

Abstract
New pseudotrisaccharide derivatives of aminoglycosides that exploit additional interaction on the shallow groove face of the decoding-site rRNA of eukaryotic ribosome were designed, synthesized and biologically evaluated. Novel lead structures (6 and 7 with an additional 7′-OH), exhibiting enhanced specificity to eukaryotic cytoplasmic ribosome, and superior nonsense mutation suppression activity than those of gentamicin, were discovered. The comparative benefit of new leads was demonstrated in four different nonsense DNA-constructs underling the genetic diseases cystic fibrosis, Usher syndrome, and Hurler syndrome.
Keywords: Aminoglycosides, genetic diseases, nonsense mutations, translational readthrough, cystic fibrosis
Nonsense mutations are caused by single nucleotide base changes within a gene that result in an in-frame premature termination codon (PTC) and lead to the formation of truncated, nonfunctional proteins.1,2 Nonsense mutations account for about 11% of all described gene lesions that cause inherited human diseases including cystic fibrosis (CF), Duchenne muscular dystrophy (DMD), Usher syndrome (USH), mucopolysaccharidosis type I–H (MPS I–H), and numerous types of cancer.3 For many of those diseases there is presently no effective treatment. In the last several years, it was shown that some aminoglycoside (AG) antibiotics (including gentamicin and G418, Figure 1) have the ability to allow the mammalian ribosome to selectively read past a false-stop signal, but not a normal termination signal, and generate full-length functional proteins.3 This unique ability of AGs, also called “PTC suppression” or “translational readthrough”, has been demonstrated in a series of in vitro and in vivo studies including clinical trials, which highlighted them as promising therapeutics for treatment of PTC underlined genetic disorders. Despite this promise, high human toxicity of conventional AGs and reduced PTC suppression efficacy at subtoxic doses have limited their clinical advantage for suppression therapy.3,4
Figure 1.

Chemical structures of natural and synthetic AGs that were investigated in this study.
To address these issues we systematically developed a series of lead compounds of NB series,5 which was found to improve readthrough activity and reduce toxicity6 as demonstrated in various in vitro, ex vivo, and in vivo models of a variety of different diseases underlined by PTC mutations.4 Note that, due to the lack of detailed structural data on the interaction of AGs within their eukaryotic target site, the design and development of all those lead structures were based mainly on the earlier reported biochemical and toxicity data on conventional AGs. For example, by initially identifying the pseudodisaccharide moiety of G418 (1) as an important fragment that induces significant read-through, we then used 1 as a common scaffold and developed the lead compounds 2(7) and 3(5) (also named NB74 and NB124, respectively, Figure 1), exhibiting improved read-through activity and reduced toxicity.
Thus, while this strategy until now has provided encouraging results, the “rational design” of synthetic AGs for suppression therapy is still far from being well established. Recently reported three-dimensional structure of 80S yeast ribosome in complex with G418,8 along with the structures of G4189 and paromomycin10 bound to their putative binding site of Leishmania ribosomes, have provided important structural insights that paved the way for structure-based rational design of novel read-through inducers. Based on these structural data, herein we report our pilot study on the design, synthesis, and evaluation of new lead structures 6 and 7 (also named NB156 and NB157, respectively, Figure 1) that differ from the previous leads (2 and 3) by the presence of an additional hydroxyl group on the side-chain of the glucosamine ring (ring I) and show substantially higher readthrough activity in comparison to those of the parent structures.
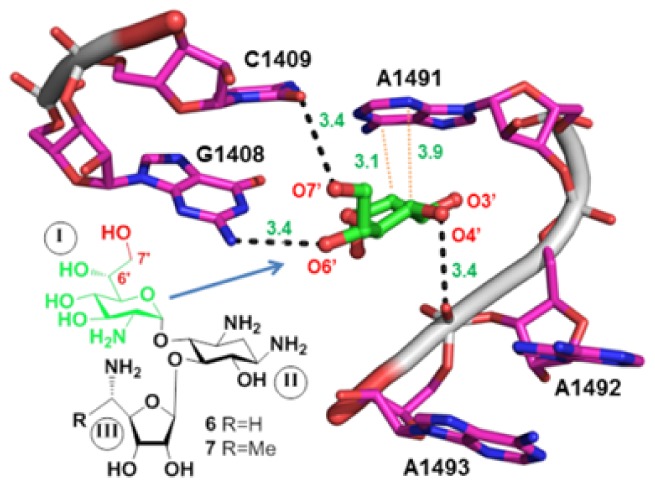
High-resolution X-ray crystal structure of 80S ribosome from Saccharomyces cerevisiae in complex with G418,8 along with our own modeling studies, suggested that extension of the side-chain methyl group at the glucosamine moiety (ring I) of G418 with a hydroxyl group to generate 6′,7′-diol might allow for additional H-bond interaction between the added 7′-OH and the O2 atom of cytosine 1409 (Figure 2). Initially, the validity of this design strategy was tested on pseudodisaccharides; we synthesized compounds 1, 4, and 5 (Figure 1) and tested their comparative in vitro readthrough efficacy.
Figure 2.
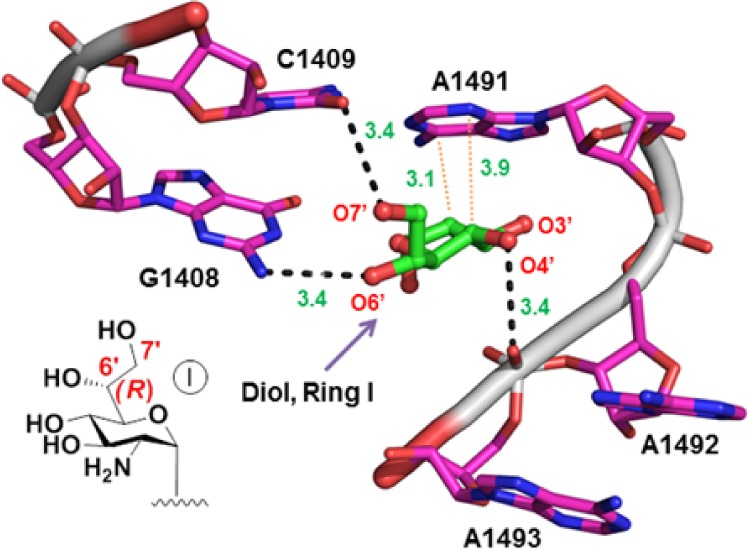
Three-dimensional structure of 80S ribosome from S. cerevisiae in complex with G418 (PDB code 4U4O),8 highlighting the interactions of G418 ring I (green) into the decoding-site rRNA (magenta, E. coli numbering). The ring I oxygen atoms (red) are specified by numbers, including the modeled O7′-oxygen (PyMol software). H-bonds (dashed black lines) and C–H···π stacking (dotted red lines) are shown. The inset shows chemical structure of ring I of G418 with the modeled additional 7′-OH.
Compound 1 was synthesized as previously reported by us.7 The syntheses of 4 and 5 are illustrated in Scheme 1. The known perazido derivative 8(11) was selectively protected by TIPSCl, and the remaining hydroxyls were protected by p-methoxybenzyl (PMB) groups to afford 9. Selective deprotection of silyl group with TBAF was followed by oxidation with 2-iodoxy benzoic acid (IBX)12 and Wittig reaction to afford the terminal alkene 10. The alkene 10 was dihydroxylated13 to provide the diol 11 as an inseparable mixture of 6′-diastereomers. Treatment of 11 with DDQ was followed by acetylation (Ac2O) and deacetylation (NaOMe) steps to afford the mixture of 6′-diastereomers (∼3:1 ratio), which was successfully separated by column chromatography to give the major diastereomer 12 and the minor diastereomer 13. The absolute configuration at 6′-position was assigned by using 1H NMR magnetic anisotropy (for the details see Supporting Information), which established 6′-(R)- and 6′-(S)-configuration for the major and minor diastereomers, respectively. The two diastereomers 12 and 13 were separately subjected to Staudinger reaction to produce the target pseudodisaccharides 4 and 5.
Scheme 1.

Reagents and conditions: (i) (a) TIPSCl, DMF, 4-DMAP, 0–25 °C, 83%; (b) PMBCl, NaH, DMF, 0–25 °C, 84%; (ii) (a) TBAF, THF, 0–25 °C, 88%; (b) IBX, EtOAc, 80 °C, 85%; (c) CH3P(Ph)3I, n-BuLi, THF, 0–25 °C, 56%; (iii) K2OsO4·2H2O, NMO, acetone/H2O/t-BuOH, 93% (3:1 ratio); (iv) (a) DDQ, CH2Cl2/H2O; (b) Ac2O, Py, 0–25 °C, 91% for 2 steps; (c) NaOMe, MeOH, 60%; (v) PMe3, THF, NaOH (0.1 M), 73% [6′-(R)-4]; 78% [6′-(S)-5].
Preliminary comparative PTC suppression activity tests in Figure 3 show that installation of C7′-hydroxyl group (compound 6′-(R)-4) on compound 1 dramatically increases its in vitro readthrough activity, whereas this effect in compound 6′-(S)-5 is comparatively small. These data, while providing proof-of-concept of our design strategy also highlights the pivotal role of stereochemistry at 6′ position in RNA target recognition; compound 4 that retains the same 6′-(R) configuration as in G418 (and in compound 1) exhibits higher activity than that of compound 5 with an opposite, 6′-(S) configuration. The observed somewhat higher activity of 5 to that of 1 suggests that the additional 7′-hydroxyl in compound 5 can overcome the configurational penalty at 6′ position. Nevertheless, the observed significantly higher PTC suppression activity of 4 compared to that of 1 suggested that the compound 4 can serve as an improved pseudodisaccharide scaffold for the generation of new lead compounds.
Figure 3.
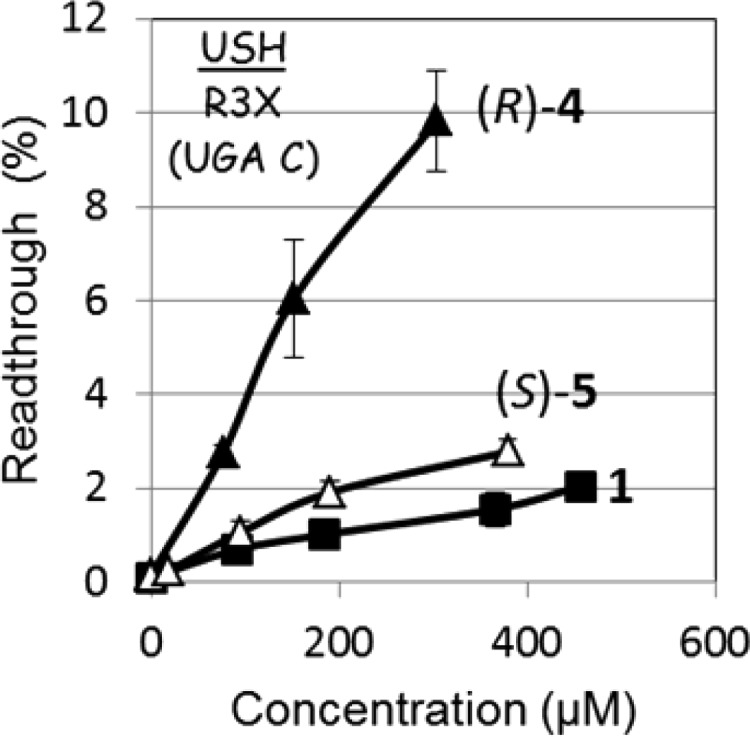
In vitro stop codon suppression levels induced by compounds 1 (-■-), (R)-4 (-▲-), and (S)-5 (-△-) in R3X nonsense mutation construct representing USH1 genetic disease.
Encouraged, we choose 6′-(R)-4 to assemble the corresponding pseudotrisaccharides 6 and 7 (Figure 1). The syntheses of 6 and 7 were accomplished from the intermediate 12 by using essentially the same chemical transformations as illustrated in Scheme 2.
Scheme 2.

Reagents and conditions: (i) Ac2O, Py., −20 °C, 53%; (ii) BF3–OEt2, CH2Cl2, −30 °C, 85% of 17, 93% of 18; (iii) (a) MeNH2, r.t., 80% (R = H), 98% (R = CH3); (b) PMe3, THF, NaOH (0.1 M), 60% of 6, 64% of 7.
Regioselective acetylation of 12 with Ac2O at low temperature gave the corresponding C5 acceptor 14. For the glycosylation of 14 we used the trichloroacemidate donors 15(14) and 16,15 which furnished the corresponding pseudotrisaccharides 17 and 18 in 85% and 93% isolated yields, respectively, exclusively as β-anomers. Treatment with methylamine was followed by Staudinger reaction to afford compounds 6 and 7.
The impact of the additional 7′-hydroxyl in compounds 6 and 7 was separately evaluated against their parent structures 2 and 3, respectively, and the observed data are illustrated in Figure 4. Since the read-through efficiency induced by AGs is highly dependent on the identity of a stop codon and the sequence contest around the stop codon,16 for broader evaluation of the structure–activity relationship, we used a collection of dual-luciferase reporter plasmids previously developed by us.17 These reporter plasmids contain different sequence contests around premature stop codons derived from the PCDH15, CFTR, and IDUA genes that underline USH1, CF, and MPS I–H, respectively. The nonsense reporters we choose were R3X and R245X for USH1, G542X for CF, and Q70X for MPS I–H. The observed data in Figure 4 clearly show that the positive impact of the C7′-hydroxyl group in scaffold 4 is largely retained in the subsequent pseudotrisaccharides 6 and 7. In all mutations tested, the in vitro readthrough activity of 6 is substantially better than that of its parent 2, and the activity of 7 is better than its parent 3. In addition, in all mutations tested, the activities of both 6 and 7 were significantly better than that of the clinical drug gentamicin.
Figure 4.

Comparative in vitro stop codon suppression levels induced by compounds 2 (-△-), 6 (-▲-), and gentamicin (--■--) (left) and by 3 (-Δ-), 7 (-▲-), and gentamicin (--■--) (right) in a series of nonsense constructs representing various genetic diseases (shown in parentheses): (A) R3X (USH1), (B) R245X (USH1), (C) Q70X (HS), and (D) G542X (CF). The assays were performed as previously described by us.17 The results are averages of at least three independent experiments.
Previous studies have demonstrated that the increased specificity toward eukaryotic cytoplasmic ribosome correlates with the increased PTC suppression activity.5 To test the validity of this trend, we examined comparative protein translation inhibition of pseudotrisaccharides 2, 3 and 6, 7 in eukaryotic system, using coupled transcription/translation assays as previously described17 (Table 1). The observed data indicate that the efficacy with which compound 7 (half-maximal inhibitory concentration value IC50Euk = 1.2 μM) inhibits eukaryotic translation is greater than that of compound 6 (IC50 = 13.9 μM) and gentamicin (IC50Euk= 62 μM), a similar trend observed for PTC suppression activity: 7 > 6 > gentamicin (Figure 5). In addition, the comparison of IC50 values reveals that 6 and 7 are 1.85-fold and 1.25-fold more specific to the eukaryotic ribosome than their parent structures 2 and 3, respectively. These data indicate that the elevated PTC suppression activities of 6 and 7 are associated with their increased specificity to the eukaryotic ribosome; these data further support the validity of our design strategy.
Table 1. Comparative Antibacterial Activity and Inhibition of Translation in Eukaryotic and Prokaryotic Systemsa.
| translation inhibitionb |
antibacterial activity MIC (μM)c |
|||
|---|---|---|---|---|
| compd | IC50Euk (μM) | IC50Pro (μM) | E. coli R477/100 | B. subtilis ATCC6633 |
| Genta. | 62 ± 9 | 0.03 ± 0.00 | 6 | <0.75 |
| G418 | 2.0 ± 0.3 | 0.01 ± 0.00 | 9 | <1.25 |
| 1 | 347.1 ± 34.3 | 6.0 ± 1.0 | ||
| (R)-4 | 120.5 ± 14.5 | 11.0 ± 1.2 | >311 | 311 |
| (S)-5 | 515.8 ± 15 | 91.9 ± 8.4 | >375 | >375 |
| 2 | 13.9 ± 1.2 | 1.0 ± 0.1 | 680 | 42 |
| 6 | 7.5 ± 0.5 | 0.7 ± 0.1 | >273 | 34 |
| 3 | 1.5 ± 0.1 | 1.1 ± 0.2 | 1267 | 156 |
| 7 | 1.2 ± 0.1 | 1.2 ± 0.1 | >257 | 64 |
In all biological tests, all AGs were in their sulfate salt forms, and the concentrations refer to the free amine form of each AG.
The eukaryotic and prokaryotic half-maximal-inhibition values (IC50Euk and IC50) were quantified in coupled transcription/translation assays by using active luciferase detection as previously described by us.5
Minimal inhibitory concentration (MIC) values were determined by using the double-microdilution method.
Figure 5.

(A) Secondary structures of rRNA A-sites of E. coli (left) and of eukaryotes (right). The conserved residues, A1492 and A1493 (E. coli numbering), are highlighted in blue and red, respectively. (B) Superimposition of G418 bound to the E. coli A-site (green, PDB code 1MWL)20 and the S. serevisiae A-site (magenta, PDB code 4U4O),8 using the PyMol software align algorithm (r.m.s.d. = 1.5 Å). (C) Expansion of the panel B highlighting different interactions of G418 ring I in the E. coli (green) and the S. serevisiae (magenta). The relevant H-bond interactions are shown by dashed lines with the bond lengths in the same color.
The measured IC50Pro and MIC values in Table 1 show that the efficacy with which 6 and 7 inhibit the prokaryotic ribosome and their subsequent antibacterial activity are very similar to those of the parent compounds 2 and 3.5 We have recently demonstrated that the observed significantly decreased prokaryotic ribosome specificity of 2(6) and 3(18) is also linked to their reduced mitochondrial translation inhibition and subsequently to their reduced ototoxic effects than gentamicin and G418. The observed similar impact on bacterial ribosome by the current (6 and 7) and previous leads (2 and 3) therefore suggest that 6 and 7 are perhaps less ototoxic than gentamicin and G418. Further studies on the impact of 6 and 7 on mitochondrial translation inhibition, along with their ototoxicity tests, are currently underway.
Finally, it is of note that a very similar rational design strategy, but for the bacterial ribosome, has been reported previously by Hermann and co-workers.19 Using 3D-structure of paromomycin bound to the bacterial decoding-site rRNA and modeling study, this group synthesized a series of C6′-modified pseudodisaccharides, including 4 but as an unassigned mixture of C6′ diastereomers, and tested them as inhibitors of bacterial in vitro translation in comparison to paromamine (rings I and II of paromomomycin, containing a primary 6′-hydroxyl at ring I). The results obtained were somewhat discouraging: all the tested 6′-modifications resulted in about 1–2-orders of magnitude decrease in inhibition potency, and the C6′-diasteromeric mixture of 4/5 (∼3:1 ratio) was a 6-fold poorer inhibitor than the parent paromamine (IC50Pro values of 24 and 3.9 μM, respectively), concluding that the 6′-position plays a pivotal role in bacterial rRNA recognition.19 Our results in regards to the bacterial ribosome are in agreement with these previously reported data. Thus, even though instead of 6′-diastereomeric mixture reported in the latter study, we used the pure 6′-diastereomers 1, 4, and 5, the data in Table 1 shows that 4 and 5 are 1.8-fold and 15.3-fold poorer inhibitors of prokaryotic translation than the parent compound 1 (IC50 values of 6.0, 11.0, and 91.9 μM for 1, 4, and 5, respectively). The scenario in eukaryotic translation is, however, different: compound 4 is 2.9-fold more potent inhibitor than its parent 1 (IC50Euk values of 120.5 and 347.1 μM, respectively), which is further supported by the observed elevated PTC suppression activity of 4 in comparison to that of 1 (Figure 3).
The observed differential selectivity of 4 can be explained by the differences between its putative binding sites in eukaryotic versus prokaryotic ribosome. A brief comparison between the secondary structures of A sites in Figure 5A clearly shows that while the bacterial A site is largely closed between the two C=G Watson–Crick base pairs, the eukaryotic A site is more open due to unpaired C1409 and A1491 (E. coli numbering), allowing higher flexibility of the eukaryotic decoding center at this region. The two major bases that differ between bacteria and eukaryotes are at positions 1408 and 1491: A1408 and G1491 in bacteria; and G1408 and A1491 in eukaryotes (Figure 5A). These differences make AGs potent antibiotics being highly selective toward the bacterial versus the eukaryotic ribosomes. For example, the data in Table 1 show that G418 is 225-fold more selective toward the prokaryotic versus the eukaryotic ribosome. The interactions of AGs ring I with the rRNA target sites have been suggested to contribute significantly to the observed selectivity of AGs.20 Superimposition of the G418–Yeast structure8 with the crystal structure of G418 bound to the bacterial A site (G418–Bact)20 indicates that the overall conformation of the binding site upon G418 binding in both structures is relatively similar (r.m.s.d. values of 1.5 Å, Figure 5B), yet there are significant differences in ligand–host interactions. Due to the lack of a strong C1409=G1491 Watson–Crick pairing that is present in G418–Bact structure, the A1491 in G418–Yeast structure is significantly shifted down toward the deep/major groove and moves all the ligand molecule to the same direction. This makes G418 relatively poorly bound in eukaryotic versus prokaryotic target site as it is highlighted only for its ring I in Figure 5C.
In the bacterial A-site (Figure 5C, green)20 ring I forms a pseudo base pair with A1408, by forming two hydrogen bonds involving the ring oxygen and the 6′-OH group. These interactions position the pyranose ring hydrogens in C–H···π stacking with G1491, whereas two other hydroxyls on the opposite side of the ring I, 3′-OH and 4′-OH, make H-bonds with the phosphate oxygen atoms. The chiral (R)-6′-Me points in the middle of the G1491=C1409 pair, thus providing some additional hydrophobic stabilization of ring-I. Because of such compact interactions of ring I in the bacterial A-site, the modifications at 6′-position could affect negatively to the binding and to the subsequent biological activity as it is observed for the compounds 4 and 5 (Table 1). In the G418–Yeast structure (Figure 5C, magenta),8 however, the ring I of G418 forms fewer contacts and is very poorly bound. Instead of a pseudo base pair presented in bacterial A site, only one H-bond between N1 of G1408 and O6′ of ring I is formed. In the opposite site of ring I, only O4′ makes H-bond contact with the phosphate oxygen atom. The chiral (R)-6′-Me is relatively far from the C1409 and makes this position prone for the modifications; addition of an extra hydroxyl (7′-OH), while keeping the same absolute configuration at 6′-postion (compound 4, Table 1), resulted in significant increase in the observed biological activity.
In summary, the recently solved X-ray crystal structure of eukaryotic ribosome in complex with the aminoglycoside G4188 provided, for the first time, a clear picture of differences between the prokaryotic and eukaryotic sites, which was exploited here for the rational design of new compounds that selectively target the eukaryotic cytoplasmic rRNA A site, a key target for PTC suppression activity. Using this tool, we discovered a new pharmacophore, 7′-hydroxyl group, as a valuable structural element of the glucosamine ring (ring I) that significantly affects eukaryotic versus prokaryotic selectivity and the subsequent PTC suppression activity. In addition, the observed data support the feasibility of using the rational design strategy employed here for the construction of new AG derivatives that may act as drug for the treatment of PTC underlined genetic disorders.
Acknowledgments
N.M.S. thanks the Fine Postdoctoral Fellowship at the Technion. V.B. acknowledges the Ministry of Science and Technology, Israel (Kamea Program).
Glossary
ABBREVIATIONS
- AG
aminoglycoside
- PTC
premature termination codon
- CF
cystic fibrosis
- DMD
Duchenne muscular dystrophy
- USH
Usher syndrome
- MPS I–H
mucopolysaccharidosis type I–H
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00006.
Biochemical assays and synthetic procedures, assignment of absolute configuration at 6′-position in 4 and 5, and copies of NMR spectra (PDF)
This work was supported by Eloxx Pharmaceutical LTD Research Fund (Grant No. 2019230).
The authors declare the following competing financial interest(s): T.B. declares that the compounds 2, 3, 6, and 7 discussed in this publication are subject to license agreement granted to a commercial third party.
Supplementary Material
References
- Keeling K. M.; Bedwell D. M. Pharmacological suppression of premature stop mutations that cause genetic diseases. Curr. Pharmacogenomics 2005, 3, 259–269. 10.2174/157016005774913149. [DOI] [Google Scholar]
- Zingman L. V.; Park S.; Olson T. M.; Alekseev A. E.; Terzic A. Aminoglycoside-induced translational read-through in disease: overcoming nonsense mutations by pharmacogenetic therapy. Clin. Pharmacol. Ther. 2007, 81, 99–103. 10.1038/sj.clpt.6100012. [DOI] [PubMed] [Google Scholar]
- Keeling K. M.; Xue X.; Gunn G.; Bedwell D. M. Therapeutics Based on Stop Codon Readthrough. Annu. Rev. Genomics Hum. Genet. 2014, 15, 371–394. 10.1146/annurev-genom-091212-153527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalev M.; Baasov T. When proteins start to make sense: fine-tuning of aminoglycosides for PTC suppression therapy. MedChemComm 2014, 5, 1092–1105. 10.1039/C4MD00081A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandasamy J.; Atia-Glikin D.; Shulman E.; Shapira K.; Shavit M.; Belakhov V.; Baasov T. Increased selectivity toward cytoplasmic versus mitochondrial ribosome confers improved efficiency of synthetic aminoglycosides in fixing damaged genes: A strategy for treatment of genetic diseases caused by nonsense mutations. J. Med. Chem. 2012, 55, 10630–10643. 10.1021/jm3012992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman E.; Belakhov V.; Wei G.; Kendall A.; Meyron-Holtz E. G.; Ben-Shachar D.; Schacht J.; Baasov T. Designer aminoglycosides that selectively inhibit cytoplasmic rather than mitochondrial ribosomes show decreased ototoxicity a strategy for the treatment of genetic diseases. J. Biol. Chem. 2014, 289, 2318–2330. 10.1074/jbc.M113.533588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudelman I.; Glikin D.; Smolkin B.; Hainrichson M.; Belakhov V.; Baasov T. Repairing faulty genes by aminoglycosides: development of new derivatives of Geneticin (G418) with enhanced suppression of diseases-causing nonsense mutations. Bioorg. Med. Chem. 2010, 18, 3735–3746. 10.1016/j.bmc.2010.03.060. [DOI] [PubMed] [Google Scholar]
- De Loubresse N. G.; Prokhorova I.; Holtkamp W.; Rodnina M. V.; Yusupova G.; Yusupov M. Structural basis for the inhibition of the eukaryotic ribosome. Nature 2014, 513, 517–522. 10.1038/nature13737. [DOI] [PubMed] [Google Scholar]
- Shalev M.; Kondo J.; Kopelyanskiy D.; Jaffe C. L.; Adir N.; Baasov T. Identification of the molecular attributes required for aminoglycoside activity against Leishmania. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 13333–13338. 10.1073/pnas.1307365110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalev M.; Rozenberg H.; Smolkin B.; Nasereddin A.; Kopelyanskiy D.; Belakhov V.; Schrepfer T.; Schacht J.; Jaffe C. L.; Adir N.; Baasov T. Structural basis for selective targeting of leishmanial ribosomes: aminoglycoside derivatives as promising therapeutics. Nucleic Acids Res. 2015, 43, 8601–8613. 10.1093/nar/gkv821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyffeler P. T.; Liang C.-H.; Koeller K. M.; Wong C.-H. The chemistry of amine-azide interconversion: catalytic diazotransfer and regioselective azide reduction. J. Am. Chem. Soc. 2002, 124, 10773–10778. 10.1021/ja0264605. [DOI] [PubMed] [Google Scholar]
- More J. D.; Finney N. S. A simple and advantageous protocol for the oxidation of alcohols with O-iodoxybenzoic acid (IBX). Org. Lett. 2002, 4, 3001–3003. 10.1021/ol026427n. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Adsool V. A.; Hale C. R. H. An expedient procedure for the oxidative cleavage of olefinic bonds with PhI(OAc)2, NMO, and catalytic OsO4. Org. Lett. 2010, 12, 1552–1555. 10.1021/ol100290a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman M.; Belakhov V.; Lee L. V.; Liang F.-S.; Wong C.-H.; Baasov T. Dual effect of synthetic amino-glycosides: Antibacterial activity against Bacillus anthracis and inhibition of anthrax lethal factor. Angew. Chem., Int. Ed. 2005, 44, 447–452. 10.1002/anie.200462003. [DOI] [PubMed] [Google Scholar]
- Kandasamy J.; Atia-Glikin D.; Belakhov V.; Baasov T. Repairing faulty genes by aminoglycosides: Identification of new pharmacophore with enhanced suppression of disease-causing nonsense mutations. MedChemComm 2011, 2, 165–171. 10.1039/c0md00195c. [DOI] [Google Scholar]
- Manuvakhova M.; Keeling K.; Bedwell D. M. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA 2000, 6, 1044–1055. 10.1017/S1355838200000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudelman I.; Rebibo-Sabbah A.; Cherniavsky M.; Belakhov V.; Hainrichson M.; Chen F.; Schacht J.; Pilch D. S.; Ben-Yosef T.; Baasov T. Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J. Med. Chem. 2009, 52, 2836–2845. 10.1021/jm801640k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X.; Mutyam V.; Tang L.; Biswas S.; Du M.; Jackson L. A.; Dai Y.; Belakhov V.; Shalev M.; Chen F.; Schacht J.; Bridges R. J.; Baasov T.; Hong J.; Bedwell D. M.; Rowe S. M. Synthetic aminoglycosides efficiently suppress cystic fibrosis transmembrane conductance regulator nonsense mutations and are enhanced by ivacaftor. Am. J. Respir. Cell Mol. Biol. 2014, 50, 805–816. 10.1165/rcmb.2013-0282OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen K. B.; Ayida B. K.; Vourloumis D.; Takahashi M.; Winters G. C.; Barluenga S.; Qamar S.; Shandrick S.; Zhao Q.; Hermann T. Novel paromamine derivatives exploring shallow-groove recognition of ribosomal-decoding-site RNA. ChemBioChem 2002, 3, 1223–1228. 10.1002/1439-7633(20021202)3:12<1223::AID-CBIC1223>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Vicens Q.; Westhof E. Crystal structure of Geneticin bound to a bacterial 16S ribosomal RNA A site oligonucleotide. J. Mol. Biol. 2003, 326, 1175–1188. 10.1016/S0022-2836(02)01435-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.