

Abstract
The impact of early childhood cigarette smoke (CS) exposure on CS-induced chronic obstructive pulmonary disease (COPD) is unknown. This study was performed to evaluate the individual and combined effects of neonatal and adult CS exposure on lung structure, function, and gene expression in adult mice. To model a childhood CS exposure, neonatal C57/B6 mice were exposed to 14 days of CS (Neo CS). At 10 weeks of age, Neo CS and control mice were exposed to 4 months of CS. Pulmonary function tests, bronchoalveolar lavage, and lung morphometry were measured and gene expression profiling was performed on lung tissue. Mean chord lengths and lung volumes were increased in neonatal and/or adult CS-exposed mice. Differences in immune, cornified envelope protein, muscle, and erythrocyte genes were found in CS-exposed lung. Neonatal CS exposure caused durable structural and functional changes in the adult lung but did not potentiate CS-induced COPD changes. Cornified envelope protein gene expression was decreased in all CS-exposed mice, whereas myosin and erythrocyte gene expression was increased in mice exposed to both neonatal and adult CS, suggesting an adaptive response. Additional studies may be warranted to determine the utility of these genes as biomarkers of respiratory outcomes.
Keywords: cigarette smoke, gene expression, lung function, newborn
Early postnatal life is a time of rapid cell growth and development. Stresses and environmental exposures during early postnatal life can have lasting effects into adulthood. Recently it has been recognized that several adult chronic diseases are associated with in utero and/or early postnatal events or exposures [1]. Intrauterine growth retardation has been associated with obesity and cardiovascular disease in adult life [2, 3] and lower respiratory tract illnesses during early childhood have been linked to a more rapid decline in adult lung function [4, 5]. Decreased lung function in adult life has also been shown to be associated with exposure to airborne pollutants during childhood [6] and over the past several decades, mortality from adult-onset chronic obstructive pulmonary disease (COPD) has increased in the United States, accounting for 42 deaths per 100,000 in 2002 [7]. It has been recognized that exposure to cigarette smoke (CS) and air-borne pollutants are major contributing factors to the worldwide COPD epidemic [8]. It has also been postulated that children with underlying lung disease may be at increased risk for developing and/or having more severe adult-onset COPD. Older children with a history of bronchopulmonary dysplasia (BPD) often have persistent airway flow obstruction and exercise intolerance in later life [9, 10] and may be an “at risk” group for developing adult-onset COPD [11]. Identifying early life exposures that influence adult-onset COPD development may be useful in distinguishing individuals at increased risk for poorer respiratory outcomes.
The mechanisms by which early environmental events or exposures can influence lung phenotype and disease expression in adult-onset COPD are not well understood and are probably multifactorial. Gene and environmental interactions have been shown to influence susceptibility to CS-induced COPD [12, 13]. Increased expression of phase I metabolism and inflammatory pathway genes has been demonstrated in adult CS-exposed lung [14]. However, little is known regarding how age of CS exposure and cumulative effect of CS exposure influences gene expression in the lung. Identifying genes that are altered by an early childhood exposure to CS may help provide insight into how early life events/exposures can alter adult lung phenotype. A recent study performed in neonatal mice found that exposure to CS caused persistent microRNA alterations in the lungs of these mice, which the authors speculated may be biomarkers of effect 15. Alternatively, cumulative exposures or re-exposure to a toxin or stress may induce stress resistance and an adaptive response [16]. It has been reported that respiratory muscle remodeling towards a more efficient and oxidative muscle response may be an adaptive response to chronic hyperinflation and COPD [17]. To this end we were interested in identifying genes that may be induced in the lungs of mice exposed to both neonatal and adult CS.
It is known that secondhand CS can have a detrimental impact on the health of infants and children by increasing the risk of middle ear infections, lower respiratory tract infections, and sudden infant death syndrome [18]. Based on our previous work in mice [19], we wanted to test whether CS exposure during neonatal life was a risk factor for adult-onset COPD changes in adult mice subsequently exposed to CS. We hypothesized that childhood and adult CS exposure would cause durable changes in lung function and structure and increase susceptibility to COPD lung changes through alterations in gene expression. We also wanted to determine if neonatal CS exposure, regardless of a second exposure in adult life, could induce durable effects on adult lung structure and function. To address these questions, we used a mouse model in which mice were exposed to CS either as neonates, adults, or both. All treated and control groups then underwent quasistatic pressure-volume curves, bronchoalveolar lavage, lung morphometry, and gene expression profiling.
Methods
Mice
C57BL/6J mice were obtained from the National Cancer Institute (NCI; Bethesda, MD). The animals were maintained on an AIN 76A diet and water ad libitum and housed at a temperature range of 20°C to 23°C under 12-hour light/dark cycles. Experiments were conducted in accordance with the standards established by the United States Animal Welfare Acts, set forth in the National Insitutes of Health (NIH) guidelines and the Policy and Procedures Manual of the Johns Hopkins University Animal Care and Use Committee.
Cigarette smoke exposure
The smoke machine (Model TE-10; Teague Enterprises, Davis, CA) was adjusted to burn 2 cigarettes (2R4F reference cigarettes—2.45 mg nicotine/cigarette; purchased from the Tobacco Health Research Institute, University of Kentucky, Lexington, KY) at one time. Cigarettes were smoked with standard puffs of 35 mL volume of 2-second duration. For the neonatal mice, the total particulate matter (TPM) in the exposure chambers was 90 mg/m3. At 1 day of age, newborn mice were placed in the smoke chamber for 1 h/day for 7 days followed by 2 h/day for 7 days. Neonatal mice were exposed to the smoke chamber for a total of 14 days (Neo CS), as previously described [19]. Mice were then removed and kept in room air. At 10 weeks of age, one half of these mice were placed in the smoke chamber and exposed to 150 mg/m3 of TPM for 3 hours a day, 5 days a week, for 4 months (Neo CS + Adult CS). A third group of mice of similar age, not previously exposed to neonatal smoke, were also placed in the smoke chamber for 4 months and received the same exposure (Adult CS). A fourth group of mice served as age-matched room air controls (Ctr).
Pulmonary function
Quasistatic pressure-volume curves were performed. Each animal was weighed and anesthetized with an intraperitoneal ketamine/xylazine mixture (100 mg/kg and 15 mg/kg, respectively). After anesthesia, the trachea was cannulated, connected to a ventilator, and the animal was ventilated with 100% O2 After determination of baseline resistance and elastance using an inspiratory occlusion [20], ventilation was stopped, and the tracheal cannula was occluded for 4 minutes, which led to complete degassing of the lungs by absorption atelectasis. Quasistatic pressure-volume (PV) curves were performed as previously reported [21]. Briefly, air was initially infused with syringe pump at a rate of 1 mL/min, and airway pressure and volume were recorded on a PowerLab digital data acquisition system (ADInstruments, Castle Hill, Australia). Once a maximal pressure of 35 cm H2O was reached, lungs were deflated to −10 cm H2O at a rate of 3 mL/min. Two sequential pressure-volume loops between −10 and 35 cm H2O, were then acquired at this rate. Residual volume (RV) was measured at a pressure of −10 cm H2O after the first deflation. Total lung capacity was defined as the volume of lung at 35 cm H2O.
Bronchoalveolar lavage (BAL)
Samples were collected with 2 × 1 mL of sterile phosphate-buffered saline (PBS) through a tracheal cannula. Total cell counts were measured and cell differential determined with Diff-Quik (Andwin Scientific, Tyron, NC).
Lung fixation and morphometry
Lungs were infused through the trachea with 0.5% low-melting agarose and then fixed overnight in 4% paraformaldehyde, paraffin-embedded, and cut into 5-μm sections. For morphometry, lung sections were stained with hematoxylin and eosin. Ten randomly chosen areas from each lung section were photographed with the 10× objective of a Nikon Eclipse 80i microscope (Nikon Instruments, Melville, NY). Mean airspace chord length (MCL) was measured from each image using NIS-Elements AR (Nikon Instruments). The software allowed for manual identification and exclusion of large airways and vessels prior to MCL calculations.
Purification and preparation of RNA
Total RNA was obtained from lung of mice exposed to Neo CS only (n = 6), Adult CS only (n = 4), Neo CS + Adult CS (n = 4), and age-matched control lung (n = 5). Total RNA was extracted using the Trizol Reagent method (Invitrogen, Carlsbad, CA; catalog no. 15596–026). Additional purification was performed on RNeasy columns (Qiagen, Valencia, CA; catalog no. 74104). The quality of total RNA samples as assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). RNA samples were labeled according to the chip manufacturer's recommended protocols. In brief, for Illumina, 0.5 μg of total RNA from each sample was labeled by using the Illumina TotalPrep RNA Amplification Kit (Ambion, Austin, TX; catalog no. IL1791) in a 2-step process of cDNA synthesis followed by in vitro RNA transcription. Single-stranded RNA (cRNA) was generated and labeled by incorporating biotin-16-UTP. Biotin-labeled cRNAs were hybridized (16 hours) to Illumina Sentrix Mouse Ref8_v2 Bead-Chips (Illumina, San Diego, CA; catalog no. BD-202–0202). The hybridized biotinylated cRNA was detected with streptavidin-Cy3 and quantified using Illumina's BeadStation 500GX Genetic Analysis Systems scanner.
Data analysis
Preliminary analysis of the scanned data was performed using Illumina BeadStudio software, which returns single intensity data values/gene following the computation of a trimmed mean average for each probe type represented by a variable number of bead probes/gene on the array. Z-transformation for normalization was performed on each Illumina sample/array on a stand-alone basis [22], and significant changes in gene expression between class pairs was calculated by Z test [23]. Significant gene lists were calculated by selecting genes that satisfy the significance threshold criteria of Z test P values less than or equal to .001 (10−3), a false discovery rate less than or equal to 0.1, and a fold change ±1.5 or greater.
Quantitative reverse transcriptase–polymerase chain reaction (qRT-PCR) analysis
Reverse transcription was performed using total RNA isolated from lung and processed with the Super-Script III first-strand synthesis system according to the manufacturer's protocol (Invitrogen). qRT-PCR was performed on an ABI Prism 7700 Sequence Detection System using Applied Biosystems (Foster City, CA) TaqMan Gene Expression assay system. Probes and primers were designed and synthesized by Applied Biosystems. Relative gene expressions was calculated using the 2−ΔΔCt method [24]. Glutaraldehyde-3-phosphate dehydrogenase (GADPH) was used an internal endogenous control.
Statistics
Statistical calculations were performed using the SPSS 8.0 statistical package for Windows (Chicago, IL) Differences in measured variables between experimental and control groups were determined using comparison of the means using 1-way analysis of variance (ANOVA) (Bonferroni and Tukey) and Student's t tests (2-tailed, equal variance).
Results
Effect of CS exposure on weight and airspace size
At 6 months of age, mice exposed to both Neo CS + Adult CS weighed significantly less than control mice (P < .04; Table 1). Total BAL cell counts were increased in all groups of mice that were exposed to CS (P < .04; Figure 1B). The percentage of macrophages in the BAL was 98% or greater in both CS-exposed and control mice.
Table 1. Mean Weights by CS Exposure.
| Groups | n | Weight (g) Mean ± SD | |
|---|---|---|---|
| Population by exposure | Control | 14 | 28.1 ± 5.7* |
| Neo CS | 13 | 28.5± 2.1 | |
| Adult CS | 14 | 25.6± 3.7 | |
| Neo CS + Adult CS | 13 | 24.2± 3.0 |
Weights of control mice were significantly greater than Neo CS +Adult CS (P < .04).
Figure 1.
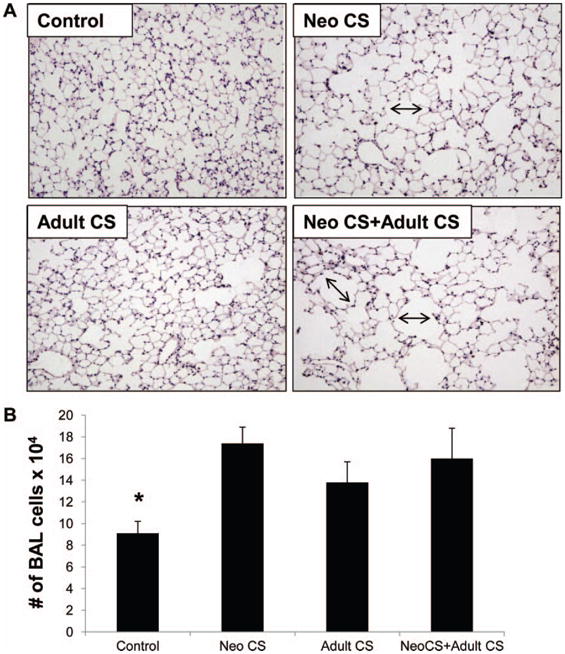
(A) Representative lung sections from Control, Neo CS, Adult CS, Neo CS + Adult CS mice. Black arrows illustrate areas of airspace enlargement in the adult lungs of mice exposed to neonatal CS (Neo CS) and both a neonatal and adult CS exposure (Neo CS +Adult CS). (B) Control lung had significantly fewer BAL cells compared to the CS-exposed groups (*P < .04), n = 5–8. (Color figure available online.)
To determine the effect of CS exposure on lung parenchyma, mean airspace chord lengths (MCLs) were measured in all groups of mice (representative lung sections from each group in Figure 1A, Table 2). Neo CS, Adult CS, and Neo CS +Adult CS mice had significantly larger MCLs compared to age-matched Ctr mice. Mice exposed to Neo CS alone and Neo CS +Adult CS had significantly larger MCLs than Adult CS mice but the mean MCLs of the Neo CS alone and Neo CS +Adult CS mice were not significantly different. In our model this finding suggested that a neonatal CS exposure, given during a period of postnatal lung development, had a greater influence on MCL size than a 4-month exposure to CS during adult life. TUNEL (terminal deoxynucleotidyl transferase-mediated dUDP nick-end labeling) staining was not significantly different in the lungs of any CS-treated mice at 6 months of age compared to Ctr mice (data not shown).
Table 2. Mean Chord Length by CS Exposure.
| Groups | n | MCL (microns) Mean ± SD | |
|---|---|---|---|
| Population by exposure | Control | 14 | 34.5 ± 3.3* |
| Neo CS | 13 | 41.0 ± 5.0 | |
| Adult CS | 14 | 37.5 ± 4.5† | |
| Neo CS + Adult CS | 13 | 41.3 ± 3.6 |
Mean chord length of control mice were significantly less than Neo CS, Adult CS, and Neo CS +Adult CS (P < .05).
Mean chord length of Adult CS were significantly less than Neo CS + Adult CS (P < .02).
Exposure to CS increased lung volumes
Quasistatic pressure-volume curves were measured in mice at 6 months of age. Lung volumes were measured at defined pressures along the expiratory curve (Figure 2A). Significantly larger lung volumes were found in Neo CS +Adult CS mice compared to Ctr and Neo CS mice, at all lung volumes except −10 cm H2O (Table 3). At pressures between 5 and 35 cmH2O, the Neo CS mice had significantly larger lung volumes compared to Ctr mice (P < .04). Although the mean lung volumes were larger in the Neo CS + Adult CS mice at all pressures compared to the Adult CS mice, these differences did not reach significance. Lung elastance was significantly decreased in the Neo CS and Neo CS +Adult CS mice compared to Ctr mice (P < .002 and P < .004, respectively; Figure 2B). These findings suggest that an early neonatal lung insult can alter lung elastance in adult mice. Decreased lung elastance has been reported in adult mice previously exposed to neonatal hyperoxia [25].
Figure 2.

(A) Expiratory limbs from PV curves obtained from mice that underwent quasistatic pressure-volume pulmonary function tests, n = 6–8. (B) Lung elastance was significantly decreased in the Neo CS and Neo CS +Adult CS mice compared to Ctr mice (*P < .002, **P < .004). n = 5–6 (error bars represent SEM).
Table 3. Lung Volumes at Fixed Pressures.
| Groups | Volume at −10 cm H2O (mL) | Volume at 0 cm H2O (mL) | Volume at 5 cm H2O (mL) | Volume at 10 cm H2O (mL) | Volume at 20 cm H2O (mL) | Volume at 35 cm H2O (mL) |
|---|---|---|---|---|---|---|
| Ctr | 0.16 ± 0.02 | 0.38 ± 0.03 | 0.72 ± 0.04* | 1.03 ± 0.06* | 1.22 ± 0.06* | 1.32 ± 0.06* |
| Neo CS | 0.14 ± 0.01 | 0.42 ± 0.02 | 0.84 ± 0.02 | 1.19 ± 0.03 | 1.37 ± 0.03** | 1.48 ± 0.03** |
| Adult CS | 0.23 ± 0.04 | 0.51 ± 0.04† | 0.92 ± 0.05† | 1.32 ± 0.07† | 1.60 ± 0.08† | 1.72 ± 0.09† |
| Neo CS + Adult CS | 0.20 ± 0.03 | 0.62 ± 0.07‡ | 1.03 ± 0.07‡ | 1.46 ± 0.07‡ | 1.76 ± 0.08‡ | 1.88 ± 0.08‡ |
Note. Quasistatic pressure-volume curves were performed and volumes (mL) were measured at −10, 0, 5, 10, 20, and 35 cm H2O (± SEM). n = 5–8 per group.
Ctr had significantly lower volumes at 5, 10, 20, and 35 cm H2O compared to Neo CS (P < .04).
Neo CS had significantly lower volumes at 20 and 35 cm H2O compared to Adult CS (P < .03).
Adult CS had significantly higher volumes at 0–35 cm H2O compared to Ctr (P < .04).
Neo CS + Adult CS had significantly higher volumes at 0 to 35 cm H2O compared to both Ctr and Neo CS (P < .01).
Gene expression influenced by CS exposure and age
Using gene expression profiling, we found significant induction of the cytochrome P450 genes CYP2A5 and CYP1B1 in lungs of mice exposed to 4 months of CS as adults. PLUNC, a host defense airway gene, was also induced in the lungs of mice exposed to adult CS but not neonatal CS exposure alone [26] (Table 4). CXCR1, a receptor for interleukin-8 (IL-8), a neutrophil chemoattractant, was found to be induced in the lungs of all groups of mice exposed to CS. Expression of both PLUNC and CXCR1 was validated by real-time PCR (Figure 3A, B).
Table 4. Genes That Are Differentially by Cigarette Smoke Exposure Compared to Control Mice.
| Name | Gene | Neo CS only | Adult CS only | Neo CS + Adult CS |
|---|---|---|---|---|
| Cytochrome P450, family 2, subfamily a | CYP2A5 | 1.04 | 3.64 | 4.15 |
| Cytochrome P450, family 1, subfamily b, polypeptide 1 | CYP1B1 | −1.17 | 1.84 | 2.11 |
| Palate lung and nasal epithelium associated | PLUNC | −1.85 | 3.51 | 1.85 |
| Macrophage receptor with collagenous structure | MARCO | −1.23 | 2.43 | 2.83 |
| Chemokine (C-X-C motif) receptor 1 | CXCR-1 | 2.78 | 3.21 | 3.9 |
| Interleukin-1β | IL1- β | 2.84 | −1.46 | −1.25 |
| Thrombospondin 1 | THBS1 | 3.04 | −1.36 | 1.15 |
| Matrix metallopeptidase 9 | MMP9 | 2.33 | −1.44 | −1.44 |
| Chemokine (C-X-C motif) ligand 1 | CXCL1 | 1.67 | 1.26 | 1.31 |
Note. Numbers indicate fold change compared to control lung. Fold changes of 1.5 and above/below are considered signifcant.
Figure 3.

Gene expression by real-time PCR reported as log fold changes above/below control lung. (A) PLUNC gene expression of Adult CS and Neo CS +Adult CS compared to control lung, *P < .0001, **P < .005; Adult CS compared to Neo CS, †P < .01. (B) CXCR1 gene expression compared to control lung, *P < .02, **P < .0001, ***P < .0001. (C) LOR gene expression compared to control lung, *P < .001, **P < .002, ***P < .05. (D) CRCT1 gene expression compared to control lung, *P < .004, **P < .003, ***P < .03. (E) MYH2 gene expression compared to control lung, *P < .05, **P < .001, ***P < .02, and Neo CS +Adult CS compared to Neo CS only and Adult CS only, †P < .01. (F) GYPA gene expression compared to control lung, *P < .005, **P < .0001. n = 4–6 (error bars represent SEM).
In lungs of mice exposed to neonatal CS alone, we found significant induction of several genes reported to be induced in models of bronchopulmonary dysplasia by both gene expression profiling and real-time PCR. These genes included IL-1β [27, 28], CXCL1 [29], matrix metalloproteinase-9 (MMP-9), and thrombospondin-1 [30–32]. Using real-time PCR, we found that IL-1β was induced 9.4 ± 4.2-fold, P < .05, in Neo CS lung compared to control lung, but was not significantly induced above control in the other CS-exposed mice. These changes in gene expression, in addition to the structural and functional changes found in the Neo CS mice, suggest that exposure to CS in the neonatal period may cause durable changes in the adult lung similar to that found in lung models of BPD [25].
In humans, cigarette smoking can cause down-regulation of airway epithelial apical junctional complex genes in smokers with and without COPD [33]. In our study we found down-regulation of cornified envelope protein gene expression in the lungs of all groups of mice exposed to CS. This family of genes is part of the epithelial differentiation complex (EDC) and some of these genes have been associated with innate immune defense [34] (Table 5). Real-time PCR validated down-regulation of loricrin (LOR) and cysteine-rich C-terminal 1 (CRCT1) expression, 2 genes located in the EDC of chromosome 3 (Figure 3C, D).
Table 5. Epidermal Differentiation Complex Genes Down-regulated in All Groups of Mice Exposed to CS.
| Name | Gene | Neo CS only | Adult CS only | Neo CS + Adult CS |
|---|---|---|---|---|
| Loricrin | LOR | −3.27 | −247.44 | −1.9 |
| Late Cornified envelope 3f | LCE3F | −4.47 | −126.43 | −3.63 |
| Cystein rich C-terminal 1 | CRCT1 | −3.85 | −119.32 | −3.22 |
| Repetin | RPTN | −4.65 | 56.87 | −3.95 |
| Late Cornified envelope 3C | LCE3c (SPRRL1) | −4.53 | −55.32 | −2.99 |
| Late Cornified envelope 3B | SPRRLA6 | −4.2 | −53.37 | −2.81 |
| Late Cornified envelope 1B | SPRRLA | −3.82 | −30.47 | −2.61 |
| Esophagin (small proline-rich protein 3) | SPRR3 | −4.45 | −29.35 | −3.4 |
| Late Cornified envelope 1D | LCE1D (SPRRL7) | −3.76 | −24.45 | −2.5 |
| Late Cornified envelope 1A1 | SPRRL3 (LCE1A1) | −3.91 | −16.1 | −2.8 |
| Late Cornified envelope 1F | LCE1F | −2.94 | −15.28 | −2.16 |
| Late Cornified envelope 1A2 | SPRRL2 (LCE1A2) | −3.02 | −12.2 | −2.32 |
| Late Cornified envelope 1G | LCE1G | −3.57 | −11.83 | −2.16 |
| Late Cornified envelope 3A | LCE3A | −3.74 | −11.71 | −3.85 |
Note. Numbers indicate fold change compared to control lung. Fold changes of 1.5 and above/below are considered significant.
In the lungs of mice exposed to both neonatal and adult CS, expression of several genes involved in myosin and erythrocyte differentiation was found to be induced (Table 6). Expression of GYPA, a gene found on the erythrocyte surface [35], and MYH2, a gene essential for muscle contraction [36], was validated by real-time PCR (Figure 3E, F).
Table 6. Muscle and Erythrocyte Genes Up-regulated by a Combination of Neonatal and Adult CS.
| Name | Gene | Neo CS only | Adult CS only | Neo CS + Adult CS |
|---|---|---|---|---|
| Myosin heavy polpeptide 2 | MYH2 | −1.62 | −2.71 | 35.7 |
| Myosin, light chain 2, regulatory, cardiac, slow | MYL2 | 1.24 | −1.33 | 19.51 |
| Creatine kinase, mitochondrial 2 | CKMT2 | 1.19 | −1.43 | 5.64 |
| Troponin T type 1 | TNNT1 | 1.11 | 1.13 | 5.48 |
| Myosin | MYL1 | −1.79 | −3.65 | 3.24 |
| Parvalbumin | P VALB | −2.25 | −11.09 | 2.9 |
| Tropomyosin 3 | TPM3 | −1.32 | −1.74 | 2.86 |
| Tropomodulin 4 | TMOD4 | −1.27 | −2.17 | 2.38 |
| Alpha hemoglobin stabilizing protein | ERAF | 1.01 | 1.49 | 8.54 |
| Solute carrier family 4, anion exchanger, member 1 | SLC4A1 | 1.73 | −4.75 | 7.1 |
| Glycophorin A | GYPA | 1.55 | −2.97 | 5.26 |
| Tripartite motif-containing 10 | Trim 10 | 1.24 | −2.39 | 5.1 |
Numbers indicate fold change compared to control lung. Fold changes of 1.5 and above/below are considered significant.
Discussion
In this study, we evaluated the impact of neonatal CS exposure and adult CS exposure, individually and in combination, on adult lung structure, function, and gene expression. We found that mice exposed to neonatal CS alone developed larger and more simplified alveoli and modest increases in lung volumes, findings similar to injury models of BPD [25]. Mice that were exposed only to adult CS had significantly larger lung volumes compared to Neo CS and Ctr mice, with a modest increase in MCL. Interestingly, we found that neonatal exposure to CS did not additively or synergistically contribute to CS-induced COPD changes in the adult lung. However, differences in gene expression in the lung were found with different CS exposures, indicating that gene expression in the adult lung could be altered by a prior exposure to neonatal CS. The significance of these gene expression changes as biomarkers of respiratory outcomes in adult lung may warrant further investigation.
We previously reported that neonatal CS exposure inhibited alveolar growth in neonatal mice [19]. Impaired alveolar growth was associated with increased alveolar apoptosis and transforming growth factor-β (TGF-β) signaling. These mice also had increased mean linear intercepts and decreased lung surface areas at 8 weeks of age [19]. Taken together, our previous and current studies indicate that neonatal CS exposure can permanently alter lung structure and function in the adult mouse. It has been previously shown that prenatal and early postnatal CS exposures can also cause durable effects on adult lung function. A recent study by Wu and colleagues found that prenatal and early postnatal exposure to side stream smoke resulted in increased airway reactivity in adult mice [37].
We did not, however, find that exposure to neonatal CS augmented COPD changes in the lungs of adult mice exposed to CS. The Neo CS +Adult CS mice had airspace changes similar to the neonatal CS-exposed mice and pulmonary function changes similar to adult CS-exposed mice. However, the combination of both neonatal and adult CS exposures did not cause additive or multiplicative changes with respect to MCL or lung volumes. This is in contrast to what we recently found in adult mice previously exposed to neonatal hyperoxia. In that study, we found that neonatal hyperoxia exposure contributed additively to CS-induced COPD-lung changes in the adult mouse [38]. Several reasons may explain the differences. First, it is possible that if the exposure to neonatal CS had been shorter or if the Neo CS mice had been subjected to a longer period of adult CS exposure, an additive or synergic effect may have been found in the adult lung. Alternatively, 4 months of cigarette smoke exposure in the adult mice may have caused a “ceiling effect,” preventing us from observing a dose-response that may have been discernible if a shorter duration of CS exposure was given.
We did find induction of several inflammatory genes in the lungs of mice exposed to neonatal CS alone. IL-1-β, MMP-9, and thrombospondin-1 were induced only in the lungs of mice exposed to neonatal CS alone, suggesting ongoing inflammation from a remote environmental exposure. MMP-9 and thrombospondin-1 genes have been shown to be induced in models of BPD, indicating that neonatal CS and neonatal hyperoxia may induce similar pathways in response to lung injury in the developing lung. Alternatively, expression of CXCR2/IL8Rα, a receptor for IL-8, was increased in all groups of mice exposed to CS. Induction of CXCR2/IL8Rα has previously been found in animal models of CS-induced COPD, and Streptococcus pneumoniae– and rhinovirus-induced airway inflammation [39–42].
In this study, we also found that CS exposure in all CS treatments groups down-regulated the expression of several epithelial differentiation complex (EDC) genes. In humans, EDC genes are located on chromosome 1q21. An association between decreased expression of the EDC genes CRCT-1 and SPRR3 with eosinophilic esophagitis and chemoradiotherapy resistance has been reported [43–45]. Decreased expression of Cornified envelope protein genes located in the EDC has also been associated with ultraviolet radiation exposure in the eye [46]. Furthermore, the Cornified envelope protein gene, SPRR2A, have been linked to innate immunity in the intestine, with differential expression depending on the type of colonizing bacteria in the intestines [34]. The decreased expression of these Cornified envelope protein genes in the lungs of CS-exposed mice may be markers of an impaired innate immune response and/or reflect a loss of epithelial cell differentiation by CS exposure.
In contrast to the lungs of mice exposed to only neonatal or adult CS, we found a marked increase in the expression of several muscle contraction and erythrocyte genes in the lungs of mice exposed to both early and late postnatal CS. These findings suggest that a neonatal CS exposure can cause a stress resistance response leading to induction of muscle contraction and erythrocyte differentiation genes with re-exposure or cumulative exposure to CS. Stress resistance induced by low-dose exposures has been previously described [16]. The increased expression of myosin genes in our mice exposed to both neonatal and adult CS may indicate respiratory muscle remodeling, which in turn may be an adaptive response of the adult lung to a second CS exposure.
There are limitations to our study. The C57BL/6J mice developed only mild lung changes in response to CS exposures. These mild changes, however, allowed us to discern differences in lung structure and function between different CS exposures. Strain specificity may also determine the injury response to neonatal CS and/or adult CS exposure. If exposures had been performed in the CS-sensitive AKR/J mice, more pronounced changes in lung structure and function may have been appreciated. We also used both males and females in our analysis and gender differences may be a factor in COPD susceptibility.
In conclusion, our study demonstrated that CS exposure during neonatal life can cause durable changes in airspace structure and lung function in the adult lung but does not potentiate CS-induced COPD changes in later life. Down-regulation of Cornified envelope protein gene expression in all groups of mice exposed to CS may indicate an altered innate immune response and/or loss of epithelial cell differentiation from CS exposure, whereas increased expression of myosin and erythrocyte genes in mice exposed to both neonatal and adult CS may indicate an adaptive response to CS and respiratory muscle remodeling. The impact of neonatal CS exposure on gene expression and its influence on adult lung phenotype requires further elucidation but additional studies may be warranted to determine the utility of these genes as biomarkers of respiratory outcomes in adult lung.
Acknowledgments
This work was funded by Flight Attendant Medical Research Institute Clinical Innovator Award (S.M.), COPD SCCOR grant, P50HL084945, and the Grace Anne Dorney Fund. The authors thank Chris Cheadle for his help in analysis of the gene profiling data.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.de Boo HA, Harding JE. The developmental origins of adult disease (Barker) hypothesis. Aust N Z J Obstet Gynaecol. 2006;46:4–14. doi: 10.1111/j.1479-828X.2006.00506.x. [DOI] [PubMed] [Google Scholar]
- 2.Eriksson JG, Forsen TJ, Kajantie E, Osmond C, Barker DJ. Childhood growth and hypertension in later life. Hypertension. 2007;49:1415–1421. doi: 10.1161/HYPERTENSIONAHA.106.085597. [DOI] [PubMed] [Google Scholar]
- 3.Yliharsila H, Kajantie E, Osmond C, Forsen T, Barker DJ, Eriksson JG. Body mass index during childhood and adult body composition in men and women aged 56–70 y. Am J Clin Nutr. 2008;87:1769–1775. doi: 10.1093/ajcn/87.6.1769. [DOI] [PubMed] [Google Scholar]
- 4.Barker DJ, Godfrey KM, Fall C, Osmond C, Winter PD, Shaheen SO. Relation of birth weight and childhood respiratory infection to adult lung function and death from chronic obstructive airways disease. BMJ. 1991;303:671–675. doi: 10.1136/bmj.303.6804.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaheen SO, Barker DJ, Holgate ST. Do lower respiratory tract infections in early childhood cause chronic obstructive pulmonary disease? Am J Respir Crit Care Med. 1995;151:1649–1651. doi: 10.1164/ajrccm/151.5_Pt_1.1649. [DOI] [PubMed] [Google Scholar]
- 6.Gauderman WJ, Vora H, McConnell R, Berhane K, Gilliland F, Thomas D, Lurmann F, Avol E, Kunzli N, Jerrett M, Peters J. Effect of exposure to traffic on lung development from 10 to 18 years of age: a cohort study. Lancet. 2007;369:571–577. doi: 10.1016/S0140-6736(07)60037-3. [DOI] [PubMed] [Google Scholar]
- 7.Mannino DM, Kiriz VA. Changing the burden of COPD mortality. Int J Chron Obstruct Pulmon Dis. 2006;1:219–233. doi: 10.2147/copd.2006.1.3.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Viegi G, Maio S, Pistelli F, Baldacci S, Carrozzi L. Epidemiology of chronic obstructive pulmonary disease: health effects of air pollution. Respirology. 2006;11:523–532. doi: 10.1111/j.1440-1843.2006.00886.x. [DOI] [PubMed] [Google Scholar]
- 9.Bader D, Ramos AD, Lew CD, Platzker AC, Stabile MW, Keens TG. Childhood sequelae of infant lung disease: exercise and pulmonary function abnormalities after bronchopulmonary dysplasia. J Pediatr. 1987;110:693–699. doi: 10.1016/s0022-3476(87)80004-5. [DOI] [PubMed] [Google Scholar]
- 10.Smith LJ, van Asperen PP, McKay KO, Selvadurai H, Fitzgerald DA. Reduced exercise capacity in children born very preterm. Pediatrics. 2008;122:e287–e293. doi: 10.1542/peds.2007-3657. [DOI] [PubMed] [Google Scholar]
- 11.Bourbon JR, Boucherat O, Boczkowski J, Crestani B, Delacourt C. Bronchopulmonary dysplasia and emphysema: in search of common therapeutic targets. Trends Mol Med. 2009;15:169–179. doi: 10.1016/j.molmed.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 12.Kabesch M, Hoefler C, Carr D, Leupold W, Weiland SK, von Mutius E. Glutathione S-transferase deficiency and passive smoking increase childhood asthma. Thorax. 2004;59:569–573. doi: 10.1136/thx.2003.016667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hunninghake GM, Cho MH, Tesfaigzi Y, Soto-Quiros ME, Avila L, Lasky-Su J, Stidley C, Melen E, Soderhall C, Hallberg J, Kull I, Kere J, Svartengren M, Pershagen G, Wickman M, Lange C, Demeo DL, Hersh CP, Klanderman BJ, Raby BA, Sparrow D, Shapiro SD, Silverman EK, Litonjua AA, Weiss ST, Celedon JC. MMP12, lung function, and COPD in high-risk populations. N Engl J Med. 2009;361:2599–2608. doi: 10.1056/NEJMoa0904006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rangasamy T, Misra V, Zhen L, Tankersley CG, Tuder RM, Biswal S. Cigarette smoke-induced emphysema in A/J mice is associated with pulmonary oxidative stress, apoptosis of lung cells, and global alterations in gene expression. Am J Physiol Lung Cell Mol Physiol. 2009;296:L888–L900. doi: 10.1152/ajplung.90369.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Izzotti A, Larghero P, Longobardi M, Cartiglia C, Camoirano A, Steele VE, De Flora S. Dose-responsiveness and persistence of microRNA expression alterations induced by cigarette smoke in mouse lung. Mutat Res. 2010 doi: 10.1016/j.mrfmmm.2010.12.008. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 16.Gems D, Partridge L. Stress-response hormesis and aging: “that which does not kill us makes us stronger”. Cell Metab. 2008;7:200–203. doi: 10.1016/j.cmet.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 17.Clanton TL, Levine S. Respiratory muscle fiber remodeling in chronic hyperinflation: dysfunction or adaptation? J Appl Physiol. 2009;107:324–335. doi: 10.1152/japplphysiol.00173.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carlsen KH, Carlsen KC. Respiratory effects of tobacco smoking on infants and young children. Paediatr Respir Rev. 2008;9:11–19. doi: 10.1016/j.prrv.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 19.McGrath-Morrow S, Rangasamy T, Cho C, Susson T, Neptune E, Wise R, Tuder RM, Biswal S. Impaired Lung Homeostasis in Neonatal Mice Exposed to Cigarette Smoke. Am J Respir Cell Mol Biol. 2008;38(4):393–400. doi: 10.1165/rcmb.2007-0104OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ewart S, Levitt R, Mitzner W. Respiratory system mechanics in mice measured by end-inflation occlusion. J Appl Physiol. 1995;79:560–566. doi: 10.1152/jappl.1995.79.2.560. [DOI] [PubMed] [Google Scholar]
- 21.Soutiere SE, Mitzner W. On defining total lung capacity in the mouse. J Appl Physiol. 2004;96:1658–1664. doi: 10.1152/japplphysiol.01098.2003. [DOI] [PubMed] [Google Scholar]
- 22.Cheadle C, Cho-Chung YS, Becker KG, Vawter MP. Application of z-score transformation to Affymetrix data. Appl Bioinformatics. 2003;2:209–217. [PubMed] [Google Scholar]
- 23.Nadon R, Woody E, Shi P, Rghei N, Hubschle H, Susko E, Ramm P. Statistical inference in array genomics. In: Geschwind J, Gregg J, editors. Microarrays for the Neurosciences. Cambridge: MIT Press; 2002. pp. 109–140. [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Yee M, Chess PR, McGrath-Morrow SA, Wang Z, Gelein R, Zhou R, Dean DA, Notter RH, O'Reilly MA. Neonatal oxygen adversely affects lung function in adult mice without altering surfactant composition or activity. Am J Physiol Lung Cell Mol Physiol. 2009;297:L641–L649. doi: 10.1152/ajplung.00023.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gakhar L, Bartlett JA, Penterman J, Mizrachi D, Singh PK, Mallampalli RK, Ramaswamy S, McCray PB., Jr PLUNC is a novel airway surfactant protein with anti-biofilm activity. PLoS ONE. 2010;5:e9098. doi: 10.1371/journal.pone.0009098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bry K, Hogmalm A, Backstrom E. Mechanisms of inflammatory lung injury in the neonate: lessons from a transgenic mouse model of bronchopulmonary dysplasia. Semin Perinatol. 2010;34:211–221. doi: 10.1053/j.semperi.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 28.Ryan RM, Ahmed Q, Lakshminrusimha S. Inflammatory mediators in the immunobiology of bronchopulmonary dysplasia. Clin Rev Allergy Immunol. 2008;34:174–190. doi: 10.1007/s12016-007-8031-4. [DOI] [PubMed] [Google Scholar]
- 29.Bry K, Whitsett JA, Lappalainen U. IL-1beta disrupts postnatal lung morphogenesis in the mouse. Am J Respir Cell Mol Biol. 2007;36:32–42. doi: 10.1165/rcmb.2006-0116OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lukkarinen H, Hogmalm A, Lappalainen U, Bry K. Matrix metalloproteinase-9 deficiency worsens lung injury in a model of bronchopulmonary dysplasia. Am J Respir Cell Mol Biol. 2009;41:59–68. doi: 10.1165/rcmb.2008-0179OC. [DOI] [PubMed] [Google Scholar]
- 31.Harijith A, Choo-Wing R, Cataltepe S, Yasumatsu R, Aghai ZH, Janer J, Andersson S, Homer RJ, Bhandari V. A Role for MMP9 in IFN{gamma}-mediated injury in developing lungs: relevance to bronchopulmonary dysplasia. Am J Respir Cell Mol Biol. 2011;44(5):621–630. doi: 10.1165/rcmb.2010-0058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Paepe ME, Greco D, Mao Q. Angiogenesis-related gene expression profiling in ventilated preterm human lungs. Exp Lung Res. 2010;36:399–410. doi: 10.3109/01902141003714031. [DOI] [PubMed] [Google Scholar]
- 33.Shaykhiev R, Otaki F, Bonsu P, Dang DT, Teater M, Strulovici-Barel Y, Salit J, Harvey BG, Crystal RG. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell Mol Life Sci. 2011;68:877–892. doi: 10.1007/s00018-010-0500-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host-microbial relationships in the intestine. Science. 2001;291:881–884. doi: 10.1126/science.291.5505.881. [DOI] [PubMed] [Google Scholar]
- 35.Young MT, Tanner MJ. Distinct regions of human glycophorin A enhance human red cell anion exchanger (band 3; AE1) transport function and surface trafficking. J Biol Chem. 2003;278:32954–32961. doi: 10.1074/jbc.M302527200. [DOI] [PubMed] [Google Scholar]
- 36.Tajsharghi H, Darin N, Rekabdar E, Kyllerman M, Wahlstrom J, Martinsson T, Oldfors A. Mutations and sequence variation in the human myosin heavy chain IIa gene (MYH2) Eur J Hum Genet. 2005;13:617–622. doi: 10.1038/sj.ejhg.5201375. [DOI] [PubMed] [Google Scholar]
- 37.Wu ZX, Hunter DD, Kish VL, Benders KM, Batchelor TP, Dey RD. Prenatal and early, but not late, postnatal exposure of mice to sidestream tobacco smoke increases airway hyperresponsiveness later in life. Environ Health Perspect. 2009;117:1434–1440. doi: 10.1289/ehp.0800511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McGrath-Morrow SA, Lauer T, Collaco JM, Yee M, O'Reilly M, Mitzner W, Neptune E, Wise R, Biswal S. Neonatal hyperoxia contributes additively to cigarette smoke-induced COPD changes in adult mice. Am J Respir Cell Mol Biol. 2011 doi: 10.1165/rcmb.2010-0259OC. Epub head of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thatcher TH, McHugh NA, Egan RW, Chapman RW, Hey JA, Turner CK, Redonnet MR, Seweryniak KE, Sime PJ, Phipps RP. Role of CXCR2 in cigarette smoke-induced lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2005;289:L322–L328. doi: 10.1152/ajplung.00039.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stevenson CS, Coote K, Webster R, Johnston H, Atherton HC, Nicholls A, Giddings J, Sugar R, Jackson A, Press NJ, Brown Z, Butler K, Danahay H. Characterization of cigarette smoke-induced inflammatory and mucus hypersecretory changes in rat lung and the role of CXCR2 ligands in mediating this effect. Am J Physiol Lung Cell Mol Physiol. 2005;288:L514–L522. doi: 10.1152/ajplung.00317.2004. [DOI] [PubMed] [Google Scholar]
- 41.Nagarkar DR, Wang Q, Shim J, Zhao Y, Tsai WC, Lukacs NW, Sajjan U, Hershenson MB. CXCR2 is required for neutrophilic airway inflammation and hyperresponsiveness in a mouse model of human rhinovirus infection. J Immunol. 2009;183:6698–6707. doi: 10.4049/jimmunol.0900298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herbold W, Maus R, Hahn I, Ding N, Srivastava M, Christ-man JW, Mack M, Reutershan J, Briles DE, Paton JC, Winter C, Welte T, Maus UA. Importance of CXC chemokine receptor 2 in alveolar neutrophil and exudate macrophage recruitment in response to pneumococcal lung infection. Infect Immun. 2010;78(6):2620–2630. doi: 10.1128/IAI.01169-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luthra MG, Ajani JA, Izzo J, Ensor J, Wu TT, Rashid A, Zhang L, Phan A, Fukami N, Luthra R. Decreased expression of gene cluster at chromosome 1q21 defines molecular subgroups of chemoradiotherapy response in esophageal cancers. Clin Cancer Res. 2007;13:912–919. doi: 10.1158/1078-0432.CCR-06-1577. [DOI] [PubMed] [Google Scholar]
- 44.Maru DM, Luthra R, Correa AM, White-Cross J, Anandasabapathy S, Krishnan S, Guha S, Komaki R, Swisher SG, Ajani JA, Hofstetter WL, Rashid A. Frequent loss of heterozygosity of chromosome 1q in esophageal adenocarcinoma: loss of chromosome 1q21.3 is associated with shorter overall survival. Cancer. 2009;115:1576–1585. doi: 10.1002/cncr.24122. [DOI] [PubMed] [Google Scholar]
- 45.Blanchard C, Stucke EM, Burwinkel K, Caldwell JM, Collins MH, Ahrens A, Buckmeier BK, Jameson SC, Greenberg A, Kaul A, Franciosi JP, Kushner JP, Martin LJ, Putnam PE, Abonia JP, Wells SI, Rothenberg ME. Coordinate interaction between IL-13 and epithelial differentiation cluster genes in eosinophilic esophagitis. J Immunol. 2010;184:4033–4041. doi: 10.4049/jimmunol.0903069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tong L, Corrales RM, Chen Z, Villarreal AL, De Paiva CS, Beuerman R, Li DQ, Pflugfelder SC. Expression and regulation of Cornified envelope proteins in human corneal epithelium. Invest Ophthalmol Vis Sci. 2006;47:1938–1946. doi: 10.1167/iovs.05-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]