

Abstract
Aims
Two phase 1 studies evaluated the pharmacokinetics (PK), safety and biological activity of tabalumab, a human monoclonal antibody against B‐cell activating factor (BAFF), administered intravenously (i.v.) or subcutaneously (s.c.) in subjects with rheumatoid arthritis (RA) or systemic lupus erythematosus (SLE).
Methods
In study A, subjects with RA (n = 23) or SLE (n = 6) received a single i.v. dose of tabalumab (RA 0.01, 0.04, 0.125, 0.5, 2.0, and 8.0 mg kg–1 and SLE 0.125 or 2.0 mg kg–1) or placebo. In study B, subjects with RA received a single tabalumab dose i.v. (10 mg) (n = 12) or s.c. (20 mg) (n = 12). Serum tabalumab and CD20+ B cells were evaluated and safety was assessed throughout both studies.
Results
Tabalumab PK were non‐linear across the 0.01 to 8.0 mg kg–1 dose range. Clearance (CL) decreased from 2.9 to 0.1 l day–1 and terminal half‐life (t 1/2) increased from about 1.6 to 25 days. Subjects with RA or SLE had similar PK. After s.c. dosing, tabalumab time to maximal concentration (t max) was 5.5 days. Absolute bioavailability (F) was approximately 62%. Following tabalumab dosing, CD20+ B cells transiently increased from baseline followed by a progressive decrease below baseline.
Conclusion
A single tabalumab dose administered i.v. or s.c. was well tolerated and had non‐linear CL over the dose range investigated in subjects with RA and SLE. The non‐linearity likely reflects target‐mediated CL due to binding to BAFF. Tabalumab showed biological activity based on changes in peripheral CD20+ lymphocyte numbers in both subjects with RA and SLE.
Keywords: B‐cell activating factor, pharmacokinetics, rheumatoid arthritis, systemic lupus erythematosus, tabalumab
What is Already Known about this Subject
Compounds that diminish B‐cell activity are in development for treating RA and SLE.
A human monoclonal antibody (belimumab) reduces B‐cell activity by neutralizing soluble B‐cell activating factor (BAFF).
Tabalumab is a human monoclonal antibody that neutralizes both membrane and soluble forms of BAFF.
What this Study Adds
These phase 1 studies characterized the non‐linear PK of tabalumab, bioavailability after s.c. dosing, effects on B cells showing biological activity and no notable differences in PK, biological activity or safety between subjects with RA and SLE.
Tabalumab was well‐tolerated and no increases in anti‐tabalumab antibodies were detected post‐treatment.
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease that predominately affects women and can result in organ damage and death. Patients with SLE have an increased mortality rate compared with the general population 1, 2, 3. Treatment options include corticosteroids, antimalarials, non‐steroidal anti‐inflammatory drugs (NSAIDs) and immunosuppressants. However, these therapies are not effective or well tolerated in some patients 4. Biologic disease modifying antirheumatic drugs (bDMARDs), such as tumour necrosis factor (TNF) inhibitors, abatacept and rituximab, which are effective in treating rheumatoid arthritis (RA), have not been shown to be effective in blinded clinical trials in the treatment of SLE 5. SLE is a chronic condition for which there is a substantial need for more effective, better targeted and well‐tolerated therapies that can be safely administered with standard therapies.
B cells are implicated in the pathogenesis of autoimmune diseases including SLE 6, 7, 8, 9. These act as antigen‐presenting cells and stimulate T‐cell activation and proliferation 10. Both autoantibody production and B‐cell anomalies are characteristics of SLE and RA 11. The roles of B cells in SLE and RA pathogenesis are thought to include loss of tolerance,defective signalling pathways and abnormal apoptosis 12, 13.
Due to the involvement of B cells in autoimmune disease, compounds that diminish B‐cell activity are being developed for treating SLE and RA 5, 10. One such treatment for SLE that received market authorization in 2011 is belimumab. Belimumab is a human monoclonal antibody that diminishes B‐cell activity by binding to and neutralizing the soluble form of B‐cell activating factor (BAFF) 14, a member of the tumour necrosis factor superfamily 14, 15. BAFF exists in two forms, a membrane‐bound form, expressed by immune cells such as activated monocytes, and a soluble form released from the cell surface that can circulate in the blood 15, 16. Both forms are biologically active and bind to receptors expressed on B cells. The biological effects of BAFF on B cells include survival, co‐stimulation, activation, proliferation, differentiation, immunoglobulin (Ig) class switching and Ig production 9, 14, 17.
Tabalumab is a human immunoglobulin G subclass 4 (IgG4)‐variant monoclonal antibody that binds and neutralizes both membrane and soluble forms of BAFF 18. The initial phase 1 study of tabalumab included patients both with RA and SLE, as it was felt that tabalumab could be effective in both populations. The development plan included additional studies in RA and SLE. Based on the results from phase 2 studies in RA patients 19, 20, 21, phase 3 studies were initiated to establish the efficacy and safety of tabalumab in patients with RA. However, these studies did not demonstrate sufficient efficacy to warrant continued development in RA (data not shown). The compound is currently in phase 3 testing in SLE.
This paper presents the findings of two tabalumab phase 1 studies. In study A, single intravenous (i.v.) doses of tabalumab were administered across a broad dose range in subjects with RA or SLE, whereas study B evaluated absolute bioavailablity (F) of a single dose administered subcutaneously (s.c.) in subjects with RA. The aims of the studies were to evaluate the safety, pharmacokinetics (PK) and biological activity of tabalumab in subjects with RA or SLE.
Methods
Study A (H9B‐LC‐BCDB) was performed in 23 subjects with RA and six SLE subjects, whereas study B (H9B‐MC‐BCDE) was performed in 24 subjects with RA. Both studies were conducted according to the principles of the Declaration of Helsinki and Good Clinical Practice guidelines. Appropriate review boards approved the protocol (Quorum Review, Seattle, Washington, USA for study A and Western Institutional Review Board, Olympia, Washington, USA and Klinik Reumatolgii Institute Reuma, Warsaw, Poland for study B), and all subjects gave their written informed consent.
Study populations
In both studies, eligible subjects were 18 to 75 years of age. Subjects with RA had to meet the 1988 American College of Rheumatology (ACR) criteria for the diagnosis of RA 22 for at least 1 year and have stable disease. Subjects with SLE fulfilled at least four of 11 criteria for classification of SLE according to the ACR 23 for at least 1 year. Eligible subjects were on stable doses of all prior medication for at least 1 month (study A) or 2 months (study B) prior to enrolment. They were expected to maintain a stable regimen of allowed medications (including low dose corticosteroids) during the study, unless a disease flare mandated dose adjustment or medication changes. In study A, subjects with RA had to receive methotrexate (MTX) treatment for at least 2 months prior to study enrolment.
For both studies, subjects were excluded if they had a history of tuberculosis, herpes zoster infection or other chronic or recurrent serious infection that in the opinion of the investigator posed an unacceptable risk for subject participation in the study. Subjects with a history or presence of systemic disorders capable of significantly altering the absorption, metabolism or elimination of tabalumab or interfering with interpretation of the data were excluded, as were subjects with a history of hypogammaglobulinaemia, IgA deficiency or serum IgG, IgM or IgA concentrations below the lower limit of normal at screening. Subjects from each study were also excluded if, prior to enrolment, they were exposed to anakinra, etanercept, adalimumab, rituximab, infliximab or any agent targeting B cells, such as belimumab or cyclophosphamide.
Subjects with SLE in study A were not eligible if they had used MTX within 2 months of enrolment, had SLE that was considered organ threatening or, in the 3 months prior to enrolment, had caused first episodes of seizures, mononeuritis, motor deficits, significant decreases in renal function or other disease manifestations that were treated with cyclophosphamide or ≥1 mg kg–1 prednisone or equivalent glucocorticoids. SLE subjects were also excluded if they had a hypercoagulable state resulting in venous thrombosis, arterial thrombosis or spontaneous abortion. In study A, subjects with RA were excluded if they had previous treatment with total lymphoid irradiation or agents that produced persistent CD4 lymphopenia, a history of non‐RA acute inflammatory joint disease, or Felty's syndrome. Rheumatoid arthritis subjects in study B were excluded if they had been recently exposed to abatacept, IL‐1 TRAP or AMG108.
Study A
Study A was a two centre, subject‐blind, placebo‐controlled, single dose, dose escalation study that assessed the safety and PK of tabalumab administered by i.v. infusion in subjects with RA or SLE.
Study design
All subjects were admitted to the clinical research unit (CRU) to acquire baseline clinical data (control day) up to 7 days before dosing. Subjects were again admitted to the CRU to receive a single i.v. infusion of tabalumab or placebo and to acquire serial blood samples for PK analysis (pre‐dose and 0, 1, 3, 6, 12 and 24 h [day 2]), and vital sign measurements and 12‐lead digital electrocardiograms within 60 min before dosing and at 0, 6 and 24 h post‐dosing. Following the single dose administered on day 1, safety, PK and pharmacodynamic (PD) data were collected at various time points for at least 36 weeks (days 3, 5, 8, 15 ± 1, 22 ± 1, 43 ± 2, 64 ± 2, 127 ± 7, 190 ± 7 and 253 ± 7). Whole blood was obtained for assessment of CD20+ B cells by flow cytometry on days 1 (pre‐dose), 8, 22 ± 1, 43 ± 2, 64 ± 2, 127 ± 7, 190 ± 7 and 253 ± 7. Serum, to test for the presence of anti‐tabalumab antibodies, was collected pre‐dose and at days 22 ± 1, 43 ± 2, 64 ± 2, 127 ± 7 and 253 ± 7.
Subjects with RA were allocated to one of six tabalumab dosing groups. For each dose group, three subjects were randomized to receive tabalumab and one to receive placebo. Subjects with RA either received a single dose of tabalumab ranging from 0.01 to 8.0 mg kg–1 (0.01 mg kg–1, 0.04 mg kg–1, 0.125 mg kg–1, 0.5 mg kg–1, 2.0 mg kg–1 or 8.0 mg kg–1) or placebo. Subjects with SLE received a single dose of placebo or tabalumab at a dose of 0.125 or 2.0 mg kg–1. Doses were selected to allow for an appropriate margin of safety below doses that were tested in non‐clinical toxicology studies and, in the case of the RA cohort, to provide a sufficiently wide range for full exploration of dose–response relationships. The selected dose range was expected to allow for characterization of potential non‐linear PK of tabalumab. Study drug was administered as a single i.v. infusion given over a period of time ranging from 6 min for the lower dose levels to 1 h for the highest dose level. Tabalumab was dosed in ascending fashion in the respective RA and SLE populations with at least two individuals in the populations tolerating the dose before administering the next higher scheduled dose. The decision to increase the dose to the next level was made jointly by the site investigator(s) and sponsor.
Study B
Study B was a multicentre, open label, randomized, parallel design, single dose study to evaluate absolute bioavailability of tabalumab following s.c. administration in subjects with RA.
Study design
Subjects were screened within 30 days prior to dosing and remained under observation on the day of dosing (day 1) for at least 1 h after tabalumab administration. Subjects were randomized to receive either a 10 mg dose of tabalumab by a 30 min, constant rate i.v. infusion or a 20 mg dose of tabalumab by two 10 mg s.c. injections (administered in the peri‐umbilical area). After dosing, subjects were followed for 12 weeks during which time blood samples for PK and safety measurements were obtained on days 1, 2, 3, 5, 8, 15 ± 1, 22 ± 1, 43 ± 2, 64 ± 2 and 85 ± 2. If B‐cell depletion was observed and recovery of B cells was not sufficient at the last scheduled visit (week 12), further visits were arranged as appropriate to monitor B‐cell recovery. The presence of anti‐tabalumab antibodies was assessed using blood samples obtained before and 22 ± 1, 43 ± 2 and 85 ± 2 days following dosing.
Bioanalytical methods
For both studies, serum levels of tabalumab and the presence of anti‐tabalumab antibodies were analyzed at PPD located in Richmond, Virginia, USA, using a validated enzyme‐linked immunosorbent assay (ELISA) method. The lower and upper limits of quantification were 25 ng ml–1 and 800 ng ml–1, respectively. The inter‐assay accuracy (% relative error) during validation ranged from −15.2% to 5.97%. The interassay precision (% relative standard deviation) during validation ranged from 4.41% to 19.8%. The presence of anti‐tabalumab antibodies was examined using a bridging format ELISA. Total CD20+ peripheral B cells were assessed by flow cytometry.
Pharmacokinetic and pharmacodynamic analysis
Tabalumab PK parameters for both studies were computed by standard non‐compartmental methods of analysis using WinNonlin® (Version 4.1). Standard non‐compartmental PK parameters, including maximum plasma concentration (C max), time to C max (t max), serum half‐life (t 1/2), and area under the concentration–time curve from zero to infinity (AUC(0,∞)) were estimated for both studies. For the i.v. infusion doses in studies A and B, clearance (CL) and volume of distribution (V ss) were estimated. For the s.c. dose in study B, apparent clearance (CL/F) was estimated. Absolute bioavailability (F) was determined for s.c. dosing in study B.
Changes in peripheral CD20+ B‐cell numbers from baseline were determined for both studies.
Statistical analysis
Study A
Study A was neither designed nor powered to test explicitly an a priori hypothesis. Summary statistics of PK parameters were calculated separately for the RA and SLE subjects for visual comparison of the PK between disease states. The dose proportionality assessment for AUC(0,∞) and C max was examined using the power model approach 24.
Changes from baseline in B‐cell count and safety laboratory parameters were analyzed using a repeated measure model. Treatment, time and the treatment by time interaction were included as dependent variables. Baseline value was included as a covariate. Subject and the interactions of subject with treatment and time were included as random effects. Least squares means with their 95% confidence intervals (CIs) were reported for every treatment and every time point. P values associated with the treatment effect were reported for each timepoint.
Study B
Study B was powered for the PK parameter estimates and absolute bioavailability of the s.c. formulation to be estimated with adequate precision, based on variability estimates from the literature 25, 26. Descriptive statistics for PK parameters were tabulated by route of administration, and F was calculated as ratio of geometric mean AUC(0,∞) for s.c. and i.v. dosing, after adjustment for dose.
Results
Study A
Demographics and disposition
A total of 35 subjects, six males and 29 females, enrolled (signed informed consent) into the study and 29 subjects met entry criteria and received study drug. Five were not randomized because of entry exclusion criteria and one because of the subject's decision. Of the subjects receiving study drug, 23 had RA (17 received tabalumab and six received placebo) and six had non‐organ threatening SLE (five received tabalumab and one received placebo). The mean age (± standard deviation [SD]) for the dosing group was 54.9 (10.7) years, average body mass index (BMI) (±SD) was 28 (5.5) kg m–2 and the majority of subjects were Caucasian (86%) (Table 1).
Table 1.
Subject demographics
| Study A (n = 29) | Study B (n = 24) | ||
|---|---|---|---|
| 10 mg i.v. (n = 12) | 20 mg s.c. (n = 12) | ||
| Age, years | 54.9 (±10.7)* | 52.8 (10.3)* | 54.3 (11.4)* |
| Female, n (%) | 23 (79) | 10 (83.3) | 8 (66.7) |
| Race, n (%) | |||
| Caucasian | 25 (86) | 11 (91.7) | 10 (83.3) |
| Hispanic | 3 (10) | 1 (8.3) | 0 |
| Other | 1 (3) | 0 | 2 (16.6) |
| Body mass index (kg m –2 ) | 28.0 (5.5)* | 30.1 (5.0)* | 27.0 (7.2)* |
Values indicate mean (± standard deviation).
i.v., intravenous; s.c., subcutaneous.
Pharmacokinetics and pharmacodynamics
Data for tabalumab serum concentrations were available from 22 subjects administered 0.01 to 8.0 mg kg–1 of tabalumab and PK parameters calculated for 21 subjects. One subject was excluded from the PK analysis as the profile was biologically implausible and PK parameters could not be calculated. Tabalumab concentrations displayed a biexponential decline, consistent with a two compartment model, which was more apparent at doses of ≥0.125 mg kg–1 (Figure 1A). Due to the long t 1/2, tabalumab was still measurable in some subjects 7 to 8 months after a single 8.0 mg kg–1 dose. The PK profile was adequately characterized with the PK sampling time points. Minimal AUC extrapolation beyond the last measurable data point was performed for doses of 0.04 mg kg–1 and above. At the lowest dose of 0.01 mg kg–1, moderate AUC extrapolation (up to 31%) was performed due to the dose level and the assay lower limit of quantitation.
Figure 1.
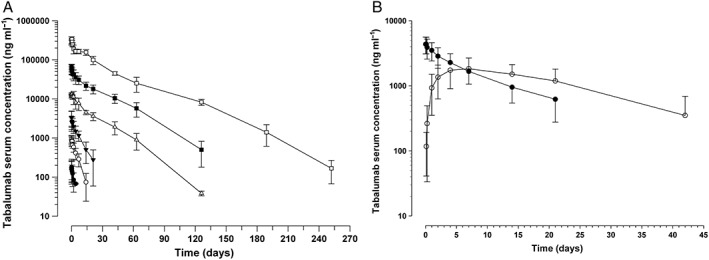
Tabalumab arithmetic mean (±SD) serum concentration–time profiles following (A) a single i.v. dose of tabalumab in subjects with stable RA or SLE (study A) (data represent the combined data of RA and SLE groups; n = 22) ( 0.01 mg kg-1 (n=3),
0.01 mg kg-1 (n=3),  0.04 mg kg-1 (n=2),
0.04 mg kg-1 (n=2),  0.125 mg kg-1 (n=6),
0.125 mg kg-1 (n=6),  0.5 mg kg-1 (n=3),
0.5 mg kg-1 (n=3),  2.0 mg kg-1 (n=5),
2.0 mg kg-1 (n=5),  8.0 mg kg-1 (n=3)) or (B) a single i.v. (n = 12) or s.c. (n = 12) dose of tabalumab in subjects with RA (study B) (
8.0 mg kg-1 (n=3)) or (B) a single i.v. (n = 12) or s.c. (n = 12) dose of tabalumab in subjects with RA (study B) ( 20 mg SC (n=12),
20 mg SC (n=12),  10 mg IV (n=12))
10 mg IV (n=12))
The PK were non‐linear over the 0.01 to 8.0 mg kg–1 dose range (Table 2). CL decreased as a function of increasing dose, with greater than dose proportional increases in exposure observed, particularly in the lower portion of the dose range. CL decreased from approximately 2.9 l day–1 at 0.01 mg kg–1 to approximately 0.1 l day–1 at 8.0 mg kg–1 and the terminal t 1/2 increased from approximately 1.6 days at 0.01 mg kg–1 to approximately 25 days at 8.0 mg kg–1. At doses of 0.5 mg kg–1 to 8.0 mg kg–1, the PK profile appears more linear and dose proportional, since CL is more consistent across this dose range (0.14–0.10 l day–1). The V ss also appears consistent across the dose range of 0.5 mg kg–1 to 8 mg kg–1.
Table 2.
Summary of pharmacokinetic parameters for tabalumab by dose group following intravenous administration for all subjects (RA and SLE) in study A*
| Tabalumab dose (mg kg –1 ) | ||||||
|---|---|---|---|---|---|---|
| 0.01 † (n = 2) | 0.04 † (n = 2) | 0.125 (n = 6) | 0.5 (n = 3) | 2.0 (n = 5) | 8.0 (n = 3) | |
| C max (ng ml–1 ) | 125, 301 | 788, 1090 | 3320 (46.6) | 13 900 (6.75) | 66 900 (16.5) | 357 000 (9.46) |
| AUC(0,∞) (ng ml −1 day) | 218, 764 | 9240, 6320 | 22 500 (39.3) | 241 000 (2.62) | 1 180 000 (22.1) | 6 730 000 (13.3) |
| CL (l day –1 ) | 4.83, 0.995 | 0.412, 0.571 | 0.370 (35.6) | 0.142 (26.9) | 0.121 (23.9) | 0.0966 (18.7) |
| V ss (l) | 8.19, 2.83 | 25.3, 3.82 | 4.28 (67.2) | 3.79 (12.7) | 3.52 (11.8) | 3.47 (28.7) |
| t 1/2 ‡ (days) | 1.17, 2.01 | 3.32, 4.64 | 5.67 (3.28–10.0) | 15.1 (13.3–17.0) | 18.2 (13.9–23.0) | 25.3 (24.0–27.4) |
Values are geometric mean (CV%) unless otherwise indicated.
Individual values.
Geometric mean (range).
AUC(0,∞), area under the concentration–time curve from zero to infinity; CL, total body clearance of drug after intravenous administration; C max, maximum observed drug concentration; CV%, coefficient of variation expressed as a percentage; t 1/2, half‐life associated with the terminal rate constant; V ss, volume of distribution at steady‐state.
The dose proportionality analysis also indicated non‐linearity and greater than proportional increases in exposure with increasing doses (Figure 2). Dose proportionality could not be established over the entire dose range. No apparent differences in PK parameters were observed between subjects with RA and SLE (Table 3), although, the small sample size limits the ability to make formal statistical comparisons.
Figure 2.
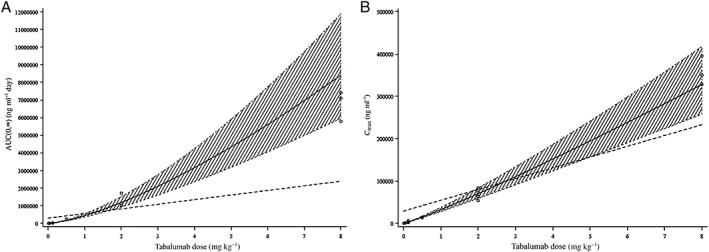
Dose proportionality assessment for (A) AUC(0,∞) and (B) C max, respectively, following intravenous administration (study A). The shaded area indicates the 95% confidence interval for the predicted line. The solid line denotes the predicted line from the fitted model. The dotted line indicates the predicted line if the PK parameters were dose proportional
Table 3.
Comparison of pharmacokinetic parameters for tabalumab by dose group in subjects with RA or SLE following intravenous administration in study A*
| Tabalumab dose | ||||
|---|---|---|---|---|
| 0.125 mg kg –1 | 2.0 mg kg –1 | |||
| RA (n = 3) | SLE (n = 3) | RA (n = 3) | SLE (n = 2) † | |
| C max (ng ml –1 ) | 3290 (19.2) | 3350 (75.8) | 63 400 (15.5) | 63 200, 83 400 |
| AUC ( 0, ∞) (ng ml –1 day) | 21 300 (38.5) | 23 800 (48.6) | 1 090 000 (7.57) | 1 020 000, 1 710 000 |
| CL (l day –1 ) | 0.392 (37.4) | 0.349 (41.2) | 0.112 (23.1) | 0.164, 0.110 |
| V ss (l) | 6.07 (80.0) | 3.01 (26.4) | 3.54 (16.5) | 3.60, 3.39 |
| t 1/2 ‡ (days) | 5.77 (3.28–10.0) | 5.57 (4.45–6.54) | 20.1 (18.5–23.0) | 13.9, 17.5 |
Values are geometric mean (CV%) unless otherwise indicated.
Individual values.
Geometric mean (range).
AUC(0,∞), area under the concentration–time curve from zero to infinity; CL, total body clearance of drug after intravenous administration; C max, maximum observed drug concentration; CV%, coefficient of variation expressed as a percentage; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; t 1/2, half‐life associated with the terminal rate constant; V ss, volume of distribution at steady‐state.
No apparent differences in peripheral CD20+ B cells were observed between subjects with RA and SLE. Consequently, the data from both populations were combined. No significant changes in the number of peripheral CD20+ B cells from baseline were seen in the placebo group (Table 4 and Figure 3A). At day 8, increases in CD20+ B‐cell numbers from baseline were seen for all dose levels, with increases being statistically significant for the 0.01, 0.04, 0.125 and 0.5 mg kg–1 dose groups (P < 0.05). At subsequent time points, there were numerical decreases in cell numbers at all dose groups, returning to baseline values or lower than baseline values, which were generally maintained for the rest of the study. Significant decreases in CD20+ cells from baseline following tabalumab administration were observed (P < 0.05) starting at day 22 in the 2.0 mg kg–1 group and day 190 in the 8.0 mg kg–1 group and were maintained throughout the remainder of the study. CD20+ cells also significantly decreased from baseline in the 0.01 mg kg–1 group at day 253. The maximal mean decrease in CD20+ B cells from baseline was approximately 79% (n = 2) and occurred approximately 252 days following the 8.0 mg kg–1 dose.
Table 4.
Summary of changes from baseline in peripheral CD20+ B cells (cells μl–1) (study A)*
| Study day | Placebo n = 7 | 0.01 mg kg –1 n = 3 | 0.04 mg kg –1 n = 2 | 0.125 mg kg –1 n = 6 | 0.5 mg kg –1 n = 3 | 2.0 mg kg –1 n = 5 | 8.0 mg kg –1 n = 3 | P value † |
|---|---|---|---|---|---|---|---|---|
| Day 8 | −28.19 (−96.75, 40.38) | 104.72 (0.39, 209.05) | 186.56 (58.96, 314.16) | 81.22 (7.36, 155.09) | 135.06 (30.04, 240.08) | 65.83 (−17.15, 148.82) | 72.59 (−31.82, 177.00) | 0.047 |
| Day 22 | −46.47 (−115.03, 22.09) | −17.95 (−122.28, 86.38) | −5.44 (−133.04, 122.16) | −71.94 (−145.80, 1.92) | 63.72 (−41.30, 168.74) | −99.77 (−182.75, −16.78) | 56.59 (−47.82, 161.00) | 0.139 |
| Day 43 | −54.19 (−122.75, 14.38) | 35.05 (−69.28, 139.38) | −20.94 (−148.54, 106.66) | −12.61 (−86.47, 61.25) | 71.39 (−33.63, 176.41) | −118.81 (−207.63, −30.00) | 2.26 (−102.15, 106.67) | 0.140 |
| Day 64 | −45.71 (−117.92, 26.51) | −6.28 (−110.61, 98.05) | 5.06 (−122.54, 132.66) | −41.53 (−120.10, 37.03) | 5.39 (−99.63, 110.41) | −173.17 (−256.15, −90.18) | −28.74 (−133.15, 75.67) | 0.096 |
| Day 127 | −13.97 (−86.07, 58.13) | −36.28 (−140.61, 68.05) | −18.44 (−146.04, 109.16) | −14.48 (−109.34, 80.37) | –‡ | −220.77 (−303.75, −137.78) | −76.41 (−180.82, 28.00) | 0.006 |
| Day 190 | −10.04 (−78.61, 58.52) | −96.61 (−200.95, 7.72) | −18.44 (−146.04, 109.16) | −9.94 (−83.80, 63.92) | −72.28 (−177.30, 32.74) | −222.17 (−305.15, −139.18) | −111.07 (−215.48, −6.67) | 0.005 |
| Day 253 | −82.95 (−234.67, 68.78) | −179.19 (−299.11, −59.26) | –‡ | −15.18 (−110.04, 79.69) | −60.28 (−165.30, 44.74) | −157.57 (−240.55, −74.58) | −182.16 (−302.09,‐62.22) | 0.120 |
There was no difference in the change in the number of peripheral CD20+ B cells between RA and SLE subjects. Consequently, the data from both populations were combined. Values represent LS mean (95% confidence interval). Negative change indicates a reduction in B‐cell counts.
Overall F test.
No useable samples were available for this time point.
Figure 3.
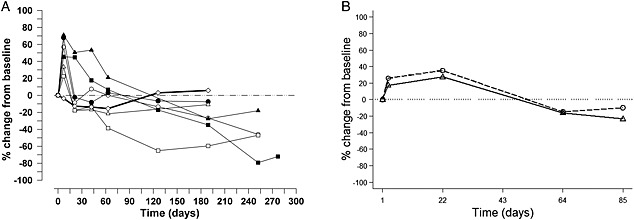
Mean percent change from baseline CD20+ B‐cell response (A) following placebo and doses ranging from 0.01 mg kg–1 to 8.0 mg kg–1 tabalumab in subjects with RA or SLE (study A) ( 0.01 mg kg-1 (n=3),
0.01 mg kg-1 (n=3),  0.04 mg kg-1 (n=2),
0.04 mg kg-1 (n=2),  0.125 mg kg-1 (n=6),
0.125 mg kg-1 (n=6),  0.5 mg kg-1 (n=3),
0.5 mg kg-1 (n=3),  2 mg kg-1 (n=5),
2 mg kg-1 (n=5),  8 mg kg-1 (n=3),
8 mg kg-1 (n=3),  Placebo (n=7),
Placebo (n=7),  baseline) and (B) single i.v. or s.c. dose of tabalumab in subjects with RA (study B) (
baseline) and (B) single i.v. or s.c. dose of tabalumab in subjects with RA (study B) ( 10 mg i.v. (n=12),
10 mg i.v. (n=12),  20 mg s.c. (n=12))
20 mg s.c. (n=12))
Study B
Demographics and disposition
Twenty‐four subjects were enrolled in the study and received study drug (n = 12 for i.v. and n = 12 for s.c. dosing) (Table 1). The majority of subjects were female (75%) and Caucasian (87.5%). The mean (SD) age was 53.5 (10.7) years and mean BMI was 28.6 (6.3) kg m–2. Two subjects discontinued early from the study, one subject in the s.c. group due to subject decision (day 44) and one in the i.v. group was lost to follow‐up (during extended B‐cell follow‐up, having completed to day 85).
Pharmacokinetics and pharmacodynamics
Similar to study A, tabalumab concentration showed a biexponential decline after i.v. dosing (Figure 1B), suggesting the PK profile is consistent with a two compartment model. After s.c. dosing, tabalumab had a slow and prolonged absorption phase resulting in a flatter PK profile, as expected. After s.c. dosing, measurable tabalumab concentrations were maintained at 42 days post‐dose (vs. 21 days post‐dose for i.v.). The t max was at the end of infusion for the 10 mg i.v. cohort, as expected, while the t max for the 20 mg s.c. cohort was approximately 5.5 days post‐dose (131 h, range of 96–334 h) (Table 5 and Figure 1B). The terminal phase t 1/2 was approximately 8 days and was similar between i.v. and s.c. routes of administration. The absolute bioavailability of a single dose of tabalumab administered s.c. was 62% (95% CI 41, 93%) (Table 5).
Table 5.
Summary of pharmacokinetic parameters from non‐compartmental analysis for i.v. and s.c. routes of tabalumab administration (study B)
| Tabalumab dose | ||
|---|---|---|
| Parameter * | 10 mg i.v. (n = 12) | 20 mg s.c. (n = 12) |
| C max (ng ml −1 ) | 4180 (32.3) | 1760 (46.2) |
| t max (h) † | 0.500 (0.480–0.920) | 131 (96–334) |
| AUC(0, ∞) (ng ml ‐1 day) | 36 200 (44.4) | 44 700 (51.8) |
| CL or CL/F (l day −1 ) | 0.276 (44.4) | 0.447 (51.8) |
| V ss (l) | 3.12 (34.1) | NC |
| Terminal phase t 1/2 (days) ‡ | 7.95 (4.73–11.7) | 8.17 (4.48–12.0) |
| F, % (95% CI) | 62 (41, 93) | |
All parameter values presented as geometric mean (percent coefficient of variation, CV%) unless otherwise noted.
Median (range).
Geometric mean (range).
CI, confidence interval; CL, clearance; C max, maximum observed concentration; F, absolute bioavailability; i.v., intravenous; NC, not calculated; t max, time of maximum observed concentration; t 1/2, half‐life; s.c., subcutaneous.
Consistent with observations from study A, the number of circulating CD20+ B cells initially increased and then decreased following both i.v. and s.c. administration of tabalumab (Figure 3B). The increase was earlier for the i.v. cohort than the s.c. cohort (median time to reach maximum increase, day 3 vs. day 22, respectively) and more pronounced (median % change from baseline, 46.2% vs. 31.7%). The maximum CD20+ B‐cell decrease appeared earlier and was less pronounced for the i.v. compared with the s.c. cohort (median time to reach maximum B‐cell decrease, day 73 vs. day 85 and median % change from baseline, −18.8% vs. –49.1%). Ten subjects (n = 3 in the i.v. group and n = 7 in the s.c. group) showed evidence of continued reduced CD20+ B‐cell counts and required additional follow‐up visits after day 85 to monitor recovery towards baseline values.
Safety
No deaths occurred during either study. Five serious adverse events (SAEs) were reported in two subjects (one subject in each study) and are described below. Most of the treatment‐emergent adverse events (TEAEs) were mild to moderate in severity. In study A and B there were 48 and 26 TEAEs, respectively, with three and four of the TEAEs being considered severe in intensity. The number of subjects reporting TEAEs was the same for the i.v. and s.c. groups in study B (n = 6 each).
In study A, eight TEAEs occurred that were considered related to study drug by the investigator. These were headache (n = 1), back pain (n = 1), dry mouth (n = 1), dysgeusia (n = 1), dysphagia (n = 1) and nausea (n = 3). For these eight TEAEs, two events occurred in the 0.01–mg kg–1 group (dysgeusia and nausea), three events in the 2 mg kg–1 group (headache, back pain, and nausea) and three events in the 8 mg kg–1 group (dry mouth, dysphagia and nausea). One SLE subject who received placebo experienced an SAE. The subject was briefly hospitalized with chest pain and was diagnosed with a lupus flare, which was considered unrelated to study drug by the investigator.
In study B, the most common TEAEs were pharyngolaryngeal pain (n = 3) and headache (n = 2). Two mild TEAEs, reported in the 20 mg s.c. group were considered related to study drug by the investigator, flushing and injection site pain. One subject in the 20 mg s.c. group was hospitalized for four SAEs (encephalopathy, urinary tract infection, pyrexia and delirium), approximately 4 months after receiving tabalumab, which were considered possibly related to tabalumab treatment by the investigator based on an infection occurring after previous immunosuppression (B‐cell depletion).
There was no evidence of an increase in titre of anti‐tabalumab antibodies post‐treatment in either study.
Discussion
Study A and study B were phase 1 studies designed to evaluate the safety, PK and biological activity of tabalumab in subjects with RA or SLE. The PK were non‐linear over the dose range investigated, with serum tabalumab declining more rapidly over time at lower doses compared with higher doses. In study A, as the dose level increased, tabalumab CL decreased and exposure was not proportional to dose over the evaluated dose range. However, at i.v. doses ≥0.5 mg kg–1, the PK became more linear and CL was similar for the 0.5, 2.0 and 8.0 mg kg–1 doses. PK parameters seemed similar between subjects with RA and SLE, although this comparison was limited by sample size. Generally, comparison of the PK parameters for i.v. dosing across these two studies showed similarity at comparable dose levels (study A, 0.125–mg kg–1 dose compared with study B, 10 mg dose). Absolute bioavailability, after s.c. administration, was 62%, which is in the range (50% to 100%) generally reported for monoclonal antibodies in humans 27, and indicates s.c. dosing is feasible for future studies. S.c. administration resulted in an extended absorption phase and flatter PK profile, which is ideal for dose administration every 2 or 4 weeks.
We speculate that a major contributing factor for the non‐linearity observed in tabalumab PK is target‐mediated CL resulting from tabalumab binding to cell membrane‐associated BAFF. Binding to membrane‐bound targets often leads to receptor‐mediated CL and non‐linear pharmacokinetics for antibodies that bind cellular targets 27. This receptor‐mediated CL becomes saturated at high doses and the PK become linear. Binding to soluble targets can also result in apparent non‐linear PK for an antibody, if the antibody–ligand complex clears at a different rate from the antibody alone, or if the assay format is such that only unbound antibody is measured and antibody‐ligand complexes comprise a significant fraction of the total antibody pool 28. Tabalumab binds to both soluble and membrane‐associated BAFF, confounding an assessment of the impact of binding to the soluble form has on the non‐linear CL observed in these studies. Interestingly, belimumab is reported to have linear CL and also to bind only soluble BAFF 29, 30, 31. However, the narrower dose range but higher maximal doses reported for belimumab (1–20 mg kg–1) makes it difficult to assess whether target binding differences play a role in the PK differences reported between tabalumab and belimumab. At these high doses of belimumab, target‐mediated CL, if present, is possibly saturated for belimumab.
Tabalumab dosing showed a biphasic effect on peripheral CD20+ B‐cell counts. Following dosing, CD20+ B‐cell counts increased transiently followed by a progressive decrease below baseline levels, with the decrease being significant at several time points (P < 0.05) for the 2 mg kg–1 and 8 mg kg–1 i.v. doses. The mechanism for the transient increase in CD20+ B cells is unclear.
The time course of CD20+ B‐cell effect may also be affected by mode of administration, as CD20+ B‐cell depletion occurred slightly earlier and was less pronounced in the i.v. compared with the s.c. cohort (median day 73 vs. day 85 and median % change from baseline −18.8% vs.−49.1%, respectively). However, given there was considerable between and within subject variability of B‐cell counts (data not shown), it is difficult to conclude whether there are differences in the B‐cell profiles between the i.v. and s.c. routes of administration.
The majority (>84%) of TEAEs observed in these tabalumab studies were mild to moderate in severity. Based on study B, the mode of administration did not appear to affect the rate of occurrence of TEAEs, as the percentage of subjects reporting a TEAE for both dose groups was equivalent (50% each). There were no apparent allergic reactions or immune responses to tabalumab treatment and there was no association of increased titre of anti‐tabalumab antibodies with tabalumab administration. Despite a long t 1/2 and a prolonged reduction in the number of peripheral CD20+ B cells, tabalumab was not associated with serious or severe infections. The only tabalumab‐related SAE was in study B. An RA subject experienced encephalopathy, urinary tract infection, pyrexia and delirium approximately 4 months after receiving 20 mg tabalumab. The events were associated with an infection, which the investigator considered possibly related to study drug due to the post‐treatment reduction in B‐cell counts.
As is typical for phase 1 studies, the size of the PK populations evaluated in both studies were small (<30 subjects) and only six SLE subjects were included in study A (five assigned to tabalumab). Large phase 3 studies in subjects with SLE will further explore the PK and safety of tabalumab in this patient population. For a rigorous bioavailability assessment, a crossover study is often preferred in which subjects would have served as their own controls. However, the crossover design is not practical for tabalumab due to its long PK half‐life and extended biological action. In study B, F was calculated as the ratio between dose‐normalized AUC between the 20 mg s.c. cohort and 10 mg s.c. cohort. An important assumption of this method is that the systemic clearance is the same between the two cohorts. With the dose‐dependent clearance observed in study A and assuming F may be approximately 50% for the s.c. injection, we intentionally chose 10 mg and 20 mg for the i.v. and s.c. dose, respectively. The resulting 62% estimate for F is close to the assumed value. Therefore, the selection of doses for study B was reasonable. On the other hand, an estimate of F based on a PK model that fits both dose‐dependent CL and F at the same time may be slightly different from what is reported here. Exploratory compartmental analyses (incorporating both linear and non‐linear clearance parameters) were performed for these studies and were the basis for later modelling and analyses performed for larger studies 32. The PK results from these exploratory compartmental analyses were consistent with those reported herein from the non‐compartmental analysis.
In summary, these two phase 1 studies show that tabalumab has non‐linear PK in subjects with RA or SLE. The non‐linearity likely reflects target‐mediated CL due to binding to BAFF. Administration of tabalumab either i.v. or s.c. resulted in an initial transient increase in CD20+ B cells, which subsequently decreased. There were no significant differences in PK, biological activity or safety between RA and SLE subjects after tabalumab administration.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare RH, LH, DR, JV, JMcC and JW are all employees and minor stockholders in Eli Lilly and Company. RF (principal investigator) has received study grants and consulting fees from Eli Lilly and VC (principal investigator) has nothing to declare.
Funding sources
The studies were supported by Eli Lilly and Company.
Author contributions
The following authors were involved in the analysis and/or interpretation of the study results as well as critical review of the manuscript: Jennifer Witcher, Ryan Hansen, Leijun Hu, David Radtke, Jim Voelker and Elisa Gomez. Juliet McColm was involved in clinical study design, implementation and medical monitoring, as well as critical review of the manuscript. The following authors were primary investigators and involved in the conduct of these studies and also critical review of this manuscript: Roy Fleischmann and Vishala Chindalore.
The authors would like to acknowledge, from Eli Lilly and Company, Karen Schneck for contributions to the PK analysis and Wendy Komocsar for valuable discussion of the findings and the manuscript. They also thank Gina Moore and Cindi Wood from inVentiv Health Clinical for writing and editorial support, respectively.
Witcher, J. , Fleischmann, R. , Chindalore, V. L. , Hansen, R. J. , Hu, L. , Radtke, D. , Voelker, J. , Gomez, E. , and McColm, J. (2016) Pharmacokinetics and safety of single doses of tabalumab in subjects with rheumatoid arthritis or systemic lupus erythematosus. Br J Clin Pharmacol, 81: 908–917. doi: 10.1111/bcp.12860.
References
- 1. Jacobsen S, Petersen J, Ullman S, Junker P, Voss A, Rasmussen JM, Tarp U, Poulsen LH, van Overeem Hansen G, Skaarup B, Hansen TM, Podenphant J, Halberg P. Mortality and causes of death of 513 Danish patients with systemic lupus erythematosus. Scand J Rheumatol 1999; 28: 75–80. [DOI] [PubMed] [Google Scholar]
- 2. Bernatsky S, Boivin JF, Joseph L, Manzi S, Ginzler E, Gladman DD, Urowitz M, Fortin PR, Petri M, Barr S, Gordon C, Bae SC, Isenberg D, Zoma A, Aranow C, Dooley MA, Nived O, Sturfelt G, Steinsson K, Alarcon G, Senecal JL, Zummer M, Hanly J, Ensworth S, Pope J, Edworthy S, Rahman A, Sibley J, El‐Gabalawy H, McCarthy T, St. Pierre Y, Clarke A, Ramsey‐Goldman R. Mortality in systemic lupus erythematosus. Arthritis Rheum 2006; 54: 2550–7. [DOI] [PubMed] [Google Scholar]
- 3. Lim SS, Bayakly AR, Helmick CG, Gordon C, Easley KA, Drenkard C. The incidence and prevalence of systemic lupus erythematosus, 2002–2004: the Georgia lupus registry. Arthritis Rheumatol 2014; 66: 357–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yildirim‐Toruner C, Diamond B. Current and novel therapeutics in the treatment of systemic lupus erythematosus. J Allergy Clin Immunol 2011; 127: 303–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fattah Z, Isenberg DA. Recent developments in the treatment of patients with systemic lupus erythematosus: focusing on biologic therapies. Expert Opin Biol Ther 2014; 14: 311–26. [DOI] [PubMed] [Google Scholar]
- 6. Chan OT, Madaio MP, Shlomchik MJ. The central and multiple roles of B cells in lupus pathogenesis. Immunol Rev 1999; 169: 107–21. [DOI] [PubMed] [Google Scholar]
- 7. Hostmann A, Jacobi AM, Mei H, Hiepe F, Dorner T. Peripheral B cell abnormalities and disease activity in systemic lupus erythematosus. Lupus 2008; 17: 1064–9. [DOI] [PubMed] [Google Scholar]
- 8. Combe B, van Vollenhoven R. Novel targeted therapies: the future of rheumatoid arthritis? Mavrilumab and tabalumab as examples. Ann Rheum Dis 2013; 72: 1433–5. [DOI] [PubMed] [Google Scholar]
- 9. Davidson A. Targeting BAFF in autoimmunity. Curr Opin Immunol 2010; 22: 732–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Conti F, Ceccarelli F, Massaro L, Cipriano E, di Franco M, Alessandri C, Spinelli FR, Scrivo R. Biological therapies in rheumatic diseases. Clin Ter 2013; 164: e413–28. [DOI] [PubMed] [Google Scholar]
- 11. Eisenberg R, Albert D. B‐cell targeted therapies in rheumatoid arthritis and systemic lupus erythematosus. Nat Clin Pract Rheumatol 2006; 2: 20–7. [DOI] [PubMed] [Google Scholar]
- 12. Traczewski P, Rudnicka L. Treatment of systemic lupus erythematosus with epratuzumab. Br J Clin Pharmacol 2011; 71: 175–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med 2011; 365: 2205–19. [DOI] [PubMed] [Google Scholar]
- 14. Stohl W. Therapeutic targeting of the BAFF/APRIL axis in systemic lupus erythematosus. Expert Opin Ther Targets 2014; 18: 473–89. [DOI] [PubMed] [Google Scholar]
- 15. Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer JL, Holler N, Ambrose C, Lawton P, Bixler S, Acha‐Orbea H, Valmori D, Romero P, Werner‐Favre C, Zubler RH, Browning JL, Tschopp J. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med 1999; 189: 1747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Melchers F. Actions of BAFF in B cell maturation and its effects on the development of autoimmune disease. Ann Rheum Dis 2003; 62 (Suppl 2): ii25–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cancro MP, D'Cruz DP, Khamashta MA. The role of B lymphocyte stimulator (BLyS) in systemic lupus erythematosus. J Clin Invest 2009; 119: 1066–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kikly K, Manetta J, Smith H, Wierda D. Characterization of LY2127399, a neutralizing antibody for BAFF [abstract]. Arthritis Rheum 2009; 60 (Suppl 10): 693. [Google Scholar]
- 19. Genovese MC, Bojin S, Biagini IM, Mociran E, Cristei D, Mirea G, Georgescu L, Sloan‐lancaster J. Tabalumab in rheumatoid arthritis patients with an inadequate response to methotrexate and naive to biologic therapy: a phase II, randomized, placebo‐controlled trial. Arthritis Rheum 2013; 65: 880–9. [DOI] [PubMed] [Google Scholar]
- 20. Genovese MC, Fleischmann RM, Greenwald M, Satterwhite J, Veenhuizen M, Xie L, Berclaz PY, Myers S, Benichou O. Tabalumab, an anti‐BAFF monoclonal antibody, in patients with active rheumatoid arthritis with an inadequate response to TNF inhibitors. Ann Rheum Dis 2013; 72: 1461–8. [DOI] [PubMed] [Google Scholar]
- 21. Genovese MC, Lee E, Satterwhite J, Veenhuizen M, Disch D, Berclaz PY, Myers S, Sides G, Benichou O. A phase 2 dose‐ranging study of subcutaneous tabalumab for the treatment of patients with active rheumatoid arthritis and an inadequate response to methotrexate. Ann Rheum Dis 2013; 72: 1453–60. [DOI] [PubMed] [Google Scholar]
- 22. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988; 31: 315–24. [DOI] [PubMed] [Google Scholar]
- 23. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, Schaller JG, Talal N, Winchester RJ. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982; 25: 1271–7. [DOI] [PubMed] [Google Scholar]
- 24. Smith BP, Vandenhende FR, DeSante KA, Farid NA, Welch PA, Callaghan JT, Forque ST. Confidence interval criteria for assessment of dose proportionality. Pharm Res 2000; 17: 1278–83. [DOI] [PubMed] [Google Scholar]
- 25. Korth‐Bradley JM, Rubin AS, Hanna RK, Simcoe DK, Lebsack ME. The pharmacokinetics of etanercept in healthy volunteers. Ann Pharmacother 2000; 34: 161–4. [DOI] [PubMed] [Google Scholar]
- 26. Weisman MH, Moreland LW, Furst DE, Weinblatt ME, Keystone EC, Paulus HE, Teoh LS, Velagapdi RB, Noertersheuser PA, Granneman GR, Fischkoff SA, Chartash EK. Efficacy, pharmacokinetic, and safety assessment of adalimumab, a fully human anti‐tumor necrosis factor‐alpha monoclonal antibody, in adults with rheumatoid arthritis receiving concomitant methotrexate: a pilot study. Clin Ther 2003; 25: 1700–21. [DOI] [PubMed] [Google Scholar]
- 27. Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci 2004; 93: 2645–68. [DOI] [PubMed] [Google Scholar]
- 28. Davda JP, Hansen RJ. Properties of a general PK/PD model of antibody‐ligand interactions for therapeutic antibodies that bind to soluble endogenous targets. MAbs 2010; 2: 576–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Struemper H, Chen C, Cai W. Population pharmacokinetics of belimumab following intravenous administration in patients with systemic lupus erythematosus. J Clin Pharmacol 2013; 53: 711–20. [DOI] [PubMed] [Google Scholar]
- 30. Shida Y, Takahashi N, Sakamoto T, Ino H, Endo A, Hirama T. The pharmacokinetics and safety profiles of belimumab after single subcutaneous and intravenous doses in healthy Japanese volunteers. J Clin Pharm Ther 2014; 39: 97–101. [DOI] [PubMed] [Google Scholar]
- 31. Furie R, Stohl W, Ginzler EM, Becker M, Mishra N, Chatham W, Merrill JT, Weinstein A, McCune WJ, Zhong J, Cai W, Freimuth W; Belimumab Study Group . Biologic activity and safety of belimumab, a neutralizing anti‐B‐lymphocyte stimulator (BLyS) monoclonal antibody: a phase I trial in patients with systemic lupus erythematosus. Arthritis Res Ther 2008; 10: R109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ji Y, Radtke D, Satterwhite J, Berclaz PY. Population pharmacokinetic modeling of sparse data of tabalumab in phase II studies of rheumatoid arthritis patients using a Bayesian approach (T‐031). J Pharmacokinet Pharmacodyn 2013; 40: S81–1. [Google Scholar]