

Abstract
Aims
γ‐Hydroxybutyrate (GHB) is used as a treatment for narcolepsy and alcohol withdrawal and as a recreational substance. Nevertheless, there are limited data on the pharmacokinetics and pharmacokinetic‐pharmacodynamic relationships of GHB in humans. We characterized the pharmacokinetic profile and exposure‐psychotropic effect relationship of GHB in humans.
Methods
Two oral doses of GHB (25 and 35 mg kg−1) were administered to 32 healthy male subjects (16 for each dose) using a randomized, placebo‐controlled, cross‐over design.
Results
Maximal concentrations of GHB were (geometric mean and 95% CI): 218 (176–270) nmol ml−1 and 453 (374–549) nmol ml−1 for the 25 and 35 mg kg−1 GHB doses, respectively. The elimination half‐lives (mean ± SD) were 36 ± 9 and 39 ± 7 min and the AUC∞ values (geometric mean and 95% CI) were 15 747 (12 854–19 290) and 40 113 (33 093–48 622) nmol∙min ml−1 for the 20 and 35 mg kg−1 GHB doses, respectively. Thus, plasma GHB exposure (AUC0‐∞) rose disproportionally (+40%) with the higher dose. γ‐Hydroxybutyrate produced mixed stimulant‐sedative effects, with a dose‐dependent increase in sedation and dizziness. It did not alter heart rate or blood pressure. A close relationship between plasma GHB exposure and its psychotropic effects was found, with higher GHB concentrations associated with higher subjective stimulation, sedation, and dizziness. No clockwise hysteresis was observed in the GHB concentration effect plot over time (i.e., no acute pharmacological tolerance).
Conclusion
Evidence was found of a nonlinear dose‐exposure relationship (i.e., no dose proportionality) at moderate doses of GHB. The effects of GHB on consciousness were closely linked to its plasma exposure and exhibited no acute tolerance.
Keywords: GHB, liquid ecstasy, pharmacokinetic, sodium oxybate
What is Already Known about this Subject
γ‐Hydroxybutyrate (GHB) presents nonlinear elimination kinetics at higher doses in humans.
γ‐Hydroxybutyrate produces mixed stimulant‐sedative subjective effects.
What this Study Adds
At moderate doses, GHB exhibited first‐order elimination kinetics but a nonlinear dose‐plasma exposure relationship.
A close relationship was found between the plasma GHB concentration and subjective effects of GHB over time, with no evidence of acute tolerance.
Introduction
γ‐Hydroxybutyrate (GHB) is a GHB and γ‐aminobutyric acid (GABA) receptor agonist 1, 2, 3 that is used for the treatment of narcolepsy and alcohol withdrawal but also as a recreational substance 3, 4, 5, 6. Moreover, GHB has recently been proposed as an experimental therapeutic for depressive disorders because of its effects on sleep, vigilance, and neuroendocrine parameters 7.
Several previous small studies investigated the pharmacokinetics of GHB 8, 9, 10, 11, 12, 13, 14. These studies provided partly conflicting findings regarding the linearity of the kinetics of GHB and mostly did not assess the psychotropic effects of GHB or the relationship between plasma GHB exposure and its psychotropic effects. Brenneisen et al. and Brailsford et al. both used single doses of GHB (25 mg kg−1) in 8 and 12 subjects, respectively, without a placebo control or assessments of pharmacodynamic effects 10, 13. Thai et al. characterized the pharmacokinetics of a single dose of 50 mg kg−1 GHB in 16 subjects and included a placebo group, but subjective effects were not assessed 14. The aforementioned studies 10, 13, 14 reported first‐order kinetics of GHB. In contrast, Palatini et al. documented nonlinear kinetics of GHB (12.5, 25, and 50 mg kg−1) in eight subjects but did not include a placebo condition or evaluate dynamic measures 9. Similar nonlinear elimination kinetics and capacity‐limited disposition were noted by other studies 15, 16. Abanades et al. assessed both the pharmacokinetic and pharmacodynamics of GHB (40, 50, 60, and 72 mg kg−1) in a total of eight subjects (n = 2–5/dose) using a placebo‐controlled design and reported dose‐dependent and GHB concentration‐related mixed sedative‐stimulant effects 11, 12. Normalized GHB area‐under‐the‐curve (AUC) values were not significantly different, except for the 72 mg kg−1 dose, indicating nonlinear kinetics only at higher GHB doses 11. However, the study was too underpowered to draw valid conclusions. Therefore, we examined the pharmacokinetics of GHB together with the subjective and cardiovascular effects of GHB in a larger sample using two doses of GHB and a placebo condition in 32 healthy male subjects. γ‐Hydroxybutyrate is endogenously produced, and the placebo condition also served as a control condition to correct for endogenous levels of GHB in plasma.
The aims of the present study were (1) to characterize the plasma pharmacokinetics of GHB using moderate doses of 20 and 35 mg kg−1 in a large study sample (n = 32), extending the previous small studies that used 25, 40, 50, 60, and 72 mg kg−1 doses of GHB, (2) to use a double‐blind, placebo‐controlled design to correct for endogenous GHB levels and validly determine the psychotropic and cardiovascular effects of GHB, and (3) to assess the plasma exposure‐psychotropic effect relationship for GHB.
Methods
Subjects and study design
The effects of oral GHB were assessed in a randomized, double‐blind, placebo‐controlled, balanced crossover study in 32 healthy, non‐smoking, male subjects. Sixteen subjects, 24 ± 3 years of age (mean ± SD) and weighing 74 ± 8 kg, were administered GHB in an oral dose of 20 mg kg−1 and placebo. Another 16 subjects, 25 ± 5 years of age and weighing 75 ± 9 kg, were administered GHB in a dose of 35 mg kg−1 and placebo. The range of GHB doses that are typically abused is 25–75 mg kg−1 (~2–6 g) 6, 11. The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of the Canton of Zurich and Swiss Agency for Therapeutic Products (Swissmedic) and registered at ClinicalTrials.gov (NCT02342366). The subjects were recruited via online advertisement at the University of Zurich and provided written informed consent before participating in the study and were paid for their participation. On the experimental days, a peripheral venous catheter for blood sampling was placed at 08:30, and GHB (Xyrem® solution in juice) or placebo (salted juice) was given orally at 09:00. Each experimental session lasted 225 min, and the intersession interval was 7 days. The subjects had to be fasting during the morning of the experiments. The exclusion criteria were a medical diagnosis, psychiatric disorders, drug dependence, or regular illicit drug use (lifetime use of illegal substances >5 times with the exception of occasional cannabis use) as reported previously 17. In order to ensure drug abstinence on the test days, urine drug screening tests were performed using Dimension RXL Max (Siemens, Erlangen, Germany) immunoassays. The present study also evaluated the behavioural and endocrine effects of GHB for the lower 20 mg kg−1 dose as reported elsewhere 17.
Outcome measures
Pharmacokinetics
Blood samples (in EDTA‐coated tubes) were collected on ice 20 min before and 35, 60, 100, 135, and 190 min after drug administration. The samples were centrifuged, and plasma was stored at 80 °C prior to analysis.
Quantification of GHB in human plasma
We determined the plasma concentrations of GHB using gas chromatography–mass spectrometry as previously reported 17, 18. GHB (Xyrem®) as analytical standard was purchased from UCB Pharma GmbH, Monheim, Germany. GHB‐d6 (internal standard) was obtained from Sigma‐Aldrich, Steinheim, Germany.
Standard solutions: Standard solutions were prepared in methanol covering a concentration range of 0.001–0.5 mg ml−1. For constructing calibration curves and to evaluate method performance, 10 or 20 μl of standard solutions were applied to plasma samples and followed by sample preparation (see below) to reach the analytical range of 4.5–4500 pg on column. The calibration curve used for the analysis of plasma samples was built using eight calibration points. An aqueous GHB‐d6 solution of 0.2 μg ml−1 was used as internal standard (900 pg on column).
Sample preparation and GC‐MS analysis: Details about instrumental conditions used for the analysis are shown in 17. The performance of the method was evaluated using external calibrations (i.e., spiking calibration solutions (see standard solutions) into plasma and processing as stated above (n = 6)) and control samples (n = 6). The results were analysed based on the peak area ratio between analyte and internal standard. The calibration curves had a correlation coefficient (R 2) of 0.9999 ± 0.0001, a slope of 0.0017 ± 0.0002 (CV 12%) and an intercept of −0.0156 ± 0.0062. The inter‐day variation coefficient (CV) evaluated at 90 and 900 pg on column (38 and 380 nmol ml−1) were 13% and 15%, respectively. Accuracy was between 90% and 115% at 90 and 900 pg on column, respectively. The recovery of control samples was in the range of 80–100%. The limit of detection (LOD) was 2.5 pg on column (1 nmol ml−1) and the lower limit of quantification (LLOQ) was 4.5 pg on column (2 nmol ml−1). The specificity of the method was evaluated on blank samples (distilled water) where no GHB was found (<LOD). Using our method, the endogenous GHB concentrations after placebo administration or before GHB administration were below the LLOQ in three subjects and therefore set to 0.
The pharmacokinetic data were analysed using non‐compartmental models (WinNonlin 6.4, Pharsight, Mountain View, CA, USA). Prior to analysis, the plasma concentrations of GHB after placebo administration (endogenous GHB production) were subtracted from the GHB levels after GHB administration for each of the time points and for each subject. Maximal plasma concentration (C max) and the time to maximal plasma concentration (t max) were obtained directly from the observed concentration–time curves. The terminal elimination rate constant (λz) was estimated by log‐linear regression after semilogarithmic transformation of the data using at least three data points of the terminal linear phase of the concentration–time curve, and the terminal elimination half‐life (t 1/2) was calculated using λz and the equation t 1/2 = ln2/λ z.
Subjective effects
For the measurement of acute subjective drug effects, we used four visual analogue scales (VASs), ranging from 0 to 10, to assess general drug effect, sedation, stimulation, and dizziness 15 min before and 40, 60, 100, 120, and 180 min after drug administration as similarly used by others 11.
Vital signs
Blood pressure and heart rate were repeatedly measured 10 min before and 40, 60, 100, 135 and 190 min (± 10 min) after drug administration using a Boso Medicus Uno device (Jungingen, Germany). Occasionally missing values because of interfering psychometric testing were entered as last observations carried forward.
Pharmacokinetic‐pharmacodynamic relationship
To illustrate the pharmacokinetic‐pharmacodynamic relationship, the mean subjective changes after GHB administration for each time point were plotted against the respective plasma concentrations of GHB and graphed as hysteresis curves. The GHB‐induced effect was determined for each dose as the difference from placebo in the same subject at the corresponding time point to control for circadian changes and endogenous GHB production (placebo condition).
Statistical analysis
Statistical analyses were performed using Statistica 12 (StatSoft, Tulsa, OK, USA). Repeated subjective and vital sign measures are expressed as peak effects (E max). E max values were then compared using repeated‐measures analysis of variance (ANOVA), with drug (placebo vs. GHB) as the within‐subjects factor and dose (low dose vs. high dose) as the between‐subjects factor. A significant main effect of drug indicates a significant difference between drug and placebo in the pooled study sample. A significant dose × drug interaction indicates a significant difference between the low and high doses (significant dose–response). Tukey post hoc tests were based on significant main effects of drug or dose × drug interactions. The criterion for significance was P < 0.05.
Results
Pharmacokinetics
The GHB plasma concentration–time curves for both doses of GHB are shown in Figure 1A. The elimination of GHB followed linear kinetics as shown in Figure 1B. The pharmacokinetic parameters for both doses of GHB are shown in Table 1. t max and t 1/2 values did not differ between the two doses. As expected, the 35 mg kg−1 GHB dose produced significantly higher C max, AUC190, and AUC∞ values compared with the 20 mg kg−1 GHB dose (independent t‐tests: t 30 = 5.08, 6.76, and 6.66, respectively, all P < 0.001). Although the 35 mg kg−1 dose was 1.75‐fold higher than the 20 mg kg−1 dose, it produced two‐fold higher C max values and a 2.5‐fold higher AUC∞ than the 20 mg kg−1 dose, indicating a nonlinear dose–response relationship. Statistical comparisons of the dose‐normalized AUC∞ values revealed a significant difference in exposure to GHB in the two dose groups (t 30 = 2.86, P = 0.008), which would not be the case if the usual linear dose‐exposure relationship existed. No significant difference in GHB C max values was found between the two dose groups (t 30 = 1.16, P = 0.26). Thus, although elimination was relatively linear and no statistically different t 1/2 values were observed for both doses, greater than expected plasma exposure (AUC∞) following administration of the higher dose was found compared with the lower dose (AUC∞ observed vs. AUC∞ expected). Plasma levels of GHB after placebo administration (endogenous GHB) were very low (typically between the level of detection and quantification, 2–6 nmol ml−1) and stable over the six time points (Figure 1A). GHB C max levels after placebo administration were (mean ± SD) 3.8 ± 2.5 nmol ml−1 (range: 0–8.9) and thus more than 100 times lower than after administration of the 35 mg kg−1 GHB dose.
Figure 1.
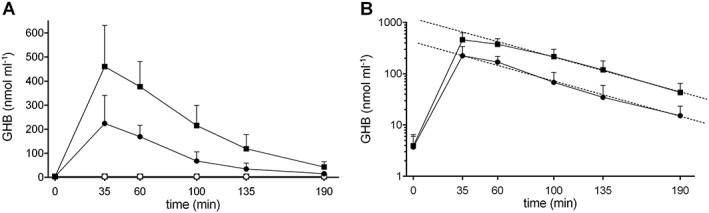
Pharmacokinetics of GHB. (A) The data represent the plasma concentrations of GHB (mean and SD) after GHB administration at doses of 20 or 35 mg kg−1 in 16 subjects for each dose group. (B) The dashed lines in the semi‐log concentration–time plot represent the terminal linear elimination rate (λz). γ‐Hydroxybutyrate was administered at t = 0 min
Table 1.
Pharmacokinetic parameters for GHB in 16 healthy subjects per dose group
| C max (nmol ml −1 ) geometric mean (95% CI) | C max (nmol ml −1 ) (range) | t max (min) median (range) | t 1/2 (min) mean ± SD | t 1/2 (min) (range) | AUC 0–190 (nmol·min ml −1 ) geometric mean (95% CI) | AUC 0–∞ (nmol·min ml −1 ) geometric mean (95% CI) | |
|---|---|---|---|---|---|---|---|
| GHB (20 mg kg −1 ) | 218 (176–270) | 97–481 | 35 (35–60) | only 36 ± 9 | 25–58 | 15 126 (12 331–18 555) | 15 747 (12 854–19 290) |
| GHB (35 mg kg −1 ) | 453 (374–549) ### | 219–843 | 35 (35–60) | 39 ± 7 | 28–53 | 38 039 (31 646–45 723) ### | 40 113 (33 093–48 622) ### |
| GHB (35 mg kg −1 ) dose normalized to 20 mg kg −1 | 259 (214–314) | 125–482 | 21 736 (18 083–26 127) ## | 22 922 (18 911–27 784) ## |
AUC, area under the plasma concentration–time curve; AUC0–∞, AUC from time zero to infinity; AUC0–190, AUC from time 0–190 min; C max, maximum observed plasma concentration; t 1/2, plasma elimination half‐life; t max, time to C max.
for P < 0.01 and
for P < 0.001 indicate significant differences compared with the 20 mg kg−1 dose (independent t‐test).
Subjective effects
The subjective effects of GHB over time are shown in Figure 2. E max values and statistics are shown in Table 2. γ‐Hydroxybutyrate produced a significant general subjective drug effect and significant sedation, stimulation, and dizziness that lasted 2 h. The higher GHB dose produced significantly greater overall effects, subjective sedation, and dizziness but not more stimulation than the lower dose.
Figure 2.
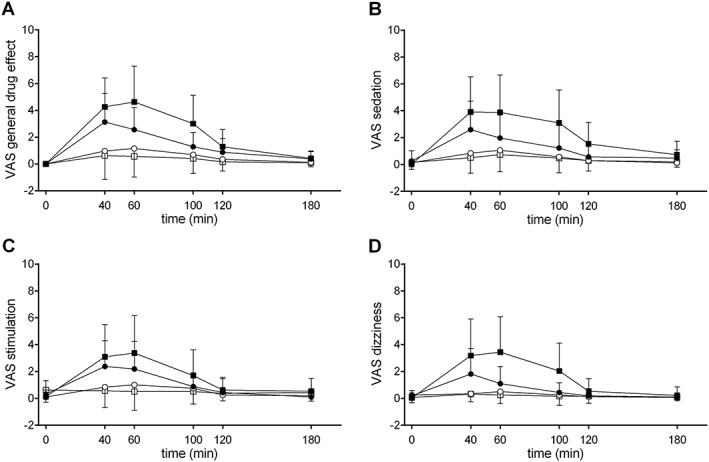
Subjective effects of GHB. (A) γ‐Hydroxybutyrate produced significant general subjective drug effects and increased VAS scores of (B) sedation, (C) stimulation, and (D) dizziness. The higher dose of GHB produced significantly greater sedation and dizziness compared with the lower dose. γ‐Hydroxybutyrate or placebo was administered at t = 0 min. The data are expressed as mean and SD in 16 subjects per group
Table 2.
Values and statistics of maximal drug effects (geometric means and 95% CIs) in 16 subjects per group
| Placebo 1 | GHB (20 mg kg −1 ) | Placebo 2 | GHB (35 mg kg −1 ) | Main effect of drug | Drug × dose interaction | |||
|---|---|---|---|---|---|---|---|---|
| F1,30 | P = | F1,30 | P = | |||||
| Subjective effects (Visual Analog Scale, VAS scores) | ||||||||
| General drug effect | 2.7 (1.2–6.0) | 3.5 (2.6–4.8) * | 2.0 (1.2–3.6) | 4.8 (3.7–6.3) | 46.22 | <0.001 | 5.14 | <0.05 |
| Sedation | 2.1 (1.0–4.4) | 2.7 (1.6–4.5) | 1.2 (0.5–2.9) | 4.1 (2.7–6.2) *** , # | 34.39 | <0.001 | 6.45 | <0.05 |
| Stimulation | 2.1 (1.0–4.4) | 2.7 (1.6–4.5) | 3.3 (1.3–8.4) | 4.2 (2.9–6.0) ** | 85.56 | <0.001 | 1.51 | NS |
| Dizziness | 1.6 (0.9–2.9) | 3.0 (2.1–4.5) | 1.2 (0.5–2.9) | 3.9 (2.7–5.6) *** , ## | 28.63 | <0.001 | 7.06 | <0.01 |
| Vital signs | ||||||||
| Systolic blood pressure (mm Hg) | 132 (126–138) | 140 (132–150) | 139 (130–149) | 146 (136–157) | 3.96 | 0.06 | 0.04 | NS |
| Diastolic blood pressure (mm Hg) | 81 (78–84) | 84 (77–90) | 87 (81–94) | 91 (85–98) | 1.73 | NS | 0.00 | NS |
| Heart rate (beats min −1 ) | 65 (59–73) | 63 (58–69) | 69 (63–77) | 66 (59–72) | 2.02 | NS | 0.13 | NS |
| Rate pressure product (mg Hg ∙ beats min −1 ) | 8484 (7492–9427) | 8760 (7969–9630) | 9406 (8411–10 520) | 9144 (8138–10 275) | 0.00 | NS | 0.38 | NS |
E max values were compared using repeated‐measures analysis of variance (ANOVA), with drug (placebo vs. GHB) as the within‐subjects factor and dose (low dose vs. high dose) as the between‐subjects factor. A significant main effect of drug indicates a significant difference between drug and placebo in the pooled study sample. A significant dose × drug interaction indicates a significant difference between the low and high doses (significant dose–response). Tukey post hoc tests were based on significant main effects of drug or dose × drug interactions.
P < 0.05,
P < 0.01,
P < 0.001 indicate a significant difference between GHB and placebo (Tukey post hoc test).
P < 0.05,
P < 0.01 indicate a significant difference between the 20 mg kg−1 and the 35 mg kg−1 GHB dose (Tukey post hoc test). NS indicates not significant.
Vital signs
No significant effects of GHB on vital signs were observed (Figure 3, Table 2).
Figure 3.
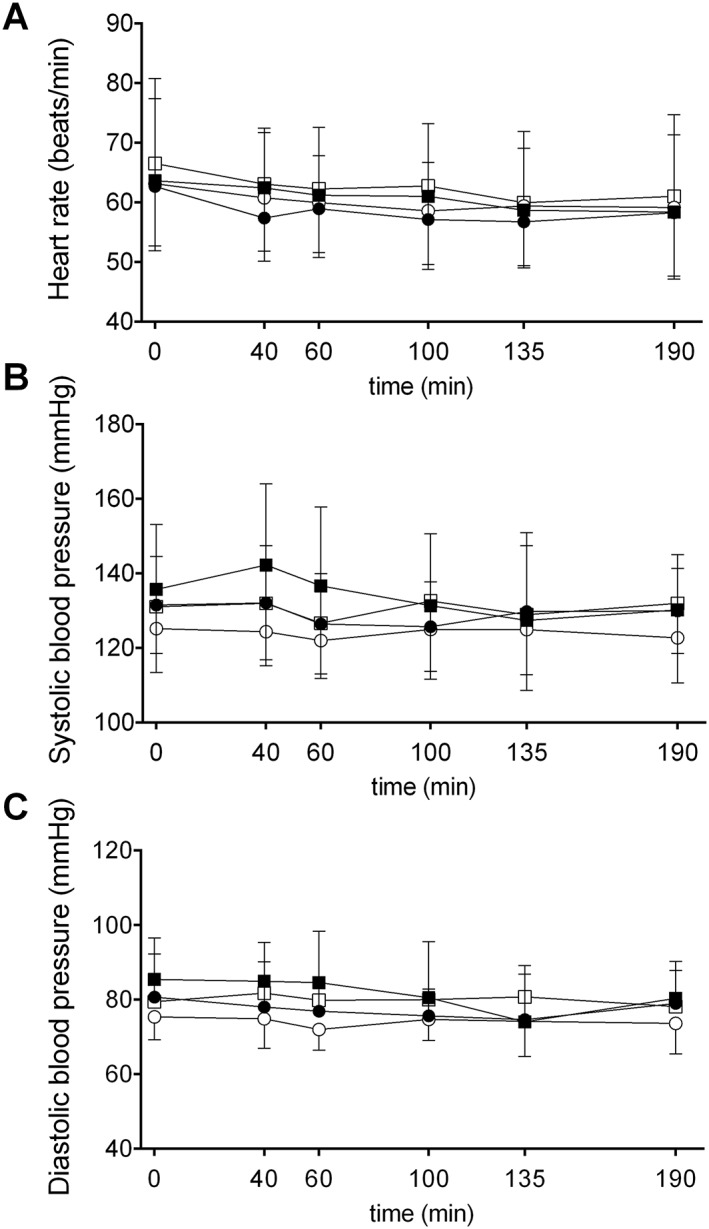
Effects of GHB on vital signs. γ‐Hydroxybutyrate had no significant effects on (A) heart rate, (B) systolic blood pressure, or (C) diastolic blood pressure. γ‐Hydroxybutyrate or placebo was administered at t = 0 min. The data are expressed as mean and SEM in 16 subjects per group
Pharmacokinetic‐pharmacodynamic relationship
The GHB plasma exposure–effect relationships over time (hysteresis curves) for the subjective responses to GHB are shown in Figure 4. No acute pharmacological tolerance to the effects of GHB was observed (i.e., no clockwise hysteresis). Higher concentrations of GHB were generally associated with higher subjective effects of GHB (Figure 4A–D), and maximal subjective sedation (Figure 4B) coincided with maximal plasma levels of GHB 35–60 min after drug administration. Maximal stimulation (Figure 4C) and dizziness (Figure 4D) occurred at 60 min when plasma GHB levels were already declining. This slight counterclockwise hysteresis was consistent with the drug distribution.
Figure 4.
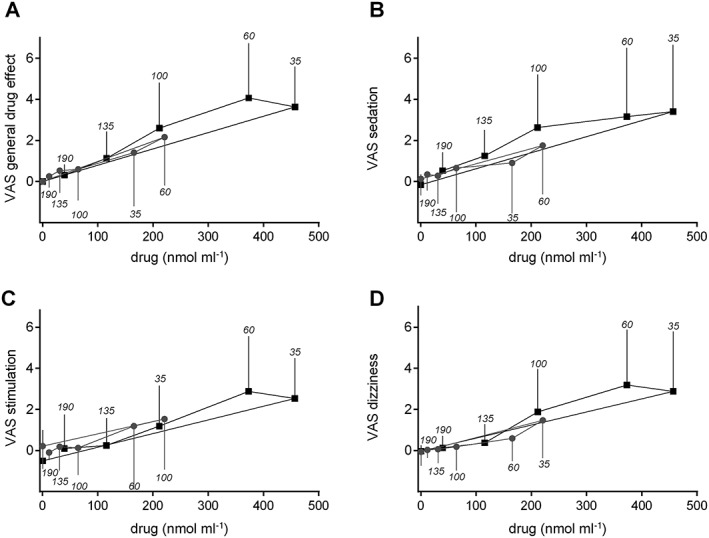
Effects of GHB plotted against GHB plasma concentrations (hysteresis curves). The values are expressed as the mean and SD in 16 subjects per dose group. Separate plots are shown for the two doses of GHB. The time of sampling is noted next to each point in minutes after drug administration. Close relationships were observed between the plasma levels of GHB and its subjective drug effects. (A–D) Overall, higher concentrations of GHB were associated with higher subjective effects of GHB. (B) Maximal subjective sedation coincided with maximal plasma levels of GHB 35–60 min after drug administration. A slight delay was observed for VAS (C) stimulation and (D) dizziness, in which the maximal responses occurred at 60 min when plasma levels were already declining. This slight counterclockwise hysteresis was likely attributable to drug distribution to the effect compartment. No evidence of acute pharmacological tolerance to the effects of GHB was found (i.e., no clockwise hysteresis)
Discussion
The present study assessed the pharmacokinetics and pharmacodynamics of two acute oral GHB doses using a placebo‐controlled study design in a relatively large sample of healthy subjects. The study showed first‐order elimination for two different moderate doses of GHB in healthy subjects, which was consistent with a previous study that similarly used a rather low dose (25 mg kg−1) 10. Elimination was rapid, with an elimination half‐life of approximately 40 min, which was consistent with previous data 10, 11, 13, 14, 15. In contrast, other studies found evidence of nonlinear elimination kinetics, particularly at higher GHB doses (50–60 mg kg−1), with slower early elimination and more rapid later elimination and convex log‐concentration vs. time profiles 9, 16. It has been suggested that the metabolism of GHB becomes saturated if concentrations exceed 25–30 μg ml−1 (243–291 nmol ml−1), thus resulting in zero‐order kinetics 16. In the present study, such higher concentrations were observed only at and shortly after t max, and the concentration–time curves may also reflect continuing absorption and distribution at that time. However, we found that the GHB plasma exposure (AUC∞) rose disproportionally with the higher dose compared with the lower dose, indicating nonlinear dose‐exposure, with approximately 40% greater plasma exposure at the higher dose than expected and as similarly described previously 9. In the present study, the dose‐normalized C max and t 1/2 values non‐significantly increased but were 17% and 8% greater at the higher dose compared with the lower dose, respectively, indicating that the higher plasma exposure to GHB was a result of both disproportionally higher C max and slower elimination. Similarly, a previous study that evaluated doses of 12.5, 25, and 50 mg reported significant increases in dose‐normalized AUCs and t 1/2 9. However, in contrast to our findings, t max values increased and C max values decreased with increasing doses, suggesting the capacity‐limited absorption of GHB 9, an observation that we could not confirm. Notably, however, we obtained only a few samples around t max because the subjects then performed neurocognitive and behavioural tests 17, not allowing for more frequent sampling. This is a limitation of the present study and the C max and t max values obtained are therefore not fully reliable.
Altogether, the clinically most relevant finding of the present study and a previous study 9 was the evidence of nonlinear kinetics already at low to moderate doses of GHB in humans. We and all other previous controlled studies that used relatively low doses of GHB (25–72 mg kg−1) indicate that the rate‐limited elimination process of GHB likely becomes even more relevant in cases of GHB intoxication at much higher doses 16. γ‐Hydroxybutyrate is metabolized to succinic semialdehyde by GHB dehydrogenase 19. Succinic semialdehyde is then further converted to succinic acid by succinic semialdehyde/aldehyde dehydrogenase 20. The nonlinear kinetics of GHB in humans is consistent with capacity‐limited GHB metabolism by these dehydrogenases, as similarly described for ethanol 21, 22, suggesting similar metabolic pathways for GHB and ethanol. Ethanol exposure resulted in a 16% higher C max and 29% longer t 1/2 of GHB, consistent with enhanced bioavailability and reduced clearance caused by ethanol 14, which was also observed with the higher GHB dose in the present study. Thus, concomitant ethanol use has similar effects on the pharmacokinetics of GHB as an increase in dose, thus partially explaining the severe intoxication seen when GHB is used in combination with alcohol 4, 6. Finally, aldehyde dehydrogenase is a polymorphic enzyme that is well known to influence the pharmacokinetics and pharmacodynamics of ethanol in humans 21, 22. Similar genetically determined alterations in exposure to GHB and the drug response remain to be studied in humans.
In the present study, we also newly assessed both the psychotropic and cardiovascular effects of GHB under placebo‐controlled conditions, thereby also determining endogenous GHB levels, and described the pharmacokinetic‐pharmacodynamic relationship. GHB produced weak stimulant effects but also sedation and dizziness that lasted approximately 2 h. Notably, sedation and dizziness were dose‐dependent, whereas the stimulant effects of the higher dose of GHB did not increase compared with the lower dose. Dizziness was also reported in other studies that used controlled GHB administration 9, 11, 12. A reduction of the level of consciousness and deep coma rather than stimulant effects are the consistent hallmarks of GHB intoxication 4, 6, 23, 24. It was previously reported that GHB‐induced feelings of euphoria and stimulation were predominant in the first hour, followed by sedating effects in the second hour after GHB administration 11, 12. We did not corroborate these results in the present study. Consistent with previous studies in humans 10, 11, 14, we found no significant effects of GHB on blood pressure or heart rate. This contrasts with the sympathomimetic stimulant effects of GHB in rats 25. Bradycardia has typically been reported in cases of severe GHB intoxication 6. In the present study, all of the subjective pharmacodynamics effects were closely related to plasma concentrations over time and exhibited no acute pharmacological tolerance over the time of acute intoxication. In contrast, other GABA agonists, such as benzodiazepines, typically show acute tolerance to their sedating and psychomotor effects 26, 27, 28. Determining whether the lack of tolerance to GHB is also present with repeated GHB administration, such as with the long‐term treatment of narcolepsy or GHB dependence, will be an interesting avenue for future studies.
Altogether, we observed a nonlinear dose‐exposure relationship (i.e., no dose proportionality) already at moderate doses of GHB. Additionally, the subjective effects of GHB on consciousness were closely linked to its plasma exposure and exhibited no acute tolerance.
Competing Interest
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: no support from any organization for the submitted work; no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work.
Boris B. Quednow and Matthias E. Liechti were supported by grants from the Swiss National Science Foundation (SNSF; grant nos PP00P1‐123 516/1 and PP00P1‐146 326/1 to BBQ and 32323B_144996 and 320030_149493/1 to MEL).
Liechti, M. E. , Quednow, B. B. , Liakoni, E. , Dornbierer, D. , von Rotz, R. , Gachet, M. S. , Gertsch, J. , Seifritz, E. , and Bosch, O. G. (2016) Pharmacokinetics and pharmacodynamics of γ‐hydroxybutyrate in healthy subjects. Br J Clin Pharmacol, 81: 980–988. doi: 10.1111/bcp.12863.
References
- 1. Snead OC III. Evidence for a G protein‐coupled γ‐hydroxybutyric acid receptor. J Neurochem 2000; 75: 1986–96. [DOI] [PubMed] [Google Scholar]
- 2. Hu RQ, Banerjee PK, Snead OC III. Regulation of γ‐aminobutyric acid (GABA) release in cerebral cortex in the γ‐hydroxybutyric acid (GHB) model of absence seizures in rat. Neuropharmacology 2000; 39: 427–39. [DOI] [PubMed] [Google Scholar]
- 3. Wong CG, Gibson KM, Snead OC III. From the street to the brain: neurobiology of the recreational drug γ‐hydroxybutyric acid. Trends Pharmacol Sci 2004; 25: 29–34. [DOI] [PubMed] [Google Scholar]
- 4. Chin RL, Sporer KA, Cullison B, Dyer JE, Wu TD. Clinical course of γ‐hydroxybutyrate overdose. Ann Emerg Med 1998; 31: 716–22. [PubMed] [Google Scholar]
- 5. Liechti ME, Kupferschmidt H. Gamma‐hydroxybutyrate (GHB) and gamma‐butyrolactone (GBL): analysis of overdose cases reported to the Swiss toxicological information centre. Swiss Med Wkly 2004; 134: 534–7. [DOI] [PubMed] [Google Scholar]
- 6. Liechti ME, Kunz I, Greminger P, Speich R, Kupferschmidt H. Clinical features of gamma‐hydroxybutyrate and gamma‐butyrolactone toxicity and concomitant drug and alcohol use. Drug Alcohol Depend 2006; 81: 323–6. [DOI] [PubMed] [Google Scholar]
- 7. Bosch OG, Quednow BB, Seifritz E, Wetter TC. Reconsidering GHB: orphan drug or new model antidepressant? J Psychopharmacol 2012; 26: 618–28. [DOI] [PubMed] [Google Scholar]
- 8. Ferrara SD, Zotti S, Tedeschi L, Frison G, Castagna F, Gallimberti L, Gessa GL, Palatini P. Pharmacokinetics of gamma‐hydroxybutyric acid in alcohol dependent patients after single and repeated oral doses. Br J Clin Pharmacol 1992; 34: 231–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Palatini P, Tedeschi L, Frison G, Padrini R, Zordan R, Orlando R, Gallimberti L, Gessa GL, Ferrara SD. Dose‐dependent absorption and elimination of gamma‐hydroxybutyric acid in healthy volunteers. Eur J Clin Pharmacol 1993; 45: 353–6. [DOI] [PubMed] [Google Scholar]
- 10. Brenneisen R, Elsohly MA, Murphy TP, Passarelli J, Russmann S, Salamone SJ, Watson DE. Pharmacokinetics and excretion of gamma‐hydroxybutyrate (GHB) in healthy subjects. J Anal Toxicol 2004; 28: 625–30. [DOI] [PubMed] [Google Scholar]
- 11. Abanades S, Farre M, Segura M, Pichini S, Barral D, Pacifici R, Pellegrini M, Fonseca F, Langohr K, De La Torre R. γ‐Hydroxybutyrate (GHB) in humans: pharmacodynamics and pharmacokinetics. Ann NY Acad Sci 2006; 1074: 559–76. [DOI] [PubMed] [Google Scholar]
- 12. Abanades S, Farre M, Segura M, Pichini S, Pastor A, Pacifici R, Pellegrini M, de la Torre R. Disposition of gamma‐hydroxybutyric acid in conventional and nonconventional biologic fluids after single drug administration: issues in methodology and drug monitoring. Ther Drug Monit 2007; 29: 64–70. [DOI] [PubMed] [Google Scholar]
- 13. Brailsford AD, Cowan DA, Kicman AT. Pharmacokinetic properties of γ‐hydroxybutyrate (GHB) in whole blood, serum, and urine. J Anal Toxicol 2012; 36: 88–95. [DOI] [PubMed] [Google Scholar]
- 14. Thai D, Dyer JE, Benowitz NL, Haller CA. Gamma‐hydroxybutyrate and ethanol effects and interactions in humans. J Clin Psychopharmacol 2006; 26: 524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Borgen LA, Okerholm R, Morrison D, Lai A. The influence of gender and food on the pharmacokinetics of sodium oxybate oral solution in healthy subjects. J Clin Pharmacol 2003; 43: 59–65. [DOI] [PubMed] [Google Scholar]
- 16. Jones AW, Eklund A, Kronstrand R. Concentration‐time profiles of gamma‐hydroxybutyrate in blood after recreational doses are best described by zero‐order rather than first‐order kinetics. J Anal Toxicol 2009; 33: 332–5. [DOI] [PubMed] [Google Scholar]
- 17. Bosch OG, Eisenegger C, Gertsch J, von Rotz R, Dornbierer D, Gachet MS, Heinrichs M, Wetter TC, Seifritz E, Quednow BB. Gamma‐hydroxybutyrate enhances mood and prosocial behavior without affecting plasma oxytocin and testosterone. Psychoneuroendocrinology 2015; 62: 1–10. [DOI] [PubMed] [Google Scholar]
- 18. Meyer MR, Weber AA, Maurer HH. A validated GC‐MS procedure for fast, simple, and cost‐effective quantification of glycols and GHB in human plasma and their identification in urine and plasma developed for emergency toxicology. Anal Bioanal Chem 2011; 400: 411–4. [DOI] [PubMed] [Google Scholar]
- 19. Kaufman EE, Nelson T. An overview of γ‐hydroxybutyrate catabolism: the role of the cytosolic NADP+‐dependent oxidoreductase EC 1.1.1.19 and of a mitochondrial hydroxyacid‐oxoacid transhydrogenase in the initial, rate‐limiting step in this pathway. Neurochem Res 1991; 16: 965–74. [DOI] [PubMed] [Google Scholar]
- 20. Malaspina P, Picklo MJ, Jakobs C, Snead OC, Gibson KM. Comparative genomics of aldehyde dehydrogenase 5a1 (succinate semialdehyde dehydrogenase) and accumulation of gamma‐hydroxybutyrate associated with its deficiency. Hum Genomics 2009; 3: 106–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pastino GM, Flynn EJ, Sultatos LG. Genetic polymorphisms in ethanol metabolism: issues and goals for physiologically based pharmacokinetic modeling. Drug Chem Toxicol 2000; 23: 179–201. [DOI] [PubMed] [Google Scholar]
- 22. Quertemont E. Genetic polymorphism in ethanol metabolism: acetaldehyde contribution to alcohol abuse and alcoholism. Mol Psychiatry 2004; 9: 570–81. [DOI] [PubMed] [Google Scholar]
- 23. Liechti ME, Kupferschmidt H. Gammahydroxybutyrate (GHB) and gammabutyrolactone (GBL) poisoning. J Toxicol Clin Toxicol 2004; 42: 758–9. [Google Scholar]
- 24. Galicia M, Nogue S, To‐Figueras J, Echarte JL, Iglesias ML, Miro O. Poisoning by liquid ecstasy (GHB) in hospital emergency departments of Barcelona: a 2‐years study. Med Clin (Barc) 2008; 130: 254–8. [DOI] [PubMed] [Google Scholar]
- 25. Hicks AR, Kapusta DR, Varner KJ. Mechanisms underlying the sympathomimetic cardiovascular responses elicited by γ‐hydroxybutyrate. J Cardiovasc Pharmacol 2004; 44: 631–8. [DOI] [PubMed] [Google Scholar]
- 26. Barbanoj MJ, Urbano G, Antonijoan R, Ballester MR, Valle M. Different acute tolerance development to EEG, psychomotor performance and subjective assessment effects after two intermittent oral doses of alprazolam in healthy volunteers. Neuropsychobiology 2007; 55: 203–12. [DOI] [PubMed] [Google Scholar]
- 27. Kroboth PD, Smith RB, Erb RJ. Tolerance to alprazolam after intravenous bolus and continuous infusion: psychomotor and EEG effects. Clin Pharmacol Ther 1988; 43: 270–7. [DOI] [PubMed] [Google Scholar]
- 28. Ellinwood EH Jr, Heatherly DG, Nikaido AM, Bjornsson TD, Kilts C. Comparative pharmacokinetics and pharmacodynamics of lorazepam, alprazolam and diazepam. Psychopharmacology (Berl) 1985; 86: 392–9. [DOI] [PubMed] [Google Scholar]