

Abstract
Despite their equal stoichiometry in spliceosomes, U1 snRNP (U1) is typically the most abundant snRNP in vertebrates. What regulates U1 over-abundance and snRNP repertoire in general is unknown. Sm core assembly is a key step in snRNP biogenesis mediated by the SMN complex. All pre-snRNAs are delivered by the snRNA-specific RNA-binding protein (RBP) Gemin5 to join SMN-Gemin2-recruited Sm proteins. Here, we find that the U1-specific RBP U1-70K bridges pre-U1 to SMN-Gemin2-Sm, establishing an additional, Gemin5-independent Sm core assembly pathway. We show that U1-70K hijacks SMN-Gemin2-Sm, enhancing U1’s and inhibiting other snRNAs’ Sm core assembly, thereby promoting U1 over-abundance and regulating snRNP repertoire. Ubiquitous SMN-Gemin2’s surprising ability to facilitate transactions between different RBPs and RNAs explains its puzzling multi-RBP valency and myriad transcriptome perturbations associated with SMN’s deficiency in neurodegenerative spinal muscular atrophy. We propose that SMN-Gemin2 is a versatile RNP exchange that functions broadly in RNA metabolism.
INTRODUCTION
U1 snRNP (U1) is needed in eukaryotes both for pre-mRNA splicing1, 2 and telescripting, suppression of premature cleavage and polyadenylation that ensures full-length transcription3–7. U1’s additional function in telescripting likely explains why human cells have a great U1 abundance compared to the other spliceosomal snRNPs (U2, U4, U5 and U6)8. All the spliceosomal snRNAs that acquire Sm cores (U1, U2, U4, U5 and the minor spliceosome’s U11, U12, U4atac) have an snRNA-defining motif, the snRNP code, consisting of the Sm site (AU5–6G), around which the Sm core forms, and an adjacent 3′ terminal stem-loop9–11. In Sm cores, Sm hetero-dimers Sm D1D2 (D1D2) and Sm BD3 (BD3) and -trimer Sm FEG (FEG) subunits are arranged in order of D1, D2, F, E, G, D3 and B, and each Sm protein interacts with a single nucleotide of the Sm site12–17. The snRNP code [~50 nucleotides (nt)] is recognized by the RBP Gemin518–20. Sm core assembly is mediated by the SMN (Survival Motor Neurons)-Gemins complex, a large oligomeric complex (30–70S) comprised of SMN, Gemins2–8 and Unrip9,21,22. The SMN-Gemins complex is ubiquitously expressed in eukaryotes (except Saccharomyces, which have a Gemin2 ortholog but no detectable SMN) and is found both in the nucleus and in the cytoplasm. SMN and its interacting protein Gemin2, the two most highly conserved SMN-Gemins complex’s components, bind Sm proteins23–25. Pre-snRNAs, including pre-U1 snRNA, are delivered to SMN-Gemin2 by Gemin5, where they encounter the Sm protein and acquire Sm cores20. Sm core assembly is a pre-requisite for the snRNP maturation, including the 5′-monomethyl guanosine cap hypermethylation (to form a trimethylguanosine), its 3′-end processing, and for snRNPs’ functions26,27. U1 in vertebrates differs from other snRNAs in two respects: it has a divergent snRNP code due to its non-canonical Sm site (AUUUGUG or AUUUCUG) that has weaker binding affinity to Gemin519, and its Sm core assembly strongly depends on stem-loop 1 (SL1), a U1-specific structure outside the snRNP code28–30. Interestingly, earlier studies have shown that SL1 alone can inhibit Sm core assembly30; however, the role of SL1 and U1’s divergent snRNP code in Sm core assembly remain unknown. Here we investigated the roles of these U1 elements in Sm core assembly aiming to understand how U1 overabundance is achieved and more generally what factors contribute to cells’ unequal snRNP repertoire.
RESULTS
U1-specific U1-70K protein bridges U1 snRNA to the SMN complex
To understand the role of U1’s SL1 and snRNP code in Sm core assembly, we compared the binding of SMN complex components (SMN, Gemin2–8 and Unrip) to biotinylated pre-U1, mature U1, U1A3 (a U1 SL1 mutant defective in the U1-snRNP specific U1-70K binding)28, U1ΔSm (a U1 Sm site mutant that cannot assemble Sm core), super-U1 (a chimeric U1 in which the Sm site was replaced by a canonical Sm site, AUUUUUG), U4 snRNA (U4), a canonical snRNP code containing snRNA, and U4ΔSm. The RNAs were incubated in human (HeLa) cell extracts in which either SMN, Gemin5 or U1-70K had been knocked down by RNAi (Fig. 1a, input). Although U1-70K has not been previously suggested to have a role in Sm core assembly, we tested its effect because it is the only protein known to bind SL128,29. For most experiments we used pre-U1, which is 50nt longer at the 3′-end than U1, because it represents the U1 substrate for Sm core assembly in cells20. For U4, whose precursor is only 6nt longer than mature U4, there is little difference between the precursor and mature forms (data not shown).
Figure 1. The U1-snRNP-specific stem-loop 1 binding protein U1-70K bridges pre-U1 or U1 snRNA to the SMN complex independent of Gemin5.
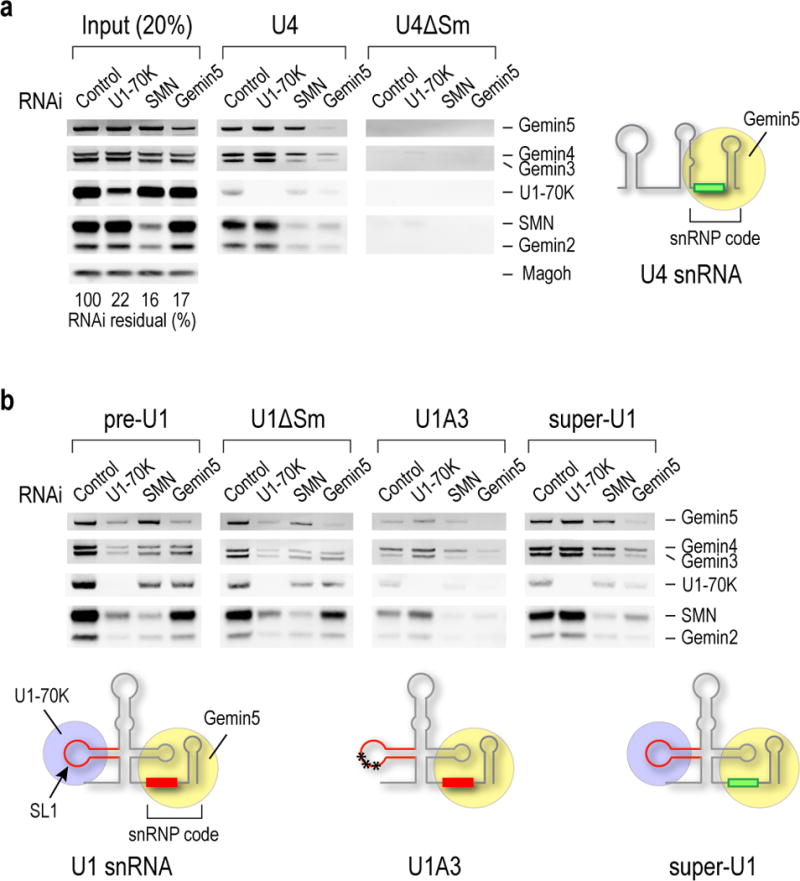
(a) Western blot analysis of SMN complex components bound to biotinylated U4 and U4ΔSm snRNAs in HeLa cells from control, U1-70K, SMN or Gemin5 siRNA knockdowns. The input lanes show 20% of each of the cell extracts used. The knockdowns’ efficiencies relative to Magoh as loading control are indicated as % of residual protein for each knockdown compared to control (100%). A schematic illustration of U4 structure and its canonical Sm site in snRNP code, which is the RNA sequence necessary and sufficient for Gemin5 binding, are indicated. (b) The same western blot analysis as in a for biotinylated U1, including U1 precursor (pre-U1), U1ΔSm, SL1 mutant (U1A3) and super-U1. A U30C) in the SL1, which abolishes U1-70K binding. Super-U1 is a mutation that replaces U1’s Sm site with a canonical Sm site. Uncropped scans of western blots are shown in Supplementary Data Set 1.
As expected, Gemin5 knockdown abolished U4 association with the SMN complex18, particularly with SMN-Gemin2, while SMN knockdown had no effect on U4 binding to Gemin5 (Fig. 1a, U4). U4ΔSm did not bind Gemin5 or any component of the SMN complex (Fig. 1a, U4 ΔSm).
In contrast, the association of pre-U1, U1 (data not shown) or U1ΔSm with SMN-Gemin2 was only partially decreased by Gemin5 knockdown, but was strongly decreased by U1-70K knockdown (Fig. 1b). However, U1-70K knockdown had no effect on U4 association with the SMN complex (Fig. 1a, U4). U1A3 binding to the SMN complex was unaffected by U1-70K knockdown, but was reduced by Gemin5 knockdown, suggesting that U1A3 can associate with the SMN complex through Gemin5, albeit more weakly than U4 due to U1’s non-canonical Sm site (Fig. 1b, U1A3). Super-U1 binding to the SMN complex was unaffected by U1-70K knockdown but was strongly decreased by Gemin5 knockdown (Fig. 1b, super-U1). Thus, U1-70K can bridge pre-U1 and U1 to the SMN complex apparently independent of and in addition to Gemin5, while the binding of U4 to the SMN complex strictly depends on Gemin5. These data demonstrate that while pre-U1 can use either U1-70K or Gemin5, U1-70K is normally the main adaptor to SMN-Gemin2-Sm proteins. These observations also suggest that U1’s Sm site may have diverged to become a weaker Gemin5 binder because this allowed U1 to use an alternate route to bind the SMN complex.
U1-70K enhances U1’s and inhibits other snRNPs’ Sm core assembly
Next, we used a quantitative snRNP assembly assay31,32 to determine the role of U1-70K and Gemin5 in Sm core assembly. (Fig. 2a and Supplementary Data Fig. 1). As expected, Sm core assembly on all tested RNAs depended on SMN-Gemin2 and required an Sm site. Gemin5 knockdown decreased Sm core assembly of canonical snRNP code-containing snRNAs (U2, U4 and U5) and to a lesser extent on SL1-containing snRNAs. Strikingly, U1-70K knockdown decreased pre-U1 assembly (by 50%), but enhanced U2, U4 and U5 Sm core assembly (up to 100%). Loss of U1-70K binding or enhancement of Gemin5 binding in U1A3 and super-U1, respectively, made them more similar to non-U1 snRNAs. These observations indicate a new role for U1-70K as a specific pre-U1 Sm core assembly factor and an inhibitor of Sm core assembly on other snRNAs.
Figure 2. U1-70K enhances U1 Sm core assembly, inhibits other snRNAs’ Sm core assembly in vitro, and regulates snRNP repertoire in cells.
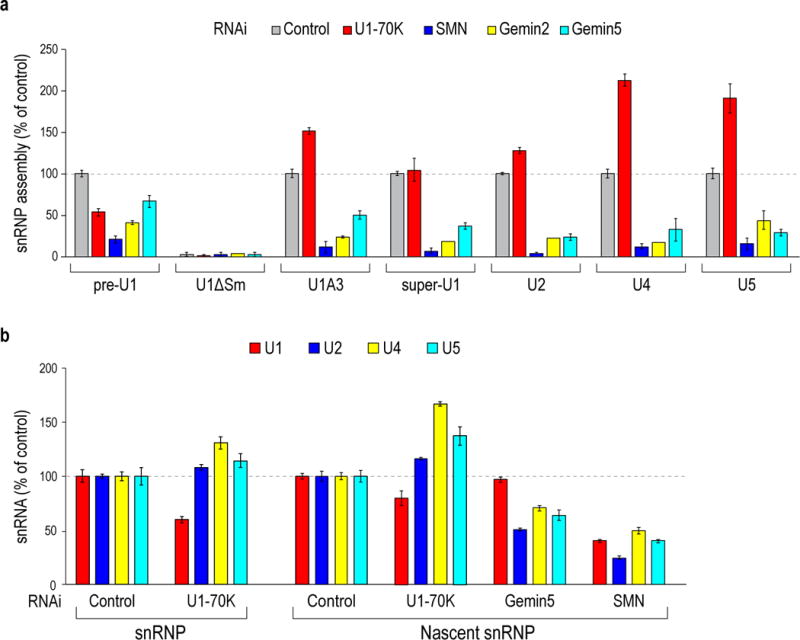
(a) Quantitative in vitro Sm core assembly activities on the indicated snRNAs in extracts from cells with control, U1-70K, SMN, Gemin2 or Gemin5 siRNA knockdowns. The Sm core assembly activities on each snRNA were compared to control RNAi extracts (100% activity), except U1ΔSm and U4ΔSm, whose relative activities were compared to pre-U1 and U4, respectively. The error bars represent standard deviation from three independent biological replicates. (b) Quantitative measurements of snRNAs from the mature and nascent snRNPs in cells with control, U1-70K, SMN or Gemin5 knockdown by real-time RT-PCR. The relative amount of snRNAs compared to control RNAi are shown. The error bars represent standard deviation from three independent biological replicates.
U1-70K regulates cells’ snRNP repertoire
We then studied the effect of U1-70K knockdown on U1 snRNP abundance in cells. U1-70K RNAi decreased steady-state U1 by 25–40% and increased other snRNPs by 10–30% (Fig. 2b, snRNP). To determine the effect on the rate of Sm core assembly, we quantitated the amount of snRNAs that assembled Sm cores in a two-hour pulse-label with 4-thiouridine33. This showed that U1-70K knockdown decreased U1 snRNP production and increased formation of other snRNPs (Fig. 2b, nascent snRNP). U1-70K knockdown had no effect on SMN or Sm proteins levels (Supplementary Data Fig. 2). In contrast to U1-70K, Gemin5 knockdown had no effect on U1’s but decreased other snRNPs’ assembly. These findings are consistent with the in vitro Sm core assembly and demonstrate that U1-70K is required for generating U1 over-abundance compared to the other snRNPs and regulates cells’ snRNP repertoire.
U1-70K domains bind SMN and mediate Sm core assembly
Delineation of U1-70K’s domains showed that U1-70K’s C-terminal arginine- and serine-rich (RS) domain (aa230–437), which is important for splicing34, was not required for bridging pre-U1 to the SMN complex (data not shown). However, recombinant U1-70K’s N-terminal 194 amino acids (N194) restored the SMN complex’s association with pre-U1 in U1-70K knockdown cell extracts (Fig. 3a). Furthermore, N194 bound SMN directly and enhanced pre-U1- and inhibited U4-Sm core assembly (Fig. 3b, c). The smallest N194 fragment that was necessary and sufficient to bind SMN was N90-194, which contains U1-70K’s SL1-specific RNA-binding domain (RBD/RRM; aa102–181)29, while the N-terminal domain lacking the RBD, N99 (aa1–99) did not bind SMN (Fig. 3c). Moreover, pre-U1 enhanced N194 binding to SMN (2.1 fold) (Fig. 3d). Thus, pre-U1 binding creates additional interactions between N194 and SMN, either by an RNA-mediated allosteric effect that increases U1-70K interaction surface with SMN or by additional binding interactions between pre-U1 with SMN. The ability of the same type of RBD as U1-70K’s to engage in protein interactions through a surface other than the RNA binding site has been described for other RBPs35,36.
Figure. 3. U1-70K bridges pre-U1 snRNA to SMN and mediates preferential Sm core assembly on U1 snRNA over other snRNAs.
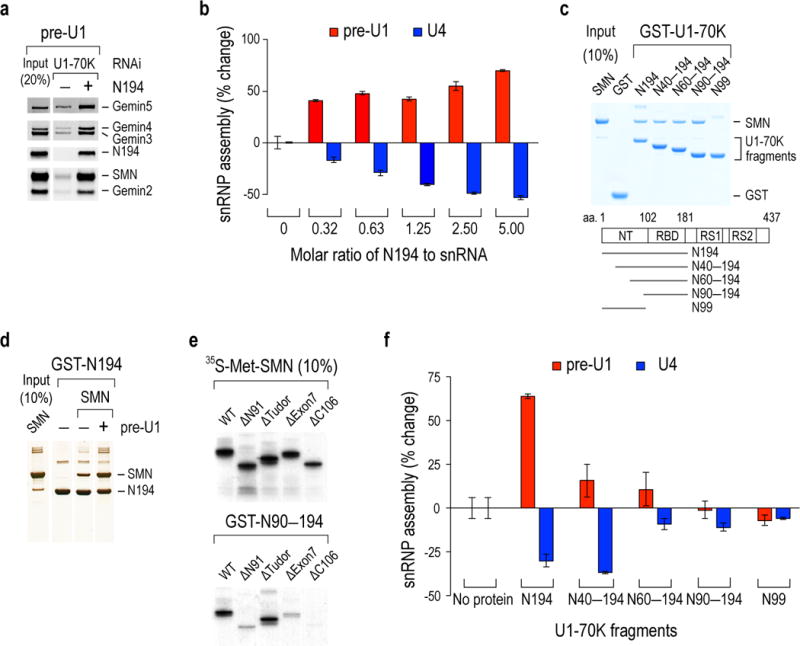
(a) Western blot of SMN complex proteins bound to the biotinylated pre-U1 snRNA in U1-70K knockdown cell extracts complemented with recombinant U1-70K N194. The input lane shows 20% of the cell extracts and the N194 used for binding. (b) In vitro Sm core assembly activities on pre-U1 and U4 with increasing concentrations of N194 in U1-70K knockdown extracts. The rescued snRNP assembly capacities on snRNAs were determined by % changes from the U1-70K knockdown without N194. The error bars represent standard deviation from three independent biological replicates. (c) Binding of GST-U1-70K proteins to recombinant SMN protein. Schematic diagram of U1-70K and its deletion fragments with corresponding residue numbers are indicated. The input lane shows 10% of the SMN protein used for binding and the gel was visualized by Simplyblue staining. (d) Binding of GST-N194 to SMN in the absences or presence of equimolar amount of in vitro transcribed pre-U1. The protein gel was visualized by silver staining. (e) Binding of GST-N90–N194 protein to in vitro-translated [35S]-methionine labeled wild-type (WT) SMN and its domain deletions. The input lanes show 10% of SMN’s deletion domains (N91, aa1–91; Tudor, aa92–158; Exon7, aa279–294; C106, aa189–294) and the gel was visualized by autoradiography. (f) snRNP assembly measurements as in b with N194 deletion fragments using 1.25 molar ratio of U1-70K proteins to snRNA. The error bars represent standard deviation from three independent biological replicates. Uncropped scans of blots, gels and autoradiographs are shown in Supplementary Data Set 1.
N90–194 binding to SMN was abrogated by deletion of SMN’s C-terminal YG box, an oligomerization domain on which many of SMN’s interactions and function depend37–39 (Fig. 3e). This included SMNΔ7, the major SMN isoform expressed in SMA patients. SMNΔ7 lacks exon7-encoded 16 amino acids that constitute much of the YG box. Deletion of SMN exons 1, 2a and 2b (ΔN91) also impaired U1-70K binding, suggesting that a peptide including exon 2b-encoded domain that engages in homotypic interactions in oligomeric SMN31, 40, is also needed. A more detailed definition of SMN’s N90–194 binding site requires additional structural information about oligomeric SMN, which is presently unknown.
Unlike N194, N90–194 did not enhance pre-U1- or inhibit U4-Sm core assembly (Fig. 3f), suggesting that peptide(s) within U1-70K’s aa1–89 are needed for these activities. Recently determined X-ray crystal structures of mature U1 snRNP showed that U1-70K aa39–58 and aa10–31 contact the Sm core’s perimeter at two apposing sides, at D2F and BD3, respectively12,14,17. Deletion of aa1–39 (N40–194) caused loss of pre-U1 Sm core assembly enhancement, but did not diminish Sm core assembly inhibition on U4 (Fig. 3f). However, a further deletion of aa40–59 (N60–194) caused loss of U4 Sm core assembly inhibition. Thus, U1-70K’s snRNA-selective effect on Sm core assembly represents two separable activities: N40–194 is necessary and sufficient for U4 Sm core inhibition, but pre-U1 Sm core assembly enhancement requires in addition to aa1–39. N99, which lacks the RBD had no effect (Fig. 3f). This suggests that the RBD, particularly when bound to pre-U1, blocks other snRNAs’ access to Sm proteins.
U1-70K and Gemin2 cooperate to recruit all Sm proteins
N99 directly bound BD3, but not D1D2, or FEG (Fig. 4a, lanes 1–7). In contrast, and as shown previously, Gemin2 did not bind BD3 nor did it bind D1D2 or FEG alone, however, it formed a stable complex with Sm pentamer D1D2-FEG (Sm5)24,25. SMN bound Gemin2-Sm5 is a key intermediate in Sm core assembly, and we therefore investigated if Gemin2 and U1-70K could collaborate in binding Sm. This showed that N99 did not bind Gemin2 (Fig. 4a, lane 8), however complexes containing N99, Gemin2 and Sm5 or all 7 Sm proteins (Sm7) readily formed (Fig. 4a, lanes 12 and 15). The Sm7-containing complex would not be expected to form a stable Sm ring without an RNA’s Sm site15. Thus, U1-70K and Gemin2 can cooperate to recruit all of the Sm core’s proteins, which neither alone could achieve.
Figure. 4. SMN-Gemin2 cooperates with U1-70K in Sm protein recruitment.
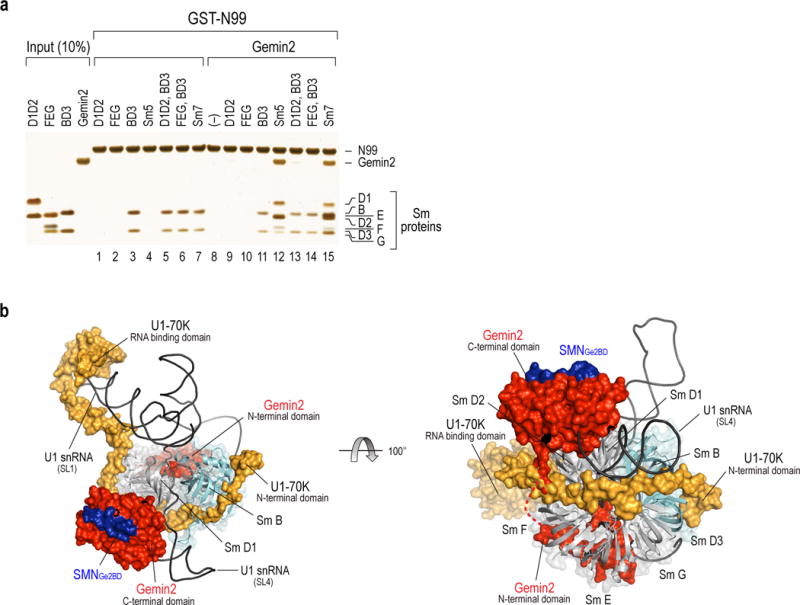
(a) In vitro binding of GST-U1-70K N99 to Gemin2 and Sm proteins. Each Sm protein subunit, combinations of the subunits (lanes 1–7), Gemin2 only (lane 8) or Gemin2 with combinations of Sm protein subunit (lanes 9–15) are indicated. The input panel shows 10% of the proteins used for binding and the gel was visualized by silver staining. Uncropped images are shown in Supplementary Data Set 1. (b) Superimposed crystal structures of U1 snRNP12,14,17 and SMN-Gemin2-Sm525, showing cooperative roles in Sm core assembly intermediate of U1. Left, This Gemin2-Sm5-U1-70K intermediate contacts with pre-U1 bound by U1-70K’s SL1 RBD, places pre-U1’s Sm site at the Sm5’s inner ring and prevents binding of other RNAs. U1-70K’s N-terminal domain binds directly to BD3, which helps in their recruitment and proper positioning for Sm ring closure. For clarity, two other U1 snRNP specific proteins, U1A, which binds to SL2, and U1C, which binds to U1-70K and U1 snRNA, are not shown. Right, A rotated view compared with left, showing Gemin2 and U1-70K assist Sm protein recruitment. Gemin2 binds D1D2 through its C-terminal domain and FEG via its N-terminal domain. The loop connecting these two domains shown in a dashed line is disordered and its length (aa70–82) is likely sufficient to avoid a clash with U1-70K’s D2F binding. Two patches (aa10–31 and aa39–59) of N-terminal U1-70K bind at the BD3 and D2F interface, respectively. The Figures were prepared with PyMOL (http://www.pymol.org/).
These findings are congruent with recent crystal structures of SMN-Gemin2-Sm5 and U1 snRNP12,14,17,25. Superimposition of the two structures supports the presence of an Sm core assembly intermediate in which Sm5 is bound simultaneously by Gemin2 and U1-70K (Fig. 4b). It illustrates how U1-70K binds at the junction of D1D2 and FEG, position pre-U1’s Sm site at the inner RNA-binding cleft, where N39’s (aa1–39) interactions with pre-U1 likely help position pre-U1’s Sm site in the Sm ring’s hole and recruit BD3 for Sm core closure12,14,17. U1-70K’s multiple interactions with SMN-Gemin2-Sm5, which may be initiated by pre-U1 binding, effectively hijack the Sm core assembly module, promoting U1 assembly and disadvantaging other snRNAs. SMN plays a key role by serving as a scaffold for Gemin2, U1-70K, and Sm B, D3 and D1’s C-terminal arginine- and glycine-rich [RG(G)] domains41–43, described below.
DISCUSSION
Previous studies had suggested that the SMN complex is a dedicated Sm core assembly device that operates with a single RBP, Gemin5, which binds a single RNA structure, the snRNP code, and joins the SMN-Gemin2 subunit20,25. However, our studies demonstrate that SMN-Gemin2 can receive RNAs from at least one additional and structurally unrelated RBP (U1-70K) that has a different RNA-binding specificity (SL1). In Gemin5’s case there is a complete exchange between the RBP donor and the acceptor RBPs, Sm5. In contrast, U1-70K remains part of the mature RNP. Yet both paths use SMN-Gemin2, revealing SMN-Gemin2 as a versatile platform for RBP-RNA transactions. SMN-Gemin2 RNP exchange function leverages the donor’s RNA specificity to produce a specific RNP, in this case Sm core that the Sm proteins on their own could not44. We refer to this function as RNP exchange and propose that SMN-Gemin2’s RNP exchange function provides a unifying theme that extends beyond Sm core assembly to play a central role in RNA metabolism. Our concept of SMN-Gemin2 as a general RNP exchange, represented in Fig. 5, rests on its ability to bind many RBPs and RNAs, which increases the opportunity for creating a greater RNP diversity, and explains many previous observations.
Figure. 5. Schematic representation of SMN-Gemin2’s function as a versatile hub for RNP exchange.
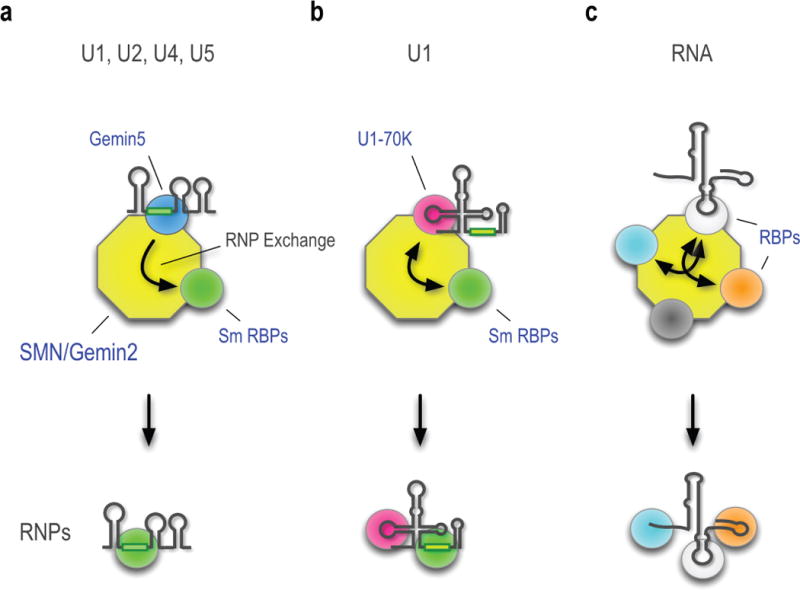
The different colored spheres represent the diverse RBPs that bind multivalent SMN-Gemin2. For simplicity, only the pre-snRNAs’ key features relevant to this pathway are shown. (a) Gemin5, which recognizes the snRNP code common to all pre-snRNAs, is a drop-and-go donor that does not remain with the fully assembled Sm core. (b) U1-70K, a pre-U1’s stem-loop 1 (SL1) binding protein associates with the SMN-Gemin2 and remains part of the completed Sm core. (c) A representative and hypothetical RNP, exemplified by FUS/TLS, associates with the SMN-Gemin2 as described in the text.
SMN and Gemin2, the SMN complex’s most highly conserved components, are ubiquitously expressed and essential for cell viability across eukaryotes21. Several features of SMN-Gemin2 make it particularly suitable for RNP exchange function. SMN is a highly oligomeric protein, making the SMN complex a large particle (30–70S), that contains Gemins 3–8 and unrip, including the putative RNA or RNP ATPase (Gemin3), whose precise functions are unknown, but likely assist RNP exchange. SMN has a relatively indiscriminate Tudor domain that binds methylarginine-modified peptides, a common post-translational modification in RG(G) domains, found in numerous RBPs41–43. Oligomeric SMN-Gemin2’s pan-RBP binding Tudor domains is thus a multivalent station that can congregate diverse RBPs and their RNA cargos thereby promoting RNP exchange. Indeed, a bewildering number of RBPs in addition to Sm and Lsm proteins bind the SMN-Gemins complex, including hnRNP proteins (e.g., hnRNP A1, A2, Q, R, U, FUS/TLS), snoRNP proteins (e.g., fibrillarin, GAR1), the SRP component SRP54, and the transcription regulator CARM141,44–47. However, the diverse RBPs that interact with the SMN complex could not be explained in terms of Sm core assembly. In addition, SMN can bind RNAs, albeit with low affinity and non-specifically48, which would increase its potential to maintain transient association with RNA intermediates. Therefore, a common theme is that SMN-Gemin2 could facilitate formation of diverse RNPs from a multitude of RBP donors and acceptors.
U1-70K’s unexpected role in Sm core assembly provides pre-U1 an exclusive path to build a great U1 abundance, allowing U1 level to be regulated separately from the other snRNPs. More generally, U1-70K emerged as a key regulator of snRNP repertoire. Other factors, including the number of snRNA genes, their transcriptional activity, and snRNPs’ stability undoubtedly also contribute to U1 abundance and snRNP repertoire. In humans, there are many (>100) U1 genes, pseudogenes and variants49 and many genes encoding other snRNAs, but they remain poorly annotated and little is known about their transcriptional activity. However, pre-snRNAs lacking Sm cores are rapidly degraded50, suggesting that the SMN complex could be a rate-limiting step in snRNP biogenesis. This makes the SMN complex and U1-70K’s ability to program its Sm core selectivity especially important factors in gene regulation.
SMN deficiency causes SMA, whose severity correlates both with the degree of SMN deficiency and a corresponding decrease in Sm core assembly32,51. However, SMA is associated with a large number of widespread perturbations in RNA metabolism, including alterations in snRNP repertoire and pre-mRNA splicing51–55. Among these are specific alternative splicing deficits that precede and can now explain important aspects of SMA’s synapto-pathology55. While splicing changes are a plausible outcome of snRNP changes, other transcriptome perturbations in SMN deficient cells, such as expression level changes, are more readily explained as resulting directly from the loss of the SMN complex’s RNP exchange capacity. Namely, that the SMN complex’s multiple interactions with RBPs indeed reflect its role as a versatile hub for chaperoning RNPs involved in diverse transcriptional and post-transcriptional processes. This perspective offers an important insight into the role of RBP mutations in other human diseases. For example, the SMN complex has recently intersected with several major neurodegenerative diseases caused by specific mutations in RBPs, FUS/TLS and TDP-43, which cause amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD)45,56–58. These pathogenic mutants also elicit many transcription, splicing and microRNA changes. Notably, FUS/TLS (and TDP-43 via FUS/TLS) binds SMN’s Tudor domain, mediated by methylated arginines in its RG(G) domain, where a cluster of pathogenic mutations are found. Impairment of the SMN-Gemins complex’s general RNP exchange function due to poisoning by mutant RBPs, small molecule inhibitors or genetic SMN deficiency provides an alternative explanation for the myriad RNA metabolism perturbations in ALS and SMA.
METHODS
Cell culture, RNA interference and preparation of cell extracts
HeLa PV cells were grown in DMEM supplemented with 10% FBS, (L)-glutamine, penicillin and streptomycin. Cells lines were tested for mycoplasma contamination. Transfection of control siRNA or siRNAs targeting U1-70K or the SMN complex components (Dharmacon, GE healthcare) into HeLa cells was performed using Lipofectamine RNAiMax (Invitrogen) as described by the manufacturer. After 40–48 hr transfection, cells were harvested and lysated to obtain cytoplasmic extracts for in vitro assays as previously described32. Total protein concentrations of various extracts were determined by the Bradford protein assay (Biorad). For nascent snRNP measurements, knockdown cells were metabolically labeled with 200 μM of 4-thiouridine (4-shU) for the last 2 hr of siRNA transfection33.
In vitro transcription and labeling of RNAs
All snRNAs used for in vitro assays were prepared by standard in vitro transcription using MEGAshortscript T7 transcription kit (Ambion) in the presence of 0.65 mM biotin-16-UTP (Roche) and 2.6 mM UTP32. Transcribed RNAs were purified by electrophoresis on 5M Urea-6% polyacrylamide gel, precipitated with ethanol and resuspended in RNase-free water. The concentrations of the labeled snRNAs were determined by UV absorbance at 260 nm.
In vitro transcription and translation
In vitro transcription and translation reactions were performed with each plasmid in the presence of [35S]-methionine using the T7 TNT Quick Coupled Transcription/Translation systems (Promega), as previously described25.
Antibodies, western blotting and immunoprecipitation
The following antibodies were used in this study; α-SMN (2B1 or 8/SMN from BD transaction laboratory), α-Gemin2 (2E17 or 3F8 from Santa Cruz biotechnology), α-Gemin3 (12H12), α-Gemin4 (64I1), α-Gemin5 (10G11), α-Sm (Y12), α-glutathione S-transferase (26H1 from Cell Signaling Technology), α-U1-70K (H111 from Synaptic Systems), α-FXR1 (6BG10) and α-Magoh (21B12). Immunoprecipitation was performed with Y12-immobilized onto magnetic Dynabeads protein G (Invitrogen) incubation in total HeLa cell extracts prepared in RSB-100 buffer (10 mM Tris-HCl, pH 7.8, 100 mM NaCl, 2.5 mM MgCl2) containing 0.1% NP-40 and protease inhibitors for 1.5 hr at 4°C. Supernatants were discarded and the beads were washed with RSB-500 containing 0.1% NP-40 five times. The beads were equilibrated with RSB-150 containing 0.02% NP-40 and collected for further experiments.
Plasmid construction, recombinant proteins expression and purification
cDNA plasmids of human U1-70K were constructed in pET42a vector (Novagen) with N-terminal GST and C-terminal His(6X) tags. Procedure for recombinant protein expression and purification were modified from a previous study6. Proteins were affinity purified using Ni-NTA beads and eluted by 500 mM imidazole in lysis buffer. Eluted proteins were further purified by glutathione beads (GE healthcare), heparin column and size exclusion chromatography. Recombinant Gemin2 and Sm proteins were expressed in E. coli and purified as previously described25. For efficient protein expression, Sm BD3 do not contain C-terminal RG(G) domains. The recombinant protein concentrations were determined by the Bradford protein assay (Biorad).
In vitro RNA pull-down
Cytoplasmic extracts (2 mg/mL) from HeLa cells with individual knockdown were incubated with biotinylated snRNAs (10 nM) in the reconstitution buffer (20 mM HEPES, pH 7.9, 50 mM KCl, 5 mM MgCl2, 0.2 mM EDTA, 0.25 mg/mL yeast tRNAs (Sigma), protease inhibitor and 0.2 U/μL RNasin RNase inhibitor (Promega)) in 96-well plates (20 μL/well). Binding experiments were performed in cytoplasmic cell extracts at 4°C without addition of ATP to avoid Sm core assembly, which would occlude the Sm site. All reactions were carried out with gentle mixing at 750 rpm using the Thermomixer (Eppendorf) for 1 hr and RNA-protein complexes were captured by the M-280 streptavidin Dynabeads (Invitrogen) in 100 μL of RSB-150 buffer containing 0.02% Triton X-100, protease inhibitor and 0.2 U/μL RNase inhibitor for an additional hour. The beads were washed in RSB-200 buffer containing 0.02% Triton X-100 five times using the Kingfisher 96 magnetic particle processor (ThermoFischer Scientific), as previously described31,32. Bound proteins on the beads were eluted by boiling in 10 μL of 1X sample buffer, resolved by SDS-PAGE and detected by western blotting. Uncropped scans of western blots are provided in Supplementary Data Set 1.
In vitro snRNP assembly
A high throughput and quantitative assay for in vitro Sm core assembly was performed as previously described31,32. In brief, cytoplasmic extracts (2 mg/mL) from HeLa cells were incubated with biotinylated snRNAs (10 nM) in the reconstitution buffer containing 2.5 mM ATP for 1 hr at 30°C. In complementation assays, 6.25–100 ng of recombinant U1-70K proteins were pre-incubated in the cytoplasmic extracts with U1-70K knockdown for 20 min at 30°C, and Sm core assembly reactions were initiated by addition of biotinylated snRNAs. Assembled snRNPs were captured by Y12-immobilized protein G magnetic beads in 100 μL of RSB-500 buffer containing 0.1% NP-40, protease inhibitor and 0.2 U/μL RNase inhibitor for an additional hour at 30°C. The beads were washed in RSB-500 buffer containing 0.1% NP-40 and 1 mg/mL heparin three times, then washed in the same buffer without heparin two times using the magnetic particle processor. The Y12-immunoprecipiated snRNPs on the beads were resuspended in 120 μL of RSB-150 buffer containing 0.02% NP-40 and 0.08 μg/mL horseradish peroxidase-conjugated NeutrAvidin (Pierce) and gently mixed for 1 hr at 30°C. The beads were washed again with the same washing buffer (RSB-500/0.1% NP-40) five times and resuspended in 150 μL of SuperSignal ELISA Femto substrate (Pierce) mixture. Chemiluminescence signals were detected at 495 nm using the Wallac Victor2 plate reader (Perkin-Elmer).
In vitro protein binding
Recombinant GST-U1-70K proteins (2.5 μg) were immobilized on the glutathione-Sepharose beads (Invitrogen) in 500 μL RSB-200 buffer containing 0.02% Triton X-100. After washing unbound proteins, 5 μg of recombinant or in vitro-translated [35S]- methionine-labeled SMN proteins were incubated for 1 hr at 4 °C and then washed with the binding buffer four times. The bound proteins were eluted by boiling in 1X sample buffer and resolved by SDS-PAGE and visualized by SimplyBlue staining (Invitrogen), silver staining, or autoradiography. The band intensities were analyzed using Bio-Rad Quantity One software. Binding experiments were performed side-by-side with the same protein preparations and run on the same gel. Uncropped images of gels, autoradiographs or western blots are provided in Supplementary Data Set 1.
Reverse transcription and RT-qPCR measurement of snRNAs
Total snRNAs were isolated using TRIzol (Invitrogen) as specified by the manufacturer. Steady-state snRNPs from HeLa cells were isolated by Y12 antibody immunoprecipitation and protease K digestion followed by phenol-chloroform extraction and ethanol precipitation. Nascent snRNPs were obtained by the same Y12 immunoprecipitation from cells that were metabolically labeled with 4-shU33. After isolation of steady-state snRNPs, free thiol groups of uridine in snRNA were biotinylated with 0.2 mg/ml of biotin-HPDP (Pierce) and captured by Dynabeads MyOne Streptavidin beads (Invitrogen). cDNA generation and quantitation of snRNA were performed as previously described54.
Supplementary Material
Acknowledgments
We thank members of our laboratory for helpful discussions and comments on the manuscript. This work was supported by the Association Française contre les Myopathies (AFM) and by the National Institutes of Health (R01 GM112923). G.D. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
AUTHOR CONTRIBUTIONS
B.R.S., L.W. and Z. Z. designed and performed experiments. B.R.S., L.W., Z. Z., P. L., E. B., J. D. and I. Y. contributed to data analysis. G.D. is responsible for the project planning and experimental design. All authors contributed to writing the paper.
References
- 1.Mount SM, Pettersson I, Hinterberger M, Karmas A, Steitz JA. The U1 small nuclear RNA-protein complex selectively binds a 5′ splice site in vitro. Cell. 1983;33:509–518. doi: 10.1016/0092-8674(83)90432-4. [DOI] [PubMed] [Google Scholar]
- 2.Will CL, Lührmann R. Spliceosome structure and function. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berg MG, et al. U1 snRNP determines mRNA length and regulates isoform expression. Cell. 2012;150:53–64. doi: 10.1016/j.cell.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaida D, et al. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature. 2010;468:664–668. doi: 10.1038/nature09479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vorlová S, et al. Induction of antagonistic soluble decoy receptor tyrosine kinases by intronic polyA activation. Mol Cell. 2011;43:927–939. doi: 10.1016/j.molcel.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Almada AE, Wu X, Kriz AJ, Burge CB, Sharp PA. Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature. 2013;499:360–363. doi: 10.1038/nature12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ntini E, et al. Polyadenylation site-induced decay of upstream transcripts enforces promoter directionality. Nat Struct Mol Biol. 2013;20:923–928. doi: 10.1038/nsmb.2640. [DOI] [PubMed] [Google Scholar]
- 8.Baserga SJ, Steitz JA. 14 The Diverse World of Small Ribonucleoproteins. Cold Spring Harbor Monograph Archive. 1993;24:359–381. [Google Scholar]
- 9.Battle DJ, et al. The SMN complex: an assembly machine for RNPs. Cold Spring Harb Symp Quant Biol. 2006;71:313–320. doi: 10.1101/sqb.2006.71.001. [DOI] [PubMed] [Google Scholar]
- 10.Guthrie C, Patterson B. Spliceosomal snRNAs. Annu Rev Genet. 1988;22:387–419. doi: 10.1146/annurev.ge.22.120188.002131. [DOI] [PubMed] [Google Scholar]
- 11.Yong J, Golembe TJ, Battle DJ, Pellizzoni L, Dreyfuss G. snRNAs Contain Specific SMN-Binding Domains That Are Essential for snRNP Assembly. Molecular and Cellular Biology. 2004;24:2747–2756. doi: 10.1128/MCB.24.7.2747-2756.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kondo Y, Oubridge C, van Roon AM, Nagai K. Crystal structure of human U1 snRNP, a small nuclear ribonucleoprotein particle, reveals the mechanism of 5′ splice site recognition. Elife. 2015;4 doi: 10.7554/eLife.04986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leung AK, Nagai K, Li J. Structure of the spliceosomal U4 snRNP core domain and its implication for snRNP biogenesis. Nature. 2011;473:536–539. doi: 10.1038/nature09956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pomeranz Krummel DA, Oubridge C, Leung AK, Li J, Nagai K. Crystal structure of human spliceosomal U1 snRNP at 5.5 A resolution. Nature. 2009;458:475–480. doi: 10.1038/nature07851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raker VA, Hartmuth K, Kastner B, Lührmann R. Spliceosomal U snRNP core assembly: Sm proteins assemble onto an Sm site RNA nonanucleotide in a specific and thermodynamically stable manner. Mol Cell Biol. 1999;19:6554–6565. doi: 10.1128/mcb.19.10.6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Urlaub H, Raker VA, Kostka S, Lührmann R. Sm protein-Sm site RNA interactions within the inner ring of the spliceosomal snRNP core structure. EMBO J. 2001;20:187–196. doi: 10.1093/emboj/20.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weber G, Trowitzsch S, Kastner B, Lührmann R, Wahl MC. Functional organization of the Sm core in the crystal structure of human U1 snRNP. EMBO J. 2010;29:4172–4184. doi: 10.1038/emboj.2010.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Battle DJ, et al. The Gemin5 protein of the SMN complex identifies snRNAs. Mol Cell. 2006;23:273–279. doi: 10.1016/j.molcel.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 19.Lau CK, Bachorik JL, Dreyfuss G. Gemin5-snRNA interaction reveals an RNA binding function for WD repeat domains. Nat Struct Mol Biol. 2009;16:486–491. doi: 10.1038/nsmb.1584. [DOI] [PubMed] [Google Scholar]
- 20.Yong J, Kasim M, Bachorik JL, Wan L, Dreyfuss G. Gemin5 delivers snRNA precursors to the SMN complex for snRNP biogenesis. Mol Cell. 2010;38:551–562. doi: 10.1016/j.molcel.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cauchi RJ. SMN and Gemins: ‘we are family’ … or are we?: insights into the partnership between Gemins and the spinal muscular atrophy disease protein SMN. Bioessays. 2010;32:1077–1089. doi: 10.1002/bies.201000088. [DOI] [PubMed] [Google Scholar]
- 22.Fischer U, Englbrecht C, Chari A. Biogenesis of spliceosomal small nuclear ribonucleoproteins. Wiley Interdiscip Rev RNA. 2011;2:718–731. doi: 10.1002/wrna.87. [DOI] [PubMed] [Google Scholar]
- 23.Chari A, et al. An assembly chaperone collaborates with the SMN complex to generate spliceosomal SnRNPs. Cell. 2008;135:497–509. doi: 10.1016/j.cell.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 24.Grimm C, et al. Structural basis of assembly chaperone- mediated snRNP formation. Mol Cell. 2013;49:692–703. doi: 10.1016/j.molcel.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 25.Zhang R, et al. Structure of a Key Intermediate of the SMN Complex Reveals Gemin2’s Crucial Function in snRNP Assembly. Cell. 2011;146:384–395. doi: 10.1016/j.cell.2011.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Will CL, Lührmann R. Spliceosomal UsnRNP biogenesis, structure and function. Curr Opin Cell Biol. 2001;13:290–301. doi: 10.1016/s0955-0674(00)00211-8. [DOI] [PubMed] [Google Scholar]
- 27.Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol. 2014;15:108–121. doi: 10.1038/nrm3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hamm J, Dathan NA, Scherly D, Mattaj IW. Multiple domains of U1 snRNA, including U1 specific protein binding sites, are required for splicing. The EMBO journal. 1990;9:1237. doi: 10.1002/j.1460-2075.1990.tb08231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nelissen RL, Will CL, van Venrooij WJ, Lührmann R. The association of the U1-specific 70K and C proteins with U1 snRNPs is mediated in part by common U snRNP proteins. EMBO J. 1994;13:4113–4125. doi: 10.1002/j.1460-2075.1994.tb06729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yong J, Pellizzoni L, Dreyfuss G. Sequence specific interaction of U1 snRNA with the SMN complex. The EMBO journal. 2002;21:1188–1196. doi: 10.1093/emboj/21.5.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wan L, Ottinger E, Cho S, Dreyfuss G. Inactivation of the SMN complex by oxidative stress. Mol Cell. 2008;31:244–254. doi: 10.1016/j.molcel.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wan L, et al. The survival of motor neurons protein determines the capacity for snRNP assembly: biochemical deficiency in spinal muscular atrophy. Mol Cell Biol. 2005;25:5543–5551. doi: 10.1128/MCB.25.13.5543-5551.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Younis I, et al. Minor introns are embedded molecular switches regulated by highly unstable U6atac snRNA. Elife. 2013;2:e00780. doi: 10.7554/eLife.00780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kohtz JD, et al. Protein-protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature. 1994;368:119–124. doi: 10.1038/368119a0. [DOI] [PubMed] [Google Scholar]
- 35.Cléry A, Blatter M, Allain FH. RNA recognition motifs: boring? Not quite. Curr Opin Struct Biol. 2008;18:290–298. doi: 10.1016/j.sbi.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 36.Cho S, et al. Interaction between the RNA binding domains of Ser-Arg splicing factor 1 and U1-70K snRNP protein determines early spliceosome assembly. Proc Natl Acad Sci U S A. 2011;108:8233–8238. doi: 10.1073/pnas.1017700108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick. Nat Rev Neurosci. 2009;10:597–609. doi: 10.1038/nrn2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lorson CL, et al. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet. 1998;19:63–66. doi: 10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- 39.Pellizzoni L, Yong J, Dreyfuss G. Essential role for the SMN complex in the specificity of snRNP assembly. Science. 2002;298:1775–1779. doi: 10.1126/science.1074962. [DOI] [PubMed] [Google Scholar]
- 40.Young PJ, et al. The exon 2b region of the spinal muscular atrophy protein, SMN, is involved in self-association and SIP1 binding. Hum Mol Genet. 2000;9:2869–2877. doi: 10.1093/hmg/9.19.2869. [DOI] [PubMed] [Google Scholar]
- 41.Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tripsianes K, et al. Structural basis for dimethylarginine recognition by the Tudor domains of human SMN and SPF30 proteins. Nat Struct Mol Biol. 2011;18:1414–1420. doi: 10.1038/nsmb.2185. [DOI] [PubMed] [Google Scholar]
- 43.Liu K, et al. Crystal structure of TDRD3 and methyl-arginine binding characterization of TDRD3, SMN and SPF30. PLoS One. 2012;7:e30375. doi: 10.1371/journal.pone.0030375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yong J, Wan L, Dreyfuss G. Why do cells need an assembly machine for RNA-protein complexes. Trends Cell Biol. 2004;14:226–232. doi: 10.1016/j.tcb.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 45.Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Piazzon N, et al. Implication of the SMN complex in the biogenesis and steady state level of the signal recognition particle. Nucleic Acids Res. 2013;41:1255–1272. doi: 10.1093/nar/gks1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng D, Côté J, Shaaban S, Bedford MT. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol Cell. 2007;25:71–83. doi: 10.1016/j.molcel.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 48.Lorson CL, Androphy EJ. The domain encoded by exon 2 of the survival motor neuron protein mediates nucleic acid binding. Hum Mol Genet. 1998;7:1269–1275. doi: 10.1093/hmg/7.8.1269. [DOI] [PubMed] [Google Scholar]
- 49.O’Reilly D, et al. Differentially expressed, variant U1 snRNAs regulate gene expression in human cells. Genome Res. 2013;23:281–291. doi: 10.1101/gr.142968.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shukla S, Parker R. Quality control of assembly-defective U1 snRNAs by decapping and 5′-to-3′ exonucleolytic digestion. Proc Natl Acad Sci U S A. 2014;111:E3277–86. doi: 10.1073/pnas.1412614111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gabanella F, et al. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS One. 2007;2:e921. doi: 10.1371/journal.pone.0000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li DK, Tisdale S, Lotti F, Pellizzoni L. SMN control of RNP assembly: from post-transcriptional gene regulation to motor neuron disease. Semin Cell Dev Biol. 2014;32:22–29. doi: 10.1016/j.semcdb.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tisdale S, et al. SMN is essential for the biogenesis of U7 small nuclear ribonucleoprotein and 3′-end formation of histone mRNAs. Cell Rep. 2013;5:1187–1195. doi: 10.1016/j.celrep.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Z, et al. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133:585–600. doi: 10.1016/j.cell.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Z, et al. Dysregulation of synaptogenesis genes antecedes motor neuron pathology in spinal muscular atrophy. Proc Natl Acad Sci U S A. 2013;110:19348–19353. doi: 10.1073/pnas.1319280110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun S, et al. ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat Commun. 2015;6:6171. doi: 10.1038/ncomms7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsuiji H, et al. Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol Med. 2013;5:221–234. doi: 10.1002/emmm.201202303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamazaki T, et al. FUS-SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep. 2012;2:799–806. doi: 10.1016/j.celrep.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.