

Abstract

Inhibition of phosphoinositide 3-kinase (PI3K) signaling is an appealing approach to treat brain tumors, especially glioblastoma multiforme (GBM). We previously disclosed our successful approach to prospectively design potent and blood–brain barrier (BBB) penetrating PI3K inhibitors. The previously disclosed molecules were ultimately deemed not suitable for clinical development due to projected poor metabolic stability in humans. We, therefore, extended our studies to identify a BBB penetrating inhibitor of PI3K that was also projected to be metabolically stable in human. These efforts required identification of a distinct scaffold for PI3K inhibitors relative to our previous efforts and ultimately resulted in the identification of GDC-0084 (16). The discovery and preclinical characterization of this molecule are described within.
Keywords: PI3K, kinase inhibitor, blood−brain barrier penetration, CNS, oncology
Owing to an associated poor prognosis and limited treatment options, glioblastoma multiforme (GBM) represents a significant unmet medical need.1 In this particular disease, aberrant PI3K signaling is associated with >80% of cases.2 While numerous PI3K inhibitors have advanced to clinical study,3 to treat GBM effectively it is anticipated that the inhibitor would need to freely cross the blood–brain barrier (BBB). To this end, we have previously reported on our initial efforts to identify BBB penetrating PI3K inhibitors.4 In those efforts molecules 1 and 2 (Table 1) were identified, which freely crossed the BBB and demonstrated a PD effect in normal mouse brain tissue as well as efficacy in intracranial mouse tumor models of GBM. Despite the evident free brain penetration of 1 and 2, they were not selected for clinical development due to poor projected clearance in humans (based on both human liver microsomal stability, Table 1, and allometric scaling, data not shown). We, therefore, extended our efforts to identify potent, BBB penetrating inhibitors of PI3K with more desirable metabolic stability that would be suitable for clinical study.
Table 1. MDR1 and Bcrp1 Transfected MDCK Cell Permeability Efflux Ratios and Human Liver Microsomal Stability for Previously Identified PI3K Inhibitors 1–4.

| Compound | PI3Kα Kiapp | mTOR Kiapp | PC3 Proliferation EC50 | B–A/A–B (MDR1) | B–A/A–B (Bcrpl) | [brain]u/[plasma]u | HLM Clhep (mL/min/kg) |
|---|---|---|---|---|---|---|---|
| 1 | 1 nM | 10 nM | 0.17 μM | 0.9 | 3.7 | 0.4a | 10.4 |
| 2 | 2 nM | 9 nM | 0.13 μM | 1.8 | 1.3 | 0.4b | 9.8 |
| 3 | 5 nM | 17 nM | 0.31 μM | 19 | 7 | <0.05c | 3.1 |
| 4 | 3 nM | 950 nM | 0.33 μM | 42 | 71 | 2.8 |
Determined 6 h after administration of 25 mg/kg orally to female CD-1 mice as an MCT suspension.
Determined 6 h after administration of 50 mg/kg orally to female CD-1 mice as an MCT suspension.
Determined 1 h after administration of 20 mg/kg to female CD-1 mice as an MCT suspension.
We continued our efforts to identify brain penetrating inhibitors of PI3K by attempting to identify analogues of thienopyrimidines 1 and 2 that attained an acceptable combination of low transporter mediated efflux, good potency, and low projected human clearance. In our attempts, we identified potent molecules that achieved good metabolic stability in human liver microsomes and many potent molecules that had low efflux ratios in MDR1 (gene coding for P-glycloprotein, P-gp) and breast cancer resistance protein (Bcrp) transfected MDCK cell permeability assays (data not shown). The combination of good human metabolic stability and low efflux ratios in this thienopyrimidine series of analogues, however, remained elusive. As a result we sought an alternative series of molecules that might allow for the desired combination of properties.
Prior to our efforts to identify BBB penetrating PI3K inhibitors, we had identified clinical inhibitors of PI3K that were suited for the treatment of peripheral disease. In our studies leading to the discovery of GDC-0980 (3, Table 1),5 a pan-PI3K and mammalian target of rapamycin (mTOR) inhibitor from the same thienopyrimidine scaffold as 1 and 2, we had also studied a related purine scaffold (e.g., 4, Table 1). Compounds within this purine series of PI3K inhibitors that had good human microsomal stability were readily identified. As an example, while it did not advance to clinical development, compound 4 was identified as a pan-PI3K inhibitor on a purine scaffold that had appealing projected human clearance based on liver microsomal stability (Table 1).
With our knowledge that desirable metabolic stability was attainable on a purine scaffold, we resolved to evaluate purine-based inhibitors for their potential to achieve the desired balance of potency, metabolic stability, and low transporter mediated efflux. The previous purine PI3K inhibitors that we had made, such as 4, were not designed to have low P-gp and Bcrp mediated efflux. Indeed, compound 4 was not expected to be freely BBB penetrating given the high number of hydrogen bond donors in the molecule. In fact, compound 4 is a significant substrate of both P-gp and Bcrp as determined by the efflux ratios in cell permeability assays (Table 1). It remained to be seen whether or not we could realize purine PI3K inhibitors that were capable of avoiding transport by P-gp and Bcrp while also maintaining good human metabolic stability.
At first we were concerned that the additional polarity of a purine scaffold, relative to a thienopyrimidine, might render such molecules more likely to be transporter substrates. To evaluate the feasibility of a purine core for brain penetration, we first studied compound 5 (Table 2). Despite a topological polar surface area (TPSA) of 107 Å2, 5 has a low efflux ratio in a P-gp transfected MDCK cell permeability assay. Additionally, compound 5 showed encouraging human microsomal stability. Unfortunately, this analogue lacked potency in a PC3 cell proliferation assay. Nevertheless, we were encouraged that the combination of low efflux and good human microsomal stability that was unattainable in the thienopyrimidine series of molecules could be obtained on a purine-based PI3K inhibitor. We were then pleased to find that substitution at R1 with either ethyl (6) or cyclobutyl (7) groups improved the cellular potency and maintained low efflux ratios (Table 2). However, the ethyl and cyclobutyl substitutions each led to reduced human liver microsomal stability. Compound 8 was even more potent than the previous purine analogues and had excellent human liver microsomal stability (Table 2). The introduction of a new hydrogen bond donor relative to compounds 5–7, however, led to very high efflux ratios for 8. This result was consistent with our experience with thienopyrimidine PI3K inhibitors and led us to synthesize compound 9, the methyl ether analogue of 8 and a direct analogue of thienopyrimidine 2. Compound 9 retained the excellent cellular potency of 8 and, as we had suspected, the conversion of the tertiary alcohol of 8 to the methyl ether of 9 ablated P-gp and Bcrp mediated efflux. While the human liver microsomal stability of 9 was moderate, we sought to further improve upon this result.
Table 2. Cellular Potency, TPSA, Efflux Ratios, and Human Metabolic Stability for Select Purine PI3K Inhibitors.
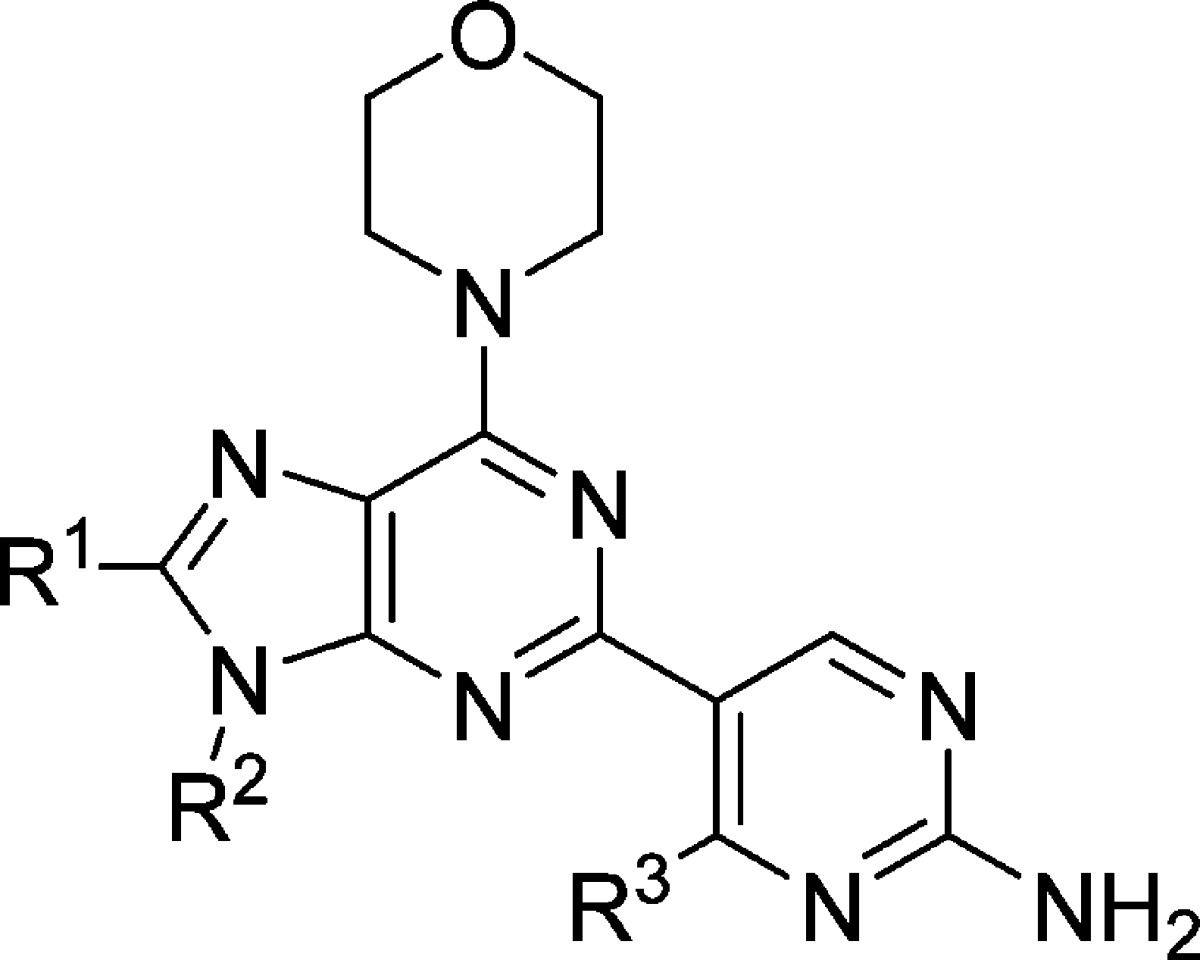

The excellent metabolic stability of compound 4 had initially incited our investigation of whether we could realize brain penetrating purine PI3K inhibitors. Its excellent potency and metabolic stability were desirable, but the efflux ratios suggested the molecule would not freely penetrate the BBB (Table 2). We, therefore, sought to eliminate the two hydrogen bond donors of the alcohols in compound 4 in an effort to reduce transporter mediated efflux. Rather than alkylation of the alcohols of 4, we investigated the effective result of ring closing condensation.
The first cyclized analogue of 4 using this approach, compound 15 (Table 3), was found to have excellent human metabolic stability and low efflux ratios but modest cellular potency (Table 3). We previously found that analogues without R1 substitution were more potent inhibitors of mTOR and generally had better cellular potency in cell lines whose proliferation were driven by aberrant PI3K signaling.6 Compound 16, where R1 is H, achieved an excellent balance of cellular potency, metabolic stability, and lack of efflux (Table 3). Additionally, compound 16 was found to be highly selective against a panel of 229 kinases where it inhibited none by >50% (1 μM 16, Supporting Information). Furthermore, 16 maintains inhibition of each of the Class I PI3K isoforms (Supporting Information) but with more potent inhibition of mTOR. Compound 16 was also tested in five different GBM cell lines and was found to have antiproliferative EC50s ranging from 0.3 to 1.1 μM.7
Table 3. Potency, Efflux Ratios, and Human Metabolic Stability of Tricyclic Purine-Based PI3K Inhibitors 15 and 16.
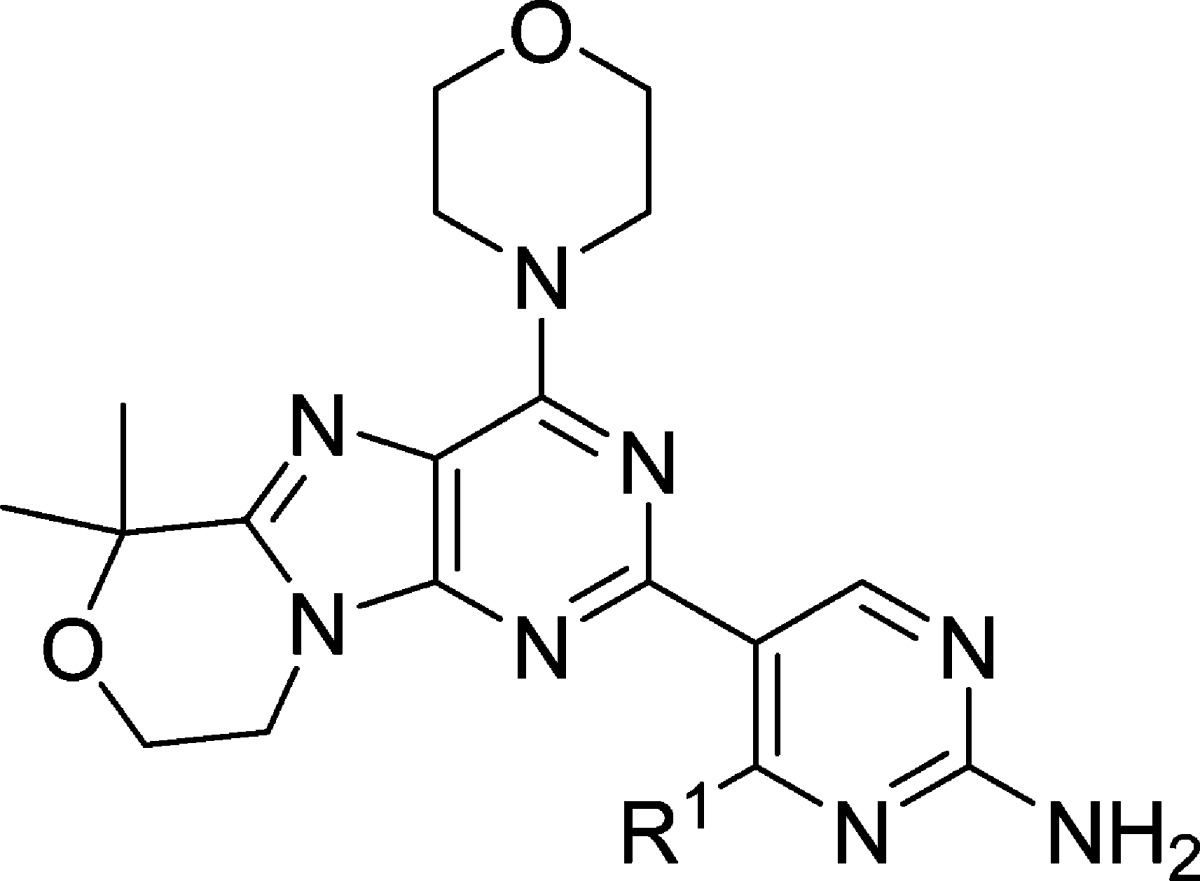
| Compound | R1 | PI3Kα Kiapp | mTOR Kiapp | PC3 Proliferation EC50 | B–A/A–B (MDR1) | B–A/A–B (Bcrpl) | HLM Clhep (mL/m in/kg) | HH Clhep (mL/min/kg) |
|---|---|---|---|---|---|---|---|---|
| 15 | Me | 2 nM | 1.2 μM | 2.0 μM | 1.7 | 3.3 | 3.1 | 1.2 |
| 16 | H | 2 nM | 0.07 μM | 0.4 μM | 0.8 | 1.6 | 5.0 | 1.0 |
The synthesis of 16 (Scheme 1) began with lithiation of purine 10(8) followed by alkylation with acetone to provide tertiary alcohol 11. The THP group was next deprotected to provide 12. Subsequent alkylation with 1,2-dibromoethane afforded the annulated product 13. Finally, Suzuki coupling of 13 with boronate ester 14 afforded the final compound 16.
Scheme 1. Synthetic Route to Obtain Tricyclic Purine-Based Brain Penetrant PI3K Inhibitor 16.

The appealing in vitro properties of 16 described in Table 3 led us to evaluate it in in vivo pharmacokinetic studies. Table 4 includes the microsomal stability of 16 as well as key in vivo pharmacokinetic parameters in rodents. The correlation between in vivo clearance and predicted clearance based on microsomal stability gave us greater confidence that human clearance would be low as predicted in a human metabolic stability assays (Table 3).
Table 4. Preclinical Species Hepatocyte Stability and in Vivo Pharmacokinetic Data for 16.
| IV (1 mg/kg)b |
POc |
||||||
|---|---|---|---|---|---|---|---|
| Species | liver microsomes Clhep (mL/min/kg)a | in vivo Cl (mL/min/kg) | Vss (L/kg) | Dose (mg/kg) | AUC (μM·h) | F% | PPB% |
| Mouse | 19 | 17 | 1.7 | 25 | 47 | 75 | 78 |
| Rat | 14 | 28 | 3.2 | 5 | 8.3 | 77 | 71 |
Hepatic clearance was predicted from liver microsomes incubations using the “in vitro t1/2 method.”9
Male Sprague–Dawley rats or female CD-1 mice were dosed intravenously with 1 mg/kg of 16 prepared in 60% PEG400/10% ethanol.
Compound 16 was administered PO at the indicated dose in 0.5% methylcellulose with 0.2% Tween 80 (MCT).
To verify that 16 was indeed capable of penetrating the BBB, we determined the brain-to-plasma ratio in rats. After a 15 mg/kg dose of 16, the total brain-to-plasma ratio was 1.9–3.3. While we did not determine brain binding for rats and therefore cannot report Bu/Pu, we determined the concentration of 16 in cerebral spinal fluid (CSF). The CSF concentration is sometimes employed as a surrogate for unbound brain concentration.10 We found that the CSF-to-free plasma concentration ratio in rats was 0.7–1.0, indicating that 16 effectively penetrates the BBB (Figure 1). In addition to demonstrating that 16 is capable of crossing the BBB in rats, we determined the unbound brain-to-unbound plasma concentration (Bu/Pu) ratio in female CD-1 mice. The Bu/Pu ratio of 0.4, at both 1 and 6 h post 25 mg/kg oral dose of 16, demonstrates the molecule is capable of substantial free brain penetration (Figure 1).
Figure 1.

CNS penetration of 16 in rat and mouse. [Brain]/[Plasma] ratios determined after oral dose of 16 to female CD-1 mice or male Sprague–Dawley rats as an MCT suspension. *[Brain]u and [Plasma]u refer to the unbound concentration measured in the brain and plasma, respectively. **[CSF] refers to the concentration measured in the cerebral spinal fluid. aDetermined to be identical at both 1 and 6 h after administration of 25 mg/kg 16 to female CD-1 mice. The [Brain]/[Plasma] ratios are the mean values from 3 animals per time point. bDetermined after administration of 15 mg/kg 16 to male Sprague–Dawley rats. [Brain]/[Plasma] determined for 1 animal at each of 0.25 and 2 h and 3 at 8 h. Data reported are the range across the three time points (average of the 3 animals at 8 h). c[CSF] determined for 1 animal at each of 0.25 and 2 h and 3 at 8 h. Data reported are the range across the three time points (average of the 3 animals at 8 h).
To further verify that 16 was indeed capable of penetrating the BBB to engage its target where intended, we evaluated the effect of 16 on pAKT in normal brain tissue. After a 25 mg/kg dose of 16 administered orally, pAKT in normal mouse brain tissue was significantly inhibited at 1 and 6 h postdose (Figure 2). The potent inhibition of pAKT at both time points in this study demonstrates that 16 inhibits its target behind a fully intact BBB.
Figure 2.

Inhibition of p-AKT by 16 in normal mouse brain tissue along with corresponding brain and unbound brain concentrations. *Significantly different from untreated control. p < 0.05, t test. [Brain] determined after 25 mg/kg oral dose of 16 female CD-1 mice as an MCT suspension. [Brain]u refers to the unbound concentration measured in the brain. Data are reported as mean values ± SD from 3 animals per time point.
In addition to the pharmacodynamic effect in normal brain tissue, 16 was studied in a subcutaneous U87 tumor xenograft model of glioblastoma in mice.11 In this study, 16 achieved significant and dose-dependent tumor growth inhibition (Figure 3). Tumor growth inhibition was first observed at a 2.2 mg/kg dose level. Higher doses led to greater tumor growth inhibition, including tumor regressions at the 17.9 mg/kg dose level. Each of these doses was well tolerated for the duration of the study.
Figure 3.
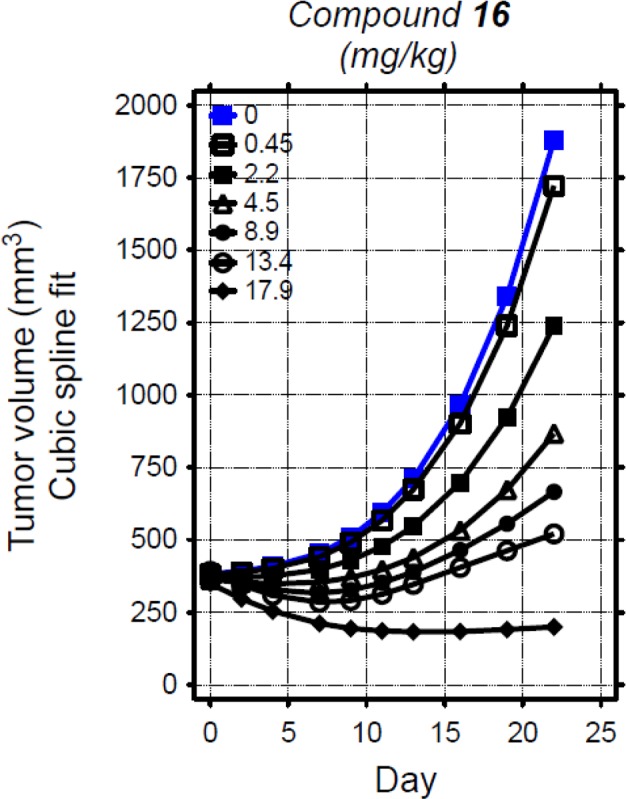
In vivo efficacy of 16 versus U87 MG/M human glioblastoma xenografts. Female NCr nude mice bearing subcutaneous tumors were administered escalating doses of 16 orally as a suspension in vehicle (0.5% methylcellulose/0.2% Tween-80) or vehicle once daily (QD) for 23 days. Changes in tumor volumes over time by dose for each compound are depicted as cubic spline fits generated via Linear Mixed Effects analysis of log-transformed volumes.
Consistent with the efficacy observed in the U87 xenograft tumor study, at similar dose levels compound 16 was found to have a significant PD effect in the U87 tumors. Dose- and concentration-dependent inhibition of pAKT was observed at both 1 and 4 h postdose, indicating that the tumor growth inhibition is the result of on-target inhibition (Figure 4).
Figure 4.
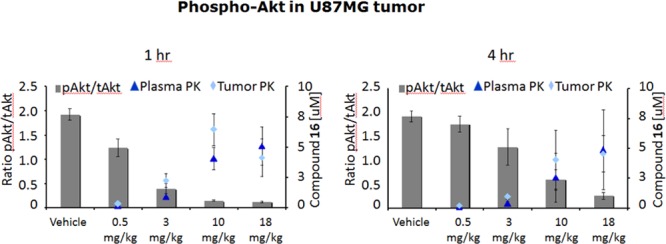
Effect of 16 on the PD marker pAKT in the U87 MG/M human glioblastoma xenograft model after 24 days of continuous dosing. Tumors were excised from animals 1 and 4 h after the last administered dose on day 24 and processed for analysis of pAKT as described in the Supporting Information. Indicated values are the means for groups of 3 animals, and error bars indicate ± standard error of the mean. Levels of pAkt (Ser473) and total Akt were measured by electrochemiluminescence using Meso Scale Discovery according to manufacturer’s instructions (Gaithersburg, MD).
The U87 subcutaneous tumor model can be considered a surrogate for the intracranial U87 model because the intracranial model has a compromised blood–brain barrier following engraftment.12 We previously studied a different BBB penetrating PI3K/mTOR inhibitor in intracranial GBM models and would expect comparable efficacy with 16 given its free brain penetration.13
To summarize, we have identified 16, a potent purine-based inhibitor of PI3K and mTOR, that is capable of penetrating the BBB. Additionally, 16 has excellent human metabolic stability in microsomal and hepatocyte incubations. Compound 16 demonstrated inhibition of pAKT, a key signal within the PI3K pathway, in both normal brain tissue and in U87 glioblastoma xenograft tumors in mice. Along with pAKT inhibition in U87 tumors, significant tumor growth inhibition was achieved. The promising preclinical profile of 16, along with the significant unmet medical need for glioblastoma treatments, led to the advancement of 16 to clinical development. Details of additional preclinical and clinical studies of 16 will be reported elsewhere.
Acknowledgments
The authors wish to thank Mengling Wong, Chris Hamman, Michael Hayes, and Steve Huhn for compound purification and determination of purity by HPLC, mass spectroscopy, and 1H NMR. We thank Krista K. Bowman, Alberto Estevez, Kyle Mortara, and Jiansheng Wu for technical assistance of protein expression and purification.
Glossary
ABBREVIATIONS
- PI3K
phosphoinositide 3-kinase
- GBM
glioblastoma multiforme
- BBB
blood–brain barrier
- P-gp
P-glycoprotein
- Bcrp
breast cancer resistance protein
- mTOR
mammalian target of rapamycin
- TPSA
topological polar surface area
- dppf
1,1′-bis(diphenylphosphino)ferrocene
Biography
Timothy P. Heffron is a Senior Scientist at Genentech. As a medicinal chemist, and Chemistry and Research Team Leader, Timothy has advanced programs directed toward treatments for oncology (including cancer immunotherapy), neurology, and ophthalmology indications. Timothy has contributed to seven molecules that have advanced to clinical development, four of which came under his leadership as a chemistry team leader, including taselisib (Phase III). Timothy completed his undergraduate studies in Chemistry at Yale University and his doctoral studies at MIT.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00005.
Assessment of inhibition of 229 kinases by 16, Class I PI3K Kiapps for 16, Western data showing inhibition of pAKT and pS6 in U87 cells by 16, synthetic details and associated analytical data for all reported compounds, experimental details for biochemical and cellular assays, in vitro transport assays, brain and plasma protein binding, PK studies in mice, pAKT/tAKT PD evaluation in mouse brain, and in vivo xenograft studies (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Rich J. N.; Bigner D. D. Development of Novel Targeted Therapies in the Treament of Malignant Glioma. Nat. Rev. Drug Discovery 2004, 3, 430–446. and references therein. 10.1038/nrd1380. [DOI] [PubMed] [Google Scholar]
- Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap T. A.; Bjerke L.; Clarke P. A.; Workman P. Drugging PI3K in cancer: refining targets and therapeutic strategies. Curr. Opin. Pharmacol. 2015, 23, 98–107. 10.1016/j.coph.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffron T. P.; Salphati L.; Alicke B.; Cheong J.; Dotson J.; Edgar J.; Goldsmith R.; Gould S. E.; Lee L. B.; Lesnick J. D.; Lewis C.; Ndubaku C.; Nonomiya J.; Olivero A. G.; Pang J.; Plise E. G.; Sideris S.; Trapp S.; Wallin J.; Zhang X. The Design and Identification of Brain Penetrant Inhibitors of Phosphoinositide 3-Kinase α. J. Med. Chem. 2012, 55, 8007–8020. 10.1021/jm300867c. [DOI] [PubMed] [Google Scholar]
- Sutherlin D. P.; Bao L.; Berry M.; Castanedo G.; Chuckowree I.; Dotson J.; Folkes A.; Friedman L.; Goldsmith R.; Gunzner J.; Heffron T.; Lesnick J.; Lewis C.; Mathieu S.; Murray J.; Nonomiya J.; Pang J.; Pegg N.; Prior W. W.; Rouge L.; Salphati L.; Sampath D.; Tian Q.; Tsui V.; Wan N. C.; Wang S.; Wei B.; Wiesmann C.; Wu P.; Zhu B.-Y.; Olivero A. Discovery of a Potent, Selective, and Orally Available Class I Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (mTOR) Kinase Inhbitor (GDC-0980) for the Treatment of Cancer. J. Med. Chem. 2011, 54, 7579–7587. 10.1021/jm2009327. [DOI] [PubMed] [Google Scholar]
- Sutherlin D. P.; Sampath D.; Berry M.; Castanedo G.; Chang Z.; Chuckowree I.; Dotson J.; Folkes A.; Friedman L.; Goldsmith R.; Heffron T.; Lee L.; Lesnick J.; Lewis C.; Mathieu S.; Nonomiya J.; Olivero A.; Pang J.; Prior W. W.; Salphati L.; Sideris S.; Tian Q.; Tsui V.; Wan N. C.; Wang S.; Wiesmann C.; Wong S.; Zhu B.-Y. Discovery of (Thienopyrimidin-2-yl)aminopyrimidines as Potent, Selective, and Orally Available Pan-PI3-Kinase and Dual-PI3-Kinase/mTOR Inhibitors for the Treatment of Cancer. J. Med. Chem. 2010, 53, 1086–1097. 10.1021/jm901284w. [DOI] [PubMed] [Google Scholar]
- In 4-day assays using CellTiter-Glo to monitor proliferation, 16 was studied in the following GBM cell lines: G111, EC50 = 0.27 μM; G96, EC50 = 0.53 μM; G112, EC50 = 0.58 μM; U87, EC50 = 0.74 μM; SF268, EC50 = 1.01 μM; G122, EC50 = 1.01 μM.
- Murray J. M.; Sweeney Z. K.; Chan B. K.; Balazs M.; Bradley E.; Castanedo G.; Chabot C.; Chantry D.; Flagella M.; Goldstein D. M.; Kondru R.; Lesnick J.; Li J.; Lucas M. C.; Nonomiya J.; Pang J.; Price S.; Salphati L.; Safina B.; Pascal P. A.; Seward E. M.; Ultsch M.; Sutherlin D. P. Potent and Highly Selective Benzimidazole Inhibitors of PI3-Kinase Delta. J. Med. Chem. 2012, 55, 7686–7695. 10.1021/jm300717c. [DOI] [PubMed] [Google Scholar]
- Obach R. S.; Baxter J. G.; Liston T. E.; Silber B. M.; Jones B. C.; MacIntyre F.; Rance D. J.; Wastall P. The prediction of humanpharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 1997, 283, 46–58. [PubMed] [Google Scholar]
- Liu X.; Van Natta K.; Yeo H.; Vilenski O.; Weller P. E.; Worboys P. D.; Monshouwer M. Unbound Drug Concentration in Brain Homogenate and Cerebral Spinal Fluid at Steady State as a Surrogate for Unbound Concentration in Brain Interstitial Fluid. Drug Metab. Dispos. 2009, 37, 787–793. 10.1124/dmd.108.024125. [DOI] [PubMed] [Google Scholar]
- Compound 16 was found to have an antiproliferation EC50 of 740 nM in U87 cells. Inhibition of pAKT in U87 cells was demonstrated qualitatively by Western and is included as Supporting Information.
- Lee J.; Kotilarova S.; Kotliarov Y.; Li A.; Su Q.; Donin N. M.; Pastorino S.; Purow B. W.; Christopher N.; Zhang W.; Park J. K.; Fine H. A. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- Salphati L.; Heffron T. P.; Alicke B.; Nishimura M.; Barck K.; Carano R. A.; Cheong J.; Edgar K. A.; Greve J.; Kharbanda S.; Koeppen H.; Lau S.; Lee L. B.; Pang; Plise E. G.; Pokorny J. L.; Reslan H. B.; Sarkaria J. N.; Wallin J. J.; Zhang X.; Gould S. E.; Olivero A. G.; Phillips H. S. Targeting the PI3K Pathway in the Brain—Efficacy of a PI3K Inhibitor Optimized to Cross the Blood-Brain Barrier. Clin. Cancer Res. 2012, 18, 6239–6248. 10.1158/1078-0432.CCR-12-0720. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.