

Abstract
Objectives:
An established theory for the pathogenesis of tardive dyskinesia is disturbed dopaminergic receptor sensitivity and/or dopaminergic intracellular signaling. We examined associations between genetic variants of neurotransmitter receptors and tardive dyskinesia.
Methods:
We assessed tardive dyskinesia in Caucasian psychiatric inpatients from Siberia (N = 431) and a long-stay population from the Netherlands (N = 168). These patients were genotyped for 43 tag single nucleotide polymorphisms in five neurotransmitter receptor genes, and the results for the two populations were compared.
Results:
Several significant associations with tardive dyskinesia were identified, but only GRIN2A (rs1345423) was found in both patient populations. This lack of agreement was probably due to the small effect size of the associations, the multiple testing and the small sample size of the Dutch patient population. After reviewing the literature, we propose that the constitutive stimulatory activity of serotonergic type 2 receptors may be relevant.
Conclusions:
Inactivity of the serotonergic, type 2C receptor or blockade of these receptors by atypical antipsychotic drugs may decrease the vulnerability to develop tardive dyskinesia.
Keywords: Tardive dyskinesia mechanism, dopaminergic receptor, serotonergic receptor, glutamatergic receptor, NMDA receptor, genetic variants
Introduction
Dyskinesia is a movement disorder characterized by involuntary, repetitive, and irregular motions that affect the mouth and face and/or the limbs and trunk.1 Tardive dyskinesia (TD) is a well-known complication of long-term treatment with dopamine (DA) blocking agents, predominantly antipsychotic drugs.2 The prevalence of TD can vary, because it depends on the influence of several risk factors like, for example, age, type, dose, and duration of antipsychotic drug usage, diagnosis, non-therapeutic risk factors, and plasma level related pharmacogenetic risk factors.3 For over six decades, antipsychotic drugs have remained the mainstay of schizophrenia treatment. Schizophrenia is a heterogeneous psychiatric disorder, with a lifetime prevalence of about 1%. TD in schizophrenic patients is associated with poor outcome and TD patients may suffer from more physical or psychological problems.3 The pathophysiological mechanism of psychotic symptoms in schizophrenia is partly explained by an increased effect of dopaminergic neurotransmission.4,5 This theory is based on the therapeutic effects of antipsychotic drugs, which are all DA receptor antagonists.6 The development of TD is also associated with dysregulation within the dopaminergic system, and is thought to be based on a structural change induced by the long-term blockade of DA receptors.7,8 These findings may explain why dopaminergic dysregulation in patients with schizophrenia is related to psychotic symptoms as well as spontaneous dyskinesia, for example, drug-naïve first-episode patients experience spontaneous dyskinesia more frequently than healthy controls.3
It is thought that TD is primarily related to DA dysregulation. Antipsychotic treatment with both serotonergic 5-HT2A (HTR2A) and 5-HT2C (HTR2C) antagonists resulted in a lower annual incidence of TD.9,10 This finding led to the hypothesis that HTR2A and HTR2C activate gamma-aminobutyric acid (GABA)ergic interneurons,11 within the cerebral cortex, basal ganglia, and mesencephalic areas, resulting in decreased dopaminergic activity. Hence, HTR2A and HTR2C antagonism increases DA release.12 Schizophrenia has substantial hereditary components and is associated with dopaminergic dysfunction; therefore, genes related to the dopaminergic neurotransmitter system are attractive candidates for studying its genetic basis.4,13 To a lesser extent, this is also true for the pathogenesis of dyskinesia.3,14 Therefore, numerous investigations have examined the association between genetic variants of DA enzymes and receptors when symptoms of dyskinesia are present. However, the results are inconsistent and even meta-analyses are not conclusive (see discussion).
In this report, we present new data on the association between genetic variants of neurotransmitter receptors and TD. We compared the results of genotyping for 43 tag single nucleotide polymorphisms (SNPs) in five receptors in two independent patient populations and related the findings with the prevalence of TD. Our results do not differ much from those found in relevant meta-analyses, which led us to propose that genetically determined variations of DA type 2 receptors and/or DA type 2 receptor signaling are unlikely to affect the sensitivity to TD. We suggest that a mechanism other than HTR2-induced augmentation of DA release is responsible for explaining the lower incidence of TD after treatment with atypical antipsychotics.
Materials and methods
Patients
The work described in this article was carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki 1975, revised in Fortaleza, Brazil, 2013) for experiments involving humans. The majority of the patients in this study were retrieved from three psychiatric hospitals: Tomsk Clinical Psychiatric Hospital, Kemerovo Regional Clinical Psychiatric Hospital, and Chita Regional Psychiatric Hospital in Siberia. Written informed consent was obtained from each subject after obtaining approval of the study protocol from the institutional bioethics committee. None of the participants had a compromised capacity/ability to consent; hence, consent from the next of kin was not necessary and not recommended by the local ethics committee. The inclusion criterion was a clinical diagnosis of schizophrenia or schizotypal disorder (according to International Classification of Diseases (ICD)-10: F20 and F21, respectively). Exclusion criteria were as follows: non-Caucasian physical appearance (e.g. Mongoloid, Buryats, or Khakassians), use of clozapine with no TD (clozapine may ameliorate TD), relevant pharmacological withdrawal symptoms, and organic brain diseases. Details on the diagnosis and clinical features for the Russian and Dutch populations were previously published.15,16 To compare antipsychotic medications, all dosages were converted into chlorpromazine (CPZ) equivalents.17 The Abnormal Involuntary Movement Scale (AIMS) was used to assess TD.1,18 The AIMS scores were transformed into a binary variable (presence or absence of dyskinesia) according to the criteria of Schooler and Kane.19 In addition, a blood sample was obtained for DNA isolation and genotyping. All genotyping was performed without knowledge of the patient’s clinical status.
DNA analysis
The DNA extraction and Veracode Assay were conducted according to standard protocols.15 We selected a subset of 43 informative SNPs, or tag SNPs, that would accurately represent the majority of SNPs for five neurotransmitter genes: the glutamate receptor, ionotropic, N-methyl D-aspartate 2A (GRIN2A) gene, glutamate receptor, ionotropic, N-methyl D-aspartate 2B (GRIN2B) gene, DRD3 gene, HTR2C gene, and DRD4 gene. The selection method was previously described by Xu and Taylor (freely available at http://snpinfo.niehs.nih.gov). The following criteria have been applied for the selection of the tag SNPs: localization in the gene including 1000 bp upstream and downstream (5′ and 3′ flanking regions), linkage disequilibrium (LD) threshold = 0.8, minor allele frequency (MAF) ⩾ 0.1, maximal distance between SNPs for calculation of LD = 250,000 bp, Genotype data = “European” (dbSNP). We selected only tag SNPs that captured at least 10 SNPs.
Statistical analysis
Different strategies were developed to account for missing data and interactions between different SNPs. Classical logistic regression and a log-linear regression approach were used to analyze the data. The Hardy–Weinberg equilibrium (HWE) of genotypic frequencies was checked by the chi-square test. For the SNPs in the X-chromosomal HTR2C gene, deviation from HWE was not calculated. The genotype prevalence of all polymorphisms was in agreement with HWE in the patient groups except for rs11866328 and rs7196095 in the GRIN2A gene and rs963468 and rs9817063 in the DRD3 gene. All calculations were performed in the R statistical environment with the SNPassoc package and basic R functions.20 The allele prevalence was first calculated separately in patients with and without dyskinesia to define the percentage of missing genotypes. The HWE test was applied with Fisher’s exact test to groups. This showed that one SNP, rs12858300, was monomorphic; therefore, it was removed from further analysis. To analyze associations between the SNPs and the phenotypes, we used logistic regression for binary response traits and log-linear regression for continuous traits. The following genetic models were tested:
Co-dominant. Both alleles of a SNP influenced the phenotype.
Dominant. Rare allele homo- and heterozygotes were tested against common allele homozygotes.
Recessive. Common allele homo- and heterozygotes were tested against rare allele homozygotes.
Over-dominant. Heterozygotes were tested against both homozygote alleles.
Log-additive. A trend test for the genotypes, similar to the allele model, but comparisons were among subjects (N) instead of chromosomes (2N). The test and estimates were based on a logistic regression model that coded the genotypes as 0, 1, or 2 to reflect the number of minor alleles.
The statistical significance of a SNP was established with a likelihood-ratio test that compared the effect of a polymorphism with the null model (only including the intercept). The Akaike information criterion was applied to identify the model that best fits the data. The SNP effects were quantified using the odds ratio (OR) and 95% confidence intervals (CIs). In the result tables, an OR for a log-additive model corresponds to an association between a rare allele and the presence of dyskinesia. The ORs for other models correspond to associations between the presence of dyskinesia and rare allele homo- and heterozygotes (dominant), common allele homo- or heterozygotes (recessive) or heterozygotes (over-dominant).
Dyskinesia was considered a qualitative trait (yes/no), but the degree of dyskinesia expression was analyzed as a quantitative trait. To avoid 0 values, 1 was added to the dyskinesia expression values, and then they were log2-transformed to obtain a log-normal distribution. There were many confounding factors in patients with schizophrenia, including gender, age, duration of disease, use of anticholinergic medicine, dose of anticholinergic medicine, and total CPZ equivalents on the day of assessment. Only gender and total CPZ equivalents were determined to be insignificant. The genetic models were adjusted for all the significant confounding factors.
The statistical power was estimated based on the prevalence of the tested phenotypes in the Siberian sample and assumptions of complete penetrance and disease allele prevalence of 0.2–0.3. For orofacial dyskinesia, the power estimates to detect associations with p-value <0.05 varied between 14%–50% for models assuming OR of 1.5; 44%–94% for models assuming OR of 2.0; and 78%–99% for models assuming OR of 2.5. For limb-truncal dyskinesia, the power estimates varied between 10%–31%, 27%–75%, and 53%–95% for these three OR values, respectively. For either type of dyskinesia, the power estimates varied between 18%–65%, 58%–99%, and 91%–99%. Overall, our study was reasonably powered to detect clinically relevant associations with an OR as little as 2.0.
Comparisons
For comparisons, we used the population described by Bakker et al.16 They included a group of 168 Caucasian patients (M/F ratio: 96/72) who were representative of a population with the most severe chronic mental illness, requiring long-stay care at a psychiatric institution. The DNA of these patients was analyzed in parallel with our patients’ samples and the results were provided to Bakker and colleagues. Genotype and allele frequency comparisons were performed using a multilevel random regression method with three measurement occasions (baseline and two follow-ups) for continuous TD and other movement disorders.
Results
Of the 431 Siberian patients (269 males/162 females) included, 401 (95.1%) had schizophrenia. The mean age was 41.5 ± 15.3 years and the average duration of illness was 16.4 ± 13.5 years. The average antipsychotic dosage (± standard deviation (SD)) at assessment was 717 ± 770.3 CPZ mg equivalents per day. The 168 Dutch patients were older (48.8 ± 12.4 years) and were admitted to the hospital for an average of 23.4 ± 12.9 years. In addition, this group was less homogenous with respect to disease characteristics and included, according to the Diagnostic and Statistical Manual of Mental Disorders 4th Edition (DSM-IV) Axis I, patients with schizophrenia (N = 112), psychosis (N = 9), affective disorders (N = 27), another Axis I diagnosis (N = 11), and no Axis I diagnosis (N = 9).
The associations between SNPs and orofacial TD (TDof) are shown in Table 1 for the Dutch and Siberian patient populations. Significant associations were found between eight SNPs encoding four receptors (GRIN2B, GRIN2A, DRD3, and HTR2C) and TDof. However, these SNPs were not the same for the two groups. The associations between SNPs and limb-truncal TD (TDlt) are displayed in Table 2 for the Dutch and Siberian patients. Significant associations were found between nine SNPs of two receptors (GRIN2A and DRD3) and TDlt. Yet, only rs1345423 in GRIN2A was significantly associated with TDlt in both patient groups: Dutch patients (B = −0.18, P = 0.0190) and Siberian patients (TD expression level, sum of scores for five to seven signs: P = 0.02504). The associations between SNPs and overall TD are displayed in Table 3 for the Dutch and Siberian patient populations. Significant associations were found in seven specific SNPs encoding three receptors (GRIN2B, GRIN2A, and DRD3). Only rs1345423 in GRIN2A was significantly associated with overall TD in both studied groups.
Table 1.
Associations between specific SNPs and orofacial TD in the Dutch and Siberian schizophrenic populations.
| Receptor | SNP | Dutch population |
Siberian schizophrenic population |
|
|---|---|---|---|---|
| TDof (B, P value) | TDof (OR (95% CI), P value) | TD expression level: sum of scores for 1–4 signs (P valuea) | ||
| GRIN2B | rs220599 | X | X | P = 0.04451 |
| GRIN2A | rs7192557 | B = 0.22, P = 0.0291 | X | X |
| rs1650420 | B = 0.16, P = 0.0336 | X | X | |
| rs11646587 | X | OR = 0.62 (0.38–1.00), P = 0.04652 | X | |
| rs7206256 | X | OR = 1.73 (1.02–2.91), P = 0.03570 | X | |
| rs1345423 | X | OR = 0.35 (0.14–0.86), P = 0.01043 | P = 0.027865 | |
| DRD3 | rs167770 | X | P = 0.01751 | X |
| HTR2C | rs4911871 | B = −0.18, P = 0.0131 | X | X |
TDof: orofacial tardive dyskinesia; OR: odds ratio; 95% CI: 95% confidence interval.
B = regression coefficient, P = chance, sum of scores = scores of AIMS scale.
Original value + 1, followed by log2-transformation.
Table 2.
Associations between specific SNPs and TDlt in the Dutch and Siberian schizophrenic populations.
| Receptor | SNP | Dutch population |
Siberian schizophrenic population |
|
|---|---|---|---|---|
| TDlt (B, P value) | TDlt (OR (95% CI), P value) | TD expression level: sum of scores for 5–7 signs (P valuea) | ||
| GRIN2A | rs1345423 | B = −0.18, P = 0.0190 | X | P = 0.02504 |
| rs7192557 | B = 0.22, P = 0.0430 | X | X | |
| rs1650420 | B = 0.16, P = 0.0471 | X | X | |
| rs11866328 | B = 0.16, P = 0.0330 | X | X | |
| rs7190619 | X | OR = 0.44[0.19–1.03], P = 0.04292 | P = 0.02510 | |
| rs9788936 | X | OR = 0.51[0.27–1.00], P = 0.04020 | X | |
| rs11644461 | X | X | P = 0.03875 | |
| DRD3 | rs963468 | X | X | P = 0.01533 |
| rs167770 | X | X | P = 0.012236 | |
TDlt: limb-truncal tardive dyskinesia; OR = odds ratio; 95% CI = 95% confidence interval.
Bold italics are statistically significant in both populations.
B = regression coefficient, P = chance, sum of scores = scores of AIMS scale.
Original value + 1, followed by log2-transformation.
Table 3.
Associations between specific SNPs and either type of TD in the Dutch and Siberian schizophrenic populations.
| Receptor | SNP | Dutch population |
Siberian schizophrenic population |
|
|---|---|---|---|---|
| TD (B, P value) | TD (OR (95% CI), P value) | TD expression level: sum of scores for 1–7 signs (P valuea) | ||
| GRIN2B | rs2192970 | X | OR = 1.93 (1.22–3.06), P = 0.005411 | X |
| GRIN2A | rs1345423 | B = −0.13, P = 0.0421 | OR = 0.42 (0.20–0.89), P = 0.015210 | P = 0.014524 |
| rs7192557 | B = 0.22, P = 0.0159 | X | X | |
| rs1650420 | B = 0.16, P = 0.0193 | X | X | |
| rs11644461 | B = −0.13, P = 0.0385 | X | X | |
| DRD3 | rs324035 | X | X | P = 0.03211 |
| rs167770 | X | X | P = 0.02197 | |
OR: odds ratio; 95% CI: 95% confidence interval.
Bold italics are statistically significant in both populations.
B = regression coefficient, P = chance, sum of scores = scores of AIMS scale.
Original value + 1, followed by log2-transformation.
Discussion
The results were inconsistent between the two populations, except for rs1345423 in GRIN2A for limb-truncal and all types of TD. The following methodological problems may explain these differences: the limited sample size (particularly of the Dutch study already published in PLoS One), multiple testing of several SNPs, age differences between the patient groups, diagnostic heterogeneity, variable influence of other risk factors, and poor clinical diagnosis of TD with the use of the AIMS.1,18 Furthermore, our study was cross-sectional, while the Dutch study was prospective with two follow-ups. This may have resulted in different outcomes, considering that dyskinesia fluctuates over time in relation to stress, anxiety, or fatigue.1 However, in our opinion, the main problem was that the magnitude of the association between the presence of a specific genetic receptor variant and the clinical phenomenon of TD was at the utmost very small and therefore irrelevant.
Glutamatergic receptors
In our current study, only rs1345423 in GRIN2A showed a significant association with TD in the Dutch and Siberian patient populations. This [G/T]-SNP is an intron variant in GRIN2A, encoding one of the glutamatergic N-methyl-d-aspartate (NMDA) receptor subunits. It is a tag SNP, which is genetically independent from the GRIN2A variants rs7192557 (rs1969060) and rs8057394 that are associated with dyskinesia’s age of onset in Huntington’s disease, as well as with levodopa-induced dyskinesia.15 The OR for rs1345423 was smaller (0.42; 95% CI: 0.20–0.89, P = 0.0152) for “all locations” TD than for rs7192557 and levodopa-induced dyskinesia in 101 patients with Parkinson’s disease (OR = 3.21; 95% CI: 1.37–7.51, P = 0.0062). The association was not found in the Siberian population when another statistical method was used.15 Therefore, this finding is not likely of significant clinical importance.
Dopaminergic receptors
The magnitude of the association between the presence of variants of the DA receptor and the presence of TD is very small. In a meta-analysis of 12 studies, Bakker et al.21 found a pooled OR (1.17, 95% CI: 1.01–1.37) for TD among carriers of the Ser9Gly allele (rs6280) in the DRD3 gene that was smaller than the OR found by Lerer et al.22 (OR = 1.33, 95% CI: 1.04–1.70) in a meta-analysis of eight studies. Tsai et al.23 found a non-significant OR after meta-analyzing 13 studies. Comparable results were found for DRD2 24,25 and DRD4.26–30 The magnitude of the association between DRD polymorphisms and TD is too weak to have a pathophysiological meaning.
A pharmacological problem should also be discussed. The Ser9Gly DRD3 variant (rs6280) has functional consequences.31 The Gly variant has a frequency of ~35% in non-African populations, and is actually the ancestral allele. The homogenous Gly variant has been associated with 4-fold greater DA binding affinity in vitro.32 However, the affinity of DA for its D2-family receptors is in the micromolar range; while, the affinity of antipsychotic drugs (when clozapine and quetiapine are excluded) is in the nanomolar range.33 Even when DA would have sufficient affinity to compete with antipsychotics for binding to the homogenous Ser9Gly variant of DRD3,32 it is difficult to understand how this activation could overcome the remaining blockade of DRD2 and DRD4 receptors within the dorsal striatum. The DRD3 receptor is characterized by an extraordinary large binding affinity for DA.34 It should be emphasized that DRD3 receptors are largely confined to the ventral striatum;34 while, the site of action of TD inducing mechanisms should be the putamen.2
It can be concluded that DA is unlikely to compete with antipsychotics for the DA type 2 receptor, even when it has far greater affinity than normal to a genetic variant of DRD2, DRD3, or DRD4. Moreover, most antipsychotics are full antagonists and dysfunctional variants of DRD2, DRD3, or DRD4 do not modify the influence of these drugs. This probably explains why genetic variants of DRD2, DRD3, or DRD4 have very little influence on the prevalence of TD in patients treated with antipsychotic drugs. Any possible influence of the genetically determined receptor inactivity on the vulnerability of TD should have taken place before treatment with DA antagonists was started. Moreover, atypical antipsychotics would be expected to increase dyskinesia instead of having protective activity, as atypical antipsychotics are known to facilitate DA release.12 Any extra DA would likely augment dyskinesia by activating excitatory DRD1, expressed by medium spiny neurons (MSNs) of the direct extrapyramidal pathway (Figure 1), which is far less potently antagonized by antipsychotic drugs. Moreover, release of extra DA would increase oxidative stress within indirect pathway MSNs, augmenting neurotoxicity.
Figure 1.
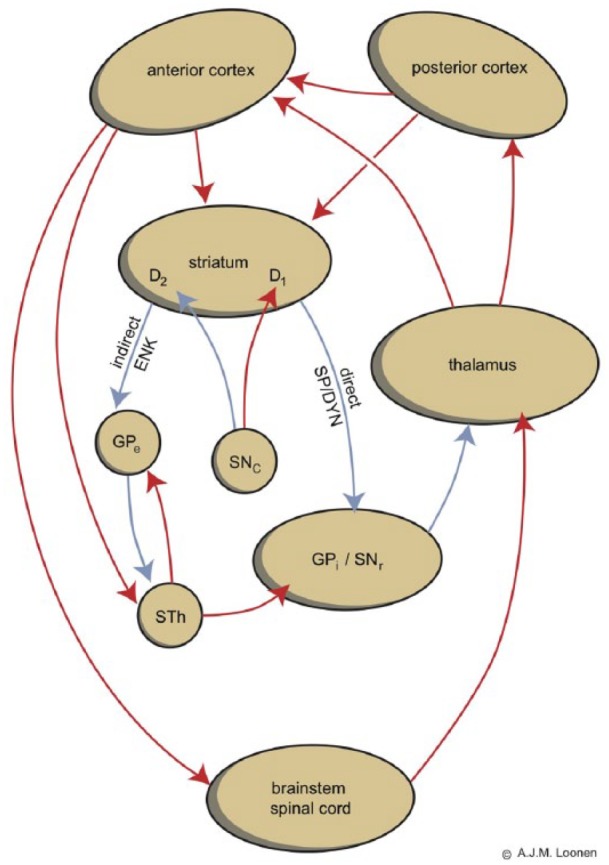
The indirect and direct extrapyramidal pathways.
ENK: enkephalin; GPe: globus pallidus, external segment; GPi: globus pallidus, internal segment; SNc: substantia nigra, pars compacta; SNr: substantia nigra, pars reticulata; SP/DYN = substance P/dynorphin; STh: subthalamic nucleus; D1, D2: medium sized spiny neurons with D1 or D2 receptors.
Red: excitatory and blue: inhibitory (colour figure in DOI: 10.1177/2050312116643673)
Serotonergic receptors
The results are also conflicting with respect to HTR2A and HTR2C. Several studies have shown an association, for example, between T102C variants of the HTR2A gene and Cys23Ser variants of HTR2C and the development of TD; while, other reports are negative.35 The affinity of serotonin itself for these binding sites is low,36,37 compared with the binding affinity of “atypical” antipsychotic drugs.12 However, these receptors spontaneously signal for cellular effector mechanisms in the absence of ligands, and HTR2C may have higher constitutive activity than HTR2A.38 In this situation, a ligand binding to the receptor may also act as an inverse agonist, that is, changing the activity of the receptor in the opposite direction instead of increasing or blocking it. This occurred when this receptor was bound by atypical antipsychotics.12,38 In these cases, the variant (of HTR2A or HTR2C) with increased constitutive activity would have greater benefit and the variant with decreased or absent constitutive activity would have less benefit due to the complete blockade of these receptors by atypical antipsychotic drugs. Thus, the affinity of previous and currently used antipsychotics for the HTR2A and HTR2C receptors should be considered when analyzing the influence of polymorphisms on the severity of dyskinesia. The difference between active and inactive receptor variants will be greater for classical antipsychotics with low binding potential for HTR2 receptors. This may explain the inconsistencies observed in studies evaluating the association between TD and genetic variations of HTR2C. The most frequently investigated 5-HT2C polymorphism, Cys23Ser (rs6318), was associated with an increased risk of TD and Parkinsonism,39–44 but the results from different studies varied. The HTR2C gene is found on the long arm of the X-chromosome (Xq24). Therefore, males are hemizygotes, and always homozygous. Al Hadithy et al. demonstrated that there was a significant association between carriers of the 23Ser allele of HTR2C and parkinsonian bradykinesia in males, but not females.40 Wilffert et al.44 did not find a correlation between TD and 23Ser male carriers when analyzed separately. However, orofacial and TDlt scores were statistically significantly higher in male patients carrying combinations of the 9Ser variant of the DRD3 gene and 23Ser allele of HTR2C.44 This may correspond with the findings of Segman et al.,39 who found no association with 23Ser male carriers, but the highest orofacial dyskinesia scores were in combined carriers of 9Gly DRD3 and 23Ser HTR2C (not specified for males and females). Al Hadithy et al.42 showed that Ser9Gly and Cys23Ser polymorphisms affect the phenotypic variability of TDlt after its occurrence, but not of TDof. Gunes et al.41 studied only male subjects with several types of extrapyramidal symptoms (Parkinsonism, dystonia, and/or dyskinesia) and observed a strong association with 23Ser HTR2C carriers. It should be emphasized that the drug treatments probably differed between studies. Gunes et al.41 excluded all patients who were using atypical drugs, while the other authors included patients who were on atypical antipsychotics.39,40,42–44 Unfortunately, Segman et al.45 did not specify the antipsychotics used by their population of Jewish patients recruited from several centers in Israel. Atypical antipsychotics are assumed to cause less Parkinsonism and TD by blocking HTR2A/2C.11,46–48 Therefore, the differences between the results of these studies may be partly explained by the use of HTR2A/2C blocking agents, which apparently decrease the differences between carriers and non-carriers. This also supports our idea that the inactive variant of HTR2C protects against developing TD.
To understand how blockade of 5-HT2A/2C receptors results in less Parkinsonism and TD, the neuronal localization of these receptors should be considered (Figure 2).49 The 5-HT2A/2C receptors not only activate GABAergic interneurons within the midbrain, striatum, and prefrontal cortex, but also stimulate MSNs within the striatum.49 Blockade in the midbrain results in DA release, but blocking the striatum directly inhibit MSNs.49 This inhibition may protect these MSNs from neurotoxicity induced by oxidative stress caused by oxidative metabolism of the excess DA.2
Figure 2.

Model for the distribution of 5-HT2A and 5-HT2C receptors in the midbrain, striatum, and prefrontal cortex.
Note: Blue: GABAergic, MSNs or fast-spiking interneurons; grey: dopaminergic neuron; purple: serotonergic interneuron; red: glutamatergic cortico-striatal neuron; and tan: striatal cholinergic interneuron. The model explains the clinical differences when 5-HT2C receptors show far more constitutive activity than 5-HT2A receptors (colour figure in DOI: 10.1177/2050312116643673).
Conclusion
Only rs1345423 in GRIN2A showed a significant association with TD in the Dutch and Siberian patient populations. It may be concluded that a direct modulation of the DA type 2 receptors is unlikely to be responsible for the protective effects of atypical antipsychotic drugs on TD. The 5-HTR2C receptor may better explain this effect, particularly when its constitutive activity is considered.
Footnotes
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics approval: Ethical approval for this study was obtained from: “Локальный этический комитет при ФГБУ «НИИПЗ» СО РАМН, Томск, Российская Федерация” (Local Ethics Committee at the Mental Health Research Institute of Siberian Branch of RAMSc, Tomsk, Russian Federation) [decision №21/2.2009 from 21.09.2009].
Funding: Part of this work (investigation of Russian patients with schizophrenia) was supported by the Russian Science Foundation [Grant #14-35-00023].
Informed consent: Written informed consent was obtained from all subjects before the study. None of the participants had a compromised capacity/ability to consent; hence, consent obtained from the next of kin was not necessary and not recommended by the Local Ethics Committee.
References
- 1. Loonen AJ, Van Praag HM. Measuring movement disorders in antipsychotic drug trials: the need to define a new standard. J Clin Psychopharmacol 2007; 27: 423–430. [DOI] [PubMed] [Google Scholar]
- 2. Loonen AJ, Ivanova SA. New insights into the mechanism of drug-induced dyskinesia. CNS Spectr 2013; 18: 15–20. [DOI] [PubMed] [Google Scholar]
- 3. Tenback DE, Van Harten PN. Epidemiology and risk factors for (tardive) dyskinesia. Int Rev Neurobiol 2011; 98: 211–230. [DOI] [PubMed] [Google Scholar]
- 4. Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III–the final common pathway. Schizophr Bull 2009; 35: 549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lau CI, Wang HC, Hsu JL, et al. Does the dopamine hypothesis explain schizophrenia? Rev Neurosci 2013; 4: 389–400. [DOI] [PubMed] [Google Scholar]
- 6. Boyd KN, Mailman RB. Dopamine receptor signaling and current and future antipsychotic drugs. In: Gross G, Geyer MA. (eds) Current antipsychotics (Handbook of experimental pharmacology). Berlin, Heidelberg: Springer-Verlag, 2012, pp. 53–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Korczyn AD. Pathophysiology of drug-induced dyskinesias. Neuropharmacology 1972; 11: 601–607. [DOI] [PubMed] [Google Scholar]
- 8. Aquino CC, Lang AE. Tardive dyskinesia syndromes: current concepts. Parkinsonism Relat Disord 2014; 20(Suppl. 1): S113–S117. [DOI] [PubMed] [Google Scholar]
- 9. Casey DE. Pathophysiology of antipsychotic drug-induced movement disorders. J Clin Psychiatry 2004; 65(Suppl. 9): 25–28. [PubMed] [Google Scholar]
- 10. Margolese HC, Chouinard G, Kolivakis TT, et al. Tardive dyskinesia in the era of typical and atypical antipsychotics. Part 2: incidence and management strategies in patients with schizophrenia. Can J Psychiatry 2005; 50: 703–714. [DOI] [PubMed] [Google Scholar]
- 11. Leysen JE. 5-HT2 receptors. Curr Drug Targets CNS Neurol Disord 2004; 3: 11–26. [DOI] [PubMed] [Google Scholar]
- 12. Meltzer HY. Serotonergic mechanisms as targets for existing and novel antipsychotics. In: Gross G, Geyer MA. (eds) Current antipsychotics (Handbook of experimental pharmacology). Berlin, Heidelberg: Springer-Verlag, 2012, pp. 87–124. [DOI] [PubMed] [Google Scholar]
- 13. Wilffert B, Zaal R, Brouwers JR. Pharmacogenetics as a tool in the therapy of schizophrenia. Pharm World Sci 2005; 27: 20–30. [DOI] [PubMed] [Google Scholar]
- 14. Koning JP, Tenback DE, Van Os J, et al. Dyskinesia and parkinsonism in antipsychotic-naive patients with schizophrenia, first-degree relatives and healthy controls: a meta-analysis. Schizophr Bull 2010; 36: 723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ivanova SA, Loonen AJ, Pechlivanoglou P, et al. NMDA receptor genotypes associated with the vulnerability to develop dyskinesia. Transl Psychiatr 2012; 2: e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bakker PR, Al Hadithy AF, Amin N, et al. Antipsychotic-induced movement disorders in long-stay psychiatric patients and 45 tag SNPs in 7 candidate genes: a prospective study. PLoS One 2012; 7: e50970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Andreasen NC, Pressler M, Nopoulos P, et al. Antipsychotic dose equivalents and dose-years: a standardized method for comparing exposure to different drugs. Biol Psychiatry 2010; 67: 255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Loonen AJ, Hovens JE. Movement disorders in clinical research. Tijdschr Psychiatr 2006; 48: 647–650. [PubMed] [Google Scholar]
- 19. Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry 1982; 39: 486–487. [DOI] [PubMed] [Google Scholar]
- 20. Gonzalez JR, Armengol L, Sole X, et al. SNPassoc: an R package to perform whole genome association studies. Bioinformatics 2007; 23: 644–645. [DOI] [PubMed] [Google Scholar]
- 21. Bakker PR, van Harten PN, van Os J. Antipsychotic-induced tardive dyskinesia and the Ser9Gly polymorphism in the DRD3 gene: a meta analysis. Schizophr Res 2006; 83: 185–192. [DOI] [PubMed] [Google Scholar]
- 22. Lerer B, Segman RH, Fangerau H, et al. Pharmacogenetics of tardive dyskinesia: combined analysis of 780 patients supports association with dopamine D3 receptor gene Ser9Gly polymorphism. Neuropsychopharmacology 2002; 27: 105–119. [DOI] [PubMed] [Google Scholar]
- 23. Tsai HT, North KE, West SL, et al. The DRD3 rs6280 polymorphism and prevalence of tardive dyskinesia: a meta-analysis. Am J Med Genet B Neuropsychiatr Genet 2010; 153B: 57–66. [DOI] [PubMed] [Google Scholar]
- 24. Zai CC, Hwang RW, De Luca V, et al. Association study of tardive dyskinesia and twelve DRD2 polymorphisms in schizophrenia patients. Int J Neuropsychopharmacol 2007; 10: 639–651. [DOI] [PubMed] [Google Scholar]
- 25. Bakker PR, van Harten PN, van Os J. Antipsychotic-induced tardive dyskinesia and polymorphic variations in COMT, DRD2, CYP1A2 and MnSOD genes: a meta-analysis of pharmacogenetic interactions. Mol Psychiatry 2008; 13: 544–556. [DOI] [PubMed] [Google Scholar]
- 26. Segman RH, Goltser T, Heresco-Levy U, et al. Association of dopaminergic and serotonergic genes with tardive dyskinesia in patients with chronic schizophrenia. Pharmacogenomics J 2003; 3: 277–283. [DOI] [PubMed] [Google Scholar]
- 27. Lattuada E, Cavallaro R, Serretti A, et al. Tardive dyskinesia and DRD2, DRD3, DRD4, 5-HT2A variants in schizophrenia: an association study with repeated assessment. Int J Neuropsychopharmacol 2004; 7: 489–493. [DOI] [PubMed] [Google Scholar]
- 28. Srivastava V, Varma PG, Prasad S, et al. Genetic susceptibility to tardive dyskinesia among schizophrenia subjects: IV. Role of dopaminergic pathway gene polymorphisms. Pharmacogenet Genomics 2006; 16: 111–117. [DOI] [PubMed] [Google Scholar]
- 29. Lee HJ, Kang SG, Choi JE, et al. No association between dopamine D4 receptor gene -521 C/T polymorphism and tardive dyskinesia in schizophrenia. Neuropsychobiology 2007; 55: 47–51. [DOI] [PubMed] [Google Scholar]
- 30. Zai CC, Tiwari AK, Basile V, et al. Association study of tardive dyskinesia and five DRD4 polymorphisms in schizophrenia patients. Pharmacogenomics J 2009; 9: 168–174. [DOI] [PubMed] [Google Scholar]
- 31. Lencz T. Pharmacogenetics of antipsychotic-induced side effects. Dialogues Clin Neurosci 2009; 11: 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lundstrom K, Turpin MP. Proposed schizophrenia-related gene polymorphism: expression of the Ser9Gly mutant human dopamine D3 receptor with the Semliki forest virus system. Biochem Biophys Res Commun 1996; 225: 1068–1072. [DOI] [PubMed] [Google Scholar]
- 33. Seeman P. Brain dopamine receptors. Pharmacol Rev 1980; 32: 229–313. [PubMed] [Google Scholar]
- 34. Schwartz JC, Levesque D, Martres MP, et al. Dopamine D3 receptor: basic and clinical aspects. Clin Neuropharmacol 1993; 16: 295–314. [DOI] [PubMed] [Google Scholar]
- 35. Arranz MJ, Rivera M, Munro JC. Pharmacogenetics of response to antipsychotics in patients with schizophrenia. CNS Drugs 2011; 25: 933–969. [DOI] [PubMed] [Google Scholar]
- 36. Farrow JT, Van Vunakis H. Characteristics of D-lysergic acid diethylamide binding to subcellular fractions derived from rat brain. Biochem Pharmacol 1973; 22: 1103–1113. [DOI] [PubMed] [Google Scholar]
- 37. Leysen JE, Gommeren W, Laduron PM. Spiperone: a ligand of choice for neuroleptic receptors. 1. Kinetics and characteristics of in vitro binding. Biochem Pharmacol 1978; 27: 307–316. [DOI] [PubMed] [Google Scholar]
- 38. Aloyo VJ, Berg KA, Spampinato U, et al. Current status of inverse agonism at serotonin 2A (5-HT2A) and 5-HT2C receptors. Pharmacol Ther 2009; 121: 160–173. [DOI] [PubMed] [Google Scholar]
- 39. Segman RH, Heresco-Levy U, Finkel B, et al. Association between the serotonin 2C receptor gene and tardive dyskinesia in chronic schizophrenia: additive contribution of 5-HT2Cser and DRD3gly alleles to susceptibility. Psychopharmacology 2000; 152: 408–413. [DOI] [PubMed] [Google Scholar]
- 40. Al Hadithy AF, Wilffert B, Stewart RE, et al. Pharmacogenetics of parkinsonism, rigidity, rest tremor, and bradykinesia in African-Caribbean inpatients: differences in association with dopamine and serotonin receptors. Am J Med Genet B Neuropsychiatr Genet 2008; 147: 890–897. [DOI] [PubMed] [Google Scholar]
- 41. Gunes A, Dahl ML, Spina E, et al. Further evidence for the association between 5-HT2C receptor gene polymorphisms and extrapyramidal side effects in male schizophrenic patients. Eur J Clin Pharmacol 2008; 64: 477–482. [DOI] [PubMed] [Google Scholar]
- 42. Al Hadithy AF, Ivanova SA, Pechlivanoglou P, et al. Tardive dyskinesia and DRD3, HTR2A and HTR2C gene polymorphisms in Russian psychiatric inpatients from Siberia. Prog Neuropsychopharmacol Biol Psychiatry 2009; 33: 475–481. [DOI] [PubMed] [Google Scholar]
- 43. Al Hadithy AF, Wilffert B, Bruggeman R, et al. Lack of association between antipsychotic-induced Parkinsonism or its subsymptoms and rs4606 SNP of RGS2 gene in African-Caribbeans and the possible role of the medication: the Curacao extrapyramidal syndromes study X. Hum Psychopharmacol 2009; 24: 123–128. [DOI] [PubMed] [Google Scholar]
- 44. Wilffert B, Al Hadithy AF, Sing VJ, et al. The role of dopamine D3, 5-HT2A and 5-HT2C receptor variants as pharmacogenetic determinants in tardive dyskinesia in African-Caribbean patients under chronic antipsychotic treatment: curacao extrapyramidal syndromes study IX. J Psychopharmacol 2009; 23: 652–659. [DOI] [PubMed] [Google Scholar]
- 45. Segman R, Neeman T, Heresco-Levy U, et al. Genotypic association between the dopamine D3 receptor and tardive dyskinesia in chronic schizophrenia. Mol Psychiatry 1999; 4: 247–253. [DOI] [PubMed] [Google Scholar]
- 46. Duinkerke SJ, Botter PA, Jansen AA, et al. Ritanserin, a selective 5-HT2/1C antagonist, and negative symptoms in schizophrenia. A placebo-controlled double-blind trial. Brit J Psychiatry 1993; 163: 451–455. [DOI] [PubMed] [Google Scholar]
- 47. Grant S, Fitton A. Risperidone. A review of its pharmacology and therapeutic potential in the treatment of schizophrenia. Drugs 1994; 48: 253–273. [DOI] [PubMed] [Google Scholar]
- 48. Owens DG. Extrapyramidal side effects and tolerability of risperidone: a review. J Clin Psychiatry 1994; 55(Suppl.): 29–35. [PubMed] [Google Scholar]
- 49. Loonen AJM, Ivanova SA. Role of 5-HT2C receptors in dyskinesia. Int J Pharm Pharm Sci 2016; 8: 5–10. [Google Scholar]