

Abstract
1,2,3,4-Tetrahydroquinolines have been identified as the most potent inhibitors of LPS-induced NF-κB transcriptional activity. To discover new molecules of this class with excellent activities, we designed and synthesized a series of novel derivatives of 1,2,3,4-tetrahydroquinolines (4a–g, 5a–h, 6a–h, and 7a–h) and bioevaluated their in vitro activity against human cancer cell lines (NCI-H23, ACHN, MDA-MB-231, PC-3, NUGC-3, and HCT 15). Among all synthesized scaffolds, 6g exhibited the most potent inhibition (53 times that of a reference compound) of LPS-induced NF-κB transcriptional activity and the most potent cytotoxicity against all evaluated human cancer cell lines.
Keywords: 1,2,3,4-Tetrahydroquinolines; NF-κB inactivation; in vitro cytotoxicity; human cancer cell lines
NF-κB is a lymphoid-specific protein that binds to the enhancer of kappa light chain in the nucleus of B cells; NF-κB was discovered by Sen and Baltimore.1 NF-κB is involved in the regulation of many immune and inflammatory responses, cellular growth, and apoptosis.2,3 At present, NF-κB and its signaling is one of the most exciting and extensively studied research fields since NF-κB dysregulation is associated with many diseases such as cancer, AIDS, asthma, arthritis, diabetes, and inflammatory bowel disease.4−6 Several natural and synthetic compounds, including some drugs, have been tested for their potential to inhibit NF-κB, but very few of them are suitable for anticancer therapy.7,8 Therefore, it has been suggested that the development of novel NF-κB inhibitors with antitumor and anti-inflammatory activities is most important. Our group has been involved in the development of novel potent NF-κB inhibitors9−11 with anticancer activity.
Besides, quinolines and tetrahydroquinolines are important ubiquitous structural motifs in biologically active natural products and pharmacologically relevant therapeutic agents.12−16 However, Khan et al.17 demonstrated that tetrahydroquinolines can be used as NF-κB inhibitors as well as anti-HIV agents, anti-Parkinson’s diseases, etc. María José Abad et al.18 also tested quinoline-based compounds as modulators of HIV transcription through NF-κB and Sp1 inhibition. These distinct inhibitory activities have encouraged us to prepare such core motifs to test our hypothesis that their derivatives would act as most potent NF-κB inhibitors and have anticancer activity. We designed and synthesized different novel derivatives of 1,2,3,4-tetrahydroquinoline-2-carboxylic acid N-(substituted)phenyl amide and tested them as potential NF-κB inhibitors and also evaluated their cytotoxicities against six human cancer cell lines (NCI-H23, ACHN, MDA-MB-231, PC-3, NUGC-3, and HCT-15). Based on our previous reports,9−11 we wish to maintain amide functionality to the core motifs, and the effect of the synthesized molecules on NF-κB transcriptional activity was measured by using a reported procedure.19In vitro cytotoxicity assay was performed using the number of cells measured indirectly by the sulforhodamine B method according to the National Cancer Institute (USA) protocol.20
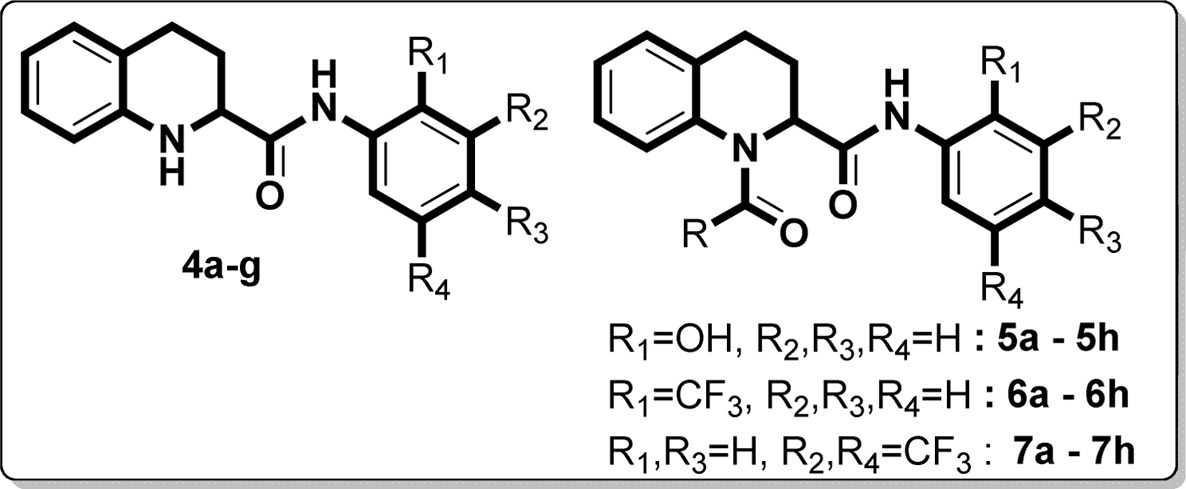
We commenced our synthesis from commercially available quinoline-2-carboxylic acid, which underwent amidation reaction with various substituted aromatic amines in the presence of the coupling reagent 1,1′-carbonyldiimidazole (CDI) in tetrahydrofuran at room temperature. This reaction afforded the 3a–g series of N-(substituted)quinoline-2-carboxamides in good yields (Scheme 1). The 3a–g derivatives were also converted to 4a–g by Pd/C hydrogenation reaction (H2 balloon) in ethanol solvent at room temperature. To obtain the 5a–h, 6a–h, and 7a–h series, we performed acylation reaction in the presence of triethylamine in anhydrous tetrahydrofuran (Scheme 2). All newly synthesized derivatives (3a–g, 4a–g, 5a–h, 6a–h, and 7a–h) were confirmed by 1H and 13C NMR and mass spectra. In evaluation studies of inhibition of LPS-induced NF-κB transcriptional activity, we compared all synthesized derivatives (4a–g, 5a–h, 6a–h, and 7a–h) with the reference compound pyrrolidine dithiocarbamate (PDTC), which acts as an antioxidant and is a potent inhibitor of NF-κB activation,21−25 and also with the lead compound KL-1156, which is an inhibitor of NF-κB translocation to the nucleus in LPS-stimulated RAW 264.7 macrophages.26
Scheme 1. Synthesis of 3a–g and 4a–g Series of Scaffolds.

Reagents and conditions: (a) CDI, anhydrous THF, RT, 1 h. (b) Pd/C, H2 balloon, EtOH, RT, 24 h.
Scheme 2. Synthesis of 5a–h, 6a–h, and 7a–h Series of Scaffolds.

Reagents and conditions: (c) triethylamine, anhydrous THF, 0 °C, 5–10 min, RT, 30–60 min.
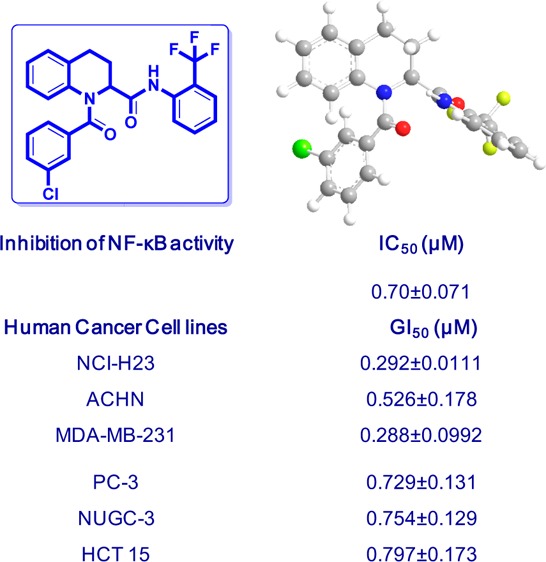
Initially, we screened the 4a–g derivatives; they exhibited marginal inhibitory effects on NF-κB transcriptional activity (Table 1). After finding that the tetrahydroquinoline core motif was less potent, we next turned our attention mainly to the substitutions of R, R1, R2, R3, and R4, which resulted in 24 derivatives (5a–h, 6a–h, and 7a–h; Table 1). Among them, 5e (IC50: 1.4 ± 0.71 μM), 6f (IC50: 0.90 ± 0.071 μM), 6g (IC50: 0.70 ± 0.071 μM), and 6h (IC50: 2.7 ± 0.42 μM) exhibited outstanding inhibitory effects (Figure 1) on LPS-induced NF-κB transcriptional activity in comparison with remaining derivatives (Table 1). After having the initial experimental results, structure–activity relationship development was initiated for the tetrahydroquinoline scaffold. The 1,2,3,4-tetrahydroquinoline-2-carboxamide motif was modified at two key positions interpreted as R, and substitutions were performed at the aromatic system (R1, R2, R3, and R4). In the first set of compounds, we performed different substitutions at R1, R2, R3, and R4, which resulted in 4a–g analogues. Subsequently R1 = R2 = R3 = R4 = H (4a, IC50 60 μM) was prepared to study the effect of the core 1,2,3,4-tetrahydroquinoline-2-carboxamide motif. We also examined the electronic influence of different groups at R1, R2, R3, and R4, including electron-deficient (−CF3) and electron-rich groups (−OCH3 and −OH). Next, we introduced various acyl and aroyl groups at the 1-position of tetrahydroquinoline-2-carboxamides, which resulted in 5a–h, 6a–h, and 7a–h analogues. Sizes of different aliphatic chains (−R) and substituents at the aromatic ring were then examined. The inhibitory effects of these compounds (4a–g, 5a–h, 6a–h, and 7a–h) depended on the nature of the substitution at the R-position and the substituents (R1, R2, R3, and R4) on the N-phenyl ring of tetrahydroquinoline-2-carboxamides. For instance, with R = an alkyl group (methyl, ethyl, propyl, or octyl), we did not notice any significant inhibitory effects, but with R = phenyl. The following compounds exhibited excellent inhibitory effects on LPS-induced NF-κB transcriptional activity (Figure 2): 5e (IC50: 1.4 ± 0.071 μM, about 26 and 37 times more potent than PDTC and KL-1156, respectively), 6f (IC50: 0.90 ± 0.071 μM, about 41 and 58 times more potent), 6g (IC50: 0.70 ± 0.071 μM, about 53 and 75 times), and 6h (IC50: 2.7 ± 0.42 μM, about 13 and 19 times). We also tested other substitutions at the R-position and different substituents at the R1, R2, R3, and R4 positions of the 1,2,3,4-tetrahydroquinoline core motif (Table 1). To explain the trend toward NF-κB inhibition interestingly, the electron-withdrawing group −CF3 at the R1 position resulted in outstanding inhibitory effects on LPS-induced NF-κB transcriptional activity in comparison with any electron-releasing group (for example, −OH). We also noticed that −I and +M effect groups (such as −Cl) at the second and third positions of the aryl group of R in combination with −CF3 at the R1 position also showed excellent inhibition of NF-κB transcriptional activity (6g, IC50: 0.70 ± 0.071 μM; 6f, IC50: 0.90 ± 0.071 μM). Overall, most of the synthesized compounds (4d, 4e, 4g, 5f, 5h, 6d, 7a–c, and 7e–h) were more potent inhibitors of NF-κB transcriptional activity than PDTC and KL-1156 (Figure 2). We next studied the in vitro cytotoxicity of the synthesized compounds and initially evaluated quinoline-2-carboxamide derivatives (3a–g) with the human lung cancer cell line (NCI-H23) and doxorubicin (ADR) as a reference compound.
Table 1. Inhibitory Effect on LPS-Induced NF-κB Transcriptional Activity for 1,2,3,4-Tetrahydroquinolines.
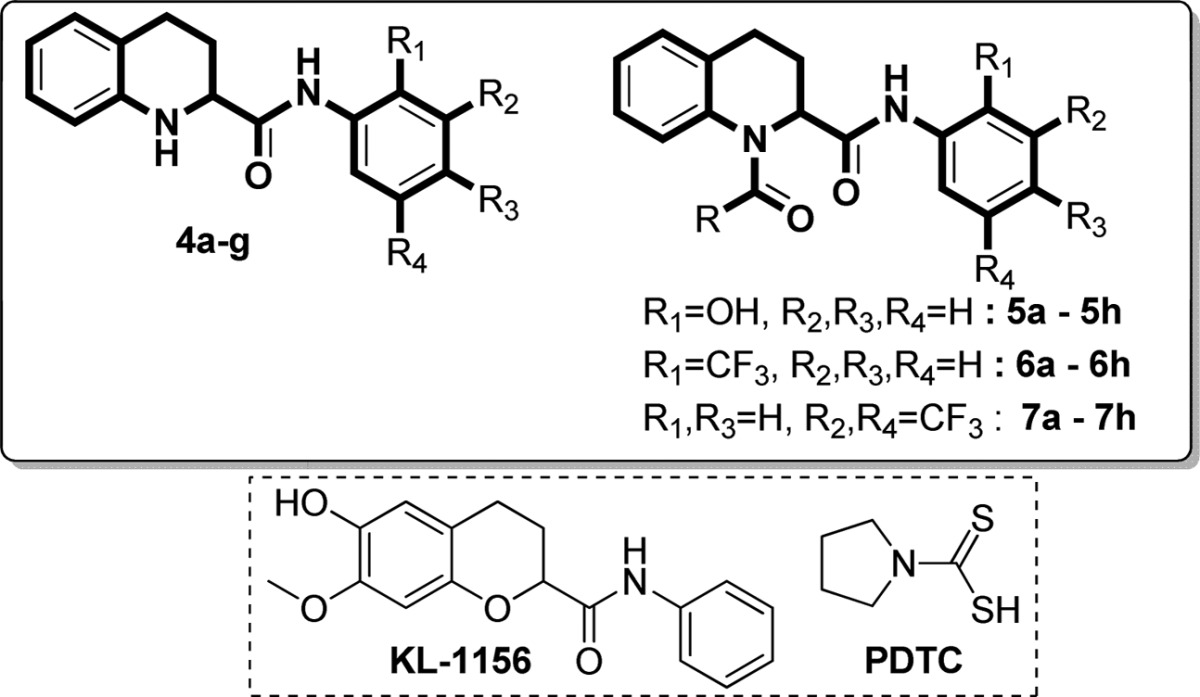
| substituents (R group) |
||||||
|---|---|---|---|---|---|---|
| No. | R1 | R2 | R3 | R4 | % inhibition at 100 μM | IC50c (μM) |
| PDTC | 37.2 | |||||
| KL-1156 | 72 | 53 ± 15 | ||||
| 4a | H | H | H | H | 85 | 60 ± 10 |
| 4b | OH | H | H | H | 57 | 83 ± 9.9 |
| 4c | H | H | OH | H | 64 | 65 ± 0.71 |
| 4d | H | H | OCH3 | H | >100 | 34 ± 12 |
| 4e | CF3 | H | H | H | >100 | 26 ± 5.0 |
| 4f | H | H | CF3 | H | 79 | 68 ± 16 |
| 4g | H | CF3 | H | CF3 | >100 | 26 ± 1.4 |
| No. | substituents (R group) | % inhibition at 100 μM | IC50c (μM) | No. | % inhibition at 100 μM | IC50c (μM) | No. | % inhibition at 100 μM | IC50c (μM) |
|---|---|---|---|---|---|---|---|---|---|
| 5a | methyl | 19 | 6a | 32 | 7a | 85 | 23 ± 0.0 | ||
| 5b | ethyl | 31 | 6b | 43 | 7b | 91 | 20 ± 0.71 | ||
| 5c | propyl | 41 | 6c | 51 | 95 ± 0.71 | 7c | 88 | 20 ± 0.71 | |
| 5d | octyl | 22 | 6d | 63 | 27 ± 0.71 | 7d | 58 | 51 ± 21 | |
| 5e | phenyl | >100 | 1.4 ± 0.071 | 6e | 44 | 7e | 66 | 29 ± 0.71 | |
| 5f | 2-chlorophenyl | 78 | 20 ± 0.71 | 6f | 82 | 0.90 ± 0.071 | 7f | 90 | 23 ± 0.0 |
| 5g | 3-chlorophenyl | 58 | 78 ± 2.1 | 6g | 90 | 0.70 ± 0.071 | 7g | 68 | 27 ± 0.71 |
| 5h | 4-chlorophenyl | 82 | 20 ± 2.8 | 6h | 92 | 2.7 ± 0.42 | 7h | 92 | 20 ± 2.1 |
IC50 values are means of the concentration (μM) exhibiting 50% inhibition of LPS-induced NF-κB transcriptional activity.
Figure 1.
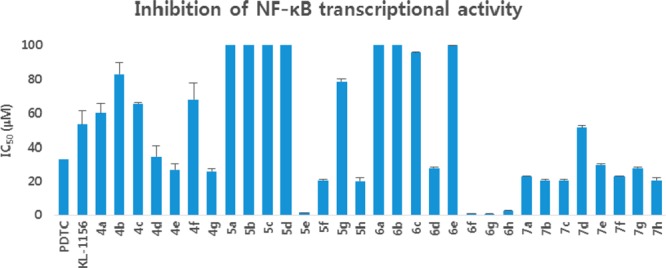
Efficacy of 4a–g, 5a–h, 6a–h, and 7a–h analogues in inhibition of NF-κB transcriptional activities.
Figure 2.
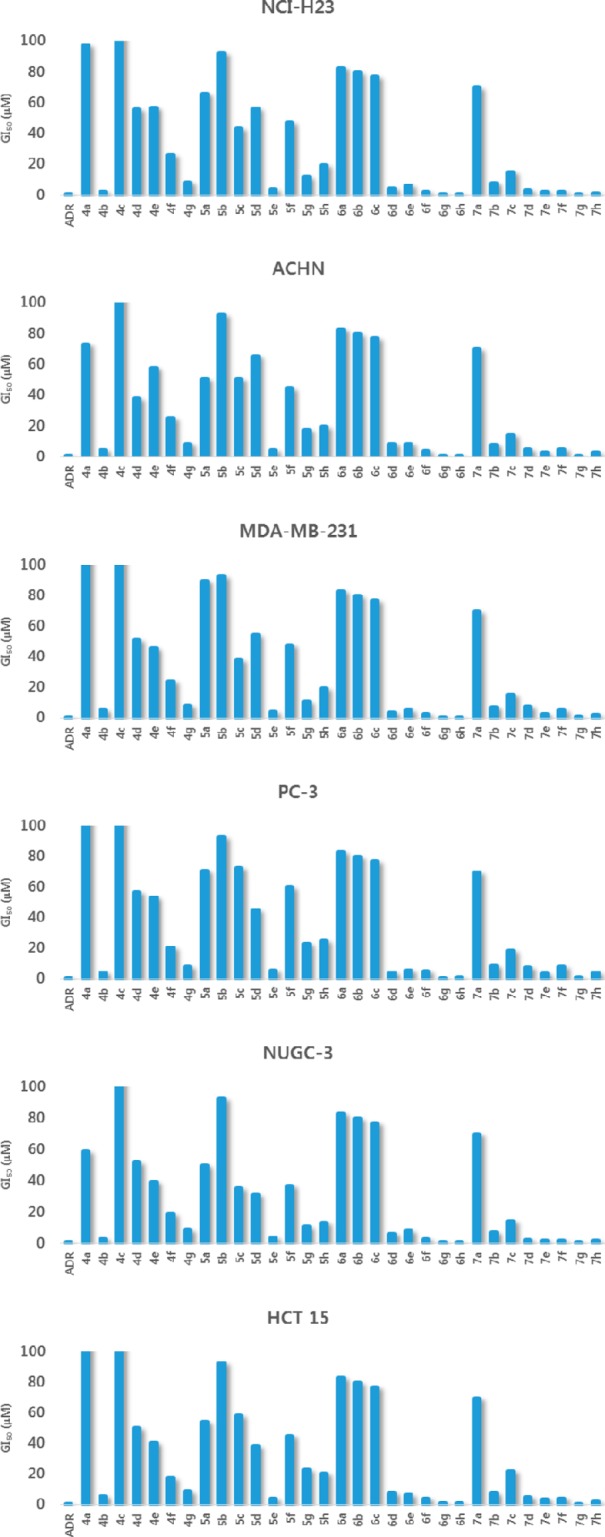
In vitro efficacy of 4a–g, 5a–h, 6a–h, and 7a–h analogues in inhibiting growth of human cancer cell lines.
As shown in Table 2, electron-withdrawing substituents such as −CF3 at the R2 and R4 positions of the phenyl ring (3g, GI50 1.44 ± 0.463 μM) resulted in the most potent cytotoxicity. The series 4a–g, 5a–h, 6a–h, and 7a–h were also evaluated for in vitro cytotoxicity against six human cancer cells: NCI-H23, ACHN (renal), MDA-MB-231 (breast), PC-3 (prostate), NUGC-3 (gastric), and HCT15 (colon) (Table 3). Any substitution on the phenyl ring was not beneficial, and only 4b (GI50 2.23 ± 0.455 μM) exhibited better cytotoxic activities against all tested cell lines than other analogues of the 4a–g series (Table 3). To further confirm that the tetrahydroquinoline motif is beneficial for cytotoxicity, we executed acylation reaction with triethyl amine in tetrahydrofuran with 4b, 4e, and 4g; introduction of electron-rich or electron-withdrawing substituents at the R1 position afforded 5a–h, 6a–h, and 7a–h analogues. As expected, these analogues had improved cytotoxicity against all tested cell lines (Figure 2), suggesting that substitutions at the R1 position and the first position of the tetrahydroquinoline motif are most important (Table 3). Compound 5e exhibited the highest cytotoxicity (Table 3) against all evaluated cell lines (NCI-H23, GI50 3.49 ± 0.999 μM; NUGC-3, GI50 3.78 ± 0.618 μM; HCT 15, GI50 3.83 ± 0.994 μM). The importance of an electron-withdrawing group was also confirmed by 6g and 6h derivatives, which have −CF3 and −Cl on both phenyl rings and exhibited outstanding cytotoxicity (6g, GI50 0.292 ± 0.111–0.797 ± 0.173 μM; 6h, GI50 0.307 ± 0.0.0941–0.839 ± 0.0610 μM) against all tested cell lines (Figure 2, Table 3). The −CF3 group at the R2 and R4 positions of the phenyl ring in 7g also resulted in potent cytotoxicity against all tested cell lines (0.420–1.19 μM; Table 3). Compound 7h also exhibited potent cytotoxicity against lung (NCI-H23 (GI50 0.889 ± 0.102 μM) and gastric (NUGC-3, GI50 1.66 ± 0.406 μM) cancer cell lines and moderate cytotoxicity against the other four cell lines (Table 3).
Table 2. Cytotoxicity of 3a–g Series against Lung Cancer (NCI-H23) Cell Lines.
| No. | R1 | R2 | R3 | R4 | GI50 (μM)d |
|---|---|---|---|---|---|
| ADR | 0.101 ± 0.00602 | ||||
| 3a | H | H | H | H | 108 ± 8.74 |
| 3b | OH | H | H | H | 87.5 ± 9.77 |
| 3c | H | H | OH | H | 102 ± 11.4 |
| 3d | H | H | OCH3 | H | 7.65 ± 1.93 |
| 3e | CF3 | H | H | H | >90 |
| 3f | H | H | CF3 | H | >90 |
| 3g | H | CF3 | H | CF3 | 1.44 ± 0.463 |
GI50 values are taken as a mean from three experiments and correspond to the agent’s concentration causing a 50% decrease in net cell growth.
Table 3. Cytotoxicity against NCI-H23, ACHN, MDA-MB-231, PC-3 NUGC-3, and HCT-15 Cancer Cell Lines.
| GI50 (μM)d |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | R | R1 | R2 | R3 | R4 | NCI-H23 | ACHN | MDA-MB-231 | PC-3 | NUGC-3 | HCT 15 |
| ADR | ref | 0.0726 ± 0.00940 | 0.0995 ± 0.00848 | 0.112 ± 0.0132 | 0.0923 ± 0.0103 | 0.125 ± 0.0104 | 0.0854 ± 0.00329 | ||||
| 4a | H | H | H | H | 97.1 ± 15.3 | 72.9 ± 11.4 | 108 ± 17.8 | 110 ± 15.6 | 59.0 ± 9.88 | 109 ± 15.0 | |
| 4b | OH | H | H | H | 2.23 ± 0.455 | 3.99 ± 1.33 | 5.26 ± 1.77 | 4.31 ± 1.34 | 2.68 ± 0.824 | 5.61 ± 1.31 | |
| 4c | H | H | OH | H | >100 | >100 | >100 | >100 | >100 | >100 | |
| 4d | H | H | OCH3 | H | 55.7 ± 9.80 | 38.0 ± 11.7 | 51.4 ± 11.2 | 56.9 ± 17.6 | 51.9 ± 11.0 | 50.2 ± 8.10 | |
| 4e | CF3 | H | H | H | 56.8 ± 11.4 | 57.6 ± 11.7 | 45.5 ± 13.1 | 53.3 ± 9.65 | 39.3 ± 8.58 | 40.4 ± 4.68 | |
| 4f | H | H | CF3 | H | 25.8 ± 4.12 | 25.0 ± 8.17 | 24.2 ± 5.10 | 20.3 ± 7.35 | 18.6 ± 5.76 | 17.6 ± 3.45 | |
| 4g | H | CF3 | H | CF3 | 8.41 ± 1.01 | 8.19 ± 3.78 | 8.27 ± 2.74 | 8.12 ± 5.09 | 8.76 ± 1.36 | 8.70 ± 3.02 | |
| GI50 (μM)d |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | R | R1 | R2 | R3 | R4 | NCI-H23 | ACHN | MDA-MB-231 | PC-3 | NUGC-3 | HCT 15 |
| 5a | methyl | OH | H | H | H | 65.7 ± 8.84 | 50.7 ± 6.32 | 89.6 ± 9.29 | 70.7 ± 14.6 | 49.7 ± 10.9 | 54.5 ± 7.60 |
| 5b | ethyl | OH | H | H | H | >90 | >90 | >90 | >90 | >90 | >90 |
| 5c | propyl | OH | H | H | H | 43.1 ± 6.24 | 50.6 ± 11.2 | 38.0 ± 3.55 | 72.5 ± 9.36 | 35.4 ± 7.10 | 58.3 ± 9.89 |
| 5d | octyl | OH | H | H | H | 55.8 ± 8.08 | 65.0 ± 6.13 | 54.3 ± 10.0 | 45.2 ± 7.03 | 31.1 ± 5.10 | 38.4 ± 6.27 |
| 5e | phenyl | OH | H | H | H | 3.49 ± 0.999 | 4.26 ± 1.21 | 4.21 ± 0.892 | 5.69 ± 1.21 | 3.78 ± 0.618 | 3.83 ± 0.994 |
| 5f | 2-chlorophenyl | OH | H | H | H | 46.9 ± 2.99 | 44.2 ± 9.55 | 47.4 ± 5.04 | 60.0 ± 14.3 | 36.8 ± 6.41 | 44.9 ± 8.04 |
| 5g | 3-chlorophenyl | OH | H | H | H | 11.6 ± 2.75 | 17.2 ± 3.54 | 10.6 ± 1.60 | 23.3 ± 4.39 | 11.0 ± 2.34 | 23.1 ± 5.28 |
| 5h | 4-chlorophenyl | OH | H | H | H | 19.2 ± 3.77 | 19.6 ± 5.06 | 19.6 ± 3.54 | 25.1 ± 4.62 | 13.4 ± 2.20 | 20.2 ± 2.97 |
| GI50 (μM)d |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | R | R1 | R2 | R3 | R4 | NCI-H23 | ACHN | MDA-MB-231 | PC-3 | NUGC-3 | HCT 15 |
| 6a | methyl | CF3 | H | H | H | >80 | >80 | >80 | >80 | >80 | >80 |
| 6b | ethyl | CF3 | H | H | H | >80 | >80 | >80 | >80 | >80 | >80 |
| 6c | propyl | CF3 | H | H | H | >70 | >70 | >70 | >70 | >70 | >70 |
| 6d | octyl | CF3 | H | H | H | 4.22 ± 1.26 | 8.33 ± 3.09 | 3.35 ± 0.482 | 4.32 ± 1.04 | 6.18 ± 1.30 | 7.70 ± 1.11 |
| 6e | phenyl | CF3 | H | H | H | 6.34 ± 2.52 | 8.09 ± 1.68 | 5.29 ± 0.846 | 5.56 ± 0.990 | 8.12 ± 1.44 | 6.50 ± 1.14 |
| 6f | 2-chlorophenyl | CF3 | H | H | H | 2.15 ± 0.680 | 3.90 ± 0.946 | 2.53 ± 0.625 | 5.14 ± 1.02 | 2.73 ± 0.554 | 4.22 ± 0.303 |
| 6g | 3-chlorophenyl | CF3 | H | H | H | 0.292 ± 0.111 | 0.526 ± 0.178 | 0.288 ± 0.0992 | 0.729 ± 0.131 | 0.754 ± 0.129 | 0.797 ± 0.173 |
| 6h | 4-chlorophenyl | CF3 | H | H | H | 0.307 ± 0.0941 | 0.824 ± 0.0708 | 0.533 ± 0.0824 | 0.786 ± 0.0996 | 0.551 ± 0.126 | 0.839 ± 0.0610 |
| GI50 (μM)d |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | R | R1 | R2 | R3 | R4 | NCI-H23 | ACHN | MDA-MB-231 | PC-3 | NUGC-3 | HCT 15 |
| 7a | methyl | H | CF3 | H | CF3 | >70 | >70 | >70 | >70 | >70 | >70 |
| 7b | ethyl | H | CF3 | H | CF3 | 7.82 ± 1.62 | 7.72 ± 2.40 | 7.28 ± 0.965 | 8.54 ± 1.99 | 7.22 ± 1.35 | 7.62 ± 0.902 |
| 7c | propyl | H | CF3 | H | CF3 | 15.0 ± 2.67 | 13.9 ± 4.03 | 15.2 ± 2.44 | 18.6 ± 2.87 | 14.4 ± 3.49 | 21.9 ± 3.11 |
| 7d | octyl | H | CF3 | H | CF3 | 3.23 ± 0.397 | 4.52 ± 1.13 | 7.53 ± 1.69 | 7.43 ± 1.89 | 2.23 ± 0.579 | 4.97 ± 1.09 |
| 7e | phenyl | H | CF3 | H | CF3 | 2.13 ± 0.638 | 2.92 ± 0.640 | 2.43 ± 0.632 | 3.50 ± 0.630 | 1.68 ± 0.238 | 3.54 ± 1.20 |
| 7f | 2-chlorophenyl | H | CF3 | H | CF3 | 2.08 ± 0.482 | 4.70 ± 0.733 | 5.25 ± 1.08 | 8.04 ± 2.55 | 1.93 ± 0.404 | 4.05 ± 0.820 |
| 7g | 3-chlorophenyl | H | CF3 | H | CF3 | 0.420 ± 0.0607 | 0.813 ± 0.271 | 1.19 ± 0.313 | 1.05 ± 0.254 | 0.560 ± 0.189 | 0.625 ± 0.0674 |
| 7h | 4-chlorophenyl | H | CF3 | H | CF3 | 0.889 ± 0.102 | 2.57 ± 0.981 | 2.00 ± 0.683 | 4.23 ± 1.67 | 1.66 ± 0.406 | 2.22 ± 0.216 |
GI50 values are taken as a mean from three experiments and correspond to the agent’s concentration causing a 50% decrease in net cell growth.
In summary, we identified a new class of 1,2,3,4-tetrahydroquinolines as the most potent NF-κB inhibitors and potent cytotoxic agents against human cancer cell lines.
The first round of screening starting from the initial lead scaffold 4a led to the discovery of 6f, 6g, and 6h analogues. These new lead analogues inhibited LPS-induced NF-κB transcriptional activity 41, 53, and 13 times more potently, respectively, than the reference compound PDTC. Analogues 6f, 6g, and 6h have also shown the most potent in vitro cytotoxicity against all evaluated human cancer cell lines. Thus, 6f, 6g, 6h, and related analogues provide new chemical tools for development of pathway-selective NF-κB inhibitors with anticancer activity. Work on the enhancement of potency and pharmacological profiles of these probe molecules are underway.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2013R1A1A2009381), and Medical Research Center Program (2008-0062275).
Glossary
ABBREVIATIONS
- LPS
lipopolysaccharide
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00004.
Synthetic procedures, characterization of final products, biological assay protocols, and data and pharmacology profiles (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Sen R.; Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. 10.1016/0092-8674(86)90346-6. [DOI] [PubMed] [Google Scholar]
- Hayden M. S.; Ghosh S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebenlist U.; Franzoso G.; Brown K. Structure, regulation and function of NF-κB. Annu. Rev. Cell Biol. 1994, 10, 405–455. 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Takada Y.; Boriek A. M.; Aggarwal B. B. Nuclear factor-κB: Its role in health and disease. J. Mol. Med. 2004, 82, 434–448. 10.1007/s00109-004-0555-y. [DOI] [PubMed] [Google Scholar]
- Arepalli S. K.; Choi M.; Jung J. K.; Lee H. Novel NF-κB inhibitors: A patent review (2011–2014). Expert Opin. Ther. Pat. 2015, 25, 319–334. 10.1517/13543776.2014.998199. [DOI] [PubMed] [Google Scholar]
- Kwak J. H.; Jung J. K.; Lee H. Nuclear factor-kappa B inhibitors; A patent review (2006 - 2010). Expert Opin. Ther. Pat. 2011, 21, 1897–1910. 10.1517/13543776.2011.638285. [DOI] [PubMed] [Google Scholar]
- Srinivasan B.; Johnson T. E.; Lad R.; Xing C. Structure - Activity relationship studies of chalcone leading to 3-hydroxy-4,3′,4′,5′-tetramethoxychalcone and its analogues as potent nuclear factor κB inhibitors and their anticancer activities. J. Med. Chem. 2009, 52, 7228–7235. 10.1021/jm901278z. [DOI] [PubMed] [Google Scholar]
- Chen H.; Yang Z.; Ding C.; Chu L.; Zhang Y.; Terry K.; Liu H.; Shen Q.; Zhou J. Discovery of O-alkylamino-tethered niclosamide derivatives as potent and orally bioavailable anticancer agents. ACS Med. Chem. Lett. 2013, 4, 180–185. 10.1021/ml3003082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M.; Hwang Y. S.; Kumar A. S.; Jo H.; Jeong Y.; Oh Y.; Lee J.; Yun J.; Kim Y.; Han S. B.; Jung J. K.; Cho J.; Lee H. Design and synthesis of 3,4-dihydro-2H-benzo[h]chromene derivatives as potential NF-κB inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 2404–2407. 10.1016/j.bmcl.2014.04.053. [DOI] [PubMed] [Google Scholar]
- Choi M.; Jo H.; Park H. J.; Sateesh Kumar A.; Lee J.; Yun J.; Kim Y.; Han S. B.; Jung J. K.; Cho J.; Lee K.; Kwak J. H.; Lee H. Design, synthesis, and biological evaluation of benzofuran- and 2,3-dihydrobenzofuran-2-carboxylic acid N-(substituted)phenylamide derivatives as anticancer agents and inhibitors of NF-κB. Bioorg. Med. Chem. Lett. 2015, 25, 2545–2549. 10.1016/j.bmcl.2015.04.050. [DOI] [PubMed] [Google Scholar]
- Kwak J. H.; Kim Y.; Park H.; Jang J. Y.; Lee K. K.; Yi W.; Kwak J. A.; Park S. G.; Kim H.; Lee K.; Kang J. S.; Han S. B.; Hwang B. Y.; Hong J. T.; Jung J. K.; Kim Y.; Cho J.; Lee H. Structure-activity relationship of indoline-2-carboxylic acid N-(substituted)phenylamide derivatives. Bioorg. Med. Chem. Lett. 2010, 20, 4620–4623. 10.1016/j.bmcl.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Sridharan V.; Suryavanshi P. A.; Menéndez J. C. Advances in the chemistry of tetrahydroquinolines. Chem. Rev. 2011, 111, 7157–7259. 10.1021/cr100307m. [DOI] [PubMed] [Google Scholar]
- Katritzky A. R.; Rachwal S.; Rachwal B. Recent progress in the synthesis of 1,2,3,4-tetrahydroquinolines. Tetrahedron 1996, 52, 15031. 10.1016/S0040-4020(96)00911-8. [DOI] [Google Scholar]
- Asolkar K. N.; Schröder D.; Heckman R.; Lang S.; Wagner-Döbler I.; Laatsch H. Helquinoline, a new tetrahydroquinoline antibiotic from Janibacter limosus Hel 1. J. Antibiot. 2004, 57, 17–23. 10.7164/antibiotics.57.17. [DOI] [PubMed] [Google Scholar]
- Pitta M. G. R.; Pitta M. G. R.; Rêgo M. J. B. M.; Galdino S. L. The evolution of drugs on schistosoma treatment: Looking to the past to improve the future. Mini-Rev. Med. Chem. 2013, 13, 493–508. 10.2174/1389557511313040003. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Carling R. W.; Moore K. W.; Moseley A. M.; Smith J. D.; Stevenson G.; Chan T.; Baker R.; Foster A. C.; Grimwood S.; Kemp J. A.; Marshall G. R.; Hoogsteen K. 4-Amido-2-carboxytetrahydroquinolines. Structure-activity relationships for antagonism at the glycine site of the NMDA receptor. J. Med. Chem. 1992, 35, 1954–1968. 10.1021/jm00089a004. [DOI] [PubMed] [Google Scholar]
- Khan P. M.; Correa R. G.; Divlianska D. B.; Peddibhotla S.; Sessions E. H.; Magnuson G.; Brown B.; Suyama E.; Yuan H.; Mangravita-Novo A.; Vicchiarelli M.; Su Y.; Vasile S.; Smith L. H.; Diaz P. W.; Reed J. C.; Roth G. P. 2011. Identification of inhibitors of NOD1-induced nuclear factor-κB activation. ACS Med. Chem. Lett. 2011, 2, 780–785. 10.1021/ml200158b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedoya L. M.; Abad M. J.; Calonge E.; Saavedra L. A.; Gutierrez C M.; Kouznetsov V. V.; Alcami J.; Bermejo P. 2010. Quinoline-based compounds as modulators of HIV transcription through NF-κB and Sp1 inhibition. Antiviral Res. 2010, 87, 338–344. 10.1016/j.antiviral.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Moon K. Y.; Hahn B. S.; Lee J.; Kim Y. S. A cell-based assay system for monitoring NF-κB activity in human HaCaT transfectant cells. Anal. Biochem. 2001, 292, 17–21. 10.1006/abio.2001.5059. [DOI] [PubMed] [Google Scholar]
- Greten F. R.; Eckmann L.; Greten T. F.; Park J. M.; Li Z. W.; Egan L. J.; Kagnoff M. F.; Karin M. IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Nurmi A.; et al. Pyrrolidine dithiocarbamate inhibits translocation of nuclear factor kappa-B in neurons and protects against brain ischaemia with a wide therapeutic time window. J. Neurochem. 2004, 91, 755–765. 10.1111/j.1471-4159.2004.02756.x. [DOI] [PubMed] [Google Scholar]
- Hayakawa M.; Miyashita H.; Sakamoto I.; Kitagawa M.; Tanaka H.; Yasuda H.; Karin M.; Kikugawa K. Evidence that reactive oxygen species do not mediate NF-κB activation. EMBO J. 2003, 22, 3356–3366. 10.1093/emboj/cdg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S. F.; Ye X.; Malik A. B. In Vivo Inhibition of Nuclear Factor-κB Activation Prevents Inducible Nitric Oxide Synthase Expression and Systemic Hypotension in a Rat Model of Septic Shock. J. Immunol. 1997, 159, 3976–3983. [PubMed] [Google Scholar]
- Ziegler-Heitbrock H. W. L.; Sternsdorf T.; Liese J.; Belohradsky B.; Weber C.; Wedel A.; Schreck R.; Bäuerle P.; Ströbel M. Pyrrolidine dithiocarbamate inhibits NF-κB mobilization and TNF production in human monocytes. J. Immunol. 1993, 151, 6986–6993. [PubMed] [Google Scholar]
- Schreck R.; Meier B.; Männel D. N.; Dröge W.; Baeuerle P. A. Dithiocarbamates as potent inhibitors of nuclear factor κB activation in intact cells. J. Exp. Med. 1992, 175, 1181–1194. 10.1084/jem.175.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B. H.; Reddy A. M.; Lee K. H.; Chung E. Y.; Cho S. M.; Lee H.; Min K. R.; Kim Y. Inhibitory mechanism of chroman compound on LPS-induced nitric oxide production and nuclear factor-κB activation. Biochem. Biophys. Res. Commun. 2004, 325, 223–228. 10.1016/j.bbrc.2004.10.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.