

Abstract

Cannabidiol is the nonpsychoactive natural component of C. sativa that has been shown to be neuroprotective in multiple animal models. Our interest is to advance a therapeutic candidate for the orphan indication hepatic encephalopathy (HE). HE is a serious neurological disorder that occurs in patients with cirrhosis or liver failure. Although cannabidiol is effective in models of HE, it has limitations in terms of safety and oral bioavailability. Herein, we describe a series of side chain modified resorcinols that were designed for greater hydrophilicity and “drug likeness”, while varying hydrogen bond donors, acceptors, architecture, basicity, neutrality, acidity, and polar surface area within the pendent group. Our primary screen evaluated the ability of the test agents to prevent damage to hippocampal neurons induced by ammonium acetate and ethanol at clinically relevant concentrations. Notably, KLS-13019 was 50-fold more potent and >400-fold safer than cannabidiol and exhibited an in vitro profile consistent with improved oral bioavailability.
Keywords: neuroprotection, cannabidiol, hepatic encephalopathy
Hepatic encephalopathy (HE) is a neuropsychiatric disorder that includes learning deficits and impairment of long-term memory. If left unchecked, HE can progress to hepatic coma and ultimately death.1 The pathogenesis of HE includes damage to the prelimbic cortex, striatum, and the hippocampus.2 Hepatic encephalopathy is caused by accumulation of toxic substances in the bloodstream that are normally removed by the liver. It has been previously demonstrated that impairment of hepatocytes by ethanol is associated with the production of free radical and oxidative stress.3 The accumulation of these free radicals and oxidative stress contribute to cognitive impairment, learning deficits, memory impairment, as well as damage and death of neuronal tissue. An emerging concept is that blockade of free radical mediated stress and oxidative stress will prevent the neural damage associated with hepatic encephalopathy and prevent cognitive impairment, learning deficits, memory impairment, as well as damage and death of neuronal tissue associated with HE.
Cannabidiol (CBD) has been shown to be neuroprotective by blocking the damage caused by free radicals and oxidative stress.4 This effect was independent of cannabinoid receptors because it could not be blocked by a cannabinoid antagonist. CBD is effective in two animal models of HE, thioacetamide induced5 and bile duct ligation induced6 liver damage, at a dose of 5 mg/kg ip. Importantly, CBD treated animals in the first study exhibited improvements in both liver and brain function as compared to untreated control animals. Free radical mediated stress and oxidative stress are also known to contribute to additional pathological conditions including epilepsy, neuropathic pain, traumatic head injury, stroke, chronic traumatic encephalopathy (CTE), and neurodegenerative diseases such as Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, and amyotrophic lateral sclerosis (ALS). Other examples of neuroprotection by CBD include use in hypoxia-ischemia7 and stroke8 models. A wide range of possible mechanisms have been attributed for CBD’s neuroprotective effects including antioxidant, anti-inflammatory, adenosine signaling, cannabinoid receptor GPR55, and serotonin mediated pathways;9 however, mitochondrial calcium modulation is fundamental.10 To hedge our bets, we chose to interrogate the hippocampal neuron, as a phenotypic screen that will measure neuroprotection independent of a mechanism. We chose to use either ammonia (300 μM) or ethanol (30 mM) as toxins at therapeutic relevant concentrations. Blood levels of ammonia in grade IV HE patients range from 150 to 300 μM,11 while heavy drinkers can reach 50 mM ethanol levels (legal limit is 17 mM). Neuronal viability and cell death are monitored in each experiment using the dyes carboxyfluorescein diacetate (CFDA) and propidium iodide (PI), respectively. Any of our new molecules that demonstrated increased potency relative to cannabidiol were evaluated for CB1 affinity to rule out potential for psychotropic side effects.
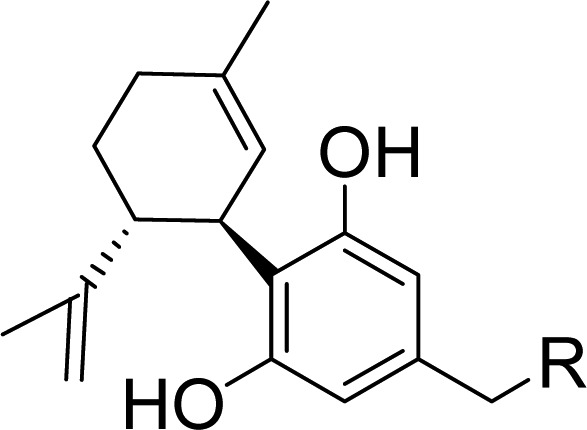
CBD is a small molecule that violates only one of Lipinski’s rules; it has excessive lipophilicity (cLogP = 6.3). Our design concept was to identify a region of the molecule that could provide novelty and greater hydrophilicity without restoring CB1 affinity. It is known that even minor structural changes to (−)-CBD (natural form) can impart potent CB1 affinity (Figure1). In fact, CBD’s (+)-enantiomer 2,12 cyclohexyl resorcinol 3, and aromatic phenol counterpart 4 are CB1 ligands.13 For these reasons we chose to leave the resorcinol and cyclohexene rings alone and introduce hydrophilicity on the five carbon side chain of CBD. Very little chemistry had been reported with heteroatoms in this region, and it was hoped that more water-soluble molecules would have advantages in terms of permeability and bioavailability.
Figure 1.

Cannabidiol, key literature compounds, and new design.
A group of target molecules were chosen for synthesis (Table 1), which had greater hydrophilicity and contained a variety of heteroatom or heterocycle substituted side chains to replace the five carbon side chain of CBD. Care was taken to introduce as little steric bulk as possible, while exploring diversity of functional groups and pKas. The cLogP values for the target compounds in Table 1 are all lower than 5 (calculated in the Collaborative Drug Discovery Vault database). Polar surface area for CBD (40 Å2) is low, while the other target molecules had PSA (43 to 78 Å2) in an acceptable range for CNS penetration.14 Compounds 7 and 8 are known metabolites15−17 of CBD that we wanted to test side by side with CBD because the metabolites could contribute to CBD’s neuroprotective effects in vivo.
Table 1. cLog P, Yield, and in Vitro Testing of Reference Standards and New Molecules.

The synthesis of target molecule 16 is described in detail for illustrative purposes (Scheme1). The synthesis began by alkylating triphenylphosphine with benzyl bromide 19. Reaction of the Wittig ylide of 20 with the azetidine carbonyl 21 afforded the olefin 22, which was conveniently reduced with hydrogen under atmospheric pressure. The Boc-group was removed with TFA and replaced with an acetyl group to deliver 25 in good yield. The methyl ethers were removed with boron tribromide with careful reaction monitoring to generate the key resorcinol intermediate 26. The substituted resorcinol 26 was coupled to (1S,4R)-4-isopropenyl-1-methyl-2-cyclohexen-1-ol (6) under acidic (BF3-Et2O) conditions similar to what was described in the literature.18 Each coupling reaction was optimized using various acids (p-TsOH, BF3-Et2O, CF3SO3H), solvents, and reaction times to maximize production of desired product by alkylation of resorcinol at the 2-position. In most cases, the reaction was terminated prior to consumption of starting material as to minimize byproducts and to simplify purification. The desired product 16 was obtained in low yield (13%) accompanied by a roughly equivalent amount of the 4-regioisomer 27 (17%). The desired product was obtained in variable yields (2–25%, Table 1). The resorcinols were derived from 3,5-dimethoxybenzaldehyde (7, 8, 14), 2-(3,5-dimethoxyphenyl)-acetic acid (9, 10, 11, 12, 13), or 3,5-dihydroxybenzoic acid (15, 17, 18). CBD (1) was prepared from olivitol using the same method, and its optical rotation was consistent with the literature ([α]D = −125),19 [α]D22 = −121.4 (c 1.00, EtOH). This gave us confidence that our couplings were proceeding with high stereoselectivity. Compound 17 was made by reduction of 18 with lithium aluminum hydride. Complete experimental details and analytical data (LC/MS, 1H NMR) for the compounds are included in the Supporting Information.
Scheme 1. Synthesis of Compound 16.

Dissociated hippocampal cultures derived from embryonic day 18 rats were employed as the primary screening system to test for toxicity responses as well as neuroprotective actions. All compounds were assessed in both ethanol- and ammonium acetate-induced neurotoxicity assays and monitored with both dyes (CFDA and PI), as measures of viability and cell death, respectively. Full efficacy was defined as the fluorescence measure of a neuron in absence of toxin. All compounds were screened with N = 5 at each test concentration and their EC50 value determined (Table 1). For compounds of highest interest (Table 2), the EC50 ± SE was determined from three replicate experiments with N = 5 for each experiment. Cell viability and cell death were typically affected at similar concentrations. For example CBD (1) showed EC50 values in the 1 to 4 μM range in the two assays using two readouts (Table 1). However, CBD has a narrow therapeutic index in that it is neurotoxic by itself at 33 μM. It was hoped that therapeutic index (CBD TI, TC50/EC50 = 16, Table 2) could be improved in new compounds, while showing broad neuroprotection like CBD. The hydroxyl metabolite 7 was mostly inactive at 100 μM, while the carboxylic acid 8 was two- to 10-fold less potent than CBD. The ether (9) was expected to behave similarly to CBD, in that it varies only in one heavy atom. It had similar potency in viability assessment after ethanol (EC50 = 0.3–3 μM) but was variably effective against ammonium acetate (EC50 = 0.2–36 μM). The triazole 10 demonstrated a more consistent efficacy across all conditions conditions (EC50 = 1 to 5 μM), with similar potency to CBD. Interestingly, the regioisomer 11 was inactive up to 100 μM. The morpholino amide (12), morpholino alkyl (13), and acetyl amide (14) derivatives were largely ineffective. Dramatic results were shown with the four-membered ring derivatives 15 to 18. Whereas the oxetane (15) was weakly effective, the methyl azetidine (17) was toxic. However, the acyl (16) and ethoxylformyl (18) azetidines were unexpectedly potent with EC50 values ranging from 20 to 110 nM. We did evaluate some of the 4-regioisomers and found little or no activity (e.g., 27, EC50 > 100 μM).
Table 2. In Vitro Profiling of Active Lead Molecules20,21.

The three most active molecules 10, 16, and 18 were further profiled in comparison to the CBD (1) in Table 2. CBD has a limited therapeutic index of 16 when comparing its toxic dose with its effective dose in the ammonium acetate toxicity model with CFDA readout. Therefore, the new molecules were evaluated for any decrease in neuronal viability at concentrations above their EC50. None of the test agents exhibited toxicity up to 100 μM, and compound 16 was only toxic at 300 μM giving it a therapeutic index of 7500. Compound 18 also demonstrated a much improved safety margin. None of the new molecules exhibited CB1 affinity at 10 μM, but 16 and 18 showed modest affinity for CB2. Kinetic aqueous solubility was improved for the triazole 10 and azetidine 16, but not for 18. All three new molecules showed great improvements in permeability in both CACO and MDCK MDR1 cell lines with no efflux potential.
Compound 16 (KLS-13019) was selected for further characterization based on its overall improved properties relative to CBD (1) in potency, safety, aqueous solubility, and permeability. The complete in vitro and in vivo work up on this lead candidate will reported in due course.
Glossary
ABBREVIATIONS
- AmAc
ammonium acetate
- CFDA
carboxyfluorescein diacetate
- CB1 CB2
cannabinoid receptor 1 or 2
- CBD
cannabidiol
- cLogP
calculated log P
- PI
propidium iodide
- PSA
polar surface area
- TI
therapeutic index
- EC50
50% efficacy concentration
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00009.
Complete experimental details on synthetic and bioassay methods (PDF)
Author Present Address
§ Fox Chase Chemical Diversity Center, 3805 Old Easton Road, Doylestown, Pennsylvania 18902, United States.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Córdoba J. New assessment of hepatic encephalopathy. J. Hepatol. 2011, 54, 1030–1040. 10.1016/j.jhep.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Arias N.; Méndez M.; Fidalgo C.; Aller M. A.; Arias J.; Arias J. L. Mapping metabolic brain activity in three models of hepatic encephalopathy. Int. J. Hypertens. 2013, 2013, 1–7. 10.1155/2013/390872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey S. M.; Cunningham C. C. Acute and chronic ethanol increases reactive oxygen species generation and decreases viability of isolated rat hepatocytes. Hepatology 1998, 28, 1318–1326. 10.1002/hep.510280521. [DOI] [PubMed] [Google Scholar]
- Hampson A. J.; Grimaldi M.; Axelrod J.; Wink D. Cannabidiol and (−)Δ9-tetrahydrocahnnnabinol are neuroprotective antioxidants. Proc. Natl. Acad. Sci. U. S. A. 1998, 95 (14), 8268–8273. 10.1073/pnas.95.14.8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avraham Y.; Grigoriadis N. C.; Poutahidis T.; Vorobiev L.; Magen I.; Ilan Y.; Mechoulam R.; Berry E. M. Cannabidiol improves brain and liver function in a fulminant hepatic failure-induced model of hepatic encephalopathy in mice. Br. J. Pharmacol. 2011, 162, 1650–1658. 10.1111/j.1476-5381.2010.01179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magen I.; Avraham Y.; Ackerman Z.; Vorobiev L.; Mechoulam R.; Berry E. M. Cannabidiol ameliorates cognitive and motor impairments in mice with bile duct ligation. J. Hepatol. 2009, 51, 528–534. 10.1016/j.jhep.2009.04.021. [DOI] [PubMed] [Google Scholar]
- Pazos M. R.; Cinquina V.; Gómez A.; Layunta R.; Santos M.; Fernández-Ruiz J.; Martínez-Orgado J. Cannabidiol administration after hypoxia-ischemia to newborn rats reduces long-term brain injury and restores neurobehavioral function. Neuropharmacology 2012, 63, 776–783. 10.1016/j.neuropharm.2012.05.034. [DOI] [PubMed] [Google Scholar]
- Hampson A. J.; Grimaldi M.; Lolic M.; Wink D.; Rosenthal R.; Axelrod J. Neuroprotective antioxidants from marijuana. Ann. N. Y. Acad. Sci. 2000, 899, 274–282. 10.1111/j.1749-6632.2000.tb06193.x. [DOI] [PubMed] [Google Scholar]
- Hayakawa K.; Mishima K.; Fujiwara M. Therapeutic potential of non-phschotropic cannabidiol in ischemic stroke. Pharmaceuticals 2010, 3, 2197–2212. 10.3390/ph3072197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan D.; Drysdale A. J.; Lafourcade C.; Pertwee R. G.; Platt B. Cannabidiol targets mitochondria to regulate intracellular Ca2+ levels. J. Neurosci. 2009, 29 (7), 2053–2063. 10.1523/JNEUROSCI.4212-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong J. P.; Aggarwal A.; Krieger D.; Easley K. A.; Karafa M. T.; Van Lente F.; Arroliga A. C.; Mullen K. D. Correlation between ammonia levels and the severity of hepatic encephalopathy. Am. J. Med. 2003, 114, 188–193. 10.1016/S0002-9343(02)01477-8. [DOI] [PubMed] [Google Scholar]
- Bisogno T.; Hanuš L.; De Petrocellis L.; Tchilibon S.; Ponde D. E.; Brandi I.; Moriello A. S.; Davis J. B.; Mechoulam R.; Di Marzo V. Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 2001, 134, 845–852. 10.1038/sj.bjp.0704327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley J. L.; Beletskaya I. D.; Ng E. W.; Dai Z.; Crocker P. J.; Mahadevan A.; Razdan R. K.; Martin B. R. Resorcinol derivatives: a novel template for the development of cannabinoid CB1/CB2 and CB2-selective agonists. J. Pharmacol. Exp. Ther. 2002, 301, 679–689. 10.1124/jpet.301.2.679. [DOI] [PubMed] [Google Scholar]
- Hitchcock S. A.; Pennington L. D. Structure - Brain Exposure Relationships. J. Med. Chem. 2006, 49, 7559–7583. 10.1021/jm060642i. [DOI] [PubMed] [Google Scholar]
- Jiang R.; Yamaori S.; Takeda S.; Yamamoto I.; Watanabe K. Identification of cytochrome P450 enzymes responsible for metabolism of cannabidiol by human liver microsomes. Life Sci. 2011, 89, 165–170. 10.1016/j.lfs.2011.05.018. [DOI] [PubMed] [Google Scholar]
- Harvey D. J.; Martin B. R.; Paton W. D. M. Comparative in vivo metabolism of Δ1-tetrahydrocannabinol (Δ1-THC), cannabidiol (CBD) and cannabinol (CBN) by several species. Recent Dev. Mass Spectrom. Biochem. Med. 1978, 1, 161–84. 10.1007/978-1-4613-3991-5_13. [DOI] [Google Scholar]
- Crombie L.; Crombie W. M. L.; Kilbee G. W.; Tuchinda P. Synthesis of 4″-carboxylated cannabinoids: stereospecific processes involving ethylidenemalonic ester. Tetrahedron Lett. 1979, 49, 4773–4776. 10.1016/S0040-4039(01)86707-1. [DOI] [Google Scholar]
- Papahatjis D. P.; Nahmias V. R.; Nikas S. P.; Andreou T.; Alapafuja S. O.; Tsotinis A.; Guo J.; Fan P.; Makriyannis A. C1′-Cycloalkyl side chain pharmacophore in tetrahydrocannabinols. J. Med. Chem. 2007, 50, 4048–4060. 10.1021/jm070121a. [DOI] [PubMed] [Google Scholar]
- Ben-Shabat S.; Hanuš L. O.; Katzavian G.; Gallily R. New cannabidiol derivatives, synthesis, binding to cannabinoid receptor, and evaluation of their anti-inflammatory activity. J. Med. Chem. 2006, 49, 1113–1117. 10.1021/jm050709m. [DOI] [PubMed] [Google Scholar]
- CB1 and CB2 affinity assays were executed at Cerep Eurofins, Le Bois l’Evêque, B.P. 30001, 86 600 Celle l’Evescault, France. Methods are contained in Supporting Information.
- In vitro ADME assays were performed at the DMPK Group at Shanghai ChemPartner Co., Ltd., No. 5 Building, 998, Ha Lei Rd, Pudong, Shanghai 201203, P. R. China.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.