.M112.352229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li M, Khambu B, Zhang H, Kang JH, Chen X, Chen D, et al. Suppression of lysosome function induces autophagy via a feedback down-regulation of MTOR complex 1 (MTORC1) activity. J Biol Chem. 2013;288:35769–80. doi: 10.1074/jbc.M113.511212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20:3852–66. doi: 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- 62.Harhaji-Trajkovic L, Arsikin K, Kravic-Stevovic T, Petricevic S, Tovilovic G, Pantovic A, et al. Chloroquine-mediated lysosomal dysfunction enhances the anticancer effect of nutrient deprivation. Pharm Res. 2012;29:2249–63. doi: 10.1007/s11095-012-0753-1. [DOI] [PubMed] [Google Scholar]
- 63.Labuzek K, Liber S, Gabryel B, Adamczyk J, Okopien B. Metformin increases phagocytosis and acidifies lysosomal/endosomal compartments in AMPK-dependent manner in rat primary microglia. Naunyn Schmiedebergs Arch Pharmacol. 2010;381:171–86. doi: 10.1007/s00210-009-0477-x. [DOI] [PubMed] [Google Scholar]
- 64.Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nature medicine. 2011;17:55–63. doi: 10.1038/nm.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chavez JA, Holland WL, Bar J, Sandhoff K, Summers SA. Acid ceramidase overexpression prevents the inhibitory effects of saturated fatty acids on insulin signaling. J Biol Chem. 2005;280:20148–53. doi: 10.1074/jbc.M412769200. [DOI] [PubMed] [Google Scholar]
- 66.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell metabolism. 2007;5:167–79. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 67.Wu Y, Song P, Xu J, Zhang M, Zou MH. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J Biol Chem. 2007;282:9777–88. doi: 10.1074/jbc.M608310200. [DOI] [PubMed] [Google Scholar]
- 68.Lin CF, Chen CL, Chiang CW, Jan MS, Huang WC, Lin YS. GSK-3beta acts downstream of PP2A and the PI 3-kinase-Akt pathway, and upstream of caspase-2 in ceramide-induced mitochondrial apoptosis. Journal of cell science. 2007;120:2935–43. doi: 10.1242/jcs.03473. [DOI] [PubMed] [Google Scholar]
- 69.Vitner EB, Platt FM, Futerman AH. Common and uncommon pathogenic cascades in lysosomal storage diseases. J Biol Chem. 2010;285:20423–7. doi: 10.1074/jbc.R110.134452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sag D, Carling D, Stout RD, Suttles J. Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol. 2008;181:8633–41. doi: 10.4049/jimmunol.181.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shirwany NA, Zou MH. AMPK: a cellular metabolic and redox sensor. A minireview. Front Biosci (Landmark Ed) 2014;19:447–74. doi: 10.2741/4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Saha A, Blando J, Tremmel L, DiGiovanni J. Effect of Metformin, Rapamycin, and Their Combination on Growth and Progression of Prostate Tumors in HiMyc Mice. Cancer Prev Res (Phila) 2015;8:597–606. doi: 10.1158/1940-6207.CAPR-15-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pusceddu S, de Braud F, Concas L, Bregant C, Leuzzi L, Formisano B, et al. Rationale and protocol of the MetNET-1 trial, a prospective, single center, phase II study to evaluate the activity and safety of everolimus in combination with octreotide LAR and metformin in patients with advanced pancreatic neuroendocrine tumors. Tumori. 2014;100:e286–9. doi: 10.1700/1778.19298. [DOI] [PubMed] [Google Scholar]
- 74.Chicago Uo. A Pharmacodynamic Study of Sirolimus and Metformin in Patients With Advanced Solid Tumors. 2015 doi: 10.1007/s00280-018-3619-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang X, Chen Y, Abdelkader D, Hassan W, Sun H, Liu J. Combination therapy with oleanolic acid and metformin as a synergistic treatment for diabetes. J Diabetes Res. 2015;2015:973287. doi: 10.1155/2015/973287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hugle M, Fulda S. Dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 synergizes with chloroquine to induce apoptosis in embryonal rhabdomyosarcoma. Cancer Lett. 2015;360:1–9. doi: 10.1016/j.canlet.2014.12.016. [DOI] [PubMed] [Google Scholar]