

Abstract

The combination of photonics and spintronics opens new ways to transfer and process information. It is shown here that in systems in which organic molecules and semiconductor nanoparticles are combined, matching these technologies results in interesting new phenomena. We report on light induced and spin-dependent charge transfer process through helical oligopeptide–CdSe nanoparticles’ (NPs) architectures deposited on ferromagnetic substrates with small coercive force (∼100–200 Oe). The spin control is achieved by the application of the chirality-induced spin-dependent electron transfer effect and is probed by two different methods: spin-controlled electrochemichemistry and photoluminescence (PL) at room temperature. The injected spin could be controlled by excitation of the nanoparticles. By switching the direction of the magnetic field of the substrate, the PL intensity could be alternated.
Keywords: Charge transfer, chirality, spin transport, oligopeptide, spin selectivity, ferromagnet
The concept of controlling magnetism by light has attracted much attention in recent years.1−3 Specifically, the controlling of spin transport by light combines two interesting technologies, photonics and spintronics.4,5 Having organic material makes the coupling of the two technologies easier6−8 because the light-absorbing properties of organic molecules can be modified relatively easily.9,10 Recently, it was found that the transmission of electrons through chiral molecules depends on the electrons’ spin orientation; this effect is referred to as chiral-induced spin selectivity (CISS).11,12 The CISS effect makes it possible to construct spintronic devices without ferromagnetic spin injectors because the chiral molecules themselves serve to select a specific spin to transport across the molecules.13−16 In the hybrid system studied herein, helical oligopeptide molecules were attached on one end to CdSe nanoparticles (NPs) and on the other end to ferromagnetic substrates. Two experimental configurations were applied; in the first, the photoemission intensity from the NPs was monitored as a function of the magnetization direction of the substrate (Figure 1A). In addition, electrochemical measurements were conducted (Figure 1B) when the ferromagnetic working electrode was coated with the oligopeptide–CdSe NP system. The faradic current through the adsorbed layer was measured as a function of the direction of the magnetic field when the system was illuminated. It was established that when an electron is transmitted through a chiral molecule, a specific spin orientation is preferred. Here, we utilized the effect to control the photoluminescence (PL) intensity from the NPs by controlling the direction of the magnetic field of the substrate. We also demonstrated control by light of the preferred spin transmitted through the structures of hybrid chiral molecules–NPs.
Figure 1.

(A) Schematic presentation of the experimental setup for the spin-dependent photoluminescence measurements. (B) Schematic presentation of the light-induced spin-dependent electrochemical measurements recorded in a homemade electrochemical cell in the presence of tris buffer containing 5 mM K4[Fe(CN)6]/K3[Fe(CN)6]. The ferromagnetic substrate was the working electrode (WE), whereas platinum wire and KCl-saturated calomel electrode (SCE) were used as the counter (CE) and reference (RE) electrodes, respectively. The working electrode was based on multiple ferromagnetic layers, and it was coated with the Ala8–CdSe NP assemblies. In both cases, a permanent magnet (H = 0.35 T) was placed underneath the modified ferromagnetic substrate and the direction of the magnetic dipole was flipped either pointing “UP” or “DOWN” (white and yellow arrows, respectively). In all cases, a green laser (λexc = 514 nm) was used for exciting the CdSe NPs.
Results and Discussion
The hybrid system studied is based on a self-assembled monolayer (SAM) of a helical oligopeptide,17 HS–CH2–CH2–NHCO(Ala-AiB)8–NH2, (known as Ala8, where Ala = Alanine, AiB = 2-aminoisobutyric acid) adsorbed on a specially prepared ferromagnetic (FM) crystalline multilayer (Co–Pt) (see Figure 1). The soft ferromagnetic multilayers with perpendicular magnetization were prepared either on glass or on a 300 nm thermally grown SiO2 substrate. In all cases, a 5 nm Ta adhesive layer was deposited on the substrates, followed by a 2 nm Pt layer on it. Co and Pt were repetitively (three or five times) deposited onto the substrates. The substrate exhibits soft magnetization with a perpendicular easy axis (Figure S1). The FM materials become readily oxidized under aerobic conditions and specifically in an electrochemical cell having an aqueous solution as the electrolyte.18 To address this problem, we coated the FM substrates with a very thin and smooth overlayer of Au (1 nm). More details on the layer preparation and characterization are given in Methods and Supporting Information.
CdSe NPs of 6–7 nm diameter are covalently attached to the SAM through its free primary amine (−NH2) of Ala8. The NPs absorb light at 514 nm, whereas the excited NPs emit at about 650 nm.19 The Ala8–CdSe NP assembly was characterized using polarization modulation-infrared reflection–absorption spectra (PM-IRRAS), atomic force microscopy (AFM) images, and X-ray photoelectron spectroscopy (XPS) spectroscopy. It was found to be stable under ambient conditions and did not deteriorate upon mild sonication (see Figures S2–S6 and Tables S1–S3 in the Supporting Information). An external static magnetic field of 0.35 T was applied to switch the magnetization of the ferromagnetic substrate. Two different experimental setups were employed. The first one is based on measuring fluorescence from the NPs under ambient conditions (Figure 1A), whereas in the second one electrochemical measurements were performed in a custom-made electrochemical cell (Figure 1B).
In the fluorescence setup, upon photoexcitation of the NPs either electron or hole transfer to the substrate may occur and their rates determine the intensity of the fluorescence. As it was established, whereas the lifetime of the isolated CdSe NPs is of the order of 20 to 30 ns, when the NPs are able to transfer electrons from the oligopeptide to a metal substrate, the lifetime is shortened to below 1 ns.20 Hence, the fast charge transfer results in lowering the fluorescence intensity because it hampers the radiative electron–hole recombination process. As was determined by surface photovoltage (SPV) measurements, in the current Ala8–CdSe hybrid system, hole transfer from the excited NPs to the substrate across the hybrid structures is more efficient than by electron transfer. Figure 2A shows that photoluminescence intensity is affected by the direction of the magnetization of the substrate. Because the quenching of the fluorescence is due to the transfer of electrons from the substrate to the NPs (hole transfer from NPs to substrate), when the electron’s spin is parallel to the adsorbed molecule axis (when the magnet was pointing “UP”), its transfer is more efficient and as a result photoluminescence intensity get quenched. This observation is consistent with former data showing the same spin preference also in spin-dependent photoelectron transmission through oligopeptides.21 The average ratio of the PL intensity, as estimated by the area under the curve measured for two different directions (the magnet pointing “DOWN” vs “UP”), was found to be 1.4 ± 0.2. These results are highly reproducible among five different samples prepared under identical conditions. Several control experiments were performed where Ala-8 was replaced by achiral 1,16-hexadecane dithiol (DT), biphenyl-4,4′-dithiol (BD), and 4-aminothiophenol; an external magnetic field was applied, keeping the experimental setup identical. The achiral SAM–CdSe NP assembles did not exhibit any effect of the external magnetic field on the PL intensity, as shown in Figure 2B and Figure S7, corroborating that the CISS effect arises exclusively from chiral structures.4
Figure 2.

(A) Photoluminescence spectra of CdSe NPs attached to an oligopeptide monolayer (Ala-8) adsorbed onto magnetic substrates, as measured in the presence of an external magnetic field of 0.35 T pointing either UP (blue curve) or DOWN (red curve). (B) Photoluminescence spectra of CdSe NPs attached to an achiral 1,16-hexadecane dithiol monolayer adsorbed onto the magnetic substrate measured under identical conditions. The nanoparticles were excited with a green laser at 514 nm.
Spin-dependent electrochemical measurements were performed
using both the oligopeptide and oligopeptide-NP structures assembled
on the ferromagnetic substrates in the presence of a chemically robust
redox probe such as K4[Fe(CN)6]/K3[Fe(CN)6] in tris(hydroxymethyl)aminomethane) (TRIS) buffer
(see Figure 1B). The
SAM-modified ferromagnetic substrate was used as the working electrode
(WE), platinum wire as the counter electrode (CE), and a KCl-saturated
calomel electrode as the reference electrode (RE). A permanent magnet
having a field of 0.35 T was placed underneath the ferromagnetic working
electrode, which can be flipped by changing its direction by 180°
so that the magnetic field points either toward the surface (UP) or
away from it (DOWN). The NPs in the hybrid structures could be excited
at 514 nm during the electron conduction measurements. The dependence
of the CV curves on the direction of the magnetic field and on the
illumination is shown in Figure 3. In the dark, the substrate was coated only with the
SAM (Figure 3A) and
one was coated both with SAM and NPs (Figure 3B); both show magnetic field-dependent CV
curves with a higher current when the magnet points DOWN. The spin
polarization (SP) is defined as 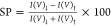 , where I(V)↓ and I(V)↑ represent the currents
obtained through monolayers with the magnetic field pointing “DOWN”
or “UP”, respectively, at a specific applied potential
(V). The SP for transmission through the chiral Ala8 monolayers is
+11 ± 1% at +50 mV and +7 ± 0.5% at +0.32 V. These values
are similar to those obtained before with different chiral molecules.10,22
, where I(V)↓ and I(V)↑ represent the currents
obtained through monolayers with the magnetic field pointing “DOWN”
or “UP”, respectively, at a specific applied potential
(V). The SP for transmission through the chiral Ala8 monolayers is
+11 ± 1% at +50 mV and +7 ± 0.5% at +0.32 V. These values
are similar to those obtained before with different chiral molecules.10,22
Figure 3.

Spin-dependent cyclic voltammograms recorded for (A) Ala8 monolayers adsorbed over ferromagnetic substrates, (B) Ala8–CdSe hybrid structures under dark conditions, and (C) under light conditions. The voltammograms were obtained when the ferromagnetic cobalt (used as the working electrode) is magnetized either with its magnetic moment pointing UP (the solid blue line), or pointing DOWN (the solid red line). Voltammograms were recorded in Tris buffer containing 5 mM K4[Fe(CN)6]/K3[Fe(CN)6] at pH 9 at a scan rate of 50 mV s–1, keeping the area of the ferromagnetic working electrode the same in all cases.
Figure 3B,C presents CV curves obtained with the CdSe NPs attached to SAM. In addition to the PMIRRAS, AFM images, and XPS data, the attachment of the NPs was also confirmed by the voltammetry measurements, which showed an increase in the oxidation potential of the redox probe, Fe2+, by 60 mV. In the dark (Figure 3B), the hybrid system has an SP value of +9 ± 1% at the reduction potential (at 0 V) and +8 ± 0.5% at the oxidation potential (+0.38 V). Upon illumination of the CdSe NPs, the sign of the SP is reversed and it is −3 ± 0.5% at +25 mV (Figure 3C). Namely, illumination causes a flip in the sign and a reduction in the absolute value of the spin polarization.
To verify the observations, we performed chronoamperometry experiments in which the current at a given potential is recorded as a function of time. The electron conduction across the SAMs (of Ala8) themselves and through illuminated oligopeptide–CdSe structures was measured (Figure 4A,B, respectively). Whereas for the SAMs themselves or for the SAM–CdSe NPs in the dark (data not shown), the Faradaic current was higher when the magnet was pointing DOWN; however, in the illuminated sample the current was higher when the magnetic field direction was pointing UP. This effect was observed for both the oxidation or reduction processes. For the chiral SAMs, only when the SAM–CdSe NPs were in the dark the SP was +17 ± 1% and +35 ± 2% at 0.32 and 0 V, respectively. When the SAM–CdSe NP sample was illuminated at 514 nm, the SP is −11 ± 1% at +0.32 V, and −30 ± 2% at the reduction potential, 0 V. The voltammograms recorded on a bare ferromagnetic substrate as a working electrode did not exhibit any magnetic field effect on the Faradaic current when the working electrode was magnetized either with its magnetic moment pointing UP or DOWN (Figure S8).
Figure 4.

Chronoamperometric measurements (current versus time) for (A) Ala8 and (B) Ala8–CdSe structures on a magnetic cobalt working electrode in the presence of an external static magnetic field either pointing UP (solid blue line) or DOWN (solid red line) in a Tris buffer containing 5 mM K4[Fe(CN)6]/K3[Fe(CN)6] at pH 9. The samples were illuminated with a green laser having a 514 nm wavelength. Magnetic field-dependent current versus time measurements were recorded at +0.32 V (oxidation process) and 0 V (reduction process) versus a saturated calomel electrode (SCE).
It is known that in the CISS effect, the alignment of the preferred spin in the electron transport process depends on several factors, among them (i) the handedness of the molecules, (ii) the electric field along them, and (iii) the electrons’ velocity.12 For example, electrons moving from right to left along molecules with a preferred spin for a given electric field will move with the opposite spin if the external field is reversed. However, if the electrons move from left to right with the opposite sign of field on the molecule then the same spins of the electrons will be preferred in both directions. In the ferromagnetic substrate cobalt, the high density of state below and above the Fermi level is of the same spin. Hence, this is why in the oxidation and reduction part of the electrochemical cycle, the same spin is preferred.
Figure 5 presents a scheme that explains the experimental observations. Before photoexcitation (Figure 5A), the NPs are positively charged, as observed by the XPS measurements that indicate that the work function of the surface decreases by 105 meV upon the NPs attachment to the functional polypeptide monolayers (see Table S3). The PL studies are consistent with the electrons being transferred with their spin aligned parallel to their velocity. This observation is consistent with former studies on spin-dependent electron transmission through the same oligopeptides.10 In the reduction process of the electrochemical measurements, electrons move from the cobalt substrate (Co) through the helical molecule to the NPs and thereafter to the redox couple in solution. In the oxidation process a reverse field is applied and the electrons are transferred in the opposite direction from the redox couple to the NPs and from there to Co; as a result, the same spin is preferred for electrons transferred from the NPs to the Co. When the NPs are photoexcited, a hole is transferred from the NPs to the Co substrate and the NPs are now negatively charged (Figure 5B). Chemically resolved electrical measurements (CREM) data show a prominent response of the peptide–CdSe NP structures to light illumination at 630 nm. A decrease in the binding energies of the Cd line by 120 meV under light, as compared with the same value recorded in the dark, results in a clear manifestation of the excited state hole transfer to the substrate and creates a negative charge over the NPs (Figure S6 and Table S3). Hence, the electric field on the chiral molecule is of the opposite direction and now electrons with spin antiparallel to their velocity are transferred. The electrochemical process is similar as in the dark with the electrons transmitted between the substrate and the redox couple in solution through the NPs.
Figure 5.

A scheme describing the effect of the light on the spin selectivity. Before excitation (A), the NPs are positively charged and electrons are transferred with their spin aligned parallel to their velocity. In the reduction part of the electrochemical process electrons move from the FM cobalt substrate (Co) through the helical molecule to the NPs and from there to the redox couple present in solution. In the oxidation stage, the electrons are transferred from the redox couple to the NPs and from there to the Co. When NPs are photoexcited (B), because of the hole being transferred from the NPs to the Co the NP is now negatively charged. Hence, the electric field on the chiral molecule is in the opposite direction and now electrons with spin antiparallel to their velocity are transferred preferentially. The electrochemical process is similar to the electrons transmitted through the NPs to the redox couple in solution.
Conclusions
The present study demonstrates the interaction between light and electrons’ spin as manifested when light controls the spin-selective electron transport through chiral molecules. By attaching a fluorophore, the NPs, to the chiral molecule the actual spin orientation being transmitted can be flipped with significant efficiency. Thus, when combining light and a magnetic field the chirality-induced electron transfer process may find increased interest in the field of light-controlled biospintronic research.23 It is important to appreciate that the effect observed here relates to weak magnetic fields applied on the ferromagnetic substrate and hence differ significantly from the known magneto-chiral anisotropy in charge transfer, in which very high fields (many Tesla) are applied.24 The combination of systems, like those presented here, and molecular magnets25,26 may result is a new type of photospintronics devices that are all molecular.
Methods
Preparation of Soft Ferromagnetic Substrates
The ferromagnetic films with a perpendicular easy axis were prepared on soda lime glass slides and on silicon substrate ⟨100⟩ on which 5 nm of tantalum and 2 nm of platinum were deposited using a magnetron sputtering system with a base pressure of <3 × 10–8 Torr. Cobalt and platinum deposition were repeated thrice on top of the 2 nm platinum layer. A thin layer of cobalt, followed by 1 nm gold, was grown on top. The stacking structure of the magnetic substrates was Si/SiO2/Ta 5/Pt 2/[Co 0.28/Pt 0.3]3/Co 0.28/Au 1 (in nm). All metal layers were deposited at a working pressure of Ar 1.5 mTorr at room temperature.
Preparation of Oligopeptide Monolayer and Oligopeptide–CdSe Nanoparticle Structures
Magnetic substrates with a 1 nm Au overlayer were cleaned with boiling acetone followed by ethanol for 15 min in each. The substrates were dried carefully under N2. Subsequently, the substrates were plasma cleaned in argon (at ∼0.4 Torr) for 1 min and immediately immersed into a solution containing 0.1 mM polyalanine (Ala8) in TFE/H2O (6:4, v/v, deoxygenated with Ar for 45 min) for 48 h in order to achieve maximum surface coverage. The functionalized substrates were rinsed with the same solvent mixture and mildly sonicated for 5 s to remove any unreacted materials. Consequently, the monolayers were dried carefully under N2 and used for IR, XPS, and electrochemical measurements.
Self-assembled monolayers of Ala8, added onto the magnetic substrates, were immersed into a solution containing CdSe in anhydrous toluene for 4 h under dark conditions. In order to remove unreacted NPs, the substrates were lightly sonicated for 5 s in toluene and dried gently under N2 flow and immediately used for measurements.
Characterization
The magnetic properties of perpendicular magnetized [Co/Pt] films were characterized by using a vibrating sample magnetometer (VSM, VersaLab Quantum Design). Saturation magnetization (Ms) is 827 ± 96.7 emu/cm3 (measured moment/volume of Co and Pt) and the coercively is 145 and 270 Oe for substrate 1 and substrate 2, respectively (Figure S1).
The formation of monolayers in the polypetide and its CdSe NPs structures were characterized by recording the polarization modulation-infrared reflection–absorption spectra (PM-IRRAS) using a Nicolet 6700 FTIR equipped with a PEM-90 photoelastic modulator (Hinds Instruments, Hillsboro, OR) at an incidence angle of 80°. In all cases, vibrational stretching frequencies of amide I and amide II bonds in the SAMs were monitored.
X-ray Photoelectron Spectroscopy (XPS) and Chemically Resolved Electrical Measurements (CREM)
Freshly prepared SAMs of oligopeptide (Ala8) and peptide–CdSe architectures on the ferromagnetic substrates were analyzed by XPS measurements using a Kratos Axis Ultra DLD spectrometer equipped with a monochromatic Al Kα X-ray source (hν = 1486.6 eV) operating at a power of 75 W. Two emission angles (65° and 0° with respect to the surface normal) were compared. Detection pass energies ranged between 20 and 80 eV. Elemental concentrations and layer thicknesses were deduced from the relative intensities of overlayer (N, C, O, and S 2p) versus substrate (Ta, Pt, Co, Au, and O) signals. Special care was devoted to eliminate beam-induced effects and damage to molecules in particular. This was achieved by conducting repeated measurements, starting with very fast scans at fresh spots, in order to evaluate the exposure-dependent spectral changes.
The coverage by CdSe nanoparticles (NPs) was estimated based on the NP average size, 6–7 nm in diameter, which was determined independently. Work function measurements were performed prior to any XPS scan, by inspecting the photoelectron onset at low kinetic energies under a source power of 0.3W. Site-specific photovoltages were extracted by evaluating the energy shifts of representative lines (Cd, N, C, and Au) under a 630 nm diode source.27 The advantage of these measurements over standard SPV experiments stems from the fact that one can resolve local electrostatic potential changes and in particular those developing specifically on the CdSe NPs.
The formation of Ala8–CdSe nanoparticle structures on ferromagnetic substrates was further confirmed by AFM images recorded on a Multimode/Nanoscope (Bruker-Nano, Santa Barbara, CA, U.S.A.). The AFM images were acquired in noncontact mode and a Si probe having a resonance frequency of 70–90 kHz was used. The topographic images were recorded at a scan rate of 1 Hz. Several images (at least at four different points) of each sample were taken from different fields of view (0.5–2.0 μm) to confirm the uniformity and reproducibility of the samples. The formation of homogeneous distributions of CdSe nanoparticles in the monolayer was confirmed by AFM imaging.
The PL measurements were carried out by using a LabRam HR800-PL spectrofluorimeter microscope (Horiba Jobin-Yivon). Typically, 514 nm laser light (argon-ion CW laser at ∼15 mW/cm2) has been used for excitation of CdSe NPs. The incident light was impinged on the surface at an angle 90° to the sample surface. Prior to collecting the PL spectra, an area (typically, 20 au × 20 au) and number points (25 points) within this area were defined in order to map the surface. Afterward, the PL spectra were collected using a microscope (with a 5× high working distance lens). During the measurement, a confocal aperture (1100 μm) was fully opened and the integration time was maintained at 15 s. Finally, the spectra were presented after averaging out the PL of individual points within the defined area at two different magnetic orientations.
Cyclic voltammetry (CV) and chronoamperometric measurements were carried out using a Bio-Logic potentiostat SAS (Model SP-200) with an inbuilt software EC Lab (V 10.36) by employing a typical three-electrode electrochemical cell. A SAM-modified Co substrate, a Pt wire, and KCl-saturated calomel electrodes were used as the working, counter, and reference electrodes, respectively, in the presence of Tris buffer containing 5 mM K4[Fe(CN)6]/K3[Fe(CN)6] taken into a custom-made electrochemical cell. Spin-dependent electrochemical measurements were carried out in the presence of a permanent magnet having a magnetic field strength of 0.35 T, which was placed underneath the Co working electrode, followed by changing the magnet’s direction (UP/DOWN).
Acknowledgments
P.C.M., P.R., and R.N. acknowledge financial assistance from the ERC-Adv grant, the VW Foundation, and the Israel Science Foundation.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nanolett.6b00582.
Sample preparation and characterization and the experimental setup. (PDF)
Author Contributions
P.C.M. fabricated and characterized the monolayers, performed the electrochemical measurements, and analyzed the data. P.R. conducted the photoluminescence experiments. D.K. fabricated the soft magnetic substrates, designed the film structure, and characterized it. E.E.F. guided the preparation of the magnetic substrates and the scientific discussion. H.C. carried out XPS and CREM measurements. R.N. conceived the research plan. All authors contributed to the writing of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Lambert C.-H.; Mangin S.; Varaprasad B. S. D. C. S.; Takahashi Y. K.; Hehn M.; Cinchetti M.; Malinowski G.; Hono K.; Fainman Y.; Aeschlimann M.; Fullerton E. E. Science 2014, 345, 1337–1340. 10.1126/science.1253493. [DOI] [PubMed] [Google Scholar]
- Koopmans B.; Malinowski G.; Dalla Longa F.; Steiauf D.; Fähnle M.; Roth T.; Cinchetti M.; Aeschlimann M. Nat. Mater. 2009, 9, 259–265. 10.1038/nmat2593. [DOI] [PubMed] [Google Scholar]
- Kirilyuk A.; Kimel A. V.; Rasing T. Rep. Prog. Phys. 2013, 76, 026501. 10.1088/0034-4885/76/2/026501. [DOI] [PubMed] [Google Scholar]
- Bliokh K. Y.; Smirnova D.; Nori F. Science 2015, 348, 1448–1451. 10.1126/science.aaa9519. [DOI] [PubMed] [Google Scholar]
- Dediu V. A.; Hueso L. E.; Bergenti I.; Taliani C. Nat. Mater. 2009, 8, 707–716. 10.1038/nmat2510. [DOI] [PubMed] [Google Scholar]
- Sun D.; Ehrenfreund E.; Vardeny Z. V. Chem. Commun. 2014, 50, 1781–1793. 10.1039/c3cc47126h. [DOI] [PubMed] [Google Scholar]
- Sanvito S. Nat. Mater. 2007, 6, 803–804. 10.1038/nmat2050. [DOI] [PubMed] [Google Scholar]
- Lee S.-Y.; Paik S.-Y.; McCamey D. R.; Yu J.; Burn P. L.; Lupton J. M.; Boehme C. J. Am. Chem. Soc. 2011, 133, 2019–2021. 10.1021/ja108352d. [DOI] [PubMed] [Google Scholar]
- Vardeny Z. V.; Heeger A. J.; Dodabalapur A. Synth. Met. 2005, 148, 1–3. 10.1016/j.synthmet.2004.09.001. [DOI] [Google Scholar]
- Sugawara T.; Matsushita M. M. J. Mater. Chem. 2009, 19, 1738–1753. 10.1039/b818851n. [DOI] [Google Scholar]
- Gohler B.; Hamelbeck V.; Markus T. Z.; Kettner M.; Hanne G. F.; Vager Z.; Naaman R.; Zacharias H. Science 2011, 331, 894–897. 10.1126/science.1199339. [DOI] [PubMed] [Google Scholar]
- Naaman R.; Waldeck D. H. Annu. Rev. Phys. Chem. 2015, 66, 263–281. 10.1146/annurev-physchem-040214-121554. [DOI] [PubMed] [Google Scholar]
- Dor O. B.; Yochelis S.; Mathew S. P.; Naaman R.; Paltiel Y. Nat. Commun. 2013, 4, 2256. 10.1038/ncomms3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew S. P.; Mondal P. C.; Moshe H.; Mastai Y.; Naaman R. Appl. Phys. Lett. 2014, 105, 242408. 10.1063/1.4904941. [DOI] [Google Scholar]
- Ravi S.; Sowmiya P.; Karthikeyan A. SPIN 2013, 3, 1350003. 10.1142/S2010324713500033. [DOI] [Google Scholar]
- Alam K. M.; Pramanik S. Adv. Funct. Mater. 2015, 25, 3210–3218. 10.1002/adfm.201500494. [DOI] [Google Scholar]
- Pawlowski J.; Juhaniewicz J.; Tymecka D.; Sek S. Langmuir 2012, 28, 17287–17294. 10.1021/la302716n. [DOI] [PubMed] [Google Scholar]
- Mondal P. C.; Fontanesi C.; Waldeck D. H.; Naaman R. ACS Nano 2015, 9, 3377–3384. 10.1021/acsnano.5b00832. [DOI] [PubMed] [Google Scholar]
- Gotesman G.; Waldeck D. H.; Naaman R. J. Phys. Chem. A 2009, 113, 7213–7217. 10.1021/jp808803v. [DOI] [PubMed] [Google Scholar]
- Odoi M. Y.; Hammer N. I.; Early K. T.; McCarthy K. D.; Tangirala R.; Emrick T.; Barnes M. D. Nano Lett. 2007, 7, 2769–2773. 10.1021/nl0713068. [DOI] [PubMed] [Google Scholar]
- Kettner M.; Gohler B.; Zacharias H.; Mishra D.; Kiran V.; Naaman R.; Fontanesi C.; Waldeck D. H.; Sek S.; Pawlowski J.; Juhaniewicz J. J. Phys. Chem. C 2015, 119, 14542–14547. 10.1021/jp509974z. [DOI] [Google Scholar]
- Mondal P. C.; Kantor-Uriel N.; Mathew S. P.; Tassinari F.; Fontanesi C.; Naaman R. Adv. Mater. 2015, 27, 1924–1927. 10.1002/adma.201405249. [DOI] [PubMed] [Google Scholar]
- Einati H.; Mishra D.; Friedman N.; Sheves M.; Naaman R. Nano Lett. 2015, 15, 1052–1056. 10.1021/nl503961p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krstić V.; Roth S.; Burghard M.; Kern K.; Rikken G. L. J. A. J. Chem. Phys. 2002, 117, 11315–11319. 10.1063/1.1523895. [DOI] [Google Scholar]
- Ishikawa N.; Sugita M.; Ishikawa T.; Koshihara S.-y.; Kaizu Y. J. Am. Chem. Soc. 2003, 125, 8694–8695. 10.1021/ja029629n. [DOI] [PubMed] [Google Scholar]
- Milios C. J.; Vinslava A.; Wernsdorfer W.; Moggach S.; Parsons S.; Perlepes S. P.; Christou G.; d Brechin E. K. J. Am. Chem. Soc. 2007, 129, 2754–2755. 10.1021/ja068961m. [DOI] [PubMed] [Google Scholar]
- Cohen H.; Sarkar S. K.; Hodes G. J. Phys. Chem. B 2006, 110, 25508–25513. 10.1021/jp0648590. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.