

Less is More
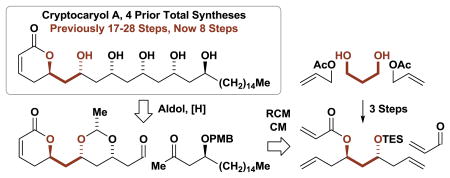
The polyketide natural product Cryptocaryol A is prepared in 8 steps via iridium catalyzed enantioselective diol double C-H allylation, which directly generates an acetate-based triketide stereodiad. In 4 previously reported total syntheses, 17-28 steps were required.
Keywords: Iridium, Transfer Hydrogenation, Polyketide, Enantioselective, Total Synthesis
Downregulation of the tumor suppressor protein PDCD4 (programmed cell death 4)[1] has been linked to diverse human cancers, including liver,[2a] colorectal, [2b] brain,[2c]; ovarian,[2d] and gastric carcinomas.[2e] Conversely, murine epidermal cells resistant to tumor promotion possess elevated levels of PDCD4.[;1a] Hence, PDCD4 has emerged as a target for the development of anticancer drugs.[3] In 2011, using a high-throughput cell-based reporter assay[4] to screen extracts of a Papua New Guinea collection of the plant Cryptocarya sp. (Lauraceae, NSC number N098347), Gustafson and coworkers identified a new class of small-molecule PDCD4-stabilizers, the amphiphilic type I polyketides cryptocaryols A-H (Figure 1).[5] In 2013, Mohapatra reported the total synthesis of the purported structure of cryptocaryol A.[6a] In elegant contemporaneous work, O'Doherty completed total syntheses of cryptocaryols A and B,[6b] revising their structural assignments and enabling the first structure-activity studies within this compound class.[7] Recently, two additional total syntheses of cryptocaryol A were reported by Cossy[6c] and Dias.[6d]
Figure 1. Initially proposed structures of cryptocaryols A-H, revised structures of cryptocaryols A and B and total syntheses of cryptocaryol A.

*For graphical summaries of prior total syntheses, see Supporting Information. Longest Linear Sequence (LLS); Total Steps (TS).
The interesting biology, recurring 1,3-polyol motif and prior synthetic work associated with cryptocaryol A made it an interesting compound to further benchmark the utility of catalytic C-C couplings developed in our laboratory.[8] These processes, which directly convert lower alcohols to higher alcohols, merge the characteristics of transfer hydrogenation with carbonyl addition, exploiting the native reducing ability of alcohols to drive generation of transient carbonyl-organometal pairs. Unlike synthetic routes involving conventional carbonyl addition chemistry, discrete alcohol-to-carbonyl redox reactions and use of premetalated carbanions are not required. This technology, which includes methods for stereo- and site-selective primary alcohol C-H allylation[9] and crotylation,[10] has been used in total syntheses of several iconic type I polyketide natural products.[8b,11] By evoking strategies beyond those accessible via conventional carbanion chemistry, our technology has provided the most concise routes reported in all cases where it has been applied.
In the context of cryptocaryol A, the double allylation of 1,3-propane diol,[9c] which directly generates a C2-symmetric acetate-based triketide stereodiad, was envisioned as a means of constructing the C3-C9 substructure (Scheme 1). This method has proven especially effective in type I polyketide construction, as illustrated in remarkably concise total syntheses of roxaticin,[11a]; bryostatin 7,[11b] neopeltolide,[11c] psymberin (irciniastatin A),[11d] cyanolide A,[11e] mandelalide,[11f,g] cryptolatifolione[11h] and cryptomoscatone E3.[11i] Another key feature of the proposed route involved concomittant ring-closing metathesis-cross metathesis[12] to convert the acrylic ester 3 to an α,β-unsaturated aldehyde, the immediate precursor to the α-pyrone, Fragment A. Stereoselective substrate directed aldol addition-ketone reduction followed by global deprotection unites Fragment A and Fragment B to deliver cryptocaryol A (Scheme 1).
Scheme 1. Retrosynthetic analysis of cyanolide A via direct generation acetate-based triketide stereodiad.

Our route to Fragment A is as follows (Scheme 2). The previously reported double C-H allylation of 1,3-propane diol provides the C2-symmetric diol 1.[9c] On gram scale, use of (R)-BINAP was preferred due to its relatively low cost, although isolated yields were diminished. Selective monoacylation of diol 1 using the method of Taylor was remarkably efficient, delivering the acrylic ester 2 in 97% yield.[13] Protection of the hydroxyl moiety of acrylate 2 to form TES-ether 3 was required to increase efficiency in the subsequent 2 steps. Treatment of compound 3 with acrolein in the presence of the Hoveyda-Grubbs-II catalyst in toluene solvent provided the desired RCM-CM product, α-pyrone 4, in 58% yield. The efficiency of the RCM-CM process was improved significantly upon use of F8-toluene as solvent,[14] which enabled acquisition of α-pyrone 4 in 82% yield. Under these conditions, but in the absence of the TES-ether, the desired RCM-CM product was formed in only 14% yield. Related metathesis reactions were explored in Pilli's recently reported synthesis of cryptolatifolione.[11h] Finally, exposure of α-pyrone 4 to Evans' conditions for oxa-conjugate addition delivered Fragment A as a single stereoisomer.[15] Attempts to purify Fragment A via flash silica gel chromatography resulted in substantial decomposition. Chromatography on florisil gave better results, providing pure Fragment A in 58% yield. However, crude Fragment A (>90% pure by 1H NMR) could be obtained in 91% yield, which served equally well in subsequent steps compared to the chromatographically purified material. Notably, in the synthesis of Fragment A, 4 of the required 5 steps are catalytic transformations and all 4 C-C bonds are forged via metal catalysis.
Scheme 2. Synthesis of Fragment A.a.

aYields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details
The synthesis of Fragment B begins with the enantioselective C-H allylation of cetyl alcohol using the iridium catalyst assembled in situ from [Ir(cod)Cl]2, (R)-BINAP, allyl acetate and 4-chloro-3-nitrobenzoic acid.[9] The homoallylic alcohol 5 was obtained in 72% isolated yield and >95% ee, as determined by Mosher ester analysis.[16] Exposure of alcohol 5 to p-methoxybenzyl trichloroacetimidate in the presence of lanthanum triflate delivered the PMB-ether 6 in 97% yield.[17] Finally, using Sigman's modification of the Wacker oxidation,[18] PMB-ether 6 is transformed into the methyl ketone in 71% isolated yield, completing the synthesis of Fragment B (Scheme 3).
Scheme 3. Synthesis of Fragment B.a.

aYields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details
The union of Fragment A and Fragment B takes advantage of substrate-directed boron-mediated aldol addition (Scheme 4). Whereas enolborinate additions to β-alkoxy aldehydes typically do not display high levels of 1,3-asymmetric induction, exceptional 1,5-anti-diastereoselectivity is observed in enolborinate additions involving β-alkoxy methyl ketones as nucleophilic partners.[19] In the event, Fragment B was exposed to dicyclohexylboron chloride in the presence of triethylamine to form the enol borinate,[20] which upon exposure to Fragment A under cryogenic conditions (-78 °C) delivered the aldol product 7 in 64% yield as a single stereoisomer. As established in the parent methodological studies,[19] this level of stereocontrol is significantly higher than that reported for stereochemically related aldol additions between corresponding silyl-protected partners.[21] Hydroxy-directed reduction of the aldol product 7 delivers the diol 8 in 94% yield with good levels of 1,3-diastereoselectivity (15:1).[22] Finally, global deprotection of the acetal and PMB ether using triflic acid/1,3-dimethoxybenzene[23] delivers cryptocaryol A in a total of 8 steps (LLS) from 1,3-propane diol, fewer than half the steps of any prior synthesis.
Scheme 4. Union of Fragment A and Fragment B and total synthesis of cryptocaryol A.a.

aYields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details
In summary, despite impressive methodological advances, the vast majority of de novo chemical syntheses remain distant from the Hendricksonian ideal.[24,25] Using step-count as the most fundamental metric to evaluate strategic efficiency,[26] it is evident that classical carbanion chemistry, which requires separate treatment of redox and C-C bond construction events, contributes significantly to this inefficiency. As borne out by an expanding body of work,[8,11] technologies that merge redox and C-C bond construction events streamline chemical synthesis. In the specific context of cryptocaryol A, the chemistry of carbonyl addition and transfer hydrogenation are united in the form of a double enantioselective alcohol C-H allylation, which directly delivers an acetate-based triketide motif that would otherwise require numerous steps to prepare.[9c] This transformation, applied in combination with recent advances in alkene metathesis (RCM/CM)[12] and catalytic diol mono-functionalization,[13] have enabled a total synthesis of cryptocaryol A in fewer than half the steps previously required, and one can easily envision application of this approach to other members of this compound class. It is our hope the present exposition in chemical synthesis (along with prior demonstrations)[11] will motivate further developments in the area of redox-economic C-C bond formation.
Supplementary Material
Footnotes
Acknowledgment is made to the Robert A. Welch Foundation (F-0038) and the NIH (RO1-GM093905) for partial support of this research. Dr. Felix Perez is an NIH-IRACDA Postdoctoral Fellow (NIH 1K12GM102745).
References
- 1.a) Yang HS, Jansen AP, Nair R, Shibahara K, Verma AK, Cmarik JL, Colburn NH. Oncogene. 2001;20:669–676. doi: 10.1038/sj.onc.1204137. [DOI] [PubMed] [Google Scholar]; b) Yang HS, Jansen AP, Komar AA, Zheng X, Merrick WC, Costes S, Lockett SJ, Sonenberg N, Colburn NH. Mol Cell Biol. 2003;23:26–37. doi: 10.1128/MCB.23.1.26-37.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Leupold JH, Yang HS, Colburn NH, Asangani I, Post S, Allgayer H. Oncogene. 2007;26:4550–4562. doi: 10.1038/sj.onc.1210234. [DOI] [PubMed] [Google Scholar]; d) Nieves-Alicea R, Colburn NH, Simeone AM, Tari AM. Breast Cancer Res Treat. 2009;114:203–209. doi: 10.1007/s10549-008-9993-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Zhang H, Ozaki I, Mizuta T, Hamajima H, Yasutake T, Eguchi Y, Ideguchi H, Yamamoto K, Matsuhashi S. Oncogene. 2006;25:6101–6112. doi: 10.1038/sj.onc.1209634. [DOI] [PubMed] [Google Scholar]; b) Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, Post S. Oncogene. 2008;27:2128–2136. doi: 10.1038/sj.onc.1210856. [DOI] [PubMed] [Google Scholar]; c) Chen Y, Liu W, Chao T, Zhang Y, Yan X, Gong Y, Qiang B, Yuan J, Sun M, Peng X. Cancer Lett. 2008;272:197–205. doi: 10.1016/j.canlet.2008.06.034. [DOI] [PubMed] [Google Scholar]; d) Wang X, Wei Z, Gao F, Zhang X, Zhou C, Zhu F, Wang Q, Gao Q, Ma C, Sun W, Kong B, Zhang L. Anticancer Res. 2008;28:2991–2996. [PubMed] [Google Scholar]; e) Yu H, Zeng J, Liang X, Wang W, Zhou Y, Sun Y, Liu S, Li W, Chen C, Jia J. PLoS One. 2014;9:e105306. doi: 10.1371/journal.pone.0105306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reviews: Lankat-Buttgereit B, Goke R. Biol Cell. 2003;95:515–519. doi: 10.1016/j.biolcel.2003.09.003.Lankat-Buttgereit B, Goeke R. Curr Top Pept Prot Res. 2005;7:63–66.Lankat-Buttgereit B, Goeke R. Biol Cell. 2009;101:309–317. doi: 10.1042/BC20080191.
- 4.Blees JS, Schmid T, Thomas CL, Baker AR, Benson L, Evans JR, Goncharova EI, Colburn NH, McMahon JB, Henrich CJ. J Biomol Screen. 2010;15:21–29. doi: 10.1177/1087057109351028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grkovic T, Blees JS, Colburn NH, Schmid T, Thomas CL, Henrich CJ, McMahon JB, Gustafson KR. J Nat Prod. 2011;74:1015–1020. doi: 10.1021/np100918z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Reddy DS, Mohapatra DK. Eur J Org Chem. 2013:1051–1057. [Google Scholar]; b) Wang Y, O'Doherty GA. J Am Chem Soc. 2013;135:9334–9337. doi: 10.1021/ja404401f. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Brun E, Bellosta V, Cossy J. J Org Chem. 2015;80:8668–8676. doi: 10.1021/acs.joc.5b01323. [DOI] [PubMed] [Google Scholar]; d) Dias LC, Kuroishi PK, de Lucca EC., Jr Org Biomol Chem. 2015;13:3575–3584. doi: 10.1039/c5ob00080g. [DOI] [PubMed] [Google Scholar]
- 7.Cuccarese MF, Wang Y, Beuning PJ, O'Doherty GA. ACS Med Chem Lett. 2014;5:522–526. doi: 10.1021/ml4005039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For recent reviews, see: Ketcham JM, Shin I, Montgomery TP, Krische MJ. Angew Chem. 2014;126:9294–9302. doi: 10.1002/anie.201403873.Angew Chem Int Ed. 2014;53:9142–9150. doi: 10.1002/anie.201403873.Dechert-Schmitt AMR, Schmitt DC, Gao X, Itoh T, Krische MJ. Nat Prod Rep. 2014;31:504–513. doi: 10.1039/c3np70076c.
- 9.Allylation: Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:6340–6341. doi: 10.1021/ja802001b.Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:14891–14899. doi: 10.1021/ja805722e.Lu Y, Kim IS, Hassan A, Del Valle DJ, Krische MJ. Angew Chem. 2009;121:5118–5121. doi: 10.1002/anie.200901648.Angew Chem Int Ed. 2009;48:5018–5021. doi: 10.1002/anie.200901648.Dechert-Schmitt AMR, Schmitt DC, Krische MJ. Org Lett. 2012;14:6302–6305. doi: 10.1021/ol3030692.Dechert-Schmitt AMR, Schmitt DC, Krische MJ. Angew Chem. 2013;125:3277–3280. doi: 10.1002/anie.201209863.Angew Chem, Int Ed. 2013;52:3195–3198. doi: 10.1002/anie.201209863.Shin I, Wang G, Krische MJ. Chem Eur J. 2014;20:13382–13389. doi: 10.1002/chem.201404065.
- 10.Crotylation: Kim IS, Han SB, Krische MJ. J Am Chem Soc 2009. 131:514–2520. doi: 10.1021/ja808857w.Gao X, Townsend IA, Krische MJ. J Org Chem. 2011;76:2350–2354. doi: 10.1021/jo200068q.Gao X, Han H, Krische MJ. J Am Chem Soc. 2011;133:12795–12800. doi: 10.1021/ja204570w.Zbieg JR, Moran J, Krische MJ. J Am Chem Soc. 2011;133:10582–10586. doi: 10.1021/ja2046028.Zbieg JR, Yamaguchi E, McInturff EL, Krische MJ. Science. 2012;336:324–327. doi: 10.1126/science.1219274.McInturff EL, Yamaguchi E, Krische MJ. J Am Chem Soc. 2012;134:20628–20631. doi: 10.1021/ja311208a.
- 11.For application of iridium catalyzed double asymmetric allylation of 1,3-diols in the total synthesis of polyketide natural products, see: Han SB, Hassan A, Kim IS, Krische MJ. J Am Chem Soc. 2010;132:15559–15561. doi: 10.1021/ja1082798. Roxaticin.Lu Y, Woo SK, Krische MJ. J Am Chem Soc. 2011;133:13876–13879. doi: 10.1021/ja205673e. Bryostatin 7.Yang Z, Zhang B, Zhao G, Yang J, Xie X, She X. Org Lett. 2011;13:5916–5919. doi: 10.1021/ol2025718. Neopeltolide:Feng Y, Jiang X, De Brabander JK. J Am Chem Soc. 2012;134:17083–17093. doi: 10.1021/ja3057612. Psymberin (Irciniastatin A)Waldeck AR, Krische MJ. Angew Chem. 2013;125:4566–4569. Cyanolide A.Angew Chem Int Ed. 2013;52:4470–4473. doi: 10.1002/anie.201300843.Willwacher J, Fürstner A. Angew Chem. 2014;126:4301–4305. Mandelalide (putative structure)Angew Chem Int Ed. 2014;53:4217–4221. doi: 10.1002/anie.201400605.Willwacher M, Heggen B, Wirtz C, Thiel W, Fürstner A. Chem Eur J. 2015;21:10416–10430. doi: 10.1002/chem.201501491. Mandelalide (revised structure)Novaes LFT, Sarotti AM, Pilli RA. RSC Adv. 2015;5:53471–53476. Cryptolatifolione.Novaes LFT, Sarotti AM, Pilli RA. J Org Chem. 2015;80:12027–12037. doi: 10.1021/acs.joc.5b01956. Cryptomoscatone E3.
- 12.For selected examples of ring-closing metathesis-cross metathesis, see: Virolleaud MA, Bressy C, Piva O. Tetrahedron Lett. 2003;44:8081–8084.Quinn KJ, Isaacs AK, Arvary RA. Org Lett. 2004;6:4143–4145. doi: 10.1021/ol040047f.Virolleaud MA, Piva O. Tetrahedron Lett. 2007;48:1417–1420.Venukadasula PKM, Chegondi R, Maitra S, Hanson PR. Org Lett. 2010;12:1556–1559. doi: 10.1021/ol1002913.Cros F, Pelotier B, Piva O. Eur J Org Chem. 2010:5063–5070.Cros F, Ruiz J, Pelotier B, Piva O. Synlett. 2010:2621–2624.Venukadasula PKM, Chegondi R, Suryn GM, Hanson PR. Org Lett. 2012;14:2634–2637. doi: 10.1021/ol301007h.Sabitha G, Shankaraiah K, Yadav JS. Eur J Org Chem. 2013:4870–4878.
- 13.Lee D, Williamson CL, Chan L, Chan MS, Taylor MS. J Am Chem Soc. 2012;134:8260–8267. doi: 10.1021/ja302549c. [DOI] [PubMed] [Google Scholar]
- 14.Samojłowicz C, Bieniek M, Pazio A, Makal A, Woźniak K, Poater A, Cavallo L, Wójcik J, Zdanowski K, Grela K. Chem Eur J. 2011;17:12981–12993. doi: 10.1002/chem.201100160. [DOI] [PubMed] [Google Scholar]
- 15.Evans PA, Grisin A, Lawler MJ. J Am Chem Soc. 2012;134:2856–2859. doi: 10.1021/ja208668u. [DOI] [PubMed] [Google Scholar]
- 16.Dale JA, Dull DL, Mosher HS. J Org Chem. 1969;34:2543–2549. [Google Scholar]
- 17.Rai AN, Basu A. Tetrahedron Lett. 2003;44:2267–2269. [Google Scholar]
- 18.a) Michel BW, Camelio AM, Cornell CN, Sigman MS. J Am Chem Soc. 2009;131:6076–6077. doi: 10.1021/ja901212h. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) McCombs JR, Michel BW, Sigman MS. J Org Chem. 2011;76:3609–3613. doi: 10.1021/jo200462a. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Michel BW, Steffens LD, Sigman MS. J Am Chem Soc. 2011;133:8317–8325. doi: 10.1021/ja2017043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Paterson I, Gibson KR, Oballa RM. Tetrahedron Lett. 1996;37:8585–8588. [Google Scholar]; b) Evans DA, Coleman PJ, Côté B. J Org Chem. 1997;62:788–789. [Google Scholar]; c) Evans DA, Côté B, Coleman PJ, Connell BT. J Am Chem Soc. 2003;125:10893–10898. doi: 10.1021/ja027640h. [DOI] [PubMed] [Google Scholar]
- 20.Brown HC, Dhar RK, Ganesan K, Singaram B. J Org Chem. 1992;57:499–504. [Google Scholar]
- 21.Paterson I, Oballa RM, Norcross RD. Tetrahedron Lett. 1996;47:8581–8584. [Google Scholar]
- 22.a) Chen KM, Hardtmann GE, Prasad K, Repic O, Shapiro MJ. Tetrahedron Lett. 1987;28:155. [Google Scholar]; b) Chen KM, Gunderson KG, Hardtmann GE, Prasad K, Repic O, Shapiro MJ. Chem Lett. 1987:1923–1926. [Google Scholar]
- 23.Jung ME, Koch P. Tetrahedron Lett. 2011;52:6051–6054. [Google Scholar]
- 24.a) Hendrickson JB. J Am Chem Soc. 1975;97:5784–5780. [Google Scholar]; b) Shin I, Montgomery TP, Krische MJ. Aldrichim Acta. 2015;47:15. [PMC free article] [PubMed] [Google Scholar]
- 25.For an excellent discussion of synthetic efficiency from a biosynthetic perspective, see: Jürgens G, Kirschning A, Candito DA. Nat Prod Rep. 2015;32:723–737. doi: 10.1039/c4np00160e.
- 26.“Given the fact that every reaction may be optimized… the total number of chemical transformations is the only variable in the determination of strategic efficiency. Obviously, the fewer the total number of reactions steps in a synthetic design, the higher the level of strategic efficiency.” Qiu F. Can J Chem. 2008;86:903–906.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.