

Abstract
Aim
Several tacrolimus population pharmacokinetic models in adult renal transplant recipients have been established to facilitate dose individualization. However, their applicability when extrapolated to other clinical centres is not clear. This study aimed to (1) evaluate model external predictability and (2) analyze potential influencing factors.
Methods
Published models were screened from the literature and were evaluated using an external dataset with 52 patients (609 trough samples) collected by postoperative day 90 via methods that included (1) prediction‐based prediction error (PE%), (2) simulation‐based prediction‐ and variability‐corrected visual predictive check (pvcVPC) and normalized prediction distribution error (NPDE) tests and (3) Bayesian forecasting to assess the influence of prior observations on model predictability. The factors influencing model predictability, particularly the impact of structural models, were evaluated.
Results
Sixteen published models were evaluated. In prediction‐based diagnostics, the PE% within ±30% was less than 50% in all models, indicating unsatisfactory predictability. In simulation‐based diagnostics, both the pvcVPC and the NPDE indicated model misspecification. Bayesian forecasting improved model predictability significantly with prior 2–3 observations. The various factors influencing model extrapolation included bioassays, the covariates involved (CYP3A5*3 polymorphism, postoperative time and haematocrit) and whether non‐linear kinetics were used.
Conclusions
The published models were unsatisfactory in prediction‐ and simulation‐based diagnostics, thus inappropriate for direct extrapolation correspondingly. However Bayesian forecasting could improve the predictability considerably with priors. The incorporation of non‐linear pharmacokinetics in modelling might be a promising approach to improving model predictability.
Keywords: adult renal transplant recipients, external evaluation, non‐linear kinetics, population pharmacokinetics, tacrolimus
What is Already Known about this Subject
Population pharmacokinetics of tacrolimus in adult renal transplant recipients are continuously published to characterize pharmacokinetics and to identify covariates for dose individualization.
Whether these models can be extrapolated to clinical centres other than those where they were established remains unclear.
What this Study Adds
For the first time, the predictive performances of all relevant models were evaluated in an independent cohort.
Models provide unsatisfactory results in the prediction‐ and simulation‐based evaluations. However, Bayesian forecasting could improve the predictability considerably.
The factors influencing model predictability included bioassays, involved covariates and whether non‐linear kinetics were employed.
Introduction
Tacrolimus (TAC) is an immunosuppressant widely used to prevent allograft rejection after solid organ transplantation. It has currently gained a predominant position as a valuable alternative to ciclosporin for recipients of renal transplants because of its superior potency and graft survival rates, which has led to increasing prescription rates 1, 2, 3.
TAC pharmacokinetics (PK) are characterized and include low oral bioavailability (approximately 25%) due to combined role of cytochrome P450 (CYP) 3A and P‐glycoprotein 4. In addition, it is extensively bound to erythrocytes 5, metabolized by CYP3A isoenzymes, and mainly eliminated by the biliary route 6.
TAC has a narrow therapeutic index 7 with poor dose–concentration correlation 5, which confounds dosing therapy. It displays considerable between and within individual PK variability. Multiple factors have been identified as contributors, such as patient race, postoperative time (POT), haematocrit (HCT) level and genetic polymorphisms of the CYP enzyme 8. However, even after considering these factors, a large degree of variability remains unexplained. Consequently, therapeutic drug monitoring (TDM) is indispensable in TAC therapy 5, 9.
Over the last decades, population pharmacokinetics (popPK), which is a superior approach to traditional PK analysis, has become an established tool and plays a pivotal role in facilitating the understanding and quantification of PK variability 10, 11. This is critical to determining how best to construct dosing regimens that will meet the individual needs of patients 12. PopPK models enable practitioners to predict the optimal initial dose prior to any available measured concentrations from a population level. Enhanced individualized dose predictions could be obtained by Bayesian forecasting using the available concentrations with the original population parameter estimates as priors 13, 14, 15.
PopPK analyses for dosing individualization are widespread and there is an increasing publication in adult renal transplant patients 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31. Nevertheless, the predictability of these models is often unclear when attempts are made to extrapolate them to other clinical centres. Therefore, it is necessary to evaluate comprehensively the cross‐centre predictabilities of published models using external datasets beforehand.
This study provides a review of currently published popPK analyses of TAC in adult recipients of renal transplants. The respective external predictability of the popPK analyses was evaluated using data from an independent cohort collected at our centre. Moreover, the factors that may influence model predictability were also analyzed. This included the structural models, of which the misspecifications may impact predictability fundamentally 32, 33.
Methods
Reviews of published popPK analyses
A literature search was conducted using PubMed, Web of Science and Scopus databases and pertinent publications up to 30 June 2015 were selected. Moreover, additional publications identified from the reference lists of selected papers would also be screened if relevant. Published studies were included in the analyses if they were (1) conducted in adult recipients of renal transplants using TAC, (2) popPK modelling and (3) written in English or Chinese. The exclusion criteria were (1) lack of required details for external evaluation and (2) studies with overlapping data or cohorts. Only the most recent or the one with the largest sample size was included.
The main covariate characteristics of the included models were compared with that of our dataset. A χ2 test and a t‐test were used to examine whether or not the populations differed in categorical and continuous variables, respectively, if applicable.
External evaluation cohort
Subjects
The whole blood trough concentrations (C 0, n = 609) of 52 adult recipients of renal transplants (from May 2009 to December 2013) who were administered oral TAC (Prograf®, Astellas Ireland Co., Ltd. Killorglin, Co., Kerry, Ireland) in Huashan Hospital (Shanghai, China) were retrospectively collected using routine TDM. To ensure that TAC concentrations were at or near steady‐state, only records of samples measured after four to five repeated oral doses administered at the same dose rate were retrieved. Patient follow‐up was conducted up to postoperative day 90. Those who switched from ciclosporin or were undergoing dialysis treatment were excluded. The study protocols were approved by the Ethics Committee of the Huashan Hospital and informed written consent was obtained from all the subjects enrolled.
Immunosuppressive therapy
A triple immunosuppressive regimen including TAC, mycophenolate and corticosteroids was administered. The TAC dosage commenced at 0.1–0.15 mg kg–1 day–1 administered twice daily immediately following surgery and adjusted according to the target C 0 as follows: 8–10 ng ml−1 in the first postoperative month, 6–8 ng ml−1 from months 1–3 and 3–7 ng ml−1 thereafter.
Mycophenolate (mycophenolate mofetil, for example) was orally administrated twice daily at 1 or 0.75 g day–1 (within or after 1 month, respectively) for patients weighing less than 50 kg, 1.5 or 1 g day–1 for those between 50 and 70 kg and 2 or 1.5 g day–1 for those who were more than 70 kg. Corticosteroids were commenced at a dose of 1 g of intravenous methylprednisolone administered during the transplant surgery and 500 mg day–1 on postoperative days 1–3. This was then switched to oral prednisone 80 mg day–1 from the day 4 and tapered by 10 mg daily to 20 mg day–1 on day 10, and then further tapered to 15, 10, and 5 mg day–1 by months 1, 3, and 6 postoperatively, respectively.
Bioassay
TAC whole blood concentrations were measured using an enzyme multiplied immunoassay technique with the SYVA Viva‐Emit 2000 kit (Siemens Healthcare Diagnostics Inc., Germany). The calibration range was 2.0–30 ng ml−1 and the intra‐run precision had coefficient variations (CVs) < 10% and inter‐run CVs < 20% 34.
The bioassays used in the investigated studies were diverse. There were systematic biases between the different methods 35 and to correct these, the concentrations in the external dataset were converted to their corresponding equivalents according to the reported bioassays with formulae below, which are from large clinical studies in adult renal transplant recipients:
| (1) |
| (2) |
| (3) |
where EMIT is the concentrations of the external dataset measured using enzyme multiplied immunoassay technique while CMIA, MEIA and LCMS are after‐conversion equivalents measured using chemiluminescent micro‐particle immunoassay, micro‐particle enzyme immunoassay and all the liquid chromatography‐mass spectrometry methods, respectively, which corresponded with the investigated studies.
Genotyping
The genotyping of five single‐nucleotide polymorphisms (SNPs), CYP3A4*1G, CYP3A5*3, ABCB1 C1236T, G2677T/A and C3435T was performed by direct sequencing 39 using an external contractor (GeneCore BioTechnologies Co., Ltd., Shanghai, China). More details are available in Appendix S1. Deviations from the Hardy–Weinberg equilibrium were examined using Pearson's χ2‐test.
External predictability evaluation
The external evaluation was conducted using nonmem 7.3 (ICON Development Solutions, MD, USA) compiled with gFortran 4.6. The output was explored using the R package (version 3.1.1, http://www.r‐project.org). Published models were re‐established and the parameters were set to the published values. The predictive performances of the model in the external dataset were assessed using prediction‐ and simulation‐ based diagnostics and Bayesian forecasting.
Prediction‐based diagnostics
The population predicted concentrations (PRED) were estimated and compared with the corresponding observations (OBS) by estimating the relative prediction error (PE%, (4)).
| (4) |
The median prediction error (MDPE, median PE%) was used to evaluate accuracy while median absolute prediction error (MDAE, median |PE|%) was used to evaluate precision. We also applied F20 and F30, which indicated the percentage of PE that fell within the ±20% and ±30% range, respectively, represent a combination index of both the accuracy and precision.
Simulation‐based diagnostics
Simulation‐based diagnostics were conducted using prediction‐ and variability‐ corrected visual predictive checks (pvcVPC) 40 and normalized prediction distribution error (NPDE) 41. The dataset was simulated 1000 and 2000 times for pvcVPC and NPDE, respectively. The pvcVPCs were generated using the PsN (v 4.2.0) toolkit. The 95% confidence intervals (CI) for the median and the 5th and 95th percentiles of the simulations at different times were calculated and compared with the prediction‐ and variability‐ corrected observations, binning automatically. The NPDE results were summarized graphically and statistically using the NPDE add‐on package in R (NPDE v 2.0, www.npde.biostat.fr). The null hypothesis of NPDE is that it obeys a standard normal distribution N (0,1).
Bayesian forecasting
Maximum a posteriori Bayesian (MAPB) forecasting was conducted to assess the influence of prior observations on model predictability. Forty‐nine patients with ≥five observations were included in the assessment. For a patient, the individual prediction (IPRED) of the last observation was predicted by the latest one, two, three and four prior observations, respectively, and then compared with the corresponding observation. The relative differences denoted by the individual PE% (IPE%, (5)) were calculated using the formula below:
| (5) |
To further highlight the model that performed best in this test, median IPE% (MDIPE), median absolute IPE% (MAIPE), F20 and F30 of IPE% (IF20 and IF30) were calculated under each circumstance (i.e. IPE% was predicted by nought to four priors) as was done to the PE% in prediction‐based diagnostics.
The impact of structural models
Since modelling strategies could largely affect the model predictive performances, the structural models employed in previous studies were reviewed at first. Then their impact on predictability was evaluated by incorporating main significant covariates or not. The evaluation methods include the predication‐ and simulation‐based diagnostics and Bayesian forecasting, as described above.
Results
Reviews of published popPK analyses
After the literature retrieval procedures (Appendix S2), 16 studies were eventually retained. They are summarized in Table 1. Among them, five were multicentre studies 16, 21, 22, 30, 31 and 11 were single centre studies 17, 18, 19, 20, 23, 24, 25, 26, 27, 28, 29. In addition, 11 were mainly conducted in Caucasians 16, 17, 19, 21, 22, 23, 24, 25, 29, 30, 31, four in East Asians 18, 26, 27, 28, and one in Native Americans 20. Furthermore, 10 were developed in a small sample size of fewer than 100 participants 16, 17, 18, 19, 20, 21, 23, 24, 27, 31. Bioassays used were EMIT in two studies 26, 31, MEIA in five 17, 18, 19, 23, 27, CMIA in three 20, 24, 29 and LCMS in six 16, 21, 22, 25, 28, 30.
Table 1.
A summary of published population pharmacokinetic studies of tacrolimus in adult renal transplant recipients.
| Study (publication year) | Country (Single/multiple sites) | Number of patients (Male/Female) | Sampling schedule (Number of samples) | Postoperative Time mean ± SD/median (range) | Bioassay | Structural model | PK parameters and formula | BSV% (IOV%) | Residual error | |
|---|---|---|---|---|---|---|---|---|---|---|
| Staatz et al. (2002) 16 | Australia (Multiple) | 70 (43/27) | C 0 (1060) | Therapy duration: 218 ± 249/128 (2–1475)days | LC–MS/MS | 1–CMT | CL/F V c/F K a | 23.6 + 76.7/AST + 31.9/POTΔ10704.48 (fixed) | 42 111/ | 3.7 ng ml−1 |
| Antignac et al. (2007) 17 | France (Single) | 83 (54/29) | C 0 (1589) | < 2 months after the initiation of TAC;38 ± 27/ 33 (1–158) days | MEIA | 1‐CMT | CLV c K a F | 1.81 × [1 + POT2.54/(POT2.54 + 3.812.54)]× (1.575, if concomitant prednisone >25 mg)98.44.5 (fixed)0.137 | 3179/32 | 18.6%0.96 ng ml−1 |
| Zhang et al. (2008) 18 | China (Single) | 41 (30/11) | C 0 (456) + C 2 (86) | 12.69 ± 20.89 months | MEIA | 1‐CMT | CL/F V c/F K a | 21.7× (0.582, if concomitant nicardipine)× (35.67/HCT)0.397 × (21.54/AST)0.244× 0.99 POT (in months )× (0.842, if concomitant diltiazem)2413.09 (fixed) | 41.649.7/ | 22.2%2.19 ng ml−1 |
| Press et al. (2009) 19 | Netherland (Single) | QD 16 (12/4) BID 15 (12/3) | IS (NA) + C 0/C 2/C 3 (NA) | IS: Week 2, 6, 12, 26 and 52; C 0/C 2/C 3: Week 4, 8, 10, 17, 21 and 39 | MEIA | 2‐CMT | CLV cQV p K a F | (3.7, if CYP3A5*3/*3) or (5.5, if CYP3A5*1/*3)(61, if QD) or (42, if BID)10(61, if QD) or (42, if BID)1.6 (fixed)0.23 × {1‐[DD/(25 + DD)]}× (0.85, if concomitant prednisolone >10 mg) | 19.528.3////(21.7) | 22.60% |
| Grover et al. (2011) 20 | USA (Single) | 24 (15/9) | IS (192): one PK profile per patient | 30 ± 23 months | CMIA | 2‐CMT | CL/F V c/FQ/F V p/F K a t lag | 10.1 ( 1 l h−1 increase with a 0.138 mg increase in DD)73.327.14621.380.573 | 43.536.950.4/46.013.3 | / |
| Woillard et al. (2011) * 21 | France (Multiple) | 32 (19/13) | IS (NA) + C12 (NA): totally 145 PK profiles | IS: Week 1 and 2, Month 1, 3, and 6;C 12: Week 1 and 2 | TFC–MS/MS | 2‐CMT | CL/F V c/FQ/F V p/F K tr | 21.2 × (HCT/35)‐1.14 × (2, if CYP3A5*1)140.94792715.11 | 28 (31)31 (75)546024 (33) | 11.3%0.71 ng ml−1 |
| Passey et al. (2011) 22 | USA and Canada (Multiple) | 681 (429/252) | C 0 (11823) | 3–180 days | 97.1% LC–MS | Steady‐state infusion model | CL/F | 38.4 × [(Age/50) ‐0.4] × (0.94, if concomitant CCB)× [(0.86, if POT 6‐10) or (0.71, if POT 11‐180)]× [(1.69, if CYP3A5*1/*3) or (2, if CYP3A5*1/*1)]× (0.70, if at a steroid sparing centre) | 40.1 | 3.19 ng ml−1 |
| Musuamba et al. (2012) 23 | Belgium (Single) | 65 (36/29) | IS (455): one PK profile per patient | Day 15 | MEIA | 2‐CMT | CL/F V c/FQ/F V p/F K a t lag | 16.3 + HCT/21 × 20.6 + (15.4, if CYP3A5*1)+ (7.6, if ABCB1 1236CC, 2677GG and 3435CC)86.458.211150.450.1 | 3255/4891 61 | 13%0.88 ng ml−1 |
| Gaïes et al. (2013) 24 | Tunisia (Single) | 20 (19/1) | IS (200): one PK profile per patient | NA | CMIA | 2‐CMT | CL/F V c/F Q/F V p/F K tr | 22.684.2 × (TBW/72)1.7750.4300 (fixed)3.77 | 41.837.69¶100/11.66 | 16%0.71 ng ml−1 |
| Ogasawara et al. (2013) 25 | USA (Single) | 102 (74/28) | IS (341) + C 0/C 2 (142) | 24 (2–123) months | LC‐ MS/MS | 2‐CMT | CL/F V c/F Q/F V p/F K a t lag | 20.7× (AGE/50)‐0.78 × (2.03, if CYP3A5*1)× (1.40, if MRP2 high‐activity group) §23470.713190.5440.183 × (2.6, if with diabetes mellitus) | 43.9157//// | 18% |
| Zuo et al. (2013) 26 | China (Single) | 161 (114/47) | C 0 (873) | 9 (0–95) days | EMIT | 1‐CMT | CL/F V c/F K a | 26.6 × (HCT/27.9)‐0.451× [(1.21, if CYP3A5*1 & CYP3A4*1)or (0.982, if CYP3A5*1 & CYP3A4*1G/*1G)or (0.77, if CYP3A5*3/*3 & CYP3A4*1)or (0.577, if CYP3A5*3/*3 & CYP3A4*1G/*1G)]10203.09 (fixed) | 24.258.5/ | 19.8%1.47 ng ml−1 |
| Han et al. (2013) 27 | Korea (Single) | 80 (51/29) | C 0 (2788) | 0–400 days | MEIA | 1‐CMT | CL/F V c/F K a | 22.9× POT‐0.00762× exp[(0.2225, if CYP3A5*1/*1)or (0.17, if CYP3A5*1/*3)or (0.0525, if CYP3A5*3/*3)]× exp[(0.297, if HCT ≤ 33) or (0.117, if HCT > 33)]716 × exp (0.355 × TBW/59.025)4.5 (fixed) | 49.848.7/ | 40% (fixed) |
| Han et al. (2014) 28 | Korea (Single) | 122 † (67/55) | IS (417)† + C 0 (1501)† | 2–20 days | LC– MS/MS | 1‐CMT | CL/F V c/F K a t lag | 21.9 × (1 + 0.0119 × (POT‐9.6))× (0.816, if CYP3A5*3/*3)2053.430.25 (fixed) | 42.664.6158.9/ | 1.94 ng ml−1 |
| Golubovic et al. (2014) 29 | Serbia (Single) | 105 (62/43) | C 0 (1999) | 60 ± 47.63 (0–206) days | CMIA | 1‐CMT | CL/F V c/F K a | 10.017 × (POT/47)‐0.0283 × (TBW/68)0.869 × (TP/63)0.161× [1–0.861 × (AST‐15)/1000]× [1–0.831 × (HCT/100–0.31)]0.68 l kg−1 (fixed)1.3 | 15.2// | 4.066 ng ml−1 |
| Størset et al. (2014) 30 | Norway and Australia (Multiple) | 242 (165/77) | IS (2068) + C 0 (1032) | 20 days (4 days ‐ 15 years); Predominantly ≤ 3 months | LC–MS/MS | 2‐CMT‡ | CLV cQV p F K a t la g | 16.1 × (FFM/60)3/4 × (1.3, if CYP3A5*1)125 × FFM/6023.8 × (FFM/60)3/4636 × FFM/601 × (2.68, if POT =1) × (0.82, if CYP3A5*1)× {1 – [0.67 × concomitant prednisolone dose (mg)]/ [35 + concomitant prednisolone dose (mg)]}1.010.41 | 40 **54 **63 **// (23)¶/ (120)/ | 14.90% |
| Andreu et al. (2015) 31 | Belgium and Spain (Multiple) | 16 (10/6) | IS (594) | Day 7, Month 1, 3, 6 and 12 | EMIT | 2‐CMT | CL/F V c/F Q/F V p/F K aMTTn K tr | 16.59.8935.56526.030.470.8333.61 | 39 (29)///3532// | 21% |
AST, aspartate transferase (U l−1); BID, twice daily; BSV, between‐subject variability; C n, concentration at n hours post‐dose; CCB, anti‐hypertensive drugs classified as calcium channel blocker; CL, clearance (l h−1); CL/F, apparent clearance (l h−1); CMIA, chemiluminescent microparticle immunoassay; CMT, compartment; CYP3A5*1, cytochrome P450 3A5 expresser (*1/*1 or *1/*3); DD, tacrolimus daily dose (mg); EMIT, enzyme multiplied immunoassay technique; exp, exponential; HCT, haematocrit (%); F, bioavailability; FFM, fat free mass (kg); IS, intensive sampling; IOV, inter‐occasion variability; K a, absorption rate constant (1 h−1); K tr, transfer rate constant (1 h−1); LC, liquid chromatography; MEIA, microparticle enzyme immunoassay; MRP2, multidrug resistance‐associated protein 2; MS, mass spectrum; MTT, mean transit time (h); n, number of transit compartments; NA, not available; POT, postoperative time (day); POTΔ, days of tacrolimus therapy; Q, inter‐compartmental clearance (l h−1); Q/F, apparent inter‐compartmental clearance (l h−1); QD, once daily; TBW, total body weight (kg); TFC, turbulent flow chromatography; t lag, lag time (h); TP, total protein (g l−1); V c, volume of distribution of central compartment (l); Vc/F, apparent volume of distribution of central compartment (l); V p, volume of distribution of peripheral compartment (l); V p/F, apparent volume of distribution of peripheral compartment (l).
Only information on immediate‐release formulation of tacrolimus is included.
The number included the data from both model development (102 patients, 1626 samples) and model evaluation (20 patients, 292 samples).
The covariate screening strategy of this study was theory‐based.
MRP2 high‐activity group (H2/H2 and H1/H2) indicates ABCC2 haplotypes H1 ‐24C, 1249G, and 3972C and H2 ‐24C, 1249 A, and 3972C.
TBW in the model by Gaies et al. 24 and POT in model by Størset et al. 30 had BSV of 41.8% and 57%, respectively.
Correlations are CL ~ V c 0.43; CL ~ Q 0.62.
The covariates included in the published final CL/F models were TAC daily dose, age, weight (total body weight (TBW) and predicted fat‐free mass (FFM)), POT (including days of TAC therapy in the model by Staatz et al. 16), aspartate aminotransferase, HCT, total protein, genetic polymorphisms such as CYP3A5*3, CYP3A4*1G, ABCB1 (C1236T, G2677T/A and C3435T), and ABCC2 (C‐24 T, G1249A and C3972T) and concomitant corticosteroids or calcium channel blockers (CCBs). CYP3A5*3, HCT, and POT were the most recognized covariates. CYP3A5*3 was identified as the most influential factor in all nine studies that tested genetic polymorphisms. HCT was screened in 13 studies and involved in seven while POT was determined to be involved in eight out of the 12 models that screened for it.
Body weight was screened in all the studies but only two published in 2014 included it in CL/F either by statistical screening 29 or theory‐based consideration 30. Most studies doubted the clinically well‐accepted body weight‐based dosing regimen.
TAC daily dose was only investigated in two studies 19, 20 but they both included it as a significant covariate of CL/F, implying possible non‐linear kinetics.
External predictability evaluation
The demographic and laboratory test data collected for evaluation were summarized according to the incorporated covariates in 16 studies. Tables 2, 3 listed the demographic characteristics, clinical data and the allele frequencies of the genetic polymorphisms of the external dataset.
Table 2.
Demographics of external dataset
| Characteristics | Mean ± SD | Median (Range) |
|---|---|---|
| Number of recipients (Samples) * | 52(609) | / |
| by Male/Female * | 35/17 (441/168) | / |
| Age (years) | 38.9 ± 8.6 | 39.4 (21.9–61.2) |
| Height (cm) | 168.3 ± 6.6 | 170.0 (150.0–180.0) |
| Total body weight (kg) | 59.7 ± 9.5 | 59.5 (39.5–86.3) |
| Fat‐free mass (kg) | 46.8 ± 8.7 | 49.3 (28.9–63.3) |
| Postoperative time (days) | 37.6 ± 25.7 | 32.0 (3.0–90.0) |
| Tacrolimus daily dose (mg day–1) | 4.6 ± 1.8 | 5.0 (1.0–8.0) |
| Tacrolimus daily dose (mg kg–1 day–1) | 0.078 ± 0.032 | 0.078 (0.017–0.203) |
| Trough concentration (ng ml−1) | 6.6 ± 2.9 | 6.3 (2.1–26.1) |
| Haemoglobin (g l−1) | 114.6 ± 20.5 | 115.0 (57.0–155.0) |
| Haematocrit (%) | 34.7 ± 6.1 | 34.9 (17.6–47.6) |
| Albumin (g l−1) | 41.3 ± 6.0 | 42.0 (22.0–52.0) |
| Globulin (g l−1) | 25.2 ± 3.8 | 25.0 (11.0–37.0) |
| Aspartate transferase (U l−1) | 22.5 ± 17.5 | 18.0 (7.0–187.0) |
| Total protein (g l−1) | 66.5 ± 7.6 | 68.0 (38.0–81.0) |
| Total bilirubin (μmol l−1) | 8.8 ± 4.4 | 7.9 (2.0–38.9) |
| Serum creatinine (μmol l−1) | 109.7 ± 43.9 | 105.0 (47.0–481.0) |
| Creatinine clearance (ml min−1) † | 69.7 ± 17.4 | 69.4 (10.9–115.8) |
| Blood uric nitrogen (mmol l−1) | 8.9 ± 4.5 | 7.8 (3.2–42.2) |
| Oral prednisone daily dose (mg day–1) | 21.6 ± 14.3 | 20.0 (0.0–80.0) |
| Concomitant calcium channel blocker * | 50 (513) | / |
| Nicardipine * | 14 (24) | / |
| Diltiazem * | 0 | / |
| Diabetes mellitus * | 4 (18) | / |
Data are expressed as number of recipients (samples).
Using Cockcroft–Gault formula: CLcr = [(140 − Age (years)) × Total body weight (kg)]/(0.818 × Scr (μmol l−1)) × (0.85, if female)
Table 3.
Allele frequencies of genetic polymorphisms in CYP3A4, CYP3A5 and ABCB1 genes of external dataset
| Genotypes | Number of recipients | Percentage (%) |
|---|---|---|
| CYP3A4*1G (G20230A, rs2242480) | ||
| GG (*1/*1) | 27 | 51.92 |
| GA(*1/*1G) | 22 | 42.31 |
| AA (*1G/*1G) | 3 | 5.77 |
| CYP3A5*3 (A6986G, rs776746) | ||
| AA (*1/*1) | 4 | 7.69 |
| GA (*1/*3) | 19 | 36.54 |
| GG (*3/*3) | 29 | 55.77 |
| ABCB1‐C1236T (rs1128503) | ||
| CC | 6 | 11.54 |
| CT | 19 | 36.54 |
| TT | 27 | 51.92 |
| ABCB1‐C3435T (rs1045642) | ||
| CC | 25 | 48.08 |
| CT | 24 | 46.15 |
| TT | 3 | 5.77 |
| ABCB1‐G2677T/A (rs2032582) | ||
| GG | 14 | 26.92 |
| GT | 19 | 36.54 |
| GA | 4 | 7.69 |
| TT | 6 | 11.54 |
| TA | 8 | 15.38 |
| AA | 1 | 1.92 |
All frequencies were in agreement with those predicted by the Hardy‐Weinberg equation (P > 0.05)
Table S1 summarizes the main characteristics of the collected data and those of the 16 studies. TBW in our dataset was less than in Caucasians (P < 0.05) but similar to that of East Asians. The CYP3A5*3 polymorphism distribution in our population and in East Asians' was similar but significantly different from Caucasians (P < 0.05).
In this study, only models that described immediate release of oral TAC were evaluated because that was the only available formulation in our data. Moreover, the ABCC2 genotype was the only uncollected covariate and, therefore, it was set at 1.24 equal to the population average in the corresponding model by Ogasawara et al. 25. To incorporate the inter‐occasion variability introduced in four studies 21, 23, 30, 31, we set every dose level for each individual as an occasion.
The prediction‐based diagnostic results are presented in Table S2 and in the box plot Figure 1. The models of Passey et al. 22 and Størset et al. 30 were superior to the others. They both had MDPE% < ±20%, MDAE% < 35%, F20% > 30% and F30% > 45%. However, none of the F30% value attained 50%.
Figure 1.
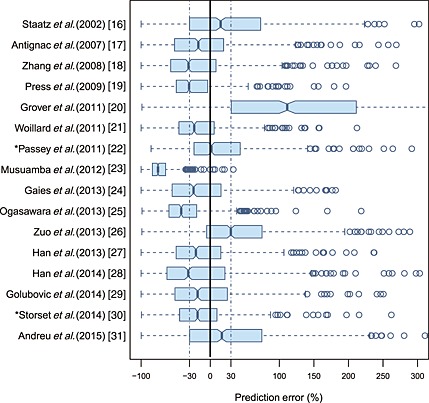
Box plots of prediction error (PE%) for 16 published population pharmacokinetic models. Black solid line and blue dotted lines are reference lines indicating PE% of 0% and ±30%, respectively. Models with asterisk (*) have the best predictability
In the simulation‐based diagnostics, the pvcVPC and NPDE were the two most common evaluation approaches. The pvcVPC provides an easy diagnosis if the model accurately predicts both the central trend as well as the variability of the data 40. The NPDE is a diagnosis with a firmer statistical background that does not require any binning of the data for a rough interpretation 41.
In our external evaluation, the pvcVPC in most studies (Figure S1) showed a large discrepancy between the observations and the model simulations. There was a trend of over or under prediction of the true between‐subject variability, except in the study by Støreset et al. 30, which showed the only misspecification in the early postoperative period (Figure 2). The conclusions arrived at with the NPDEs (Figure S2) were generally consistent with those of the pvcVPC but the results of the model by Støreset et al. 30 were not as good as that in the pvcVPC (Figure 3). Moreover, NPDEs of most studies failed to obey the normal distribution. Statistically, all models were rejected with no P values of global tests >0.01 (Table S3). The above two evaluations indicated that these models may not be appropriate for simulation‐based applications.
Figure 2.
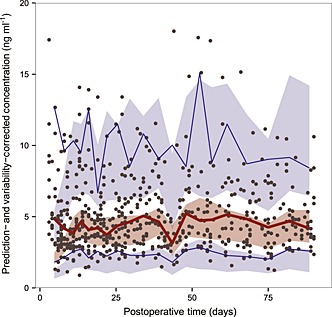
Prediction‐ and variability‐ corrected visual predictive check (pvcVPC) plot of the model by Størset et al. 30. The solid red line represents the median prediction‐ and variability‐corrected observations and semitransparent red field represents a simulation‐based 95% confidence interval (CI) for the median. Solid blue lines represent the corrected observed 5th and 95th percentiles and semitransparent blue fields represents a simulation‐based 95% CI for the corresponding model predicted percentiles. The prediction‐ and variability‐corrected observations are represented by black circles
Figure 3.
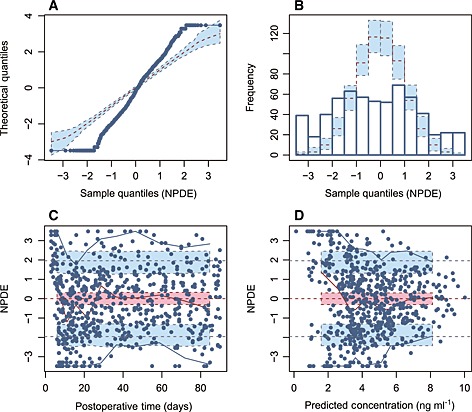
Normalized prediction distribution error (NPDE) plots of the model by Størset et al. 30. A) quantile–quantile plot of the distribution of the NPDE against theoretical distribution (semitransparent blue fields), B) histogram of the distribution of the NPDE against theoretical distribution (semitransparent blue fields), (C) NPDE vs. time after the first dose and D) NPDE vs. predicted concentrations. In plot C and D, the solid red line represents the median NPDE of the observations and the semitransparent red field represents a simulation‐based 95% confidence interval (CI) for the median. Solid blue lines represent the NPDE of the observed 5th and 95th percentiles and semitransparent blue fields represent a simulation‐based 95% CI for the corresponding model predicted percentiles. The NPDE of the observations are represented by blue circles
Bayesian forecasting demonstrated that both precision and accuracy of IPREDs could be significantly improved by MAPB as long as one prior observation was available. The individual predictability achieved the best with two to three prior observations and stabilized after that. Additional prior observations did not necessarily imply further improvement. The differences in IPEs% between the models after MAPB were much smaller than those in PEs% in prediction‐based diagnostics. The box plots are presented in Figure 4 and results are displayed in Table S4. The models by Zuo et al. 26 and Støreset et al. 30 were two of the best in this evaluation, which showed MDIPE <10%, MAIPE% < 20%, IF20% > 50% and IF30% > 60%. However, the results were not significantly different from those of the other studies.
Figure 4.
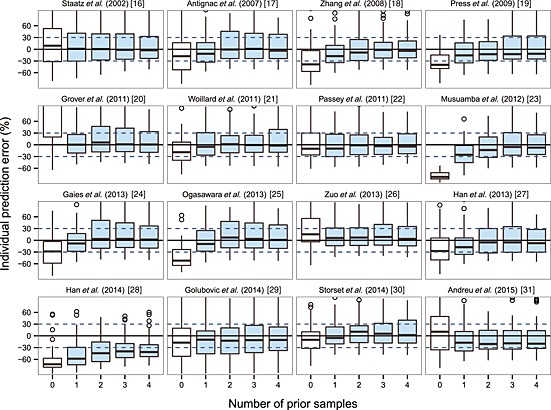
Box plots of individual prediction error (IPE%) with Bayesian forecasting for 16 published population pharmacokinetic models in different scenarios (0 represents predictions without prior information and 1‐4 represents with prior one to four observations, respectively). In scenario n, prior n observations were used to estimate the individual prediction and it was then compared with the corresponding observation
We grouped all the models with different bioassay methods together but did not find any obvious trends in the predictive performance and assay methods used. Models in the EMIT showed no superiority in any one of the above three diagnostics. The overall performances of the different bioassay groups were similar.
The impact of structural models
The structural models adopted in 16 investigated models were either two compartment (provided intensive sampling was available) or one compartment except for one study by Passey et al. 22, which used a non‐compartment steady‐state infusion model instead ((6)):
| (6) |
where C 0 indicates trough TAC concentrations and CL/F is the TAC apparent clearance.
In addition, several previous studies indicated non‐linear PK of TAC. Firstly, dose‐dependent CL/F was found in both studies that tested such a relationship 19, 20. Secondly, the model by Størset et al. 30, which was comparatively the best of all 16 published models, employed two theory‐based non‐linear kinetics to describe 1) the binding of TAC to erythrocytes and 2) the impact of prednisolone dose on TAC bioavailability (F). These two models are showed typical Michaelis–Menten (MM) kinetics, which is commonly used to describe non‐linear behaviour 42.
Given the review of all the published models, the impact of the MM structural model was explored in the same way as was successfully applied to ciclosporin 43, 44, 45, displayed as below ((7)):
| (7) |
where V m denotes the maximal dose rate (daily dose) at steady‐state and K m is an MM constant equal to the steady‐state trough concentration at half‐maximum dose rate. Comparing with the above three linear (one and two compartment and steady‐state infusion) structural models, the concentration in the MM model is an independent variable and daily dose is the dependent variable. Considering that only trough samples were available, the MM model with daily dose as a function of concentration here may mostly reflect the non‐linearity of the CL/F.
The above four covariate‐free (one and two compartment, steady‐state infusion and MM) structural models were first established and evaluated. CYP3A5*3, POT and HCT were then screened stepwise. Since there was a linear relationship between POT and HCT (r = 0.6) in our dataset, only HCT was added to the models, apart from CYP3A5*3. Daily dose was further tested in three linear models and successfully included. PopPK parameter estimates for all these models are listed in Table S5.
The prediction‐based diagnostic results were presented in Table S2 and Figure 5 simulation‐based diagnostics in Table S3 and Figure S3 and S4 and Bayesian forecasting in Table S4 and Figure 6. Compared with the predictive performance of the linear PK models, those of the non‐linear MM models with and without covariates were significantly better. The prediction‐based MDAE% in the covariate‐free and included MM model was within 35% and 25%, respectively, showing around 20% and 30% improvement over the other linear PK models. The IF20% and IF30% of the MM models after MAPB forecasting reached >90%, demonstrating their superiority compared with <60% in the linear models. However, in simulation‐based diagnostics, the MM models performed nearly the same as linear PK models did. There was no misspecification in pvcVPC while there was rejection in NPDE, mainly because of the unsatisfying normality of prediction discrepancy.
Figure 5.
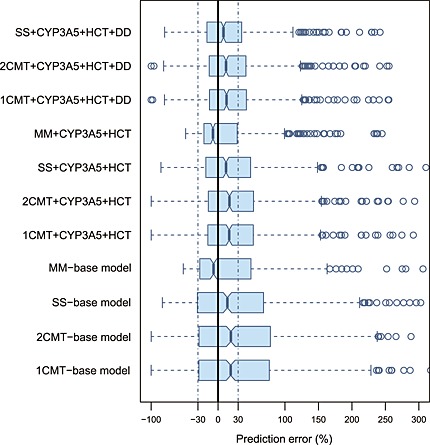
Box plots of prediction error (PE%) for four structural models with and without covariates. The black solid line and blue dotted lines are reference lines indicating PE% of 0% and ±30%, respectively. 1CMT, one compartment model; 2CMT, two compartment model; CYP3A5, CYP3A5*3 polymorphism; DD, tacrolimus daily dose; HCT, haematocrit; MM, Michaelis–Menten model; SS, steady‐state infusion model
Figure 6.
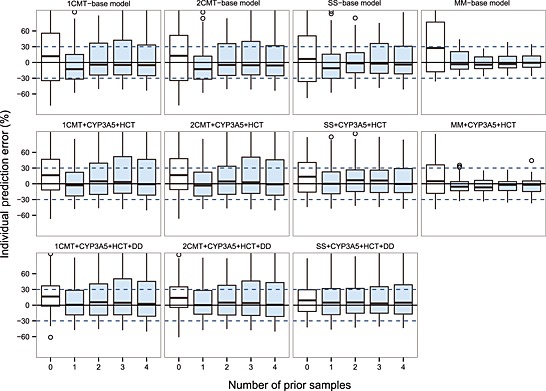
Box plots of individual prediction error (IPE%) with Bayesian forecasting for four structural models with and without covariates in different scenarios \(0 represents predictions without prior information and 1‐4 represent with prior 1‐4 observations, respectively). 1CMT, one compartment model; 2CMT, two compartment model; CYP3A5, CYP3A5*3 polymorphism; DD, tacrolimus daily dose; HCT, haematocrit; MM, Michaelis–Menten model; SS, steady‐state infusion model
Discussion
Over the past decades, more than 20 TAC popPK studies have been conducted in adult recipients of renal transplants. However, most of these were centre‐based and their predictability was unclear when extrapolated to other clinical sites. Therefore, the evaluation of their external accuracy, robustness and predictive performance is mandatory before the models can be applied 46. To the best of our knowledge, this is the first comprehensive external evaluation of published TAC popPK studies. Our evaluation was performed on three levels. Prediction‐based diagnostics are frequently used and appealing because their simplicity illustrates how well the observations and predictions are in agreement 47. Simulation‐based diagnostics, which compare the already determined simulations of a single data set with the real data can be a useful diagnostic and reveal model misspecification patterns that are not easily diagnosed by other methods 47. Bayesian forecasting is based on prior observations in each individual and is regularly used to facilitate dosage adjustments in clinical practice.
It is expected that a model developed in a population similar to that in an evaluation dataset should have superior predictive performance due to similar racial background implying similar genetics, prescribing and dietary habits of subjects. However, models developed in East Asians 18, 26, 27, 28 showed no superiority in the evaluation. The best two in prediction‐based diagnostics were performed in Caucasians. This might be attributable to their largest sample size of all the evaluated studies with 681 patients with 11 823 samples 22 and 242 patients with 3100 samples 30, indicating a more extensive representation of the studied population.
Nevertheless, the F30 value of the two superior models still failed to attain 50%. In other words, more than half of the PE% values fell outside the ±30% in all of the published models. Moreover, the simulation‐based pvcVPC and NPDE tests showed that no model fulfilled the criteria of diagnostic tests.
Limitation of model predictability could be due to multiple factors. TAC bioassay is one of the often mentioned factors. Compared with the specific LCMS results, non‐specific immunoassays are biased by some haematological parameters 48, 49 and cross‐reactivity with TAC metabolites 50, which depend on the clinical status of the patients 51. Such biases could range from −6% to 73% 9, 38, 52. After bioassay conversion, we found no obvious bioassay‐related trends in the external evaluation. However, no formula is exactly ‘correct’ 9, and the conversional method used here is likely not a perfect method of eliminating the biases, although no trend was observed. This suggests that the bioassay difference between the external dataset and the evaluated model would not be a dominant limitation after inter‐method conversion.
The inclusion of proper covariates is also an essential influencing factor. The three most recognized covariates, CYP3A5*3, HCT and POT, represent the prevailing consensus accepted in terms of covariate selection. Dose‐dependent CL/F found in the only two models screening it indicate dose as an important covariate. The exploration of the impact of structural models with and without those covariates demonstrated that these covariates could partially explain the inter‐individual variability and improve model predictability. The diagnostic results of those CYP3A5 and HCT included models were almost the same as the best published models. In addition, further inclusion of daily dose on CL/F showed even better performance, which also supported the non‐linearity of TAC PK.
CYP3A5*3 is regarded as a predominant biomarker and is associated with TAC PK, whereas the role of other genetic polymorphisms is still controversial 53, 54, 55. According to the investigated models, CL/F in patients expressing CYP3A5 (*1 allele carriers) was 1.2–2.1‐fold higher than that of non‐expressers (homozygous *3 allele carriers). This is in agreement with the previous finding of an approximately 48% (range 26–65%) higher CL/F in CYP3A5 expressers than in non‐expressers determined in classical PK studies 56. ABCB1 and ABCC2 genetic polymorphisms were screened in six of the evaluated studies 19, 23, 25, 27, 28, 31, but their impact is inconsistent and dependent on the analyzed cohort or study design, which may not have clinical relevance 57. Recently, CYP3A4*1G in East Asians 26, 58, 59, 60 and CYP3A4*22 in Caucasians 61, 62 were found to play a major role in TAC PK variability, and this phenomenon may need to be further elucidated.
TAC binds strongly to erythrocytes and plasma proteins and low HCT values increase the fraction of unbound TAC. This unbound form is more readily metabolized by the liver, leading to an increase in CL/F 6. This trend is well‐described in the HCT‐involved models and an increase in HCT of 20–40% leads to a decrease in CL/F in the range of 15.23–54.62% 18, 21, 26, 27, 29, 30. For other studies, which did not find or follow such a trend, the small sample size or proportion of patients with abnormal lower values or short follow‐up periods were the main reasons 16, 19, 23, 24, 25, 28, 31. Besides, the non‐linear binding of TAC to erythrocytes is explained by saturation of binding capacity to red blood cells 19, 63, 64. This theory was successfully applied in the model by Størset et al. 30.
POT is a surrogate for many time‐dependent factors 5, 65. Recovery of gastrointestinal function and increased food intake may alter TAC bioavailability 66. Corticosteroids dose, HCT and albumin values are known to change systematically with POT 67. The combination of these factors decreases the CL/F as POT increases, particularly in the early postoperative phase. Although the majority of the evaluated models followed this expected trend, the estimated changes were diverse. The POT‐mediated decrease in CL/F was high at 40% and the influence lasted for only 1 day postoperatively or at 4% for up to 3 years 16, 18, 22, 27, 29, 30. The complexity of the impact of POT is related to several clearly identified factors such as the HCT value and whether corticosteroids were concomitantly administered or not. It may also be related to other unidentified confounding factors as well. Their simultaneous inclusion as covariates with POT would definitely affect the description of POT in the models. Therefore, a clearer understanding of POT as a covariate is required.
It is worth noting that a dose‐dependent CL/F was found in previous studies 19, 20, 30. In our investigation of the impact of structural models, the obvious improvement of the predictability of the three linear PK models when a dose was incorporated and the superb performance of the MM models also supported the underlying non‐linear kinetics.
The observed non‐linearity may be attributed to the TDM effect. Individuals with higher CLs will have lower drug concentrations, which lead clinicians to prescribe higher doses. However, with individuals sampled at more than three to four dose levels, the TDM effect would not affect the judgment of non‐linearity 68. One PK study demonstrated a linear PK profile following a single oral dose of TAC in 18 fasted healthy volunteers 69. Nevertheless, as mentioned above, a low HCT level in patients in the early postoperative period, could lead to saturable TAC concentration‐dependent binding to erythrocytes. This would cause non‐linearity in drug distribution and elimination 19, 63, 64. POT‐associated alterations in absorption due to the recovery of gastrointestinal function, the activity of P‐glycoprotein and CYP3A enzymes and increased food intake in patients may cause non‐linear kinetics 6. High dose corticosteroids followed by gradual dose tapering may also be the reason for non‐linearity owing to the induction of P‐glycoprotein and CYP3A enzymes 17, 19, 22, 30, 67, 70, 71.
Despite the fact that the exact mechanism of non‐linearity is still unknown and impossible to elucidate with the current trough data, such interesting findings provide a new perspective for future investigation. A clear understanding may greatly contribute to the current unsatisfactory external predictability.
Although prediction‐based diagnostics provided unsatisfactory results, Bayesian forecasting affirmed the value of the MAPB and provided the practitioners with the clinical reference to collect two to three prior observations as a premise to obtain a reliable prediction.
A limitation of our study was the retrospective data collection from routine TDM. As a result, there was no way to confirm whether the patients were adherent or not. Moreover, C 0 alone limited the exploration of absorption or distribution. Nonetheless, the C 0 alone could still yield reliable estimates of CL/F in TAC 72, which is one of the aims of this study. The second limitation was that we had no data of extended‐release TAC and could not investigate during the external evaluation. Extended‐release formulations are currently gaining more acceptance and, therefore, related studies need to be conducted in future.
All the models evaluated in this study were based on a data‐driven approach except that by Størset et al. 30, which was a theory‐based model. The data‐driven approach may adequately predict a model development dataset but may not be as accurate when extrapolating outside the data range 73. In contrast, the ‘theory‐based’ or ‘mechanism‐based’ strategy including the physiologically‐based PK modelling, which incorporates anatomical and physiological knowledge 74, is likely to be another promising approach for improving extrapolation.
In conclusion, 16 published TAC popPK models used for recipients of renal transplants were externally evaluated in this study using an independent dataset from our centre. Their external predictive performances were unsatisfactory in prediction and simulation based diagnostics. However, the MAPB approach improved the predictive performance of the model considerably and, therefore, may be a useful tool for guiding dose adjustments. Multiple factors, including bioassays, involved covariates and whether non‐linear kinetics are used can influence model predictability. Further investigations still need to be conducted to confirm our findings.
Competing Interests
All authors have completed the United Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on the request from the corresponding author) and declare Dr Z. Jiao had support from National Natural Science Foundation of China (No. 81 072 702, 81 473 409 and 81 573 505) and from Major Research and Development Project of Innovative Drugs, China Ministry of Science and Technology (2012ZX09303004‐001) and Dr X.Y. Qiu had support from National Natural Science Foundation of China (No. 81 302 854), which promoted the study. There are no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years and no other relationships or activities that could appear to have influenced the submitted work. The other authors have no conflicts of interest to declare.
The authors would like to sincerely thank Ms Elisabet Størset of the department of Transplant Medicine, Oslo University Hospital Rikshospitalet (Norway), Dr Christine E. Staatz of the School of Pharmacy, University of Queensland (Australia), Dr Bojana Golubovic of the Faculty of Pharmacy, University of Belgrade (Serbia), Dr In‐Wha Kim of the College of Pharmacy and Research Institute of Pharmaceutical Sciences, Seoul National University (Korea) and Dr Xiao‐Cong Zuo of the Third Xiangya Hospital of Central South University (China) for providing details about the research and active discussions on the model code. We would like to thank Dr Emmanuelle Comets of Faculté de médecine Paris Diderot Paris 7 (France) for her valuable insight on NPDE and pcVPC. We would also thank to Li‐Xuan Qian and Chen‐Yu Wang for editing data and preparing the figures.
Contributors
CYZ, ZJ, JJM and XYQ participated in research design. XYQ contributed to acquisition of the patients' data. CYZ and JJM performed the research and analyzed the data. CYZ and ZJ drafted and all the authors revised the manuscript. No other relationships or activities that could appear to have influenced the submitted work.
Supporting information
Appendix S1
Genotyping of CYP3A4*1G, CYP3A5*3, ABCB1 C1236T, G2677T/A and C3435T5 single‐nucleotide polymorphisms.
Appendix S2
Selection process of population pharmacokinetic study of tacrolimus.
Table S1
Demographics of our dataset and 16 investigated published studies.
Table S2
Results of prediction‐based diagnostics.
Table S3
Results of normalized prediction distribution error (NPDE) diagnostics.
Table S4
Results of Bayesian forecasting.
Table S5
Parameter estimates of the four structural models with and without covariates.
Figure S1
Prediction‐ and variability‐ corrected visual predictive check (pvcVPC) plots of the 16 investigated published models.
Figure S2 A–D
Normalized prediction distribution error (NPDE) plots of the 16 investigated published models. (A) quantile–quantile plot of the distribution of the NPDE against theoretical distribution, (B) histogram of the distribution of the NPDE against theoretical distribution, (C) NPDE vs. postoperative time (days) and (D) NPDE vs. predicted concentrations (ng ml–1).
Figure S3
Prediction‐ and variability‐ corrected visual predictive check (pvcVPC) plots of the four structural models with and without covariates. Y‐axis represents prediction‐ and variability‐corrected daily dose (mg day–1) in MM models or concentrations (ng ml–1) in other models.
Figure S4 A–D
Normalized prediction distribution error (NPDE) plots of the four structural models with and without covariates. (A) quantile–quantile plot of the distribution of the NPDE against theoretical distribution, (B) histogram of the distribution of the NPDE against theoretical distribution, (C) NPDE vs. postoperative time (days) and (D) NPDE vs. predicted daily dose (mg day–1) in MM models or concentrations (ng ml–1) in other models.
Supporting info item
Supporting info item
Supporting info item
Zhao, C.‐Y. , Jiao, Z. , Mao, J.‐J. , and Qiu, X.‐Y. (2016) External evaluation of published population pharmacokinetic models of tacrolimus in adult renal transplant recipients. Br J Clin Pharmacol, 81: 891–907. doi: 10.1111/bcp.12830.
References
- 1. USRDS Annual data report National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases. 2013. Available from: http://www.usrds.org/2013/pdf/v2_ch7_13.pdf. (last accessed 25 June 2015).
- 2. Matas AJ, Smith JM, Skeans MA, Thompson B, Gustafson SK, Stewart DE, Cherikh WS, Wainright JL, Boyle G, Snyder JJ, Israni AK, Kasiske BL. OPTN/SRTR 2013 Annual Data Report: Kidney. Am J Transplant 2015; 15: 1–34. [DOI] [PubMed] [Google Scholar]
- 3. Webster AC, Woodroffe RC, Taylor RS, Chapman JR, Craig JC. Tacrolimus versus ciclosporin as primary immunosuppression for kidney transplant recipients: meta‐analysis and meta‐regression of randomised trial data. BMJ 2005; 331: 810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang Y, Benet LZ. The gut as a barrier to drug absorption: combined role of cytochrome P450 3A and P‐glycoprotein. Clin Pharmacokinet 2001; 40: 159–68. [DOI] [PubMed] [Google Scholar]
- 5. Venkataramanan R, Swaminathan A, Prasad T, Jain A, Zuckerman S, Warty V, McMichael J, Lever J, Burckart G, Starzl T. Clinical pharmacokinetics of tacrolimus. Clin Pharmacokinet 1995; 29: 404–30. [DOI] [PubMed] [Google Scholar]
- 6. Staatz CE, Tett SE. Clinical pharmacokinetics and pharmacodynamics of tacrolimus in solid organ transplantation. Clin Pharmacokinet 2004; 43: 623–53. [DOI] [PubMed] [Google Scholar]
- 7. Johnston A. Equivalence and interchangeability of narrow therapeutic index drugs in organ transplantation. Eur J Hosp Pharm Sci Pract 2013; 20: 302–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barraclough KA, Isbel NM, Kirkpatrick CM, Lee KJ, Taylor PJ, Johnson DW, Campbell SB, Leary DR, Staatz CE. Evaluation of limited sampling methods for estimation of tacrolimus exposure in adult kidney transplant recipients. Br J Clin Pharmacol 2011; 71: 207–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wallemacq P, Armstrong VW, Brunet M, Haufroid V, Holt DW, Johnston A, Kuypers D, Le Meur Y, Marquet P, Oellerich M, Thervet E, Toenshoff B, Undre N, Weber LT, Westley IS, Mourad M. Opportunities to optimize tacrolimus therapy in solid organ transplantation: report of the European consensus conference. Ther Drug Monit 2009; 31: 139–52. [DOI] [PubMed] [Google Scholar]
- 10. Ette EI, Williams PJ. Population pharmacokinetics I: background, concepts, and models. Ann Pharmacother 2004; 38: 1702–6. [DOI] [PubMed] [Google Scholar]
- 11. Sun H, Fadiran EO, Jones CD, Lesko L, Huang SM, Higgins K, Hu C, Machado S, Maldonado S, Williams R, Hossain M, Ette EI. Population pharmacokinetics. A regulatory perspective. Clin Pharmacokinet 1999; 37: 41–58. [DOI] [PubMed] [Google Scholar]
- 12. Duffull SB, Wright DF. What do we learn from repeated population analyses? Br J Clin Pharmacol 2015; 79: 40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. El DE, Meinshausen J, Buhl K, Engel G, Harings‐Kaim A, Drewelow B, Klotz U. Generation of pharmacokinetic data during routine therapeutic drug monitoring:Bayesian approach vs. pharmacokinetic studies. Ther Drug Monit 1993; 15: 281–8. [DOI] [PubMed] [Google Scholar]
- 14. Proost JH. Adaptive control of drug dosage regimens using maximum a posteriori probability Bayesian fitting. Int J Clin Pharmacol Ther 1995; 33: 531–6. [PubMed] [Google Scholar]
- 15. Del MFDG, Martin‐Suarez A, Lanao JM. Approaches for dosage individualisation in critically ill patients. Expert Opin Drug Metab Toxicol 2013; 9: 1481–93. [DOI] [PubMed] [Google Scholar]
- 16. Staatz CE, Willis C, Taylor PJ, Tett SE. Population pharmacokinetics of tacrolimus in adult kidney transplant recipients. Clin Pharmacol Ther 2002; 72: 660–9. [DOI] [PubMed] [Google Scholar]
- 17. Antignac M, Barrou B, Farinotti R, Lechat P, Urien S. Population pharmacokinetics and bioavailability of tacrolimus in kidney transplant patients. Br J Clin Pharmacol 2007; 64: 750–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang GM, Li L, Chen WQ, Bi SS, Liu X, Zhang XL, Lu W. Population pharmacokinetics of tacrolimus in Chinese renal transplant patients [in Chinese]. Acta Pharmacol Sin 2008; 43: 695–701. [PubMed] [Google Scholar]
- 19. Press RR, Ploeger BA, den Hartigh J, van der Straaten T, van Pelt J, Danhof M, de Fijter JW, Guchelaar HJ. Explaining variability in tacrolimus pharmacokinetics to optimize early exposure in adult kidney transplant recipients. Ther Drug Monit 2009; 31: 187–97. [DOI] [PubMed] [Google Scholar]
- 20. Grover A, Frassetto LA, Benet LZ, Chakkera HA. Pharmacokinetic differences corroborate observed low tacrolimus dosage in native American renal transplant patients. Drug Metab Dispos 2011; 39: 2017–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Woillard JB, de Winter BC, Kamar N, Marquet P, Rostaing L, Rousseau A. Population pharmacokinetic model and bayesian estimator for two tacrolimus formulations–twice daily Prograf and once daily Advagraf. Br J Clin Pharmacol 2011; 71: 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Passey C, Birnbaum AK, Brundage RC, Oetting WS, Israni AK, Jacobson PA. Dosing equation for tacrolimus using genetic variants and clinical factors. Br J Clin Pharmacol 2011; 72: 948–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Musuamba FT, Mourad M, Haufroid V, Demeyer M, Capron A, Delattre IK, Delaruelle F, Wallemacq P, Verbeeck RK. A simultaneous d‐optimal designed study for population pharmacokinetic analyses of mycophenolic acid and tacrolimus early after renal transplantation. J Clin Pharmacol 2012; 52: 1833–43. [DOI] [PubMed] [Google Scholar]
- 24. Gaïes E, Bacha MM, Woillard JB, Eljebari H, Helal I, Abderrahim E, Jebabli N, Saint‐Marcoux F, Marquet P, Abdallah TB, Kheder A, Gorji Y, Lakhal M, Klouz A. Tacrolimus population pharmacokinetics and bayesian estimation in Tunisian renal transplant recipients. Int J Pharm Pharm Sci 2013; 5: 108–15. [Google Scholar]
- 25. Ogasawara K, Chitnis SD, Gohh RY, Christians U, Akhlaghi F. Multidrug resistance‐associated protein 2 (MRP2/ABCC2) haplotypes significantly affect the pharmacokinetics of tacrolimus in kidney transplant recipients. Clin Pharmacokinet 2013; 52: 751–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zuo XC, Ng CM, Barrett JS, Luo AJ, Zhang BK, Deng CH, Xi LY, Cheng K, Ming YZ, Yang GP, Pei Q, Zhu LJ, Yuan H, Liao HQ, Ding JJ, Wu D, Zhou YN, Jing NN, Huang ZJ. Effects of CYP3A4 and CYP3A5 polymorphisms on tacrolimus pharmacokinetics in Chinese adult renal transplant recipients: a population pharmacokinetic analysis. Pharmacogenet Genomics 2013; 23: 251–61. [DOI] [PubMed] [Google Scholar]
- 27. Han N, Yun HY, Hong JY, Kim IW, Ji E, Hong SH, Kim YS, Ha J, Shin WG, Oh JM. Prediction of the tacrolimus population pharmacokinetic parameters according to CYP3A5 genotype and clinical factors using NONMEM in adult kidney transplant recipients. Eur J Clin Pharmacol 2013; 69: 53–63. [DOI] [PubMed] [Google Scholar]
- 28. Han N, Ha S, Yun HY, Kim MG, Min SI, Ha J, Lee JI, Oh JM, Kim IW. Population pharmacokinetic‐pharmacogenetic model of tacrolimus in the early period after kidney transplantation. Basic Clin Pharmacol Toxicol 2014; 114: 400–6. [DOI] [PubMed] [Google Scholar]
- 29. Golubovic B, Vucicevic K, Radivojevic D, Kovacevic SV, Prostran M, Miljkovic B. Total plasma protein effect on tacrolimus elimination in kidney transplant patients–population pharmacokinetic approach. Eur J Pharm Sci 2014; 52: 34–40. [DOI] [PubMed] [Google Scholar]
- 30. Storset E, Holford N, Hennig S, Bergmann TK, Bergan S, Bremer S, Asberg A, Midtvedt K, Staatz CE. Improved prediction of tacrolimus concentrations early after kidney transplantation using theory‐based pharmacokinetic modelling. Br J Clin Pharmacol 2014; 78: 509–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Andreu F, Colom H, Grinyo JM, Torras J, Cruzado JM, Lloberas N. Development of a population PK model of tacrolimus for adaptive dosage control in stable kidney transplant patients. Ther Drug Monit 2015; 37: 246–55. [DOI] [PubMed] [Google Scholar]
- 32. Merle Y, Aouimer A, Tod M. Impact of model misspecification at design (and/or) estimation step in population pharmacokinetic studies. J Biopharm Stat 2004; 14: 213–27. [DOI] [PubMed] [Google Scholar]
- 33. Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model‐based drug development‐part 2: introduction to pharmacokinetic modeling methods. CPT Pharmacometrics Syst Pharmacol 2013; 2: e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Viva‐E System Gold‐standard drug‐testing performance with EMIT technology Siemens Global Website. 2014. Available from: http://www.healthcare.siemens.com/drug‐testing‐diagnostics/viva‐drug‐testing/viva‐e‐drug‐testing‐system. (last accessed June 25 2015).
- 35. Agrawal YP, Cid M, Westgard S, Parker TS, Jaikaran R, Levine DM. Transplant patient classification and tacrolimus assays: more evidence of the need for assay standardization. Ther Drug Monit 2014; 36: 706–9. [DOI] [PubMed] [Google Scholar]
- 36. Bazin C, Guinedor A, Barau C, Gozalo C, Grimbert P, Duvoux C, Furlan V, Massias L, Hulin A. Evaluation of the architect tacrolimus assay in kidney, liver, and heart transplant recipients. J Pharm Biomed Anal 2010; 53: 997–1002. [DOI] [PubMed] [Google Scholar]
- 37. Hesse CJ, Baan CC, Balk AH, Metselaar HJ, Weimar W, van Gelder T. Evaluation of the new EMIT enzyme immunoassay for the determination of whole blood tacrolimus concentrations in kidney, heart, and liver transplant recipients. Transplant Proc 2002; 34: 2988–90. [DOI] [PubMed] [Google Scholar]
- 38. Ansermot N, Fathi M, Veuthey JL, Desmeules J, Rudaz S, Hochstrasser D. Quantification of cyclosporine and tacrolimus in whole blood. Comparison of liquid chromatography‐electrospray mass spectrometry with the enzyme multiplied immunoassay technique. Clin Biochem 2008; 41: 910–3. [DOI] [PubMed] [Google Scholar]
- 39. Geng F, Jiao Z, Dao YJ, Qiu XY, Ding JJ, Shi XJ, Li ZD, Zhong MK. The association of the UGT1A8, SLCO1B3 and ABCC2/ABCG2 genetic polymorphisms with the pharmacokinetics of mycophenolic acid and its phenolic glucuronide metabolite in Chinese individuals. Clin Chim Acta 2012; 413: 683–90. [DOI] [PubMed] [Google Scholar]
- 40. Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J 2011; 13: 143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Comets E, Brendel K, Mentre F. Computing normalised prediction distribution errors to evaluate nonlinear mixed‐effect models: the npde add‐on package for R. Comput Methods Programs Biomed 2008; 90: 154–66. [DOI] [PubMed] [Google Scholar]
- 42. Malcolm Rowland TNT. Nonlinearities In: Clinical Pharmacokinetics and Pharmacodynamics: Concepts and Applications, 4th edn. Baltimore and Philadelphia: Lippincott Williams & Wilkins, 2010; 445–82. [Google Scholar]
- 43. Grevel J. Area‐under‐the‐curve versus trough level monitoring of cyclosporine concentration: critical assessment of dosage adjustment practices and measurement of clinical outcome. Ther Drug Monit 1993; 15: 488–91. [DOI] [PubMed] [Google Scholar]
- 44. Grevel J, Post BK, Kahan BD. Michaelis‐menten kinetics determine cyclosporine steady‐state concentrations: a population analysis in kidney transplant patients. Clin Pharmacol Ther 1993; 53: 651–60. [DOI] [PubMed] [Google Scholar]
- 45. Porta Oltra B, Perez Ruixo JJ, Jimenez Torres NV, Pallardo ML. Cyclosporin pharmacokinetic modeling in renal transplant patients [in spanish]. Farm Hosp 2004; 28: 5–19. [PubMed] [Google Scholar]
- 46. Zhao W, Kaguelidou F, Biran V, Zhang D, Allegaert K, Capparelli EV, Holford N, Kimura T, Lo YL, Peris JE, Thomson A, van den Anker JN, Fakhoury M, Jacqz‐Aigrain E. External evaluation of population pharmacokinetic models of vancomycin in neonates: the transferability of published models to different clinical settings. Br J Clin Pharmacol 2013; 75: 1068–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Karlsson MO, Savic RM. Diagnosing model diagnostics. Clin Pharmacol Ther 2007; 82: 17–20. [DOI] [PubMed] [Google Scholar]
- 48. Armendariz Y, Garcia S, Lopez RM, Pou L. Hematocrit influences immunoassay performance for the measurement of tacrolimus in whole blood. Ther Drug Monit 2005; 27: 766–9. [DOI] [PubMed] [Google Scholar]
- 49. Akbas SH, Ozdem S, Caglar S, Tuncer M, Gurkan A, Yucetin L, Senol Y, Demirbas A, Gultekin M, Ersoy FF, Akaydin M. Effects of some hematological parameters on whole blood tacrolimus concentration measured by two immunoassay‐based analytical methods. Clin Biochem 2005; 38: 552–7. [DOI] [PubMed] [Google Scholar]
- 50. Murthy JN, Davis DL, Yatscoff RW, Soldin SJ. Tacrolimus metabolite cross‐reactivity in different tacrolimus assays. Clin Biochem 1998; 31: 613–7. [DOI] [PubMed] [Google Scholar]
- 51. Gonschior AK, Christians U, Winkler M, Linck A, Baumann J, Sewing KF. Tacrolimus (FK506) metabolite patterns in blood from liver and kidney transplant patients. Clin Chem 1996; 42: 1426–32. [PubMed] [Google Scholar]
- 52. Saint‐Marcoux F, Debord J, Parant F, Labalette M, Kamar N, Rostaing L, Rousseau A, Marquet P. Development and evaluation of a simulation procedure to take into account various assays for the Bayesian dose adjustment of tacrolimus. Ther Drug Monit 2011; 33: 171–7. [DOI] [PubMed] [Google Scholar]
- 53. Hesselink DA, Bouamar R, Elens L, van Schaik RH, van Gelder T. The role of pharmacogenetics in the disposition of and response to tacrolimus in solid organ transplantation. Clin Pharmacokinet 2014; 53: 123–39. [DOI] [PubMed] [Google Scholar]
- 54. Staatz CE, Goodman LK, Tett SE. Effect of CYP3A and ABCB1 single nucleotide polymorphisms on the pharmacokinetics and pharmacodynamics of calcineurin inhibitors: part I. Clin Pharmacokinet 2010; 49: 141–75. [DOI] [PubMed] [Google Scholar]
- 55. Barbarino JM, Staatz CE, Venkataramanan R, Klein TE, Altman RB. PharmGKB summary: cyclosporine and tacrolimus pathways. Pharmacogenet Genomics 2013; 23: 563–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Barry A, Levine M. A systematic review of the effect of CYP3A5 genotype on the apparent oral clearance of tacrolimus in renal transplant recipients. Ther Drug Monit 2010; 32: 708–14. [DOI] [PubMed] [Google Scholar]
- 57. Bruhn O, Cascorbi I. Polymorphisms of the drug transporters ABCB1, ABCG2, ABCC2 and ABCC3 and their impact on drug bioavailability and clinical relevance. Expert Opin Drug Metab Toxicol 2014; 10: 1337–54. [DOI] [PubMed] [Google Scholar]
- 58. Li DY, Teng RC, Zhu HJ, Fang Y. CYP3A4/5 polymorphisms affect the blood level of cyclosporine and tacrolimus in Chinese renal transplant recipients. Int J Clin Pharmacol Ther 2013; 51: 466–74. [DOI] [PubMed] [Google Scholar]
- 59. Shi XJ, Geng F, Jiao Z, Cui XY, Qiu XY, Zhong MK. Association of ABCB1, CYP3A4*18B and CYP3A5*3 genotypes with the pharmacokinetics of tacrolimus in healthy Chinese subjects: a population pharmacokinetic analysis. J Clin Pharm Ther 2011; 36: 614–24. [DOI] [PubMed] [Google Scholar]
- 60. Miura M, Satoh S, Kagaya H, Saito M, Numakura K, Tsuchiya N, Habuchi T. Impact of the CYP3A4*1G polymorphism and its combination with CYP3A5 genotypes on tacrolimus pharmacokinetics in renal transplant patients. Pharmacogenomics 2011; 12: 977–84. [DOI] [PubMed] [Google Scholar]
- 61. Elens L, Bouamar R, Hesselink DA, Haufroid V, van der Heiden IP, van Gelder T, van Schaik RH. A new functional CYP3A4 intron 6 polymorphism significantly affects tacrolimus pharmacokinetics in kidney transplant recipients. Clin Chem 2011; 57: 1574–83. [DOI] [PubMed] [Google Scholar]
- 62. Elens L, Hesselink DA, van Schaik RH, van Gelder T. The CYP3A4*22 allele affects the predictive value of a pharmacogenetic algorithm predicting tacrolimus predose concentrations. Br J Clin Pharmacol 2013; 75: 1545–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jusko WJ, D'Ambrosio R. Monitoring FK 506 concentrations in plasma and whole blood. Transplant Proc 1991; 23: 2732–5. [PubMed] [Google Scholar]
- 64. Chow FS, Piekoszewski W, Jusko WJ. Effect of hematocrit and albumin concentration on hepatic clearance of tacrolimus (FK506) during rabbit liver perfusion. Drug Metab Dispos 1997; 25: 610–6. [PubMed] [Google Scholar]
- 65. Ette EI, Ludden TM. Population pharmacokinetic modeling: the importance of informative graphics. Pharm Res 1995; 12: 1845–55. [DOI] [PubMed] [Google Scholar]
- 66. Bekersky I, Dressler D, Mekki Q. Effect of time of meal consumption on bioavailability of a single oral 5 mg tacrolimus dose. J Clin Pharmacol 2001; 41: 289–97. [DOI] [PubMed] [Google Scholar]
- 67. Undre NA, Schafer A. Factors affecting the pharmacokinetics of tacrolimus in the first year after renal transplantation. European Tacrolimus Multicentre Renal Study Group. Transplant Proc 1998; 30: 1261–3. [DOI] [PubMed] [Google Scholar]
- 68. Ahn JE, Birnbaum AK, Brundage RC. Inherent correlation between dose and clearance in therapeutic drug monitoring settings: possible misinterpretation in population pharmacokinetic analyses. J Pharmacokinet Pharmacodyn 2005; 32: 703–18. [DOI] [PubMed] [Google Scholar]
- 69. Bekersky I, Dressler D, Mekki QA. Dose linearity after oral administration of tacrolimus 1‐mg capsules at doses of 3, 7, and 10 mg. Clin Ther 1999; 21: 2058–64. [DOI] [PubMed] [Google Scholar]
- 70. Shimada T, Terada A, Yokogawa K, Kaneko H, Nomura M, Kaji K, Kaneko S, Kobayashi K, Miyamoto K. Lowered blood concentration of tacrolimus and its recovery with changes in expression of CYP3A and P‐glycoprotein after high‐dose steroid therapy. Transplantation 2002; 74: 1419–24. [DOI] [PubMed] [Google Scholar]
- 71. Christians U, Jacobsen W, Benet LZ, Lampen A. Mechanisms of clinically relevant drug interactions associated with tacrolimus. Clin Pharmacokinet 2002; 41: 813–51. [DOI] [PubMed] [Google Scholar]
- 72. Ling J, Qian LX, Ding JJ, Jiao Z. Effects of multiple‐trough sampling design and algorithm on the estimation of population and individual pharmacokinetic parameters [in Chinese]. Acta Pharmacol Sin 2014; 49: 686–94. [PubMed] [Google Scholar]
- 73. Bonate PL. Pharmacokinetic‐Pharmacodynamic Modeling and Simulation, 2nd edn New York (NY): Springer US, 2011. [Google Scholar]
- 74. Rowland M, Peck C, Tucker G. Physiologically‐based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol 2011; 51: 45–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Genotyping of CYP3A4*1G, CYP3A5*3, ABCB1 C1236T, G2677T/A and C3435T5 single‐nucleotide polymorphisms.
Appendix S2
Selection process of population pharmacokinetic study of tacrolimus.
Table S1
Demographics of our dataset and 16 investigated published studies.
Table S2
Results of prediction‐based diagnostics.
Table S3
Results of normalized prediction distribution error (NPDE) diagnostics.
Table S4
Results of Bayesian forecasting.
Table S5
Parameter estimates of the four structural models with and without covariates.
Figure S1
Prediction‐ and variability‐ corrected visual predictive check (pvcVPC) plots of the 16 investigated published models.
Figure S2 A–D
Normalized prediction distribution error (NPDE) plots of the 16 investigated published models. (A) quantile–quantile plot of the distribution of the NPDE against theoretical distribution, (B) histogram of the distribution of the NPDE against theoretical distribution, (C) NPDE vs. postoperative time (days) and (D) NPDE vs. predicted concentrations (ng ml–1).
Figure S3
Prediction‐ and variability‐ corrected visual predictive check (pvcVPC) plots of the four structural models with and without covariates. Y‐axis represents prediction‐ and variability‐corrected daily dose (mg day–1) in MM models or concentrations (ng ml–1) in other models.
Figure S4 A–D
Normalized prediction distribution error (NPDE) plots of the four structural models with and without covariates. (A) quantile–quantile plot of the distribution of the NPDE against theoretical distribution, (B) histogram of the distribution of the NPDE against theoretical distribution, (C) NPDE vs. postoperative time (days) and (D) NPDE vs. predicted daily dose (mg day–1) in MM models or concentrations (ng ml–1) in other models.
Supporting info item
Supporting info item
Supporting info item