

Abstract

X-ray crystal structural determination of FABP4 in complex with four inhibitors revealed the complex binding modes, and the resulting observations led to improvement of the inhibitory potency of FABP4 inhibitors. However, the detailed structure–activity relationship (SAR) could not be explained from these structural observations. For a more detailed understanding of the interactions between FABP4 and inhibitors, fragment molecular orbital analyses were performed. These analyses revealed that the total interfragment interaction energies of FABP4 and each inhibitor correlated with the ranking of the Ki value for the four inhibitors. Furthermore, interactions between each inhibitor and amino acid residues in FABP4 were identified. The oxygen atom of Lys58 in FABP4 was found to be very important for strong interactions with FABP4. These results might provide useful information for the development of novel potent FABP4 inhibitors.
Keywords: FABP, aP2, inhibitor, X-ray crystal structure, FMO
Fatty acid-binding proteins (FABPs) are cytoplasmic proteins that bind to hydrophobic ligands, such as long chain fatty acids, in a noncovalent and reversible manner. It has been suggested that FABPs act as chaperone proteins for fatty acids and play important roles in homeostasis of fatty acids and the lipid signaling pathway.1 The protein FABP4 (aka aFABP or aP2), a member of the FABP family, is a 14.6 kDa cytosolic protein mainly expressed in macrophages and adipocytes. Previous studies have reported that FABP4-deficiency in genetic and diet-induced obese mice show protection against the development of hyperinsulinemia and insulin resistance.2,3 An FABP4-deficiency in apolipoprotein E-deficient mice also shows protection against the development of atherosclerosis.4 Furthermore, treatments with prototype small molecular inhibitors for FABP4 attenuate fatty infiltration in livers of ob/ob mice.5 These studies have demonstrated that FABP4-deficiency is related to several diseases, including diabetes, atherosclerosis, and liver disease, and that potent, selective, small molecular inhibitors for FABP4 might be potential therapeutic agents for these diseases. Recently, a short series of FABP4 inhibitors have been reported, with most of them identified by a structure-based drug design (SBDD) method.6−10 Thus, the crystal structures of several ligand-bound human FABP 4s have been determined,5,6,11 which provided insights into the structural foundation underlying the binding modes of endogenous ligands and small molecular inhibitors in the FABP4 binding site. Structurally, FABP4 contains 10 antiparallel β-strands forming a β-clam shell structure, and its characteristic structure provides a substrate binding site.12−19 Considering these results, ligands lie in the FABP4 internal pocket and usually have polar interactions between FABP4’s Arg126 and Tyr128 side chains and the ligand carboxylic acid. In addition, the importance of a negatively charged substituent, such as a CO2–, SO3–, or PO3– group, in these inhibitors has been clarified.20
The purpose of this study was to identify an FABP4 inhibitor as a new drug candidate. Initially, our in-house compound library was screened using a human FABP4 binding assay. We identified a hit compound that possessed an indole core structure (data not shown). Furthermore, 300 compounds were designed based on this compound and assayed for their binding activity against FABP4. Novel FABP4 inhibitors were thus identified (Table 1). The complex crystal structures of FABP4 bound with four novel inhibitors were then determined, and this information led to selection of improved FABP4 inhibitors, compounds 1 and 2 (Table 1). Among the four inhibitors, compounds 1, 2, and 3 possessed activities sequentially from high to low, and compound 4 showed the lowest activity (Table 1, Ki values). As a control, the Ki value of the reference compound, BMS309403,5 was 0.16 μM using the same assay.
Table 1. Ki and IFIE Sum of FABP4 and Inhibitorsa.
| compd | Ki (uM) | IFIE sum (kcal/mol) | ES (kcal/mol) | EX (kcal/mol) | CT + mix (kcal/mol) | DI (kcal/mol) |
|---|---|---|---|---|---|---|
| 1 | 0.03 | –97.06 | –88.43 | 103.78 | –43.13 | –69.28 |
| 2 | 0.10 | –59.84 | –53.88 | 90.60 | –29.28 | –67.28 |
| 3 | 0.12 | –51.29 | –26.56 | 49.28 | –18.64 | –55.36 |
| 4 | >1.4 | –31.80 | –10.51 | 26.23 | –13.28 | –34.23 |
ES is electrostatic energy, EX is exchange repulsion energy, CT + mix is charge transfer and mixing term energy, DI is dispersion energy, IFIE is sum of ES, EX, CT + mix, and DI, and IFIE of all residues is added up to IFIE sum.
However, it was difficult to understand the structure–activity relationship (SAR) by observation of only hydrogen bond interactions. For more detailed consideration, these interactions were investigated using the fragment molecular orbital (FMO) method,21−24 which has been developed for accurate analysis of large biomolecules, such as protein–ligand complexes. Using the calculated interfragment interaction energies (IFIE)22−28 between these four inhibitors and FABP4 amino acid residues, the interaction differences were described in detail to allow the development of new FABP4 inhibitors.
Before the crystallized complex structures were determined, the question was examined as to whether FABP4 maintained its structure and binding activity to palmitate, the native lipid substrate, when bound to compound 3, the lead candidate inhibitor identified by nuclear magnetic resonance (NMR). A 1H–15N heteronuclear single quantum correlation (HSQC) spectrum (Figure S1) of apo-FABP4 showed approximately 120 signals, which equaled the number of FABP4 amino acid residues. The well-dispersed signals in the spectrum suggested that conformation was maintained and implied a β-sheet-rich structure, as has been reported for the apo-FABP4 structure (PDB code, 3RZY).29 The HSQC spectrum of FABP4 with palmitic acid showed that the addition of palmitic acid caused chemical shift perturbations in almost half of the FABP4 signals. These changes might have arisen from changes in the intramolecular hydrogen-bond network beyond the binding interface, as has been described in the NMR evaluation of FABP4 with R- and S-ibuprofen.30 This suggested that palmitic acid bound to FABP4 through the same binding mode as the ibuprofens. The lead inhibitor candidate, compound 3, was subsequently subjected to an FABP4/compound 3 complex NMR analysis. In the resulting HSQC spectrum, almost half of FABP4’s signals were shifted, which strongly suggested that compound 3 bound to FABP4 in the same manner as palmitic acid (Figure 1). From these results, FABP4/compound 1–4 complexes were prepared and used for crystallization analysis.
Figure 1.
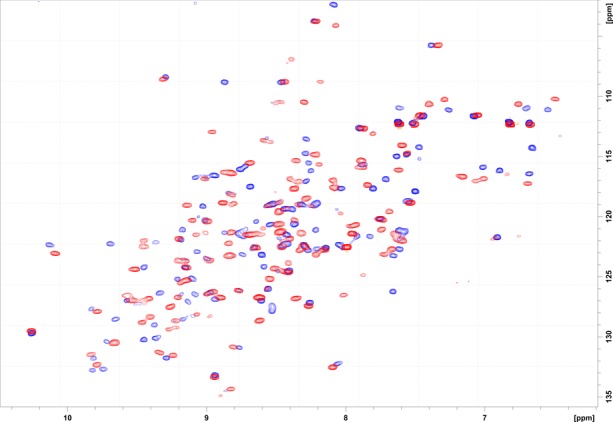
1H–15N HSQC spectrum of FABP4. Chemical shift perturbation patterns observed for apo-FABP4 (blue) and FABP4 with compound 3 (red).
Next, the crystallographic structure of FABP4 was solved as complexes with the four inhibitors (compounds 1–4), and the interactions analyzed to provide useful information for the development of novel potent FABP4 inhibitors. The 31 residues located within 5.0 Å of any atom of compound 1, the most potent inhibitor, were aligned. Among the complexes, the binding sites were found to be very similar, as demonstrated by the root-mean-square deviation value of ∼0.3 Å. All four compounds possessed a carboxyl group that generally formed hydrogen bonds with FABP4’s Arg126 and Tyr128 (Figure 2), but the other moieties also showed distinct interactions. These observations clearly indicated that FABP4 had more interactions with compounds 1 and 2 than with compounds 3 and 4. The effective interactions of FABP4 with inhibitors were further elucidated by observing in detail the interactions of FABP4 and the characteristic moieties of compounds 1 and 2. Two hydrogen bonds with Ser55 and Lys58 and four hydrophobic interactions with Ala36, Pro38, Phe57, and Ala75 were observed in compound 1’s pyrazole substructure (Figure 2a). Meanwhile, one hydrogen bond with Ser55 and five hydrophobic interactions with Ala33, Ala36, Pro38, Lys58, and Ala75 were observed in compound 2’s 2-methoxypyridine moiety (Figure 2b). Observing only the numbers of hydrogen bonds and hydrophobic interactions in these crystal structures was insufficient to explain the precise activity differences between compounds 1 and 2. More information regarding binding modes and potencies was obtained via the FMO method, which was expected to allow calculation of the precise interaction energies in these complexes.
Figure 2.

Two-dimensional diagrams of the binding sites around compounds 1 (a), 2 (b), 3 (c), and 4 (d), drawn using Discovery Studio 4.5. Green, conventional hydrogen bonds, and pink, other interactions, as pi–alkyl, pi–pi T-shaped, alkyl, and CH···O hydrogen bonds.
The validity of the FMO method was confirmed by analyzing the correlation between IFIE and Ki values. The IFIE is the sum of the interaction energy components, as electrostatic, exchange repulsion, charge transfer and mixing term, and dispersion energies (ES, EX, CT + mix, and DI, respectively), and was calculated for each amino acid residue. IFIEs of all amino acid residues were totaled to create the IFIE sum. Negative values represented effective interaction energies for binding an amino acid residue with a ligand. The IFIE sum and Ki of the four inhibitors for FABP4 are shown in Table 1. The correlation coefficient between log(Ki) and IFIE sum was 0.915, indicating that they correlated completely. Therefore, the FMO method was considered to be a valid argument for assessing the activities observed in this study.
IFIEs between each compound and FABP4 amino acid residues are shown in Figure 4. An amino acid residue was treated as a single fragment by separating the carbonyl group and α-carbon. The main interactions of residues from Lys52 to Asp77 are shown magnified in Figure 5.
Figure 4.

IFIEs between each compound and aP2 amino acid residues. Horizontal axis shows fragment numbers that represent the approximate amino acid residue numbers, and vertical axis represents the IFIE for each fragment.
Figure 5.

IFIEs magnified from Lys52 to Asp77.
Compound 1 exhibited strong interactions with Ser55, Phe57, and Lys58 (fragment 59), which enabled compound 1 to have the smallest Ki value among the four inhibitors. Notably, compound 1 formed hydrogen bonds with the Ser55 side chain oxygen atom and Lys58 main chain (Figure 3a), and the related IFIEs were the lowest observed, at −12.33 and −14.96 kcal/mol, respectively (Table S1). A hydrogen bond with Lys58 has been described in the docking model of Pimozide.31 It is notable that this interaction was detected here in the X-ray crystal structure, indicating its importance in the present molecular binding as well as the utility of the FMO method. Phe57 also formed pi–pi T-shaped interactions (Figure 2a), but the related strengths were not clear. The IFIE of Phe57 revealed strong interactions, at −9.79 kcal/mol. In addition, the IFIEs of fragment 76 (Asp76) represented the largest repulsive force of all IFIEs. Among Asp76 IFIEs, the IFIE between compound 1 and Asp76 was the smallest. The moieties of compound 1 near Asp76 were cyclopropanes, according to the X-ray crystal structure, and cyclopropane was a common moiety in compounds 1, 2, and 3. Comparing the positions of each cyclopropane, the distance between compound 1 cyclopropane and Asp76 was ∼0.4 Å longer than that of compounds 2 and 3 (Figure S2). This greater distance was probably because compound 1 was anchored by hydrogen bonds with Ser55 and Lys58 and assumed to be the main reason the repulsive energy between compound 1 and Asp76 was the lowest.
Figure 3.

X-ray crystal structures of the binding sites around compounds 1 (a) and 2 (b) (PDB IDs, 5D4A and 5D45, respectively). Four key side chains of aP2 shown in stick format and colored by atom type (gray, carbon; blue, nitrogen; and red, oxygen). Compounds 1 and 2 shown in stick format and colored by atom type (green, carbon; blue, nitrogen; and red, oxygen). Dashed line, hydrogen bond.
The situation for compound 2 was a bit complicated because this compound hydrogen-bonded with Ser55 in the crystal structure (Figure 3b) and the IFIE was −7.93 kcal/mol. However, the largest IFIE for compound 2 was −9.83 kcal/mol, with FABP4 fragment 58 (Table S1). While, the Lys58 oxygen atom formed a CH···O hydrogen bond32 with compound 2 in the crystal structure (Figure 2b), the amine group of Lys58’s main chain interacted strongly, as ES energy (−7.30 kcal/mol). Furthermore, Phe57’s IFIE was also large (−5.39 kcal/mol), as determined from FMO measurements (Figure 5), but no interactions were recognized from X-ray structure analyses (Figure 2b). As FMO analysis calculated interaction energies based on electronic states, the interactions could not be recognized using only distances and degrees. In summary, compound 1’s IFIE sum was smaller than that of compound 2.
In this way, the SARs of compounds 1 and 2 could be explained on the basis of interactions with amino acid residues and energies calculated from FMO analyses. These results showed that an FABP4 inhibitor required a strong interaction with Lys58, and thus, compound 1, which possessed strong interactions with Lys58, also possessed high inhibition activity.
Adding the feature of FMO analysis, aP2 fragment 53 (Ser53) exhibited the largest IFIE (−9.64 kcal/mol; Figure 5, green bar) with compound 3 although no interactions with Ser53 were recognized from X-ray crystal structural analysis (Figure 2c). As the main IFIE component with compound 3 and Ser53 was DI energy (−6.04 kcal/mol, Table S1), this energy could not be recognized from distances and angles of X-ray crystal structures. In addition, Ser53’s IFIE was greater than that of Arg126 and Tyr128 (−2.16 and −1.21 kcal/mol, respectively), which unexpectedly formed hydrogen bonds.
In conclusion, aP2 crystallographic structures were solved as complexes with four inhibitors and the interactions were described by FMO analyses. These analyses were very helpful in determining why compound 1 possessed the most potent inhibitory activity. Furthermore, the importance of the interaction with Lys58 was revealed, contributing to the understanding for the design of a strong inhibitor, which will be useful for future inhibitor identification efforts.
Acknowledgments
We extend our deepest appreciation to Hitoshi Wakatsuki for their technical assistance. We thank the staff of the Photon Factory (Tsukuba, Japan) for assistance during data collection. We also thank Naoyuki Fukuchi, Ajinomoto Pharmaceuticals Co., Ltd. for helpful discussions. Finally, we thank Kaori Fukuzawa, Nihon University, and Takayuki Tsukamoto, Mizuho Information & Research Institute.
Glossary
ABBREVIATIONS
- aP2
adipocyte protein 2
- FABP
fatty acid binding protein
- FMO
fragment molecular orbital
- SAR
structure–activity relationship
- IFIE
interfragment interaction energies
- NMR
nuclear magnetic resonance
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00040.
Process of four compounds, assay protocols, and experimental procedures (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Furuhashi M.; Hotamisligil G. S. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discovery 2008, 7, 489–503. 10.1038/nrd2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil G. S.; Johnson R. S.; Distel R. J.; Ellis R.; Papaioannou V. E.; Spiegelman B. M. Uncoupling of obesity from insulin resistance through a targeted mutation in aP2, the adipocyte fatty acid binding protein. Science 1996, 274, 1377–1379. 10.1126/science.274.5291.1377. [DOI] [PubMed] [Google Scholar]
- Uysal K. T.; Scheja L.; Wiesbrock S. M.; Bonner-Weir S.; Hotamisligil G. S. Endocrinology 2000, 141, 3388–3396. 10.1210/endo.141.9.7637. [DOI] [PubMed] [Google Scholar]
- Makowski L.; Boord J. B.; Maeda K.; Babaev V. R.; Uysal K. T.; Morgan M. A.; Parker R. A.; Suttles J.; Fazio S.; Hotamisligil G. S.; Linton M. F. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat. Med. 2001, 6, 699–705. 10.1038/89076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi M.; Tuncman G.; Görgün C. Z.; Makowski L.; Atsumi G.; Vaillancourt E.; Kono K.; Babaev V. R.; Fazio S.; Linton M. F.; Sulsky R.; Robl J. A.; Parker R. A.; Hotamisligil G. S. Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature 2007, 447, 959–965. 10.1038/nature05844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann F.; Haile S.; Axen E.; Medina C.; Uppenberg J.; Svensson S.; Lundbäck T.; Rondahl L.; Barf T. Discovery of inhibitors of human adipocyte fatty acid-binding protein, a potential type 2 diabetes target. Bioorg. Med. Chem. Lett. 2004, 14, 4445–4448. 10.1016/j.bmcl.2004.06.057. [DOI] [PubMed] [Google Scholar]
- Ringom R.; Axen E.; Uppenberg J.; Lundbäck T.; Rondahl L.; Barf T. Substituted benzylamino-6-(trifluoromethyl)pyrimidin-4(1H)-ones: a novel class of selective human A-FABP inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 4449–4452. 10.1016/j.bmcl.2004.06.058. [DOI] [PubMed] [Google Scholar]
- Sulsky R.; Magnin D. R.; Huang Y.; Simpkins L.; Taunk P.; Patel M.; Zhu Y.; Stouch T. R.; Bassolino-Klimas D.; Parker R.; Harrity T.; Stoffel R.; Taylor D. S.; Lavoie T. B.; Kish K.; Jacobson B. L.; Sheriff S.; Adam L. P.; Ewing W. R.; Robl J. A. Potent and selective biphenyl azole inhibitors of adipocyte fatty acid binding protein (aFABP). Bioorg. Med. Chem. Lett. 2007, 17, 3511–3515. 10.1016/j.bmcl.2006.12.044. [DOI] [PubMed] [Google Scholar]
- Barf T.; Lehmann F.; Hammer K.; Haile S.; Axen E.; Medina C.; Uppenberg J.; Svensson S.; Rondahl L.; Lundbäck T. N-Benzyl-indolo carboxylic acids: Design and synthesis of potent and selective adipocyte fatty-acid binding protein (A-FABP) inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 1745–1748. 10.1016/j.bmcl.2009.01.084. [DOI] [PubMed] [Google Scholar]
- Hertzel A. V.; Hellberg K.; Reynolds J. M.; Kruse A. C.; Juhlmann B. E.; Smith A. J.; Sanders M. A.; Ohlendorf D. H.; Suttles J.; Bernlohr D. A. Identification and characterization of a small molecule inhibitor of Fatty Acid binding proteins. J. Med. Chem. 2009, 52, 6024–6031. 10.1021/jm900720m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr E.; Tardie M.; Carty M.; Brown Phillips T.; Wang I. K.; Soeller W.; Qiu X.; Karam G. Expression, purification, crystallization and structure of human adipocyte lipid-binding protein (aP2). Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2006, 62, 1058–1060. 10.1107/S1744309106038656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ory J. J.; Kane C. D.; Simpson M. A.; Banaszak L. J.; Bernlohr D. A. Biochemical and crystallographic analyses of a portal mutant of the adipocyte lipid-binding protein. J. Biol. Chem. 1997, 272, 9793–9801. 10.1074/jbc.272.15.9793. [DOI] [PubMed] [Google Scholar]
- Ory J. J.; Banaszak L. J. Studies of the ligand binding reaction of adipocyte lipid binding protein using the fluorescent probe 1, 8-anilinonaphthalene-8-sulfonate. Biophys. J. 1999, 77, 1107–1116. 10.1016/S0006-3495(99)76961-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ory J. J.; Mazhary A.; Kuang H.; Davies R. R.; Distefano M. D.; Banaszak L. J. Structural characterization of two synthetic catalysts based on adipocyte lipid-binding protein. Protein Eng., Des. Sel. 1998, 11, 253–261. 10.1093/protein/11.4.253. [DOI] [PubMed] [Google Scholar]
- LaLonde J. M.; Bernlohr D. A.; Banaszak L. J. X-ray Crystallographic Structures of Adipocyte Lipid-Binding Protein Complexed with Palmitate and Hexadecanesulfonic Acid. Properties of Cavity Binding Sites. Biochemistry 1994, 33, 4885–4895. 10.1021/bi00182a017. [DOI] [PubMed] [Google Scholar]
- LaLonde J. M.; Levenson M. A.; Roe J. J.; Bernlohr D. A.; Banaszak L. J. Adipocyte lipid-binding protein complexed with arachidonic acid. Titration calorimetry and X-ray crystallographic studies. J. Biol. Chem. 1994, 269, 25339–25347. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Bernlohr D. A.; Banaszak L. J. The adipocyte lipid-binding protein at 1.6-A resolution. Crystal structures of the apoprotein and with bound saturated and unsaturated fatty acids. J. Biol. Chem. 1993, 268, 7874–84. [PubMed] [Google Scholar]
- Xu Z. H.; Bernlohr D. A.; Banaszak L. J. Crystal structure of recombinant murine adipocyte lipid-binding protein. Biochemistry 1992, 31, 3484–3492. 10.1021/bi00128a024. [DOI] [PubMed] [Google Scholar]
- Xu Z. H.; Buelt M. K.; Banaszak L. J.; Bernlohr D. A. Expression, purification, and crystallization of the adipocyte lipid binding protein. J. Biol. Chem. 1991, 266, 14367–14370. [PubMed] [Google Scholar]
- Maria J. P.; Jonas U.; Stefan S.; Thomas L.; Tomas Å.; Mats W.; Johan S. Structure-Based Screening As Applied to Human FABP4: A Highly Efficient Alternative to HTS for Hit Generation. J. Am. Chem. Soc. 2002, 124, 11874–11880. 10.1021/ja017830c. [DOI] [PubMed] [Google Scholar]
- Kitaura K.; Ikeo E.; Asada T.; Nakano T.; Uebayasi M. Fragment molecular orbital method: an approximate computational method for large molecules. Chem. Phys. Lett. 1999, 313, 701–706. 10.1016/S0009-2614(99)00874-X. [DOI] [Google Scholar]
- Fedorov D. G.; Kitaura K. Extending the power of quantum chemistry to large systems with the fragment molecular orbital method. J. Phys. Chem. A 2007, 111, 6904–6914. 10.1021/jp0716740. [DOI] [PubMed] [Google Scholar]
- Fedorov D. G.; Nagata T.; Kitaura K. Exploring chemistry with the fragment molecular orbital method. Phys. Chem. Chem. Phys. 2012, 14, 7562–7577. 10.1039/c2cp23784a. [DOI] [PubMed] [Google Scholar]
- Gordon M. S.; Fedorov D. G.; Pruitt S. R.; Slipchenko L. V. Fragmentation methods: a route to accurate calculations on large systems. Chem. Rev. 2012, 112, 632–672. 10.1021/cr200093j. [DOI] [PubMed] [Google Scholar]
- Nakano T.; Kaminuma T.; Sato T.; Fukuzawa K.; Akiyama Y.; Uebayasi M.; Kitaura K. Fragment molecular orbital method: use of approximate electrostatic potential. Chem. Phys. Lett. 2002, 351, 475–480. 10.1016/S0009-2614(01)01416-6. [DOI] [Google Scholar]
- Hitaoka M.; Harada T.; Yoshida T.; Chuman H. Correlation analyses on binding affinity of sialic acid analogues with influenza virus neuraminidase-1 using ab initio MO calculations on their complex structures. J. Chem. Inf. Model. 2010, 50, 1796–1805. 10.1021/ci100225b. [DOI] [PubMed] [Google Scholar]
- Watanabe C.; Fukuzawa K.; Okiyama Y.; Tsukamoto T.; Kato A.; Tanaka S.; Mochizuki Y.; Nakano T. Three- and four-body corrected fragment molecular orbital calculations with a novel subdividing fragmentation method applicable to structure-based drug design. J. Mol. Graphics Modell. 2013, 41, 31–42. 10.1016/j.jmgm.2013.01.006. [DOI] [PubMed] [Google Scholar]
- Miyanaga W.; Sugiki M.; Ejima C.; Tokumasu M.; Yoshida T.; Takeshita S. Indole derivative or salt thereof. Patent WO2014003158, 2014.
- González J. M.; Fisher S. Z. Structural analysis of ibuprofen binding to human adipocyte fatty-acid binding protein (FABP4). Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2015, 71, 163–170. 10.1107/S2053230X14027897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guoyun B.; Huaping M.; Michael S. NMR evaluation of adipocyte fatty acid binding protein (aP2) with R- and S-ibuprofen Bioorganic &. Bioorg. Med. Chem. 2008, 16, 4323–4330. 10.1016/j.bmc.2008.02.092. [DOI] [PubMed] [Google Scholar]
- Yan W.; Huang-Quan L.; Wai-Kit L.; Wei-Cheng L.; Jin-Fang Z.; Jian-Shu H.; Tsz-Ming I.; Mary M. W.; David C. W. Pimozide, a Novel Fatty Acid Binding Protein 4 Inhibitor, Promotes Adipogenesis of 3T3-L1 Cells by Activating PPARγ. ACS Chem. Neurosci. 2015, 6, 211–218. 10.1021/cn5002107. [DOI] [PubMed] [Google Scholar]
- Pierce A. C.; Sandretto K. L.; Bemis G. W. Kinase inhibitors and the case for CH···O hydrogen bonds in protein-ligand binding. Proteins: Struct., Funct., Genet. 2002, 49, 567–576. 10.1002/prot.10259. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.