

Abstract
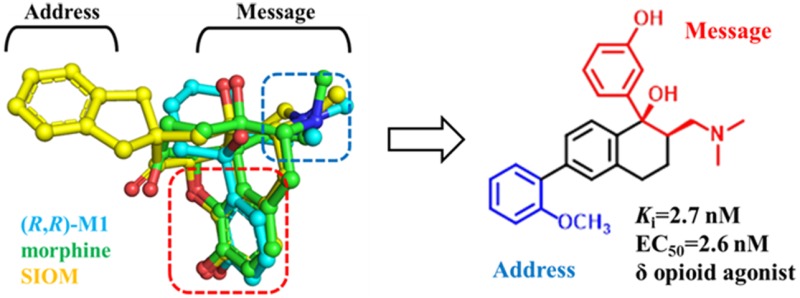
The classic “message-address” concept was proposed to address the binding of endogenous peptides to the opioid receptors and was later successfully applied in the discovery of the first nonpeptide δ opioid receptor (DOR) antagonist naltrindole. By revisiting this concept, and based on the structure of tramadol, we designed a series of novel compounds that act as highly potent and selective agonists of DOR among which (−)-6j showed the highest affinity (Ki = 2.7 nM), best agonistic activity (EC50 = 2.6 nM), and DOR selectivity (more than 1000-fold over the other two subtype opioid receptors). Molecular docking studies suggest that the “message” part of (−)-6j interacts with residue Asp1283.32 and a neighboring water molecule, and the “address” part of (−)-6j packs with hydrophobic residues Leu3007.35, Val2816.55, and Trp2846.58, rendering DOR selectivity. The discovery of novel compound (−)-6j, and the obtained insights into DOR-agonist binding will help us design more potent and selective DOR agonists.
Keywords: δ opioid receptor, agonist, affinity, selectivity, molecular docking
Opioid analgesics are widely used as treatments to relieve moderate-to-severe pain, and most of them (e.g., morphine) are μ opioid receptor (MOR) agonists (Figure 1). The observed side effects of these MOR agonists include respiratory depression, constipation, addiction, and physical dependence. Recently, the δ opioid receptor (DOR) has been confirmed as an attractive target for the development of novel analgesic drugs, with less side effects and more significant analgesia in animal models of inflammatory1−3 and neuropathic pain.3−5 It has also been shown that DOR agonists can possibly be used as potential therapeutics for depression,6,7 affective disorder,7,8 organ protection,9 and neurodegenerative diseases.10
Figure 1.

Structures of morphine, maltrindole, SIOM, (±)-cis-tramadol, and (±)-cis-M1.
The classic “message-address” concept11 was proposed to explore how the opioid receptors bind with their endogenous peptide ligands (e.g., DOR and enkephalin). Residues at the headgroup of these peptides were identified as the “message” part responsible for the recognition and affinity for the receptor, while residues at the middle or tail group of these peptides were viewed as “address” part to enhance the specificity and/or potency. This concept was then successfully applied in the discovery of the first nonpeptide DOR antagonist naltrindole (NTI) and the DOR agonist SIOM (Figure 1).12,13 The morphinan fragment of these two compounds was envisaged as the “message” part, and the aromatic group (e.g., indole group of NTI) as “address” part for DOR selectivity.
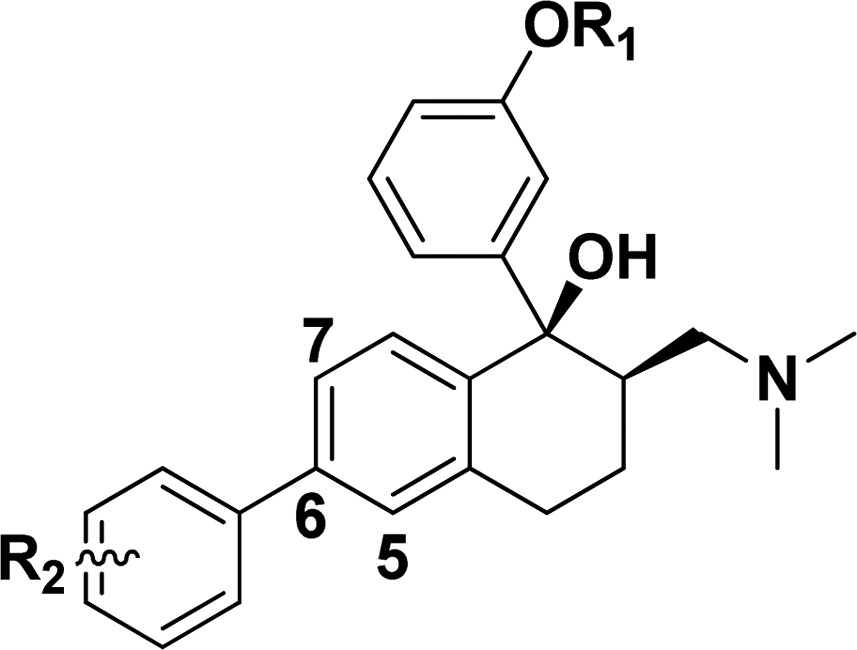
(±)-cis-Tramadol (4, Figure 1), a commonly prescribed medicine to treat moderate pain, is actually an agonist of MOR with Ki = 2400 nM.14 Its O-metabolite M1 was found to be much more potent (Ki = 11.0 nM) than tramadol. (1R,2R)-M1 stereoisomer was identified as its active form.14 We found that the nitrogen atom and phenol fragment of both (1R, 2R)-M1 and SIOM overlapped very well (Figure 2A) after superimposing them together with morphine. This indicates that the (1R,2R)-M1 shares similar pharmacophore features of morphine and SIOM. According to the “message-address” concept, the nitrogen atom and phenol fragment of these compounds could be viewed as the “message” part for DOR activity, and aromatic groups at the C ring of morphinan skeleton could function as “address” for DOR selectivity. Inspired by the discovery of NTI and SIOM, we wanted to add an “address” part to the tramadol structure, i.e., incorporating a biphenyl group to the cyclohexane group of tramadol, in order to design novel DOR agonists. We hypothesized that adding an “address” part to tramadol structure would improve DOR activity and selectivity. We also wanted to know which group (−OCH3 or – OH) at the “message” part of tramadol would possibly enhance DOR affinity. For these purposes, we designed and synthesized a series of novel DOR compounds for biological evaluation based on the structure of tramadol (Figure 2B).
Figure 2.

(A) Structural superimposition of (R,R)-M1 with the structures of morphine and SIOM. Different parts for these compounds are labeled according to the “message-address” concept.11 (B) The DOR compounds designed by adding the biphenyl substituent as the “address” part to the skeleton of tramadol structure.
The synthetic route for preparation of the designed compounds was shown in Scheme 1. Compound 7 was prepared from 6-hydroxy-1-tetralone through a novel three-step one-pot Smiles rearrangement process.15 The amino group of 7 was transformed to a bromo substituent by Sandmeyer reaction in the presence of HBr and CuBr to give compound 8. Suzuki coupling of 8 with various substituted phenylboronic acids in the presence of tetrakis(triphenylphosphine) palladium(0), and potassium carbonate led to compounds 9a–9l. The ketones 9a–9l were condensed with N-methyl-N-methylenemethanaminium to afford 10a–10l. Addition of organolithium reagent prepared by halogen-metal exchange between n-butyl lithium and 3-bromoanisole to the carbonyl group of 10a–10h provided 6a–6h. Addition of organolithium reagent prepared by halogen-metal exchange between n-butyl lithium and (3-bromophenoxy) (tert-butyl)dimethylsilane to the carbonyl group of 10a–10h gave 6i–6t after removal of the tert-butyl-dimethylsilane group with Cs2CO3 dissolved in DMF/H2O.
Scheme 1.

Reagents and conditions: (a) NaOH, N,N-dimethylacetamide, 2-bromo-2-methylpropanamide, yield: 63%; (b) HBr, CuBr, NaNO2, H2O, yield 85%; (c) Pd(PPh3)4, phenylboronic acids, K2CO3, toluene 90°C, yield 78% for 9a as example; (d) N-methyl-N-methylenemethanaminium, MeCN rt, without purification for next step; (e) n-BuLi, 3-bromoanisole, −78 °C, THF, yield 54% for 6a as example; (f) n-BuLi, (3-bromophenoxy) (tert-butyl)dimethylsilane, −78°C, THF; Cs2CO3, DMF/H2O 10:1, rt, yield 39% for 6i as example.
The binding affinities of compounds 6a–6t with opioid receptors (MOR, DOR, and κ opioid receptor (KOR)) were measured using a radio-ligand displacement assay. The radio-ligands [3H]DAMGO, [3H]DPDPE, and [3H]U69,593 were used for competitive inhibition assay on the MOR, DOR, and KOR, respectively. Then the representative compounds were evaluated for receptor activation using the standard [35S]GTP-γ-S binding assay. The results of these assays are summarized in Table 1 and Table 2, respectively.
Table 1. Binding Affinities of Compounds 6a–6t for the Opioid Receptors MOR, DOR, and KOR, Reported as Ki, or Percentage for the Displacement of Each of the Radio-Labeled Ligands at 1.0 μM, or the Testing Concentration if Totally Inactive.
|
Ki (±SEM, nM)
or inhibition (%) |
|||||
|---|---|---|---|---|---|
| compd | R1 | R2 | MORa | DORb | KORc |
| 6a | CH3 | H | >104d | >104 | 37 ± 1% |
| 6b | CH3 | o-OCH3 | >104 | 37.9 ± 0.2% | >104 |
| 6c | CH3 | m-OCH3 | >104 | >104 | >104 |
| 6d | CH3 | p-OCH3 | >104 | >104 | >104 |
| 6e | CH3 | p-CF3 | >104 | >104 | >104 |
| 6f | CH3 | p-Cl | >104 | >104 | >104 |
| 6g | CH3 | m,p-diCl | >104 | >104 | >104 |
| 6h | CH3 | o-CH3 | >104 | 13 ± 2% | 16.2 ± 0.3% |
| 6i | H | H | 20 ± 1% | 22 ± 1% | 140 ± 9 |
| 6j | H | o-OCH3 | >104 | 4.7 ± 0.1 | 17.7 ± 0.1% |
| 6k | H | m-OCH3 | >104 | 43 ± 1% | 15 ± 1% |
| 6l | H | p-OCH3 | >104 | 32 ± 2% | >104 |
| 6m | H | p-CF3 | >104 | >104 | >104 |
| 6n | H | p-Cl | >104 | 17 ± 1% | 8.9 ± 0.2% |
| 6o | H | m,p-diCl | >104 | >104 | >104 |
| 6p | H | o-CH3 | >104 | 140 ± 4 | >104 |
| 6q | H | o-F | >104 | 720 ± 89 | >104 |
| 6r | H | o-Cl | >104 | 160 ± 58 | >104 |
| 6s | H | o-CF3 | >104 | 65 ± 4 | >104 |
| 6t | H | o-OCF3 | >104 | 260 ± 26 | >104 |
| (+)-6j | H | o-OCH3 | >104 | >104 | >104 |
| (−)-6j | H | o-OCH3 | >104 | 2.7 ± 0.6 | >104 |
| ( ± )-tramadol | 9.6 ± 0.4% | >104 | >104 | ||
| ( ± )-M1 | 13.0 ± 0.5 | >104 | >104 | ||
Displacement of [3H]DAMGO from CHO cell membranes expressing human MOR.
Displacement of [3H]DPDPE from CHO cell membrane expressing human DOR.
Displacement of [3H]U69593 from CHO cells expressing human KOR.
No binding affinity could be determined up to 104 nM. The binding-assay was performed three times for each compound, and the result was reported as mean ± standard error of the mean.
Table 2. Standard [35S]GTP-γ-S Binding Assay for the Selected Compounds with Measurable Ki from Table 1.
| MOR |
DOR |
KOR |
||||
|---|---|---|---|---|---|---|
| compd | EC50 (nM) | Emax%a | EC50 (nM) | Emax% | EC50 (nM) | Emax% |
| 6i | b | 580 ± 84 | 187 ± 2 | |||
| 6j | 9.3 ± 0.5 | 230 ± 7 | ||||
| (+)-6j | ||||||
| (−)-6j | 2.6 ± 0.3 | 205 ± 3 | ||||
| 6p | 490 ± 9 | 199 ± 7 | ||||
| 6q | 1600 ± 240 | 172 ± 1 | ||||
| 6r | 950 ± 63 | 216 ± 3 | ||||
| 6s | 900 ± 190 | 217 ± 5 | ||||
| 6t | 1000 ± 120 | 215 ± 7 | ||||
| (±)-tramadol | 1000 ± 120 | 220 ± 7 | ||||
| (±)-M1 | 240 ± 24 | 220 ± 9 | ||||
| DAMGO | 30 ± 2 | 225 ± 1 | ||||
| DPDPE | 0.12 ± 0.02 | 216 ± 2 | ||||
the basal activity level of G-protein was defined as the Emax% = 100%.
[35S]GTP-γ-S binding activity was not able to be determined. The assay was performed three times for each compound, and the result was reported as mean ± standard error of the mean.
SARs of Newly Designed DOR Agonists
As shown in Table 1, tramadol has very low affinity for MOR, but its O-desmethyl metabolite M1 exhibited much higher affinity for MOR, which agrees with the reported affinity data of tramadol and M1.14 Interestingly, all the new compounds have no affinity for MOR, except that 6i showed very low MOR affinity and moderate KOR affinity (Ki = 140 nM). All the methoxyl substituent (R1 = Me) at the “message” part of compounds 6a–6h showed very low affinity for either DOR or KOR. However, the phenolic substituent (R1 = H) in 6i–6t generally exhibited higher affinities for DOR, or for KOR than those of 6a–6h. The effect of different R1 substituents on binding affinity suggests that the R1-substituted phenyl group at the “message” part is critical for DOR affinity. At the “address” part, the newly added phenyl group does help to increase the DOR affinity for these compounds. More interestingly, for the newly added phenyl group at “address” part, introduction of ortho-substitution (−F, −Cl, −CH3, −CF3, −OCF3, −OCH3) dramatically enhanced both the affinity and selectivity for compounds 6j and 6p–6t toward DOR, while the meta- or para-substitutions (e.g., compounds 6k–6o) did not exhibit any DOR affinity. Among these new compounds, 6j displayed very high DOR affinity (Ki = 4.7 nM) and high DOR/KOR selectivity (over 1000-fold). We separated the enantiomers of compound 6j and measured their affinities with DOR. The (−)-6j is 2-fold more potent than the racemic mixture 6j (Ki = 2.7 nM), while (+)-6j did not exhibit DOR affinity at all.
As shown in Table 2, compounds 6j (also (−)-6j) and 6p–6t turned out to be selective full DOR agonists in the [35S]GTP-γ-S binding assay, while compound 6i is a KOR agonist. Among them, the (−)-6j is the most potent agonist with EC50 = 2.6 nM. Thus, by adding the new “address” part to the basic structure of tramadol, we obtained highly potent and selective DOR agonists.
Structural Determinants of DOR Selectivity
In order to see how these newly designed compounds bind with DOR, molecular docking operations were performed on 6j and SIOM. We used the GOLD 5.0.1 program16 to dock these compounds into the binding site of the X-ray structure of DOR (PDB entry as 4eji with resolution of 3.40 Å).17 The results of molecular docking are shown in Figure 3. We found that (1R,2R)-isomer of 6j fits the binding site of DOR very well (Figure 3A). At the subsite of “message” part in (1R,2R)-6j, the protonated nitrogen atom formed a strong salt bridge with residue Asp1283.32 of DOR, and the distance for the −N···OD1 hydrogen bonding is 2.90 Å. The phenolic group at the “message” part of (1R,2R)-6j formed a strong hydrogen bond with the neighboring water molecule. The binding mode of (1R, 2R)-6j with DOR (Figure 3A) explains why the −OH substituted phenyl group at the “message” part of our newly designed compounds was able to significantly enhance the DOR affinity and selectivity (Table 1, Table 2). The mode of binding between DOR and (1R, 2R)-6j is consistent with reported binding modes of morphinan derivatives with opioid receptors.17−19 As shown in Figure 3A for the “address” part of our newly designed compounds, the 6-ortho-methoxyphenyl group of (1R,2R)-6j stretched into the hydrophobic pocket formed by residues Leu3007.35, Val2816.55, and Trp2846.58 of DOR and form cation−π interaction with Lys2145.39 (i.e., the distance from the center of phenyl group of (1R, 2R)-6j to the positive charged nitrogen atom at Lys2145.39 side chain is 5.5 Å) (Figure 3A). Meanwhile, the 6-ortho-methoxyphenyl group of (1R,2R)-6j packs perpendicularly with the side chain of Leu3007.35 of DOR. Based on the binding structure, we would predict that, if Leu3007.35 is replaced by a residue with a larger side chain, the hydrophobic interaction between the “address” part of (1R,2R)-6j and residues Leu3007.35, Val2816.55, and Trp2846.58 will probably be weakened. Such specific packing between the “address” part of (1R, 2R)-6j and DOR can be used to explain why the newly designed active compounds are selective agonists of DOR. Actually, residue Leu3007.35 of DOR corresponds to Trp3187.35 of MOR and to Tyr3127.35 of KOR. The much larger side chain of Trp3187.35 at the agonist-binding site of MOR will not be able to form optimal contact points with the “address” part of (1R,2R)-6j as that of Leu3007.35 of DOR. Similarly, the Tyr3127.35 side chain at the agonist-binding site of KOR is not compatible with the “address” part of (1R, 2R)-6j as that of Leu3007.35 of DOR. This is consistent with the reported observation that Leu3007.35 is responsible for the DOR selectivity of compound NTI(17) as shown in the X-ray crystal structure. The binding structure of DOR with (1S,2S)-isomer of 6j (Figure 3B) shows that (1S, 2S)-6j could not form a salt bridge with Asp1283.32 and that the “address” part of (1S,2S)-6j has much worse packing with hydrophobic residues Leu3007.35, Val2816.55, and Trp2846.58, although (1S,2S)-6j keeps hydrogen bonding with the neighboring water molecule. Overall, the limited contact points of (1S,2S)-6j with DOR must lead to very low or no obvious affinity. This docking result suggested that, like tramadol, the (1R,2R)-6j must be the (−)-6j isomer, the active compound.
Figure 3.

Binding structures of DOR-6j and DOR-SIOM obtained from molecular docking. (A) The binding mode of (1R,2R)-6j (colored in orange) at the agonist-binding site of DOR. (B) The binding mode of (1S,2S)-6j (colored in yellow) at agonist-binding site of DOR. (C) The binding mode of SIOM (blue color) at DOR binding site. (D) The superimposed binding structures of (1R,2R)-6j and SIOM with DOR, respectively.
The docked DOR-SIOM binding structure (Figure 3C) shows that the “message” part of SIOM also formed a conserved interaction with Asp1283.32 and a water molecule at the agonist-binding site of DOR. The “address” part (i.e., 7-spiroindanyl group) of SIOM interacted with Ile3047.39, Leu3007.35, and Trp2846.58 through hydrophobic packing. When superimposing these two binding structures (Figure 3D), we found that the “message” part of each of these two compounds adopts a similar orientation at the agonist-binding site of DOR. The only difference is the orientation of the “address” part of these two agonists, i.e., SIOM has hydrophobic packing with residue Ile3047.39, but no cation−π interaction with Lys2145.39 of DOR.
In summary, we designed and synthesized a series of novel compounds showing high affinity and good selectivity for δ opioid receptor. Starting from the structure of tramadol, we revisited the classic “message-address” concept and added the biphenyl group as the “address” part in our newly designed compounds. Among these compounds, we found that compound 6j was the most potent and selective DOR agonist, and we also identified that the steroisomer (−)-6j is the actual active component of the racemic 6j. Through molecular docking operations, we found that the “message” part of (−)-6j formed a salt bridge with Asp1283.32 of DOR and a hydrogen bond with the neighboring water molecule. The “address” part of (−)-6j has cation−π interaction with Lys2145.39 and is packed with hydrophobic residues Leu3007.35, Val2816.55, and Trp2846.58. The interaction with residue Leu3007.35 is responsible for the DOR selectivity of (−)-6j. Taken together, our newly discovered (−)-6j can be used as a lead compound for further pharmacological studies.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00423.
Chemical synthesis and structural characterization of the target compounds; protocols of biological assays and molecular docking study (PDF)
Author Contributions
W.F., W.L., and Q.S. designed the research; Q.S. and Y.Q. synthesized the compounds; X.X. and J.L. performed the pharmacological assay; and Q.S., X.H., and W.F. wrote the paper. Q.S. and Y.Q. equally contributed to the work.
This work was supported by the National Natural Science Foundation of China (No. 81473136 and 81172919) and Shanghai Science and Technology Development Funds (14431900500).
The authors declare no competing financial interest.
Supplementary Material
References
- Fraser G. L.; Gaudreau G. A.; Clarke P. B. S.; Menard D. P.; Perkins M. N. Antihyperalgesic effects of delta opioid agonists in a rat model of chronic inflammation. Br. J. Pharmacol. 2000, 129, 1668–1672. 10.1038/sj.bjp.0703248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt M. R.; Furness M. S.; Mello N. K.; Rice K. C.; Negus S. S. Antinociceptive effects of delta-opioid agonists in rhesus monkeys: Effects on chemically induced thermal hypersensitivity. J. Pharmacol. Exp. Ther. 2001, 296, 939–946. [PubMed] [Google Scholar]
- Petrillo P.; Angelici O.; Bingham S.; Ficalora G.; Garnier M.; Zaratin P. F.; Petrone G.; Pozzi O.; Sbacchi M.; Stean T. O.; Upton N.; Dondio G. M.; Scheideler M. A. Evidence for a selective role of the delta-opioid agonist [8R-(4bS*,8a alpha,8a beta,12b beta)]7,10-dimethyl-1-methoxy-11-(2-methylpropyl)oxycarbonyl 5,6,7,8,12,12b-hexahydro-(9H)-4,8-methanobenzofuro[3,2-e]pyrrolo[ 2,3-g]isoquinoline hydrochloride (SB-235863) in blocking hyperalgesia associated with inflammatory and neuropathic pain responses. J. Pharmacol. Exp. Ther. 2003, 307, 1079–1089. 10.1124/jpet.103.055590. [DOI] [PubMed] [Google Scholar]
- Desmeules J. A.; Kayser V.; Guilbaud G. Selective opioid receptor agonists modulate mechanical allodynia in an animal-model of neuropathic pain. Pain 1993, 53, 277–285. 10.1016/0304-3959(93)90224-D. [DOI] [PubMed] [Google Scholar]
- Mika J.; Przewlocki R.; Przewlocka B. The role of delta-opioid receptor subtypes in neuropathic pain. Eur. J. Pharmacol. 2001, 415, 31–37. 10.1016/S0014-2999(01)00814-7. [DOI] [PubMed] [Google Scholar]
- Broom D. C.; Jutkiewicz E. M.; Folk J. E.; Traynor J. R.; Rice K. C.; Woods J. H. Convulsant activity of a non-peptidic delta-opioid receptor agonist is not required for its antidepressant-like effects in Sprague-Dawley rats. Psychopharmacology 2002, 164, 42–48. 10.1007/s00213-002-1179-y. [DOI] [PubMed] [Google Scholar]
- Broom D. C.; Jutkiewicz E. M.; Rice K. C.; Traynor J. R.; Woods J. H. Behavioral effects of delta-opioid receptor agonists: Potential antidepressants?. Jpn. J. Pharmacol. 2002, 90, 1–6. 10.1254/jjp.90.1. [DOI] [PubMed] [Google Scholar]
- Jutkiewicz E. M. The antidepressant -like effects of delta-opioid receptor agonists. Mol. Interventions 2006, 6, 162–169. 10.1124/mi.6.3.7. [DOI] [PubMed] [Google Scholar]
- Gross G. J. Role of opioids in acute and delayed preconditioning. J. Mol. Cell. Cardiol. 2003, 35, 709–718. 10.1016/S0022-2828(03)00135-4. [DOI] [PubMed] [Google Scholar]
- Hill M. P.; Hille C. J.; Brotchie J. M. delta-opioid receptor agonists as a therapeutic approach in Parkinson’s disease. Drug News Perspect 2000, 13, 261–268. [PubMed] [Google Scholar]
- Schwyzer R. ACTH: a short introductory review. Ann. N. Y. Acad. Sci. 1977, 297, 3–26. 10.1111/j.1749-6632.1977.tb41843.x. [DOI] [PubMed] [Google Scholar]
- Portoghese P. S.; Sultana M.; Nagase H.; Takemori A. E. Application of the message address concept in the design of highly potent and selective non-peptide delta-opioid receptor antagonists. J. Med. Chem. 1988, 31, 281–282. 10.1021/jm00397a001. [DOI] [PubMed] [Google Scholar]
- Portoghese P. S.; Moe S. T.; Takemori A. E. A selective delta-1 opioid receptor agonist derived from oxymorphone - Evidence for separate recognition sites for delta-1 opioid receptor agonists and antagonists. J. Med. Chem. 1993, 36, 2572–2574. 10.1021/jm00069a017. [DOI] [PubMed] [Google Scholar]
- Gillen C.; Haurand M.; Kobelt D. J.; Wnendt S. Affinity, potency and efficacy of tramadol and its metabolites at the cloned human mu-opioid receptor. Naunyn-Schmiedeberg's Arch. Pharmacol. 2000, 362, 116–121. 10.1007/s002100000266. [DOI] [PubMed] [Google Scholar]
- Mizuno M.; Yamano M. A new practical one-pot conversion of phenols to anilines. Org. Lett. 2005, 7, 3629–3631. 10.1021/ol051080k. [DOI] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Granier S.; Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Weis W. I.; Kobilka B. K. Structure of the delta-opioid receptor bound to naltrindole. Nature 2012, 485, 400–404. 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Mathiesen J. M.; Sunahara R. K.; Pardo L.; Weis W. I.; Kobilka B. K.; Granier S. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H. X.; Wacker D.; Mileni M.; Katritch V.; Han G. W.; Vardy E.; Liu W.; Thompson A. A.; Huang X. P.; Carroll F. I.; Mascarella S. W.; Westkaemper R. B.; Mosier P. D.; Roth B. L.; Cherezov V.; Stevens R. C. Structure of the human kappa-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.