

Abstract
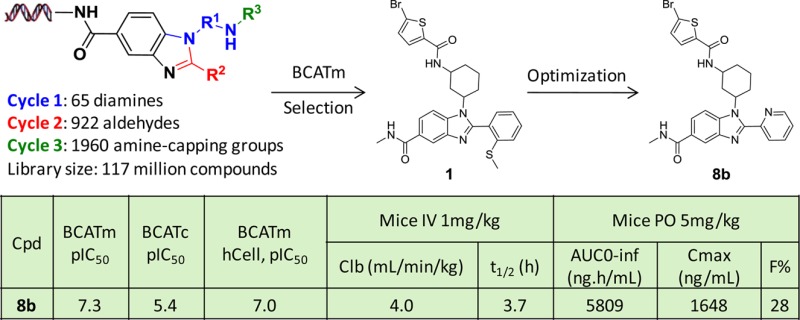
To identify BCATm inhibitors suitable for in vivo study, Encoded Library Technology (ELT) was used to affinity screen a 117 million member benzimidazole based DNA encoded library, which identified an inhibitor series with both biochemical and cellular activities. Subsequent SAR studies led to the discovery of a highly potent and selective compound, 1-(3-(5-bromothiophene-2-carboxamido)cyclohexyl)-N-methyl-2-(pyridin-2-yl)-1H-benzo[d]imidazole-5-carboxamide (8b) with much improved PK properties. X-ray structure revealed that 8b binds to the active site of BACTm in a unique mode via multiple H-bond and van der Waals interactions. After oral administration, 8b raised mouse blood levels of all three branched chain amino acids as a consequence of BCATm inhibition.
Keywords: BCATm, branched chain amino acids (BCAAs), DNA Encoded Library, ELT, 2-aryl benzimidazole
Recent studies suggest branched chain aminotransferases (BCATs) as a potential target for metabolic disorders. BCATs catalyze transamination of branched chain amino acids (BCAAs) leucine, isoleucine, and valine to their respective α-keto acids, which are further converted to succinyl-CoA and acetyl-CoA and participate in the tricarboxylic acid cycle and glycolysis.1 There are two isozymes, mitochondrial (BCATm) and cytosolic (BCATc), and the human BCAT isozymes are 58% identical in amino acid sequence.2 While BCATm is expressed in many peripheral tissues, BCATc is largely restricted to the central nervous system.1 A recent mouse BCATm knockout study demonstrated that lack of this enzyme increased the mice’s energy expenditure, associated with a futile protein turnover cycle, and protected them from obesity when subjected to a high fat diet.3 To further validate BCATm as a therapeutic target for the intervention of metabolic disorders, a suitable small molecular inhibitor with potency, selectivity, and in vivo activity is highly desirable. To develop such inhibitors, we utilized various screening technologies including HTS, fragment-based screen, and DNA Encoded Library Technology (ELT).4−6 Herein we report the discovery of a novel BCATm hit series from ELT7−12 and subsequent structure–activity relationship studies, which ultimately led to the discovery of a not only highly potent but also highly selective and in vivo efficacious compound (8b) that raised serum BCAAs levels after oral administration to mice.
In pursuit of novel small molecule inhibitors of BCATm, we screened the chemically biotinylated human BCATm protein construct against our more than 14 billion DNA encoded compounds.13 Several hit series were identified and one of them was from a three-cycle benzimidazole based novel library that contains 117 million distinct compounds. As shown in Scheme 1, the library utilized 65 monoprotected diamines at cycle 1, formed the benzimidazole ring with 922 aldehydes at cycle 2, then following amine deprotection was reacted with 1960 building blocks at cycle 3 to afford the final library of 117 million compounds. Selection for the diamines, the aldehydes, and the cycle 3 building blocks was based on availability, drug-like properties, and reactivity. The details of the library synthesis will be the subject of a different publication in the near future.
Scheme 1. Structure of DNA Encoded Benzimidazole Library.
The screening of this library suggested two preferred subscaffolds I and II for BCATm as illustrated by a Spotfire cube view in Figure 1. The most promising BCATm hit from this library screening is exemplified by compound 1 (I, R2 = 2-(methylthio)phenyl, Figure 1; also in Scheme 2) which exhibited human BCATm inhibition in both biochemical and cellular assays (pIC50, 6.6 and 6.8, respectively) with about 5-fold selectivity over BCATc (pIC50, 5.9). Mouse pharmacokinetic (PK) study suggested that 1 has a short half-life of 1.4 h and low oral bioavailability (10%). To improve potency and PK properties we conducted SAR studies around three different moieties of this benzimidazole-based scaffold including the benzimidazole 2-position (R2), the bromothiophene moiety, and the benzimidazole 5-position where DNA was originally attached. Since the subscaffold II is very similar to the subscaffold I, no exemplars were made for this series.
Figure 1.
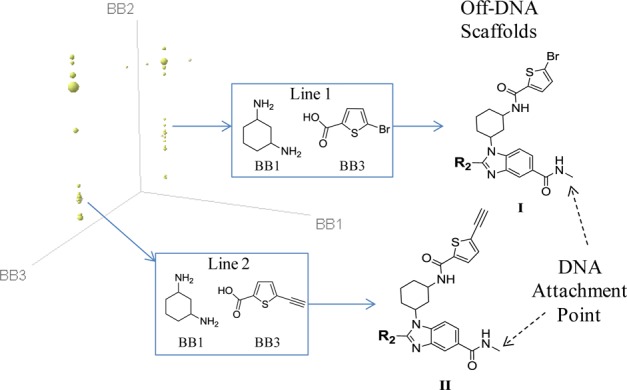
Result of BCATm screen of the DNA encoded benzimidazole library and the generic structures of the two BCATm hit series I and II, where BB1, BB2, and BB3 refer to cycle 1, 2, and 3 building blocks, respectively. Compounds on line 1 and line 2 share common BB1 and BB3 as indicated in their corresponding frames with BB2 as a variable component. The size of a dot is proportional to the DNA tag copy number. Library members with a single copy were removed to simplify visualization. An enlarged Figure 1 can be found in the SI section.
Scheme 2. Synthesis of 1,2,5-Substituted Benzimidazole Analogues.

Reagents and conditions: (a) tert-butyl (3-aminocyclo-hexyl)carbamate (mixture of diastereomers), DIEA, EtOH, 85 °C, 6 h; (b) 2-(methylthio)benzaldehyde, Na2S2O4, 1,4-dioxane/H2O (4:1), 80 °C, 24 h; (c) (i) TFA/DCM, overnight; (ii) 5-bromothiophene-2-carboxylic acid, HATU, DIEA, AcCN; (d) (i) 5-bromothiophene-2-carboxylic acid, HOBT, EDCI, TEA, DCM; (ii) TFA/DCM; (e) 4-fluoro-N-methyl-3-nitrobenzamide, DIEA, EtOH, 85 °C, 6 h.
A representative synthesis for the proposed modifications is illustrated in Scheme 2. Briefly, fluoro replacement of 4-fluoro-3-nitrobenzamide (2) with 1,3-cyclohexyldiamine followed by in situ nitro reduction and simultaneous cyclization with an aldehyde under sodium dithionite (Na2S2O4) conditions led to the 2-aryl benzimidazole intermediate (4), which after Boc deprotection was acylated by an appropriate carboxylic acid to provide the desired product 1 and its analogues. HPLC purification yielded two diastereomers (each is a racemic mixture of two enantiomers) with the cis-isomer being the more active configuration. Alternatively, the disynthon N-(3-aminocyclohexyl)-5-bromothiophene-2-carboxamide (6, cis-confirmation) was synthesized first followed by fluoro replacement of 4-fluoro-3-nitrobenzamide and cyclization with an appropriate aldehyde to afford the desired compound 1 and its analogues.
All of the synthesized compounds were evaluated for BCATm inhibition in a biochemical assay, and some selected compounds were further tested in a cellular assay.4,5 Both enzymatic and cellular activities are summarized in Tables 1–3.
Table 1. Benzimidazole 2-Position SAR.

Table 3. SAR at Benzimidazole 5-Position.

We began our structural optimization by addressing the potentially labile 2-(methylthio)phenyl group. As shown in Table 1, at the benzimidazole 2-postion, replacement of this group with unsubstituted phenyl (8a) led to increased BCATm inhibitory activity. We then further converted the phenyl group into 2-pyridinyl (8b), 3-pyridinyl (8d), and 4-pyridinyl (8e) groups in a hope to further decrease lipophilicity while maintaining adequate potency. The 2-pyridinyl analogue showed similar activity to the phenyl analogue in both enzymatic and cellular assays, whereas both the 3-pyridinyl and 4-pyridinyl analogues showed decreased potency suggesting that polar interactions in this region are poorly tolerated. Several five-membered heteroaryl rings including thiophene, thiazole, and pyrazole were also explored. While the 2- or 3-thiophene analogues (8f, 8g) achieved similar potency as the phenyl analogue in both biochemical and cellular assays, the two thiazole analogues (8h, 8i) showed moderately reduced activity. Similar to the 4-pyridinyl analogue, the pyrazole compound (8j) had much decreased enzymatic activity, confirming the early observation that polarity is not tolerated here. It is worth mentioning that for all of the compounds in Tables 1–3 the 1,3-cyclohexyldiamine is in the cis-configuration. Switching it to the trans-configuration rendered the compounds almost inactive.14
We next explored SAR around the 5-bromothiophene-2-carboxamide moiety including introducing bromo replacements, thiophene ring replacements, and modifications of the linker amide bond. Two series of compounds with either carboxamide (9A) or N-methyl carboxamide (9B) at the benzimidazole 5-position were simultaneously prepared for this purpose as shown in Table 2. In the 9A series, cyano (9Aa) and chloro (9Ab) replacements of bromo group led to moderately decreased activity in both enzymatic and cellular activities, and the methyl analogue (9Ac) produced >10-fold activity decrease highlighting the importance of the bromo substituent. Likewise, N-methylation of the amide linkage (9Ad) or conversion to a sulfonamide (9Ae) significantly abolished activity. In the 9B series, additional heteroaryl rings as thiophene replacements were explored. Switching the thiophene ring to thiazole (9Ba) led to almost 2 log loss of activity. Similarly 5-bromo-N-methyl pyrrol-2-yl (9Bb), 4-bromo-pyrro-2yl (9Bc), and 3-chloroisoxazol-5-yl (9Bd) analogues were all much less active than the thiophene ring identified from the original library screen.
Table 2. SAR for the Bromothiophene Moiety.

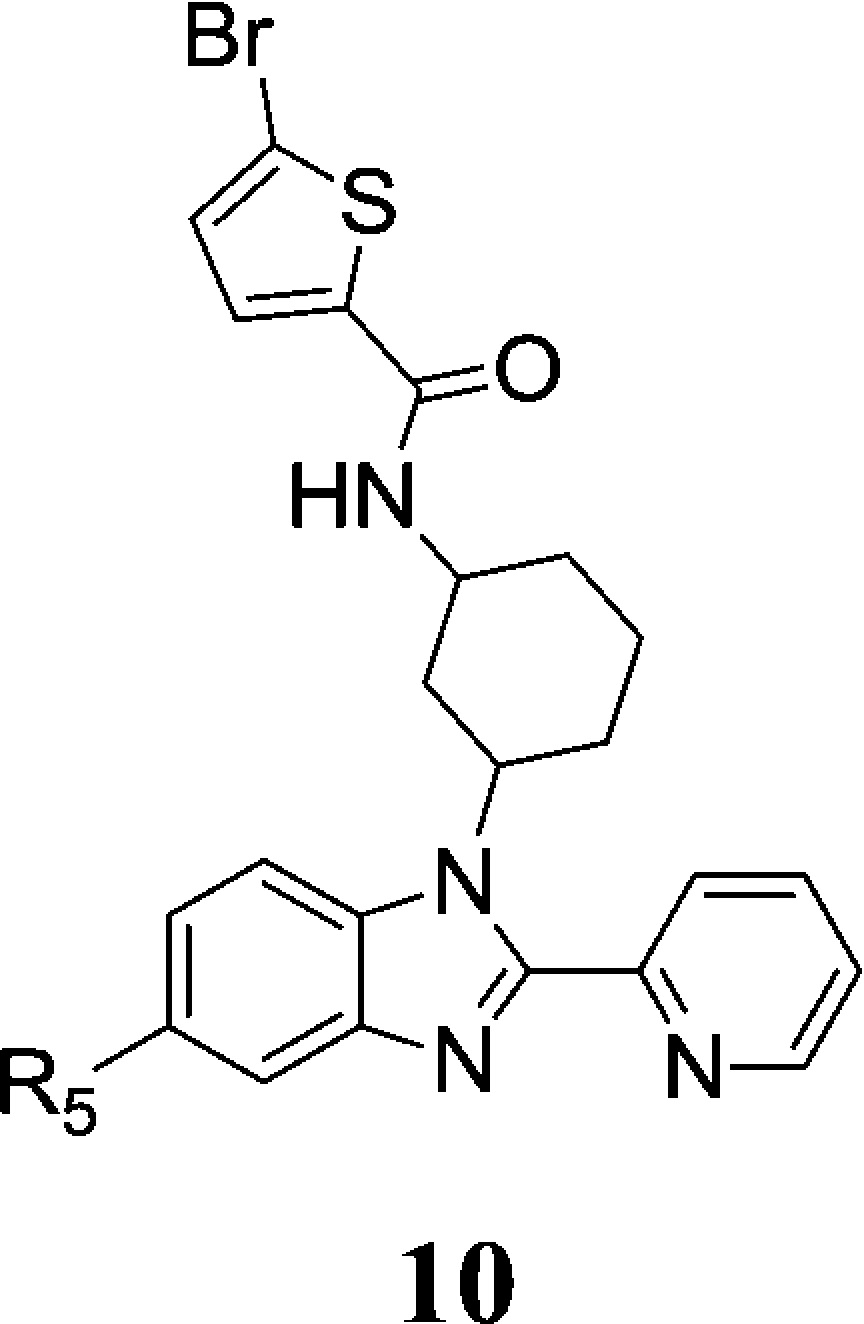
Lastly we explored substitution effect at the benzimidazole 5-position where the original DNA linker was attached. As shown in Table 3, the methyl amide (8b) and primary amide (8c) have similar enzymatic activity, but the latter showed a significant decrease in cellular activity. Removal of the carboxamide moiety (10a) led to more than 1 log loss of enzymatic activity, and its cellular activity is undetectable. Converting the carboxamide to N,N-dimethyl amide (10b) rendered the compound almost inactive. An effort to ensure the optimal substitution at this position prompted the synthesis of the cyclopropyl amide (10c) and isopropyl amide (10d) analogues, but neither exceeded the activity of 8b. Therefore, the methyl amide group seems the best fit for this position.
Numerous compounds with potent enzymatic and cellular activities were progressed to BCATc selectivity and pharmacokinetic (PK) studies. The compound 8b was found to inhibit BCATc with pIC50 = 5.4 (n = 2), which is about 100-fold selective for BCATm over BCATc, a significant improvement compared to the initial hit, 1 (pIC50, BCATm/BCATc, 6.6/5.9). Meanwhile, 8b also demonstrated a superior PK profile relative to 1 as shown in Table 4. Compound 8b has decreased clearance (4.0 mL/min/kg vs 23 mL/min/kg) and significantly increased oral bioavailability (28% vs 10%) and lower plasma protein binding (97.1% vs 99.5%). As a consequence, a longer T1/2 and much increased exposure and Cmax were achieved by 8b. Compound 8b was further found to inhibit mouse BCATm with pIC50 = 5.9 (n = 4) in the cellular assay. Therefore, 8b was selected as a candidate for in vivo studies aimed at investigating the pharmacology of BCATm inhibition in mice.
Table 4. PK Data for Representative Compounds.
| mice IV 1 mg/kg |
mice PO 5 mg/kg |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd # | BCATm pIC50 | BCATc pIC50 | hCell pIC50 | Clb (mL/min/kg) | t1/2 (h) | Vss (L/kg) | AUC0-inf (ng·h/mL) | Cmax (ng/mL) | F% | mice PPB (%) |
| 1 | 6.6 | 5.9 | 7.2 | 23 | 1.4 | 1.3 | 338 | 129 | 10 | 99.5 |
| 8b | 7.3 | 5.4 | 7.0 | 4.0 | 3.7 | 0.9 | 5809 | 1648 | 28 | 97.1 |
A high-resolution (2.0 Å) X-ray crystal structure of BCATm in complex with compound 8b was obtained as shown in Figure 2. In this structure, compound 8b binds to the active site of BCATm proximal to the pyridoxal-5′-phosphate (PLP) cofactor, which is covalently linked to Lys202 (Figure 2a). The ligand adopts a T-shaped conformation directing the 2-pyridyl toward the PLP, while the benzimidazole ring itself sits perfectly sandwiched between Phe30 and Ala314 forming extensive aromatic contacts with both (Figure 2b). The methyl amide at the benzimidazole 5-position makes H-bonds from its carbonyl to the basic nitrogen of Lys79 and to water from its NH group (Figure 2c). Increasing the size of the amide substituent from methyl to cyclopropyl (10c) or isopropyl (10d) decreases affinity due to the restricted space at this position. The cis-1,3-diaminocyclohexyl group at the benzimidazole 1-position directs the 5-bromo-thiophene ring deep into a hydrophobic pocket formed by Met241, Gln224, Val182, and Cys318 (Figure 2a). The amide linker further anchors this trajectory by forming an H-bond between its carbonyl oxygen and the backbone NH of Thr240, and a water-bridged interaction between the amide NH and Tyr173OH. The thiophene sulfur atom lies on the side of the ring next to the amide oxygen. Exchanging the bromine on the thiophene for the chlorine (9Ab) or methyl (9Ac) group results in reduced activity thus bromine here provides the best lipophilic contact with the highly hydrophobic pocket.
Figure 2.

Views of X-ray crystal structure of BCATm in complex with 8b (in cyan) and cofactor PLP (in yellow).
A comparison of binding modes between 8b and the recently published ELT series 11 (compound 15e in ref (4), PDP code 5CR5) as well as the HTS series 12 (compound 66 in ref (5), PDP code 5BWX) revealed that the two ELT series bind to BCATm in a similar orientation, while they only partially overlap with the HTS series (Figure 3). The key interactions in 8b including the extensive aromatic contacts with both Phe30 and Ala314 (Figure 2b) and hydrogen bond interactions with both Lys79 and a water (Figure 2c) are missing in both 11 and 12, which could account for the reduced potency of 11 and lack of selectivity of 12. The crystallographic analysis also confirms the outcome of the SAR work and suggests that the requirements for good binding were well met by the structural diversity of the benzimidazole library.
Figure 3.
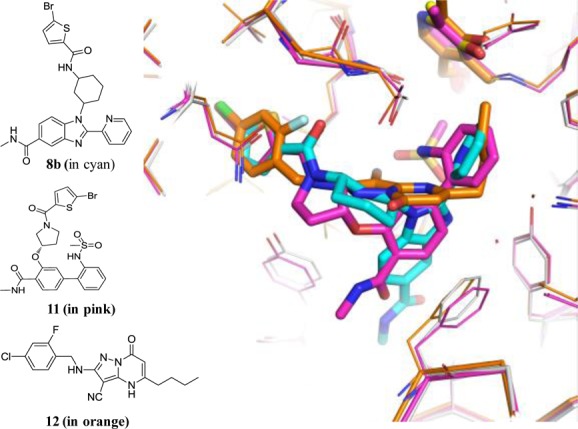
Overlay of the X-ray structures of 8b, 11, and 12.
To test whether compound 8b can increase the level of circulating branched chain amino acids, mice were given either the 300 mg/kg compound P.O. or vehicle after 5 h of fasting. After 30 min, the mice were refed with amino acid mixture (1.5 g/kg/10 mL, P.O.) followed by blood and tissue sampling at 1 h postfeeding to quantify amino acid levels. As shown in Figure 4, elevated levels of Leu, Ile, and Val were observed in plasma. Encouraged by this observation, we further tested the compound for its effect on BCAAs’ metabolism for a prolonged time period. As shown in Figure 5, the compound was given at different doses (30, 100, and 300 mg/kg, P.O.), and the amino acid mix was fed after 6 h of drug dosing. This study confirmed the initial findings and demonstrated dose-dependent enhancement of all three BCAA levels relative to serine and vehicle controls. These experiments demonstrated that the BCATm inhibitor 8b effectively raised the levels of BCAAs in an acute animal model over an extended duration.
Figure 4.

Acute dosing of 8b at 300 mg/kg followed by refeeding the mice with amino acid mix after 30 min: quantification of BCAAs serum level at 1 h postfeeding.
Figure 5.

Acute dosing of 8b at 30, 100, and 300 mg/kg P.O. followed by refeeding of amino acid mix after 6 h: quantification of BCAAs serum level at 1 h postfeeding.
In summary, affinity screening of a 117 million DNA Encoded Library identified a novel benzimidazole-based BCATm hit series, which was followed by an extensive SAR study that led to the discovery of compound 8b, a potent and selective BCATm inhibitor with reasonable PK properties. The X-ray crystal structure of BCATm in a complex with 8b revealed that the compound binds to BCATm at a site that is proximate to the catalytic residue lysine 202 and cofactor PLP binding site. Finally, compound 8b demonstrated oral efficacy in a mouse PD model, where it successfully raised the levels of all three BCAAs. Compound 8b is thus a valuable pharmacological tool for investigating the biological roles of BCATm in metabolic diseases.
Glossary
ABBREVIATIONS
- BCAAs
branched chain amino acids
- BCATm
mitochondrial branched-chain aminotransferase
- BCATc
cytosolic branched-chain aminotransferase
- ELT
Encoded Library Technology
- NTC
no-target control
- PLP
pyridoxal 5′-phosphate
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00389.
Accession Codes
The PDB code for 8b is 5HNE
Author Present Address
⊥ (J.Z.) Lilly China Research and Development Center (LCRDC), Eli Lilly and Company, 338 Jia Li Lue Road, Shanghai 201203, P. R. China.
All studies were conducted in accordance with the GSK Policy on the Care, Welfare and Treatment of Laboratory Animals and were reviewed by the Institutional Animal Care and Use Committee either at GSK or by the ethical review process at the institution where the work was performed.
The authors declare no competing financial interest.
Supplementary Material
References
- Hutson S. Structure and Function of Branched Chain Aminotransferases. Prog. Nucleic Acid Res. Mol. Biol. 2001, 70, 175–206. 10.1016/S0079-6603(01)70017-7. [DOI] [PubMed] [Google Scholar]
- Goto M.; Miyahara K.; Hirotsu K.; Conway M.; Yennawar N.; Islam M. M.; Hutson S. M. Structural Determinants for Branched-Chain Aminotransferase Isozyme-Specific Inhibition by the Anticonvulsant Drug Gabapentin. J. Biol. Chem. 2005, 280, 37246–37256. 10.1074/jbc.M506486200. [DOI] [PubMed] [Google Scholar]
- She P.; Reid T. M.; Bronson S. K.; Vary T. C.; Hajnal A.; Lynch C. J.; Hutson S. M. Disruption of BCATm in Mice Leads to Increased Energy Expenditure Associated with the Activation of a Futile Protein Turnover Cycle. Cell Metab. 2007, 6, 181–194. 10.1016/j.cmet.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H.; Zhou J.; Sundersingh F. S.; Summerfield J.; Somers D.; Messer J. A.; Satz A. L.; Ancellin N.; Arico-Muendel C. C.; Bedard K. L.; Beljean A.; Belyanskaya S. L.; Bingham R.; Smith S. E.; Boursier E.; Carter P.; Centrella P. A.; Clark M. A.; Chung C.; Davie C. P.; Delorey J. L.; Ding D.; Franklin J.; Grady L. C.; Herry K.; Hobbs C.; Kollmann C. S.; Morgan B. A.; Kaushansky L. J.; Zhou Q. Discovery, SAR, and X-Ray Binding Mode Study of BCATm Inhibitors from a Novel DNA-Encoded Library. ACS Med. Chem. Lett. 2015, 6, 919–924. 10.1021/acsmedchemlett.5b00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand S. M.; Ancellin N.; Beaufils B.; Bingham R. P.; Borthwick J. A.; Boullay A.-B.; Boursier E.; Carter P. C.; Chung C-w.; Churcher I.; Dodic N.; Fouchet M.-H.; Fournier C.; Francis P. L.; Gummer L. A.; Herry K.; Hobbs A.; Hobbs C. I.; Homes P.; Jamieson C.; Nicodeme E.; Pickett S. D.; Reid I. H.; Simpson G. L.; Sloan L. A.; Smith S. E.; Somers D. O.; Spitzfaden C.; Suckling C. J.; Valko K.; Washio Y.; Young R. J. The Discovery of in Vivo Active Mitochondrial Branched-Chain Aminotransferase (BCATm) Inhibitors by Hybridizing Fragment and HTS Hits. J. Med. Chem. 2015, 58, 7140–7163. 10.1021/acs.jmedchem.5b00313. [DOI] [PubMed] [Google Scholar]
- Borthwick J. A.; Ancellin N.; Bertrand S. M.; Bingham R. P.; Carter P. S.; Chung C.-W.; Churcher I.; Dodic N.; Fournier C.; Francis P. L.; Hobbs A.; Jamieson C.; Pickett S. D.; Smith S.; Somers D. O’N.; Spitzfaden C.; Yound R. J.. Structurally Diverse Mitochondrial Branched Chain Aminotransferase (BCATm) Leads with Varying Binding Modes Identified by Fragment Screening. J. Med. Chem., in press. [DOI] [PubMed] [Google Scholar]
- Clark A. M.; Acharya R. A.; Arico-Muendel C. C.; Belyanskaya S. L.; Benjamin D. R.; Carlson N. R.; Centrella P. A.; Chiu C. H.; Creaser S. P.; Cuozzo J. W.; Davie C. P.; Ding Y.; Franklin G. J.; Franzen K. D.; Gefter M. L.; Hale S. P.; Hansen N. JV.; Israel D. I.; Jiang J.; Kavarana M. J.; Kelley M. S.; Kollmann C. S.; Li F.; Lind K.; Mataruse S.; Medeiros P. F.; Messer J. A.; Myers P.; O’Keefe H.; Oliff M. C.; Rise C. E.; Satz A. L.; Skinner S. R.; Svendsen J. L.; Tang L.; Vloten K. V.; Wagner R. W.; Yao G.; Zhao B.; Morgan B. A. Design, Synthesis and Selection of DNA-Encoded Small-Molecule Libraries. Nat. Chem. Biol. 2009, 5, 647–654. 10.1038/nchembio.211. [DOI] [PubMed] [Google Scholar]
- Deng H.; O’Keefe H.; Davie C. P.; Lind K. E.; Acharya R. A.; Franklin G. J.; Larkin J.; Lohr T.; Matico R.; Thompson M.; Gross J. W.; Neeb M.; O’Donovan G. K.; Centrella P. A.; Mataruse S.; Bedard K. L.; Vloten K. V.; Skinner S. R.; Belyanskaya S. L.; Cuozzo J. W.; Clark M. A.; Arico-Muendel C. C.; Morgan B. A. Discovery of Highly Potent and Selective Small Molecule ADAMTS-5 Inhibitors that Inhibit Human Cartilage Degradation via Encoded Library Technology (ELT). J. Med. Chem. 2012, 55, 7061–7079. 10.1021/jm300449x. [DOI] [PubMed] [Google Scholar]
- Franzini R. M.; Neri D.; Scheuermann J. DNA-Encoded chemical libraries: Advancing beyond conventional small-molecule libraries. Acc. Chem. Res. 2014, 47, 1247–1255. 10.1021/ar400284t. [DOI] [PubMed] [Google Scholar]
- Kleiner R. E.; Dumelin C. E.; Liu D. R. Small-molecule discovery from DNA-encoded chemical libraies. Chem. Soc. Rev. 2011, 40, 5707–5717. 10.1039/c1cs15076f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnow R. A., Jr.Handbook for DNA-Encoded Chemistry; John Wiley & Sons Press: Hoboken, NJ, 2014. [Google Scholar]
- Encinas L.; O’Keefe H.; Neu M.; Remuiñán M. J.; Patel A. M.; Guardia A.; Davie C. P.; Pérez-Macías N.; Yang H.; Convery M. A.; Messer J. A.; Pérez-Herrán E.; Centrella P. A.; Álvarez-Gómez D.; Clark M. A.; Huss S.; O’Donovan G. K.; Ortega-Muro F.; McDowell W.; Castañeda P.; Arico-Muendel C. C.; Pajk S.; Rullás J.; Angulo-Barturen I.; Álvarez-Ruíz E.; Mendoza-Losana A.; Pages L. B.; Castro-Pichel J.; Evindar G. Encoded Library Technology as a Source of Hits for the Discovery and Lead Optimization of a Potent and Selective Class of Bactericidal Direct Inhibitors of Mycobacterium tuberculosis InhA. J. Med. Chem. 2014, 57, 1276–1288. 10.1021/jm401326j. [DOI] [PubMed] [Google Scholar]
- ELT screen was performed as described in ref (4).
- The corresponding trans-isomer of compounds 1 and 8b both showed pIC50 (BCATm) = 4.1 (n = 4).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.