

Abstract
Aims
Trastuzumab, an antibody binding to epidermal growth factor receptor‐2 (HER2), has been approved to treat HER2‐positive breast cancer in different settings. This study aimed at evaluating the influence of tumour size on trastuzumab pharmacokinetics (PK) in non‐metastatic breast cancer patients treated with short term pre‐operative trastuzumab.
Methods
Trastuzumab PK data were obtained from a multicentre, randomized and comparative study. This antibody was administered pre‐operatively to patients with localized HER2‐positive breast cancer as a single 4 mg kg−1 loading dose followed by 5 weekly 2 mg kg−1 doses. Trastuzumab concentrations were measured repeatedly using an ELISA technique. Tumour size was evaluated at baseline using breast echography. Trastuzumab pharmacokinetics were studied using a population approach and a two compartment model. The influence of tumour burden on trastuzumab pharmacokinetics was quantified as a covariate.
Results
A total of 784 trastuzumab concentrations were available from the 79 eligible patients. Estimated parameters (interindiviual standard deviation) were central volume of distribution =2.1 l (23%), peripheral volume of distribution =1.3 l (38%), intercompartment clearance =0.36 l day−1, with an elimination half‐life of 11.8 days. Typical clearance was 0.22 l day−1 (19%) and its value was increased with tumour size. In patients with the highest tumour size, trastuzumab clearance was 50% [18%–92%] higher than in patients with the lowest tumour size.
Conclusions
In non‐metastatic breast cancer patients, trastuzumab clearance increases with tumour size. The elimination half‐life of trastuzumab was shorter in the present population of patients than in metastatic breast cancer patients previously studied.
Keywords: breast cancer, pharmacokinetics, population PK modelling, trastuzumab
What is Already Known about this Subject
Trastuzumab pharmacokinetics were described in metastatic breast cancer patients using pharmacokinetic modelling, the trastuzumab elimination half‐life being 28 days.
Trastuzumab pharmacokinetics were reported to be influenced by the circulating part of target antigen.
The influence of tumour burden was reported for several monoclonal antibodies, as anti‐CD20 antibodies and cetuximab, but never for trastuzumab.
What this Study Adds
This study is the first to describe trastuzumab pharmacokinetics in non‐metastatic breast cancer patients.
The pharmacokinetics of trastuzumab may be different in non‐metastatic than in metastatic breast cancer patients. Notably, in this study, elimination half‐life was around 12 days.
This study suggested that tumour size increases target‐mediated clearance of trastuzumab.
Introduction
Trastuzumab (Herceptin®) is a humanized monoclonal antibody that targets the human epidermal growth factor receptor type 2 (HER2), for which overexpression is a poor prognostic factor in breast cancers 1. Trastuzumab blocks HER2 mediated signalling and induces antibody‐dependent cell cytotoxicity (ADCC) through Fc gamma receptors 2. Trastuzumab is approved for the treatment of women with HER2‐positive metastatic breast cancer as a single agent or in combination with chemotherapy and in the adjuvant setting after surgery, and HER2‐positive gastric cancer.
The pharmacokinetics of trastuzumab in HER2‐positive metastatic breast cancer patients were analyzed in five main studies which reported controversial results. An early study reported an elimination half‐life of approximately 10 days 3 but more recent studies reported half‐lives between 16 and 28 days 4, 5, 6, 7, 8. Using population pharmacokinetic modelling, trastuzumab concentrations were described using a two compartment model with first order transfer and elimination rate constants 5, 6. In patients with advanced gastric or gastroesophageal junction cancer, trastuzumab elimination was described using a two compartment model with linear and non‐linear terms 9.
The amount of target antigen was shown to influence trastuzumab pharmacokinetics as its clearance increased with the serum concentration of HER2 extra‐cellular domain (ECD) 5, 6, 9. Such an influence of target antigen on the pharmacokinetics of monoclonal therapeutic antibodies in humans was reported for several of them, including omalizumab (anti‐IgE) 10 and infliximab (anti‐TNF‐α) 11, 12. Paradoxically, there are few human studies on such an influence for anticancer antibodies. An influence of tumour volume on the pharmacokinetics of rituximab (anti‐CD20) was observed in a murine model 13. In the case of trastuzumab, concentrations of circulating HER2‐ECD may be influenced by tumour size but may not be representative of total HER2 burden. No study has reported an analysis of the influence of tumour burden on trastuzumab pharmacokinetics. In addition, trastuzumab pharmacokinetics in non‐metastatic cancer patients have never been reported.
The aim of the present study was to analyze trastuzumab pharmacokinetics in patients with localized disease. Data from the RADHER study were analyzed to determine trastuzumab pharmacokinetic parameters and to study individual factors explaining their between‐subject variability, notably tumour size.
Methods
Study design
This ancillary study was part of a prospective, phase II, open randomized and multicentre study (RADHER, ClinicalTrials.gov identifier: NCT00674414) that enrolled patients between June 2008 and February 2012. This study was approved by the regional ethics committee and all participants gave written informed consent at the time of enrolment. The primary objective of this study was to assess the interest of adding everolimus to trastuzumab as pre‐operative therapy of HER‐2 positive primary breast cancer amenable to surgery using clinical tumour evaluations and biological investigations.
Eligible patients (≥18 years old) had a histologically‐confirmed diagnosis of invasive breast cancer, previously untreated, with HER‐2 positive primary tumour, defined as IHC 3+ or IHC 2+ and fluorescence in situ hybridization (FISH) positive (centrally confirmed), clinical stage M0 (bone scan, chest X‐ray, liver ultrasound required at screening to exclude metastatic disease), a WHO performance status of 1 or less and satisfactory haematologic parameters. Main exclusion criteria were inflammatory breast cancer, metastatic disease, concomitant radiotherapy, chemotherapy and/or patient candidate for neoadjuvant chemotherapy. A central histological review of HER2 status was performed by a reference pathologist at each investigational site to confirm the eligibility of the patients before randomization. Of the 84 patients included in the RADHER trial, 79 patients were eligible for this post hoc analysis.
Trastuzumab (Herceptin® 150 mg i.v., Roche, Basel, Switzerland) was used. Patients were randomly assigned to trastuzumab alone (TE–) or to the combination of trastuzumab and everolimus (TE+) arm. In the TE+ arm, everolimus was administered at the standard dose of 10 mg once daily.
In both arms of the study, at week 0, a 4 mg kg−1 dose of trastuzumab was administered as a 90 min infusion. Between weeks 1 and 5, 2 mg kg−1 doses of trastuzumab were administered weekly as 30 min infusions.
Data
Trastuzumab concentrations
Between weeks 0 and 5, blood samples were collected to measure trastuzumab serum concentrations before each trastuzumab infusion and after 1 h, 1 day, 3 days, and at 1, 2, 4, 8 and 12 weeks after the last infusion of trastuzumab. Serum trastuzumab concentrations were determined using a validated ELISA test. The limit of detection was 0.072 mg l−1, lower (LLOQ) and upper (ULOQ) limit of quantitation were 0.24 and 15 mg l−1, respectively. Quality controls were 0.5, 4.7 and 15 mg l−1, respectively. Corresponding inter‐assay precisions and bias were <20%.
Tumour size measurements
For all patients, tumour size was evaluated using WHO criteria. Length and width were the largest diameter and greatest perpendicular diameter, measured by breast echography at baseline and within week before the surgical procedure 14.
HER2‐ECD measurements
Measurements of HER2‐ECD were performed using the Human sHER‐2 Platinum ELISA kit (reference BMS207CE), Affymetrix® eBioscience, San Diego, CA, USA.
Pharmacokinetic analysis
Population model development
Pharmacokinetic data were analyzed by a population approach using the non‐linear mixed effects program MONOLIX 4.3.2 software (Lixoft, Orsay, France) which combines the stochastic expectation‐maximization (SAEM) algorithm and a Markov Chain Monte‐Carlo procedure for likelihood maximization. The number of iterations for K1 and K2 (K1 and K2 being iteration kernels 1 and 2) were 600 and 300, respectively. Two Markov chains were used and simulated annealing was used to improve the convergence of the SAEM algorithm towards the global maximum of the likelihood. The random seed was changed between each of the three runs. Fisher information matrix was computed using stochastic approximation. The objective function (OF), which is the –2Ln likelihood (−2LL), was computed using importance sampling.
Structural PK model design
Trastuzumab concentrations were described using compartmental pharmacokinetic models. One, two and three mammillary models with first order distribution constants were tested. Linear and non‐linear (Michaelis–Menten) eliminations were also tested. Structural models were compared using Akaike's information criterion (AIC), defined as: AIC = OFV + 2.p, where OFV is OF value and p is the number of model parameters to estimate. The model with the lowest AIC was selected.
Interindividual model
The interindividual variability of pharmacokinetic parameters was described using an exponential model: θi = θTV. exp (ηi), where θi is the estimated individual parameter, θTV is the typical value of the parameter and ηi is the random effect for the ith patient. The values of ηi were assumed to be normally distributed with mean 0 and variance ω2. For each parameter, ω2 was fixed to 0 if ω2 or ηi could not be estimated with sufficient precision.
Error model
Additive, proportional and mixed additive‐proportional models were tested. For example, the combined additive‐proportional model was implemented as follows: C O,ij = C P,ij.(1+ εprop,ij) + εadd,ij where C O,ij and C P,ij are observed and predicted jth concentrations for the ith patient, respectively and εprop,ij and εadd,ij are proportional and additive errors, with mean 0 and variances σprop 2 and σadd 2, respectively.
Model goodness‐of‐fit and evaluation
The goodness–of‐fit was assessed for each model by plotting population‐predicted (PRED) and individually predicted (IPRED) concentrations vs. observed concentrations (DV) and IPRED and DV vs. time, and by evaluating the residuals by graphical inspection of population (PWRES) and individual (IWRES) weighted residual distributions, and normalized prediction distribution errors (NPDE) 15.
Covariates
Age, body weight, serum creatinine, tumour size at baseline and circulating HER2‐ECD concentrations were tested as continuous covariates, whereas everolimus cotreatment was tested as a dichotomous covariate. Since two measures of tumour size (length and width) were available, the influence of tumour size was tested using three strategies, (i) the largest measure between tumour length and width (LLW), (ii) the sum of tumour length and width (SUM) and (iii) the product of tumour length and width (AREA).
The influence of a dichotomous covariate (CAT) on θTV was implemented as ln(θTV) = ln(θCAT=0) + βCAT=1, where θCAT=0 is the value of θ for an arbitrary reference category and βCAT=1 is the value of θTV for the other category. Continuous covariates (COV) were centred on their median as follows: θi = θ0.(COV/med(COV))βcov, where θ0 is value of θ for a median subject, βCOV quantifies the influence of COV on θ and med(COV) is the median value of COV in the population.
From pairs of nested models, the one with the lowest OFV was chosen. This was assessed by a likelihood ratio test (LRT) in which the difference in OFV between two models (ΔOFV) is assumed to follow a χ2 distribution. The influence of patient characteristics (covariates) was assessed in two steps:
Univariate step. The influence of each factor on pharmacokinetic parameters associated with interindividual variability was tested. Covariates were separately included into the base model. Covariates showing a significant influence (α < 0.1) were included in the model (full model).
Multivariate step. A backward stepwise elimination was performed: the covariates of the full model were removed one by one. Covariates whose removal resulted in a statistically significant increase in the OFV (α < 0.01) were retained in the model.
Management of missing data
No values were missing for age, body weight, serum creatinine and everolimus cotreatment. For tumour size, two (3%) and 13 (16%) values were missing for length and width, respectively. No patient had both baseline tumour length and width missing. If one tumour measurement was missing, LLW was set as the available tumour size measurement, and SUM and AREA were respectively set as twice and the squared value of this measurement.
Samples for HER2‐ECD were available for 50 patients, 37% were missing. HER2‐ECD was therefore managed as a covariate into three analyses:
PK modelling in the subgroup of 50 patients for which baseline HER2‐ECD concentration measurements were available;
PK modelling of all patients, where missing HER2‐ECD values were imputed as median HER2‐ECD concentration in 50 patients;
PK modelling of all patients, where missing HER2‐ECD values were imputed using multiple imputation.
Simulations
To show the quantitative influence of tumour burden on trastuzumab pharmacokinetics, five trastuzumab pharmacokinetic profiles were simulated with increasing AREA values (50, 500, 1000, 2000 and 5000 mm2) and using typical parameters for a median‐weighted patient (61 kg). Dosing regimen was 4 mg kg−1 dose at week 0, then 2 mg kg−1 weekly between weeks 1 and 5.
Results
Of the 79 patients analyzed in this study, 784 trastuzumab concentration measurements were available (Table 1). Trastuzumab concentrations were best described using a two compartment model with first order transfer and elimination rate constants, as in previous studies in breast cancer 5, 6. Neither parameters quantifying a third compartment nor a non‐linear elimination were identifiable. The best residual model was mixed additive‐proportional. Pharmacokinetic parameters were estimated with good accuracy (Table 2). Estimated parameters (interindividual standard deviation) were central volume of distribution (V 1) = 2.1 l (23%), clearance (CL) = 0.22 l day–1 (19%) peripheral volume of distribution (V 2) = 1.3 l (38%) intercompartment clearance (Q) = 0.36 l day–1, distribution and elimination half‐lives were t ½,λ1 = 1.4 days and t ½,λz = 11.8 days, respectively (Table 2). Plots of predicted vs. observed concentrations showed that the pharmacokinetic model described the data satisfactorily (Figure 1). Population and individual residuals and normalized prediction distribution error (NPDE) plots showed that there was no obvious model misspecification (Figure 2). Notably, the distribution of NPDE was not significantly different from a Gaussian distribution (Kolmogorov–Smirnov test, KS = 3%, P = 0.076).
Table 1.
Baseline patient characteristics
| Patients (n = 79) | |
|---|---|
| Age (years) | 51 [25–78]* |
| Body weight (kg) | 61 [48–147]* |
| Everolimus comedication | 34 (44)** |
| Serum creatinine (μmol l −1 ) | 13.5 [12.8–14.4]* |
| Echographic tumour length (mm) | 19 [7–70]* |
| Echographic tumour width (mm) | 17 [7–80]* |
| LLW (mm) | 19 [7–80]* |
| SUM (mm) | 35 [11–150]* |
| AREA (mm 2 ) | 306 [49–5600]* |
LLW, largest measure between tumour length and width; SUM, sum of tumour length and width tumour measurements; AREA, product of tumour length and width tumour measurements.
Expressed as median [min, max].
Expressed as number of patients (%).
Table 2.
Estimated pharmacokinetic parameters
| Parameter (unit) | Estimate | RSE (%) |
|---|---|---|
| V 1 (l) | 2.1 | 4 |
| WT on V 1 | 0.70 | 23 |
| AGE on V 1 | −0.36 | 26 |
| CL (l day –1 ) | 0.22 | 2 |
| WT on CL | 0.56 | 19 |
| AREA on CL | 0.089 | 32 |
| V 2 (l) | 1.3 | 6 |
| Q (l day –1 ) | 0.36 | 4 |
| ω V1 (%) | 23 | 14 |
| ω CL (%) | 19 | 9 |
| ω V2 (%) | 38 | 14 |
| ω Q (%) | – | – |
| σ C,add (mg l −1 ) | 0.83 | 16 |
| σ C,prop (%) | 0.11 | 3 |
AGE, age (years); AREA, product of tumour length and width measured by breast echography; CL, clearance from the central compartment; V 1, central volume of distribution; V 2, peripheral volume; Q, intercompartment clearance; WT, body weight (kg); ω, interindividual standard deviation; σadd, additive error standard deviation; σprop, proportional error standard deviation.
Figure 1.
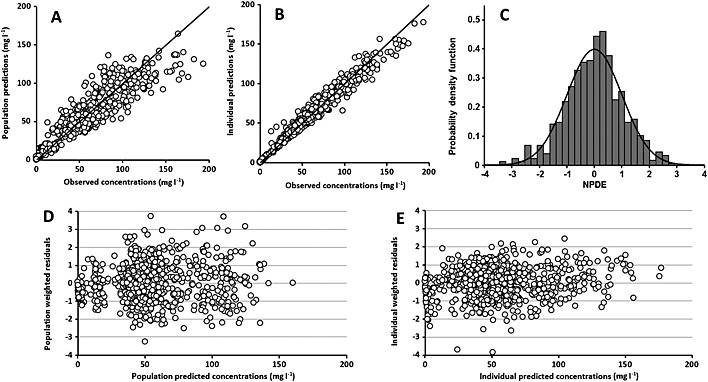
Diagnostic charts for trastuzumab concentrations. Observed values vs. population model‐predicted values (A) and individual model‐predicted values (B), distribution of normalized prediction distribution error (NPDE) vs. Gaussian distribution (C), distribution of population weighted residuals vs. population predicted concentrations (D) and distribution of individual weighted residuals vs. individual predicted concentrations (E)
Figure 2.
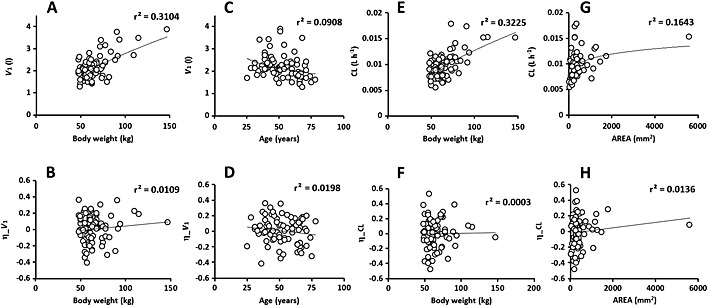
Values of individual pharmacokinetic parameters (above) and random effects (η, below), central volume of distribution (V 1) vs. body weight (A) and age (C), and η_V 1 vs. body weight (B) and age (D), clearance (CL) vs. body weight (E) and the product between tumour length and width measurements (AREA, G), and η_CL vs. body weight (F) and AREA (H)
The interindividual variability in pharmacokinetic parameters ranged between 19% and 38%. The interindividual variance of Q was not estimable and was therefore fixed to 0. During the univariate step, V 1 was found to be influenced by age, body weight and tumour size, whereas CL was influenced by body weight, serum creatinine and tumour size. Everolimus cotreatment did not significantly influence the pharmacokinetics of trastuzumab. In the final model, V 1 decreased with age (LRT = 6.9, P = 0.0085) and increased with body weight (LRT = 18.9, P = 1.4. 10−5), whereas CL increased with body weight (LRT = 27.6, P = 1.5. 10−7) and tumour size. Clearance increased with (LRT = 5.8, P = 0.016, AIC = 7154), SUM (LRT = 9.5, P = 0.0002) and AREA (LRT = 9.4, P = 0.002, AIC = 7151). The covariates SUM and AREA led to similar decrease in –2LL. Yet, AREA provided a small (even if non‐significant) advantage compared with SUM and was considered as the best tumour size covariate. Baseline circulating HER2‐ECD concentration was not significantly associated with trastuzumab pharmacokinetics in the 50 patient subgroup where these concentrations were available, or after imputation of missing values, either with median HER2‐ECD concentration or with multiple imputation. Of note, the final model in the subgroup was the same as in all patients (notably for AREA, LRT = 5.2, P = 0.023).
When body weight increased from 50 to 90 kg, V 1 and CL were increased by 50% (33% – 71%) and 40% (22% – 58%), respectively. When age increased from 30 to 60 years, V 1 was decreased by 22% (7% – 35%). When AREA increased from 50 to 5000 mm2, CL was increased by 50% (18% – 92%), and elimination half‐life was decreased from 13.7 to 11.7 days. This increase in AREA led to a decrease in trastuzumab exposure (Figure 3).
Figure 3.
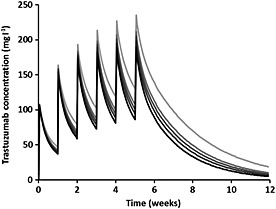
Simulations of four trastuzumab pharmacokinetic profiles using typical pharmacokinetic parameters and increasing baseline product between tumour length and width measurements (AREA): 50 ( ), 500 (
), 500 ( ), 1000 (
), 1000 ( ), 2000 (
), 2000 ( ) and 5000 (
) and 5000 ( ) mm2
) mm2
Discussion
This is the first study showing an influence of tumour size on the pharmacokinetics of trastuzumab in humans. In our study, trastuzumab concentrations were satisfactorily described by a two compartment model and pharmacokinetic parameters were reliably estimated. In addition, to our knowledge, this is the first study where trastuzumab pharmacokinetics were evaluated in non‐metastatic breast cancer patients. The observed increase of volume of distribution and clearance of trastuzumab with body weight is in agreement with previous studies in other conditions 5, 6, 9. The influence of age on clearance has never been reported for trastuzumab, but has been reported for efalizumab, an anti‐CD11a monoclonal antibody 16. However, unlike the present study, efalizumab clearance was reported to increase with age.
We observed an increase in trastuzumab clearance with tumour size. The influence of tumour burden was assessed using either the largest measure of length or width (LLW), the sum or the product of length and width (AREA). Even if SUM and AREA are difficult to interpret as a tumour burden measurement, these biomarkers brought more information about the influence of tumour burden on trastuzumab clearance than LLW. Indeed, LRT tests for the influence of SUM or AREA on CL was more significant than for LLW (for SUM and AREA, LRT tests were 9.5 and 9.4, respectively, and for LLW, LRT = 5.8, P = 0.016). These results suggest that both length and width bring information on the variability of trastuzumab pharmacokinetics.
The extracellular domain of HER2 (HER2‐ECD) is the result of a proteolytical cleavage from the tumour cell surface and is a small fraction of the total load of HER2 receptor. Bruno et al. found a modest increase of trastuzumab volume of distribution and clearance with circulating concentrations of HER2‐ECD 5. In the present study, baseline tumour size and HER2‐ECD concentrations were not correlated (data not shown) and HER2‐ECD concentrations were not significantly associated with trastuzumab pharmacokinetics. This may be explained by (i) a low amount of circulating antigenic targets: indeed, the present study was made in patients with primary non‐metastatic breast cancer, whereas Bruno et al. assessed patients with metastatic tumours with HER2‐ECD concentrations higher than in the present study (9.33 ng ml−1 and 5.20 ng ml−1, respectively) and (ii) a lack of power, since baseline HER2‐ECD concentrations were available in 50 patients (476 patients were assessed by Bruno et al. 5). Nevertheless, these results suggest that tumour size may provide more information on trastuzumab pharmacokinetic variability than circulating HER2‐ECD concentrations, even if this marker may not bring redundant information of trastuzumab pharmacokinetic variability.
The influence of tumour size on trastuzumab clearance may be explained by target‐mediated drug disposition (TMDD) 17, a mechanism of elimination frequently reported for monoclonal antibodies 18, 19, 20. In patients with large tumour burden, trastuzumab clearance is high because of its capture by HER2 expressed on tumour cells. However, TMDD models could not be tested because trastuzumab elimination was linear. Under the quasi‐steady‐state equilibrium hypothesis, the non‐linear elimination can be written using a Michaelis–Menten term as follows: Vm. C/(K m + C) where Vm is the maximum elimination rate, K m is the Michaelis constant and C is trastuzumab concentration. When C decreases, the influence of Michaelis–Menten elimination on global elimination decreases. Using this model, tumour size (measured as AREA) should influence Vm and/or K m. However, since both Vm and K m cannot be identified for linear pharmacokinetics, the influence of tumour burden may be deported on CL. This influence on CL is an approximation of its influence on Vm. Indeed, CL is a first order parameter, unlike Vm and K m.
In metastatic breast cancer patients, a previous study using population PK modelling reported an estimated elimination half‐life of 28.5 days, which was estimated using data from several studies. In addition, several studies reported variable estimations of elimination half‐life: around 10 days 3, 16 days 4, 20 days 8, 23 days 6 and 28.5 days 5. Except for the Tokuda et al. study 3, elimination half‐life estimates were similar to the value for endogenous IgG1 21. Surprisingly, in our study, this half‐life was shorter (around 12 days) than in the values reported by the two population modelling PK studies (23 days 6 and 28 days 5). Important differences in estimated half‐lives were also reported for rituximab, an anti‐CD20 monoclonal antibody used in B lymphocyte malignancies. For rituximab, the elimination half‐life was found to be around 21 days in follicular non‐Hodgkin lymphoma patients 22 and rheumatoid arthritis 23, but was 37 days in diffuse large B‐cell lymphoma patients 24. This difference may be explained by different target antigen turnover between diseases.
The PK study of trastuzumab reported by Bruno et al. 5 was performed in metastatic breast cancer patients, who had a higher tumour burden than the patients of our study who had no metastatic disease. This study reported similar elimination rate (k 10) and transfer from central to peripheral rate (k 12) constants to what we estimated in the present study (k 10 = 0.08 day−1 vs. 0.10 day−1, respectively, and k 12 = 0.17 day−1 in both studies), but the transfer from peripheral to central compartment rate constant (k 21) of the Bruno et al. study was lower than that estimated in the present study (0.10 day−1 vs. 0.28 day−1). These surprising results might be explained by TMDD kinetics. Because the full TMDD model could not be used, the estimated peripheral compartment may represent not only peripheral distribution of trastuzumab, but also its interaction with target antigen. Therefore, k 12 and k 21 may be influenced by TMDD parameters, and a lower value of k 21 may be interpreted as a lower release of unbound trastuzumab from antigen–antibody complexes. This lower release may be due to the retention of trastuzumab by a high target‐antigen burden, resulting in a longer elimination half‐life, associated with lower concentrations of unbound circulating trastuzumab. However, this hypothesis needs to be confirmed. Overall, an increase in tumour burden may result of (i) an increase in target‐mediated clearance of trastuzumab and (ii) an increase in its elimination half‐life, with decreased concentrations of trastuzumab.
In addition, Michaelis–Menten elimination of trastuzumab was reported in advanced gastric or gastroesophageal junction cancer patients by Cosson et al. 9 In this study, the non‐linear elimination pathway becomes preponderant compared with the endogenous (linear) pathway when trastuzumab concentrations are lower than 25 mg l−1, whereas, in the present study, non‐linear elimination was not identifiable, even for concentrations lower than this threshold. This could be explained by (i) a higher amount of antigenic targets in gastric or gastroesophageal cancer patients than in non‐metastatic breast cancer patients and (ii) the weekly dosing regimen of the present study which lead to higher trastuzumab trough concentrations compared with the 3‐weekly schedule used in gastric or gastroesophageal cancers. Indeed, the higher the concentrations, the lower the contribution of the non‐linear elimination pathway. In the present study, it cannot be claimed that non‐linear elimination does not exist. Indeed, approximately 30% of patients were not sampled until week 12 which may have prevented from non‐linear parameter identification.
Overall, the pharmacokinetic implications of the interaction between trastuzumab and target antigen burden may lead to target‐mediated elimination (because of the presence of a non‐linear elimination pathway 9) and/or trastuzumab sequestration (because of increased volumes of distribution and elimination half‐life in the study of Bruno et al. 5 compared with the present study). These different target‐mediated kinetic patterns may be due not only to the amount of available target antigens, but also the distribution, density and turnover of these targets, and differences in trastuzumab‐target complex clearances.
In conclusion, our study is the first to describe trastuzumab pharmacokinetics in non‐metastatic breast cancer patients and shows a potential influence of tumour size on trastuzumab pharmacokinetics. The difference in pharmacokinetic parameters as compared with previous studies might be due to differences in target‐antigen turnover.
Competing Interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf and declare MC, JLM, VGG, TB, FL, KR, MA, VD, MJ and GP had no support from any organization for the submitted work, DB was granted by Cancen, Tours, France, GB, VGG, FL, KR and DT had no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years, MC reports grants from Novartis, JLM reports speaker activity for Roche, TB reports grants, personal fees and non‐financial support from Roche and Novartis, MA reports personal fees from Novartis, VD reports honoraria for advisory boards and speaker bureau Roche Genentech, and Novartis MJ reports grants and non‐financial support from Roche and Novartis and GP reports grants from Roche Pharma and Novartis, MC, JLM, VGG, TB, FL, KR, MA, VD, MJ and DT had no other relationships or activities that could appear to have influenced the submitted work. DB reports grants from Janssen during the conduct of the study and GP reports grants from MSD, Servier and Pfizer.
This study was sponsored by UniCancer, Cancen (Tours, France) and supported by Roche® and Novartis®. Measurements of trastuzumab serum concentrations were carried out within the CePiBAc platform. CePiBAc was cofinanced by the European Union. Europe is committed to the region Centre with the European Regional Development Fund. This work was partly supported by the French Higher Education and Research ministry under the programme ‘Investissements d'avenir’ Grant Agreement: LabEx MAbImprove ANR‐10‐LABX‐53‐01. The authors thank Anne‐Claire Duveau and Caroline Brochon for technical assistance with trastuzumab assays.
Bernadou, G. , Campone, M. , Merlin, J.‐L. , Gouilleux‐Gruart, V. , Bachelot, T. , Lokiec, F. , Rezai, K. , Arnedos, M. , Diéras, V. , Jimenez, M. , Paintaud, G. , and Ternant, D. (2016) Influence of tumour burden on trastuzumab pharmacokinetics in HER2 positive non‐metastatic breast cancer. Br J Clin Pharmacol, 81: 941–948. doi: 10.1111/bcp.12875.
Mario Campone is the principal investigator
References
- 1. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER‐2/neu oncogene. Science 1987; 235: 177–82. [DOI] [PubMed] [Google Scholar]
- 2. Hudis CA. Trastuzumab–mechanism of action and use in clinical practice. N Engl J Med 2007; 357: 39–51. [DOI] [PubMed] [Google Scholar]
- 3. Tokuda Y, Watanabe T, Omuro Y, Ando M, Katsumata N, Okumura A, Ohta M, Fujii H, Sasaki Y, Niwa T, Tajima T. Dose escalation and pharmacokinetic study of a humanized anti‐HER2 monoclonal antibody in patients with HER2/neu‐overexpressing metastatic breast cancer. Br J Cancer 1999; 81: 1419–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baselga J, Carbonell X, Castaneda‐Soto NJ, Clemens M, Green M, Harvey V, Morales S, Barton C, Ghahramani P. Phase II study of efficacy, safety, and pharmacokinetics of trastuzumab monotherapy administered on a 3‐weekly schedule. J Clin Oncol 2005; 23: 2162–71. [DOI] [PubMed] [Google Scholar]
- 5. Bruno R, Washington CB, Lu JF, Lieberman G, Banken L, Klein P. Population pharmacokinetics of trastuzumab in patients with HER2+ metastatic breast cancer. Cancer Chemother Pharmacol 2005; 56: 361–9 Epub 2005 May 3. [DOI] [PubMed] [Google Scholar]
- 6. Fukushima Y, Charoin J, Brewster M, Jonsson E. Population pharmacokinetic analysis of trastuzumab (Herceptin®) based on data from three different dosing regimens. PAGE 2007; 16: Abstract 1121. Available at [www.page‐meeting.org/?abstract=21] (last accessed 09 February 2016).
- 7. Leyland‐Jones B, Colomer R, Trudeau ME, Wardley A, Latreille J, Cameron D, Cubedo R, Al‐Sakaff N, Feyereislova A, Catalani O, Fukushima Y, Brewster M, Cortes J. Intensive loading dose of trastuzumab achieves higher‐than‐steady‐state serum concentrations and is well tolerated. J Clin Oncol 2010; 28: 960–6. [DOI] [PubMed] [Google Scholar]
- 8. Leyland‐Jones B, Gelmon K, Ayoub JP, Arnold A, Verma S, Dias R, Ghahramani P. Pharmacokinetics, safety, and efficacy of trastuzumab administered every three weeks in combination with paclitaxel. J Clin Oncol 2003; 21: 3965–71 Epub 2003 Sep 24. [DOI] [PubMed] [Google Scholar]
- 9. Cosson VF, Ng VW, Lehle M, Lum BL. Population pharmacokinetics and exposure‐response analyses of trastuzumab in patients with advanced gastric or gastroesophageal junction cancer. Cancer Chemother Pharmacol 2014; 73: 737–47. [DOI] [PubMed] [Google Scholar]
- 10. Hayashi N, Tsukamoto Y, Sallas WM, Lowe PJ. A mechanism‐based binding model for the population pharmacokinetics and pharmacodynamics of omalizumab. Br J Clin Pharmacol 2007; 63: 548–61 Epub 2006 Nov 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ternant D, Berkane Z, Picon L, Gouilleux‐Gruart V, Colombel JF, Allez M, Louis E, Paintaud G. Assessment of the influence of inflammation and FCGR3A genotype on infliximab pharmacokinetics and time to relapse in patients with Crohn's disease . Clin Pharmacokinet 2014; 2014: 17. [DOI] [PubMed] [Google Scholar]
- 12. Ternant D, Ducourau E, Perdriger A, Corondan A, Le Goff B, Devauchelle‐Pensec V, Solau‐Gervais E, Watier H, Goupille P, Paintaud G, Mulleman D. Relationship between inflammation and infliximab pharmacokinetics in rheumatoid arthritis. Br J Clin Pharmacol 2013; 2013: 12313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dayde D, Ternant D, Ohresser M, Lerondel S, Pesnel S, Watier H, Le Pape A, Bardos P, Paintaud G, Cartron G. Tumor burden influences exposure and response to rituximab: pharmacokinetic ‐ pharmacodynamic modelling using a syngeneic bioluminescent murine model expressing human CD20. Blood 2008; 24: 24. [DOI] [PubMed] [Google Scholar]
- 14. Miller AB, Hoogstraten B, Staquet M, Winkler A. Reporting results of cancer treatment. Cancer 1981; 47: 207–14. [DOI] [PubMed] [Google Scholar]
- 15. Comets E, Brendel K, Mentre F. Computing normalised prediction distribution errors to evaluate nonlinear mixed‐effect models: the npde add‐on package for R. Comput Methods Programs Biomed 2008; 90: 154–66 Epub 2008 Jan 22. [DOI] [PubMed] [Google Scholar]
- 16. Sun YN, Lu JF, Joshi A, Compton P, Kwon P, Bruno RA. Population pharmacokinetics of efalizumab (humanized monoclonal anti‐CD11a antibody) following long‐term subcutaneous weekly dosing in psoriasis subjects. J Clin Pharmacol 2005; 45: 468–76. [DOI] [PubMed] [Google Scholar]
- 17. Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target‐mediated drug disposition. J Pharmacokinet Pharmacodyn 2001; 28: 507–32. [DOI] [PubMed] [Google Scholar]
- 18. Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet 2010; 49: 633–59. doi:10.2165/11535960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 19. Dostalek M, Gardner I, Gurbaxani BM, Rose RH, Chetty M. Pharmacokinetics, pharmacodynamics and physiologically‐based pharmacokinetic modelling of monoclonal antibodies. Clin Pharmacokinet 2013; 52: 83–124. [DOI] [PubMed] [Google Scholar]
- 20. Gibiansky L, Gibiansky E. Target‐mediated drug disposition model: relationships with indirect response models and application to population PK‐PD analysis. J Pharmacokinet Pharmacodyn 2009; 36: 341–51. doi:10.1007/s10928-009-9125-9 Epub 2009 July 4. [DOI] [PubMed] [Google Scholar]
- 21. Morell A, Terry WD, Waldmann TA. Metabolic properties of IgG subclasses in man. J Clin Invest 1970; 49: 673–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Regazzi MB, Iacona I, Avanzini MA, Arcaini L, Merlini G, Perfetti V, Zaja F, Montagna M, Morra E, Lazzarino M. Pharmacokinetic behavior of rituximab: a study of different schedules of administration for heterogeneous clinical settings. Ther Drug Monit 2005; 27: 785–92. [DOI] [PubMed] [Google Scholar]
- 23. Ng CM, Bruno R, Combs D, Davies B. Population pharmacokinetics of rituximab (anti‐CD20 monoclonal antibody) in rheumatoid arthritis patients during a phase II clinical trial. J Clin Pharmacol 2005; 45: 792–801. [DOI] [PubMed] [Google Scholar]
- 24. Muller C, Murawski N, Wiesen MH, Held G, Poeschel V, Zeynalova S, Wenger M, Nickenig C, Peter N, Lengfelder E, Metzner B, Rixecker T, Zwick C, Pfreundschuh M, Reiser M. The role of sex and weight on rituximab clearance and serum elimination half‐life in elderly patients with DLBCL. Blood 2012; 119: 3276–84. [DOI] [PubMed] [Google Scholar]