

Abstract
Background
Parent-of-origin effects are due to differential contributions of paternal and maternal lineages to offspring phenotypes. Such effects include, for example, maternal effects in several species. However, epigenetically induced parent-of-origin effects have recently attracted attention due to their potential impact on variation of complex traits. Given that prediction of genetic merit or phenotypic performance is of interest in the study of complex traits, it is relevant to consider parent-of-origin effects in such predictions. We built a whole-genome prediction model that incorporates parent-of-origin effects by considering parental allele substitution effects of single nucleotide polymorphisms and gametic relationships derived from a pedigree (the POE model). We used this model to predict body mass index in a mouse population, a trait that is presumably affected by parent-of-origin effects, and also compared the prediction performance to that of a standard additive model that ignores parent-of-origin effects (the ADD model). We also used simulated data to assess the predictive performance of the POE model under various circumstances, in which parent-of-origin effects were generated by mimicking an imprinting mechanism.
Results
The POE model did not predict better than the ADD model in the real data analysis, probably due to overfitting, since the POE model had far more parameters than the ADD model. However, when applied to simulated data, the POE model outperformed the ADD model when the contribution of parent-of-origin effects to phenotypic variation increased. The superiority of the POE model over the ADD model was up to 8 % on predictive correlation and 5 % on predictive mean squared error.
Conclusions
The simulation and the negative result obtained in the real data analysis indicated that, in order to gain benefit from the POE model in terms of prediction, a sizable contribution of parent-of-origin effects to variation is needed and such variation must be captured by the genetic markers fitted. Recent studies, however, suggest that most parent-of-origin effects stem from epigenetic regulation but not from a change in DNA sequence. Therefore, integrating epigenetic information with genetic markers may help to account for parent-of-origin effects in whole-genome prediction.
Background
Parent-of-origin effects are asymmetric influences that act on phenotype of offspring, depending on the sex of the parent. Genomic imprinting, manifested as differential and/or preferential gene expression that is usually caused by differential DNA methylation [1, 2] or histone modification [3] on different parental alleles, is one of the most studied epigenetic mechanisms and an important source of parent-of-origin effects. Imprinting has an impact on several human diseases [4–8] such as the Prader–Willi (PWS) and Angelman (AS) syndromes [9, 10], as well as on complex traits in livestock [11–13]. For example, mapping studies have detected presumably imprinted quantitative trait loci (QTL) that affect economically important traits in swine [14–21], beef cattle [22–24], sheep [25], mice [26, 27], and dogs [28]. In addition, genome-wide scan studies with dense single nucleotide polymorphism (SNP) chips have also suggested that imprinted loci are associated with complex traits in various mammalian species (e.g., [29–34]).
QTL mapping studies can identify genomic regions that contribute to traits of interest and to marker assisted selection (MAS, [35, 36]). However, use of QTL mapping for breeding purposes has failed to yield clear dividends (e.g., [37, 38]). A possible explanation is that QTL mapping studies require, e.g., carefully designed crossbreeding experiments and these are seldom available in livestock. Thus, artificial selection using predicted genetic merit of selection candidates is still mainly used in animal improvement programs. Breeding values have been predicted based on resemblances between relatives using pedigree information (e.g., [39, 40]). In the genomics era, however, the availability of high-throughput genotyping techniques makes it possible to interrogate genotypes of hundreds of thousands or even millions of SNPs simultaneously, resulting in what is known as “genomic selection” or “whole-genome prediction” [41–43]. With continuously decreasing genotyping costs, genomic selection has become affordable for commercial settings in some species [44], and QTL mapping is less used in animal breeding, unless the objective is to find a major gene. Even in crops, genomic selection is gradually replacing QTL-MAS. Although some debate persists [45], genomic selection will probably be the main approach used in the foreseeable future [46].
Genomic selection (GS) and whole-genome prediction (WGP) exploit associations between phenotypes and an enormous number of SNPs under certain statistical assumptions regarding the underlying trait architecture. Often, the association between phenotype and SNPs is explored by using the SNPs as covariates in a linear regression model. Since the number of covariates (p) is usually much larger than the number of observations (n), different techniques have been used to circumvent the “curse of dimensionality” in GS/WGP studies. Commonly used methods include Bayesian regression (e.g., [41, 43, 47, 48]), G-BLUP (e.g., [49, 50]), semi-parametric methods (e.g., [51–54]) and neural networks (e.g., [55–57]), among others. All these models assume that the inheritance of the complex trait is Mendelian, i.e., paternally- and maternally-inherited alleles are functionally equivalent. Under this assumption, no phenotypic difference between genotypes and is expected. SNPs are assigned codes such as 0, 1 or 2 according to genotype at the locus, and the average substitution effects of all markers in the model are estimated simultaneously. Prediction is then performed by combining the estimates of these SNP effects with a genotype matrix in an independent set of individuals. However, recent studies suggest that some traits are not strictly Mendelian. For example, Mott et al. [58] found that 91 out of 97 murine traits were subject to parent-of-origin effects. In a review, Lawson et al. [13] also suggested that parent-of-origin effects may be more prevalent than previously thought. Perhaps parent-of-origin effects may enhance WGP models, if considered appropriately.
Currently used GS models may not be suitable for parent-of-origin-effects-affected traits, for which inheritance of one allele from the father may have a different effect on the phenotype than when the same allele is inherited from the mother. This suggests that two distinct substitution effects associated with the two parental origins of an allele are needed. A one-locus quantitative genetic model that takes imprinting into account has been proposed [17, 59, 60], where genotypes , , and are assumed to have genotypic values , , and a, respectively, and paternal and maternal allele substitution effects are defined as and . In a previous study, a genome-wide association study (GWAS)-like scan conducted with this model indicated that ignoring imprinting may underestimate additive genetic variation [61], which suggested that prediction accuracy may be higher when imprinting is considered in WGP. In a recent simulation study, Nishio and Satoh [62] suggested that the unbiasedness of variance component estimation may be enhanced when imprinting is integrated under a genomic best linear unbiased prediction (G-BLUP) framework. Here, we build a prediction model that incorporates parent-of-origin effects parametrically and assess whether or not this model improves prediction of phenotypes over the additive model currently employed in WGP using real data. In addition, we evaluate the advantages and limitations of the full model using simulated data and give a detailed discussion on its application under various conditions. Our study complements that of [62] and provides insights into prediction with parent-of-origin effects from a Bayesian perspective.
Before proceeding, some clarification is necessary. In much of the epigenetic literature, the terms “imprinting effects” and “parent-of-origin effects” have been used interchangeably. In “iQTL mapping” studies, for example, the detected QTL are putatively imprinted. However, the statistical model used in iQTL mapping does not guarantee that the detected parent-of-origin effects are necessarily due to imprinting. A counter-example was presented by Hager et al. [63], where maternal effects can mimic imprinting effects in statistical analysis. Furthermore, parent-of-origin effects were detected in birds [64, 65], although no strong evidence of imprinting in birds is available [66–68]. Thus, results obtained from the model described herein and its variants should be interpreted as parent-of-origin effects but not beyond [61]. In this study, we build WGP models to incorporate parent-of-origin effects, aiming at obtaining a higher predictive accuracy when a complex trait is subject to parent-of-origin effects. We use the term “parent-of-origin effects” throughout, but in the simulations we mimicked imprinting mechanisms to simplify the source of parent-of-origin effects. The simulated data was used for model evaluation under various conditions.
This paper is organized as follows. First, a previously proposed mixed effect model that incorporates parent-of-origin effects at the lineage level is introduced. Then, a brief introduction of a one-locus quantitative genetic model that takes genomic imprinting into account is provided. We extend this model to incorporate all available SNPs simultaneously to include parent-of-origin effects at the DNA (SNP) level, and our prediction model is constructed using both pedigree and DNA information. This model is applied to real (mouse) and simulated data and its predictive performance is compared to that of an additive model. Following a section that discusses advantages and drawbacks of this model, a discussion on the possibilities and challenges of conducting whole-genome prediction models that use epigenetic information to incorporate parent-of-origin effects is provided.
Prediction model incorporating parent-of-origin effects
Consider the pedigree-based additive effects model (e.g., [39, 40]):
| 1 |
where the vector contains phenotypic records; is an effect common to all individuals; is a vector of fixed effects with associated incidence matrix ; is the vector of normally distributed infinitesimal additive effects with zero mean vector and variance–covariance matrix , where is the pedigree-based numerator relationship matrix and is the additive genetic variance; and is the residual vector whose elements are assumed to be independent and identically distributed as normal with zero mean and variance . A commonly used technique for making predictions of yet-to-be-observed data is best linear unbiased prediction (BLUP) [39], where estimation of and prediction of are performed simultaneously. Variance components can be estimated, for example, by restricted maximum likelihood (REML).
If dense markers (e.g., SNPs) are available, the following model can be used for genome-enabled prediction (e.g., [41]):
| 2 |
Here, p is the (possibly large) number of SNPs and the assumption is that the QTL that contribute to the phenotype are in linkage disequlibrium (LD) with at least one SNP. In this model, is the substitution effect of the SNP; is an vector, whose elements are the genotype code (, 1 or 2 for genotypes , or ) of SNP j for the individual. One can also write as , where is , whose column is , and is , whose element is . SNP effects can be learned in a Bayesian process (e.g., [41, 47]) by drawing samples from posterior distributions using Markov chain Monte Carlo (MCMC) techniques. Predictive performance using Model 2 is often better than for Model 1, and several studies have suggested that including both pedigree and marker information can achieve an even higher prediction accuracy [69, 70].
Models described above assume that QTL and SNPs are inherited in a Mendelian manner. However, in the presence of imprinting, or more generally, parent-of-origin effects, receiving one allele from the mother might have a different effect on than receiving the same allele from the father [59, 60, 71]. Before the genomic era, the following mixed model using pedigree information was proposed to account for parent-of-origin effects [72, 73]:
| 3 |
where , , , and are as in Model 1; is a vector of additional genetic effects expressed only when inherited from a maternal or paternal gamete, assuming that with being a gametic relationship matrix calculated from a known pedigree.
When considering SNPs, Shete and Amos [60] proposed the following one-locus model that regresses phenotype on the number of alleles received from a specific parent to account for parent-of-origin effects:
| 4 |
where and are the average effects of receiving one allele from the female and male parents (maternal and paternal allele substitution effects), respectively, and and are vectors of associated indicator variables. Both and (the element of vectors and , respectively) take values 0 or 1 so the combination of these two indicators gives the genotype codes of four genotypes. For example, indicates an genotype and , indicates an genotype (maternally inherited allele is written first). This model can be extended to include all available SNPs simultaneously as in whole-genome prediction studies. Thus, we combined Models 3 and 4, which contain both pedigree and marker information, into a WGP model (called POE model hereafter) that is suitable for traits affected by parent-or-origin effects:
| 5 |
To evaluate the performance of the POE model, it was compared with the additive model (referred to as ADD model hereafter) without parent-of-origin effects at either the pedigree or SNP levels. Model ADD is then:
| 6 |
Data and model evaluation
Mouse data
Some studies have suggested that obesity-related traits might be affected by imprinting in both humans [74] and mice [75]. An indicator of obesity, body mass index (BMI), was shown to be affected by parent-of-origin effects as well [76]. Hence, we chose BMI as the response variable in this study.
The data set used here is publicly available at http://mus.well.ox.ac.uk/mouse/HS/ and has been used in other studies (e.g., [69, 77]). It includes 1940 individuals that were obtained by crossing eight inbred strains, followed by 50 generations of approximately random mating. BMI measurements pre-corrected for body weight, season, month and day, and more than 12,000 genotyped SNPs located on 19 autosomes were collected. Additional description of this data set can be found from the data website and from [78]. In order to incorporate POE into the analysis, the two reciprocal heterozygotes and need to be distinguished from each other such that each allele of a SNP has a known parental origin. To do this, haplotype inference was performed using BEAGLE 3.3.2 [79, 80]. After this step, all SNPs with a minor allele frequency (MAF) less than 0.05 were removed, resulting in 10,021 SNPs for subsequent analyses.
Models POE and ADD (as in Eqs. 5 and 6 above, respectively) were used to perform whole-genome predictions of BMI. In this data, included sex, litter size and cage density. Regarding the polygenic effect , Legarra et al. [77] and de los Campos et al. [69] conducted whole-genome prediction studies using the same mouse data and both suggested that including pedigree information in this data set provided no benefit in terms of predictive ability because the relationships among the full-sib families were relatively weak. Therefore, we dropped the polygenic term in the mouse data analysis. For the same reason, the term was also dropped. Furthermore, a vector of random cage effects with incidence matrix was included in both models. was assumed to be normally distributed with zero mean and variance–covariance matrix .
Simulated data
We used simulated data to evaluate the performance of the POE and of the ADD models under different situations. Parent-of-origin effects were simulated using the following two-step procedure. First, we used QMSim [81] to simulate a genome of 10 pairs of chromosomes each 1 Morgan long. Each chromosome had 1000 randomly located bi-allelic SNPs, so there were 10,000 SNPs in total, as in the mouse data. Approximately 150 simulated QTL were randomly located in the genome and these were not chosen from the simulated SNPs. QTL effects were randomly drawn from a normal distribution with zero mean and variance set to the software default value. The population started from 100 males and 100 females with 1000 generations of random mating to create LD between QTL and between SNPs and QTL; mutation rates were per QTL and per SNP, respectively. All QTL and SNP genotypes were fixed in generation 1. In the three most recent generations, without mutation, the population was expanded to 2000 individuals per generation with a 1:1 sex ratio.
In step 2, parent-of-origin effects were introduced by mimicking imprinting. For a long time, imprinting has been viewed as a “full-null” phenomenon, where the silencing of the imprinted allele is complete while the expression of the allele inherited from the other parent is intact; this is usually considered as the canonical definition of imprinting [71]. However, genomic imprinting can potentially operate at any level of gene regulation (e.g., at promoters, enhancers, splicing junctions, or polyadenylation sites, etc.) to present a more complex pattern of parent-specific differential expression [82]. For example, recent studies have provided evidence that, for some imprinted loci, both alleles are differentially expressed in a parent-of-origin-preferential or parent-of-origin-dependent manner [83], indicating that the silencing is incomplete [84, 85]. Such deviation from the canonical imprinting, defined as partial imprinting [30, 86], has been incorporated in the aforementioned one-locus imprinting model [17, 59, 60], and was also considered in our simulation. Let and (given by QMSim output) be the two allele effects of QTL j in individual i obtained from a certain QMSim run. Because QMSim records the parental origin of these two alleles, and can be represented by, say, and , respectively. If this QTL is maternally imprinted, the genotypic value at this QTL for individual i can be written as:
| 7 |
where is a parameter that controls the level of imprinting. Five different values were assigned to : 0, 0.25, 0.5, 0.75, and 1, where corresponds to no imprinting, to complete imprinting, and define different levels of partial imprinting. We further assumed that a proportion , 0.3, 0.45, 0.6} of QTL were either paternally or maternally imprinted with equal frequency (a validation on the choice of these values is given in Discussion). Hence, the phenotypic value of individual i is:
| 8 |
where NI, MI and PI are sets of non-imprinted, randomly selected maternally imprinted and paternally imprinted QTL, respectively, and is the environmental effect on individual i given by QMSim. Note that the environmental effect was not changed and that a common was shared by all imprinted QTL in a particular scenario for simplification.
Equation 8 was applied to the three recent generations (1001, 1002, and 1003) in all 20 combinations of and s. In subsequent analyses, generation 1002 was the training set and generation 1003 was the testing set. This whole procedure was replicated 5 times and the average predictive performance of all replicates was used for model evaluation.
Model training and phenotype prediction
The additive relationship matrix and the gametic relationship matrix were calculated from the pedigree using the R package synbreed [87]. Both the ADD and POE models were trained with an implementation of MCMC using the R package BGLR [88, 89]. Each chain was run for 60,000 iterations, with the first 10,000 iterations discarded as burn-in and the rest were thinned by a factor of 10.
For the ADD model, the conditional prior distribution of the substitution effect of marker j was a normal distribution with zero mean and variance , where came from a scaled inverted distribution with scale and degrees of freedom set to default values in package BGLR [89]; was drawn from an exponential distribution with parameter . Hyperparameter was drawn from a Gamma distribution with shape s and rate r set to default values. This prior creates a double-exponential posterior density for marker effects, given , and is referred to as Bayesian Lasso [69, 90]. The infinitesimal additive effects had a conditional multivariate normal prior , where was drawn from a scaled inverted distribution with scale and degrees of freedom set to default values. Similarly, for cage effects, and again, the scale and degrees of freedom for the prior of were set to default values.
For the POE model, prior distributions were similar to those described above, except that two marker effects, the paternal and maternal allelic substitution effects, were included for each marker. The extra vector of gametic effects was assumed to have the distribution . Again, all hyperparameters for the scaled inverted distributions were set to package default values.
After model training, predictions were made on the testing set. Predictive correlation and predictive mean squared error (MSE) were the two metrics used for model evaluation.
Results
Mouse data analysis
The data set was randomly partitioned into training and testing sets according to the within-families approach of [77]. The cross-validation was repeated five times for stability assessment. Table 1 gives average results over the five replications. The ADD model performed slightly better than the POE model when evaluated by different metrics, but the difference was minimal. Our results with the ADD model were in agreement with those of [77] and [69], including the estimated variance components (Table 2).
Table 1.
Average of testing set results of five cross validation replicates in the mouse data ( SE standard error)
| Model | |||
|---|---|---|---|
| ADD | 0.321 (±0.067) | 0.327 (±0.071) | 0.00347 (±1.49) |
| POE | 0.309 (±0.059) | 0.318 (±0.076) | 0.00371 (±1.53) |
ADD additive model, POE parent-of-origin effects model
: Pearson’s correlation between observed and predicted value
: Spearman’s correlation between observed and predicted value
: Mean squared error
Table 2.
Estimated variance components () in the two models with all individuals included
| Model | ||
|---|---|---|
| ADD | 3.37 | 17.89 |
| POE | 3.39 | 17.74 |
ADD additive model, POE parent-of-origin effects model
Analysis of simulated data
In the simulation, five replicates were run, with each replicate resulting from an independent run of QMSim simulation. Each of the five realizations had training and testing sample sizes of 2000 individuals each; the number of SNPs was equal to 10,000 and the number of QTL in each replicate was equal to 142, 167, 158, 141 and 149, respectively.
As described in “Simulated data” section, each replicate had 20 scenarios, each corresponding to a combination of (imprinting level) and s (proportion of imprinted QTL). When , however, three scenarios were redundant because in this case, all QTL were unimprinted such that different values of s made no difference (Eq. 8). Figure 1 displays the average prediction accuracy measured by Pearson’s correlation between observed and predicted phenotypes in different simulation scenarios, and Fig. 2 shows the MSE performance of the two models. Under both evaluation metrics, the ADD model performed better than the POE model when no imprinting was simulated (). When there were no parent-of-origin effects, the p extra parameters in the POE model led to overfitting of the training data, thus sacrificing predictive ability of future data. With parent-of-origin effects, the POE model outperformed the ADD model but in a manner that depended on the s and settings. Typically, the POE model was better than the ADD model when was small and s was large.
Fig. 1.
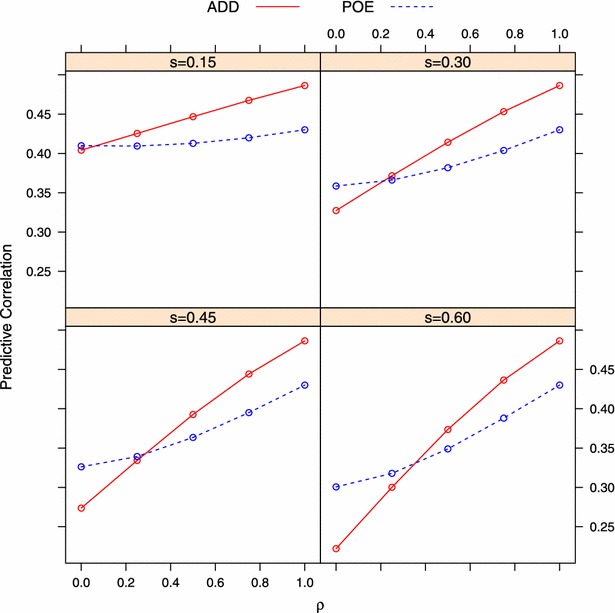
Average predictive correlation of two models measured by Pearson’s correlation between observed and predicted phenotype under different simulation settings. ADD additive model, POE parent-of-origin effects model. proportion of imprinted QTL; and denote complete imprinting and no imprinting, respectively
Fig. 2.
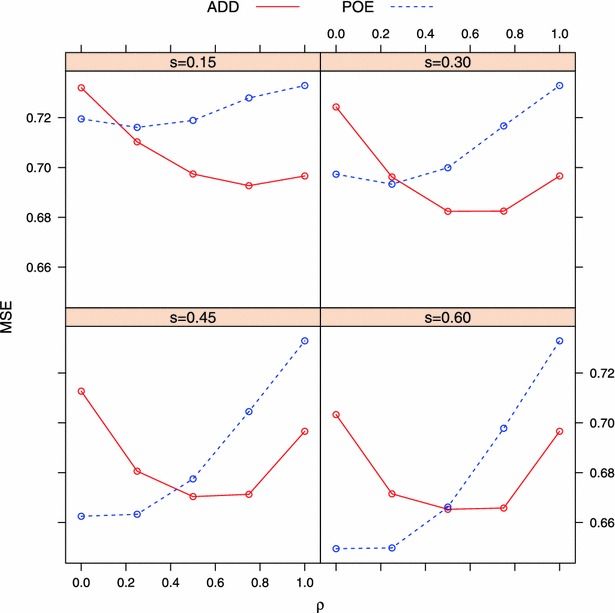
Averaged mean squared error (MSE) of two models between observed and predicted phenotype under different simulation settings. ADD additive model, POE parent-of-origin effects model. proportion of imprinted QTL; and denote complete imprinting and no imprinting, respectively
Discussion
Our results indicate that the POE model was not superior to the ADD model in terms of prediction when applied to the real mouse data. When using simulated data, however, our results showed that the POE model outperformed the ADD model under some circumstances, depending mainly on the choice of s and . This result was consistent with a recent simulation study that incorporated imprinting effects in WGP [62]. Their prediction model was adapted from the one-locus imprinting model of [17, 59, 60]. However, instead of applying Bayesian regression directly by extending the one-locus imprinting model to include all available SNPs, the authors adopted a G-BLUP framework where genetic relationship matrices were generated for the additive, dominance, and imprinting effects. In their simulation, two parameters affected the performance of a prediction model with imprinting, namely the degree of imprinting and the number of imprinted QTL, which had the same role as and s in our simulation, and produced similar results as those obtained in our study. A discussion on our simulation results and related topics is provided in the following sections.
Predictive performance of the ADD and POE models
Case 1: complete imprinting ()
When imprinting was complete (), it was not surprising that the POE model performed better than the additive ADD model. The superiority of the POE model over the ADD model depended on s, i.e., the larger the proportion of imprinted genes, the bigger the difference on predictive correlation and MSE between the two models. As s increased, a larger fraction of genetic variation was attributed to parent-of-origin effects, which cannot be captured by the ADD model. An interesting observation from Figs. 1 and 2 is that, for a given model, the predictive correlation and MSE decreased with an increase of s (Fig. 3). Recall that the data was simulated such that the allele effect was multiplied by (less imprinting as ), and fraction s of all QTL were assumed to be imprinted (Eq. 8). Suppose a QTL is maternally imprinted (the allele inherited from the mother written first), and that the values of the four genotypes (expressed as deviations from the population mean) are:
| 9 |
Let p and q be the frequencies of the and alleles. The genetic variance at this locus can be calculated as:
| 10 |
where is the genotype frequency of assuming Hardy-Weinberg equilibrium. Note that is , the allele substitution effect defined by a standard additive genetic model. From Eq. 10, when (no imprinting), the expression yields , the additive variance of a standard genetic model (e.g., [91, 92]). When , however, this variance (“signal”) decreases as approaches 0 (i.e., increased imprinting level). Hence, for a given value of that is smaller than 1 (0 in this case), the total variance of all QTL becomes smaller as s increases. Since the environmental distribution was the same in all settings, heritability decreased as s increased, producing a lower predictive ability.
Fig. 3.
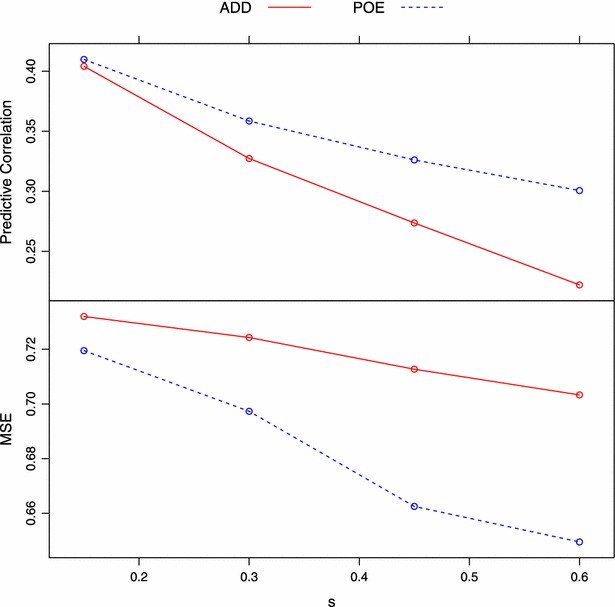
Trend of averaged predictive correlation and MSE with change of s (proportion of imprinted QTL) under (complete imprinting). Predictive correlation and MSE decrease as s goes up for both models. ADD additive model, POE parent-of-origin effects model
Case 2: no imprinting ()
As stated above, when , the value of s does not affect the simulated data. In this simpler case, the ADD model outperformed the POE model in terms of predictive correlation and MSE, since the extra parameters in the POE model captured noise only. This is because, if, instead of capturing signal in the data, the better fit is due to higher model complexity, a penalty would be given to such a model during the testing process [93]. In our Bayesian implementation, genome-wide incorporation of parent-of-origin effects approximately doubled the number of parameters relative to the ADD model. This higher complexity provided a better fit to the data, as shown in Fig. 4: the training correlation of the POE model was always higher than that of the ADD model by about 4 %. However, a lower predictive correlation of the POE model (, Fig. 1) indicated that the extra parameters in the POE model were not capturing model signal, at least when . For the same reason, the POE model was expected to have a higher prediction error than the ADD model when no parent-of-origin effects affected the trait (Fig. 2).
Fig. 4.
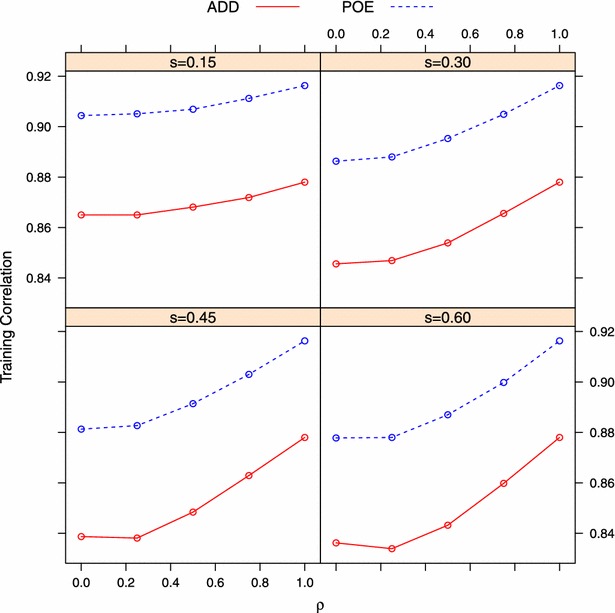
Training accuracy of two models measured by Pearson’s correlation between observed and fitted phenotype under different simulation settings. ADD additive model, POE parent-of-origin effects model. proportion of imprinted QTL; and denote complete imprinting and no imprinting, respectively
Overfitting might be a reason why the ADD model was better as observed in the mouse data analysis and here in simulation when . Technically, a more complex model would enhance prediction if true underlying signals are captured by the extra parameters, so that overfitting is not an issue. However, if the true signal is not strong enough or training sample size is not large enough, overfitting would degrade prediction performance in the testing step. Although some parent-of-origin effects seem to exist in the mouse data, as indicated by the previous study using the same data [61], these are not strong enough to overwhelm overfitting, resulting in a lower predictive performance when the POE model was used.
Case 3: partial imprinting
As imprinting changed from the highest (, complete imprinting) to the lowest level (, no imprinting), the predictive correlation of both models increased gradually for any value of s, since total additive variance (signal) increased during this course (Eq. 10; left panel of Fig. 5), so the predictive ability increased accordingly. Also, because the ADD model was better at but the POE model was better at , curves representing the two models crossed at some point, and it was interesting to note that the value of associated with the cross point increased (representing a lower level of imprinting) as s went up (Fig. 1). Intuitively, the POE model would outperform the ADD model when the proportion of signal due to parent-of-origin effects reaches some threshold. Here, the variance accounted for by parent-of-origin effects is expressed as:
| 11 |
according to the four genotypic values in Eq. 9 and the one-locus imprinting model of [59, 60], and [17]; the ratio between Eqs. 11 and 10 gives the proportion of additive variance accounted for by parent-of-origin effect at that locus. For a larger s, this threshold is reached much faster than at a smaller s as (Fig. 5, right panel), indicating that when fewer QTL are imprinted, a higher level of imprinting is needed for the POE model to gain advantage, as expected.
Fig. 5.
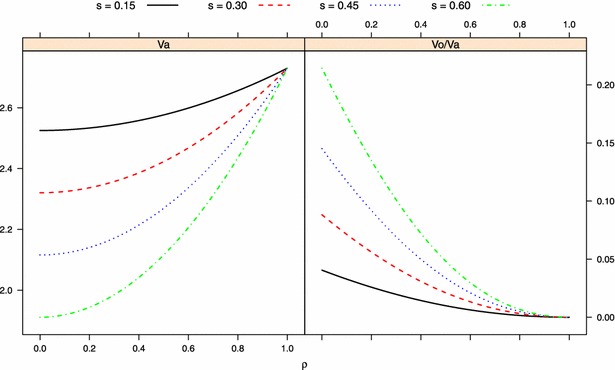
Stylized representation of the change of total additive variance across all 150 simulated QTL loci (, left panel) and proportion of total additive variance due to parent-of-origin effects (, right panel) at different values of (imprinting level, changes from 0 to 1) and s (, proportion of imprinted QTL)
Overfitting and combining the ADD and POE models
Modeling all SNPs with two substitution effects each (i.e., and ) could be problematic since not all SNPs are subject to parent-of-origin effects and this could be the cause of overfitting, as observed previously. In order to circumvent the potential overfitting problem in modeling the parent-of-origin effects, it may be worth to detect SNPs that are strongly associated with parent-of-origin effects a priori and model two substitution effects for those SNPs only. Furthermore, as an extension of [72], we assumed that parental contributions from the paternal and maternal sides are independent with equal variance at the pedigree level. However, when there are no parent-of-origin effects, these two effects are likely to be correlated [94]; in this case the overparameterization at the pedigree level may reduce the predictive ability as well [62], and it may be worth to drop the gametic relationship term from the model. To implement this analysis, we first detected such parent-of-origin-effect-associated SNPs by using the method suggested in our previous study [61]. The following model (ADD-POE model) was then evaluated in the same cross-validation approach to assess if it improved the overfitting problem:
| 12 |
where represents a set of markers with significant parent-of-origin effects at a 0.05 significance level after controlling for multiple testings using the Šidák’s correction.
Contrary to our expectation, Model 12 did not compromise prediction accuracy. Instead, the predictive performance of this model was only mildly better than the ADD model but much worse than the POE model in a simulation case where s is large and is small (Table 3). One possible reason for this result could be that although our simulation configured a relatively strong parent-of-origin case, the single-marker regression approach may still not be able to detect a large number of markers that are strongly associated with a true imprinted QTL (no replicates identified more than 50 significant SNPs), similar to all conventional GWAS studies. Therefore, the vast majority (i.e., ) of all available SNPs will be modeled as additive instead of “imprinted”, and hence the improvement of the ADD-POE model over the ADD model was very limited.
Table 3.
Comparison of predictive correlations (Pearson’s) among ADD, POE, and ADD-POE models
| Data | ||
|---|---|---|
| Mouse | 0.000 | 0.005 |
| 0.001 | −0.080 |
ADD additive model, POE parent-of-origin model, ADD-POE parent-of-origin model where two substitution effects are modeled only to markers with significant signals on parent-of-origin effects
Takes , as a benchmarking scenario
The ADD-POE model attempted to find a possible source of overfitting and tried to handle it at both the pedigree and the SNP levels. Since these two factors could be confounded, we added the term back to Model 12 and evaluated how the predictive ability changed. As a result, this model’s performance was almost identical to that of the ADD-POE model when using the simulated data, which indicated that when dense SNPs are used for prediction, modeling a relatively “rare” effect (here the parent-of-origin effects) across all SNPs may lead the model to suffer from severe overfitting, and overfitting due to this reason at the SNP level could be much larger than that due to an overparameterization at the pedigree level. Furthermore, our simulation chose QTL randomly and assigned a smaller “absolute” effect with a proportion parameter to reflect a non-equivalent contribution from the paternal and maternal genomes. Although this approach was able to introduce parent-of-origin effects, it may break some connection between the pedigree and the QTL that was established in the original QMSim simulation as well. This nearly identical predictive ability with or without the gametic relationship term as observed here could be the result of this disconnection. In order to better understand the behavior of gametic relationships, a more specific simulation approach that considers other mechanisms than imprinting would be helpful.
Proportion of imprinted QTL
In our simulation, values of 0.15, 0.3, 0.45 and 0.6 were assigned to s (proportion of imprinted QTL) in different scenarios. These values were chosen arbitrarily and are much larger than the proportion of imprinted genes with available evidence since, among the approximately 25,000 human or murine genes, only about 200 have been identified as imprinted (http://igc.otago.ac.nz/home.html), i.e., 1 % of the total number of genes. Even the smallest value of s chosen (0.15) is too large compared to this small fraction observed to date.
However, this (i.e., about 200) is the number of experimentally identified imprinted genes, approximately. This means that the function, expression profile and regulating mechanisms of such genes were assessed in well-designed experiments, with verified imprinting status. It is possible that there are more imprinted genes in the mammalian genomes that have not been discovered so far. For example, Luedi et al. [95] and Brideau et al. [96] predicted that there might be hundreds of imprinted genes in the murine genome, although no consensus estimate on the number of imprinted genes in the mammalian genome is available [97]. Furthermore, imprinting might be more prevalent than previously assumed, as argued in several review studies (e.g., [13, 98]). Specifically, among 127 detected metabolic-related QTL, about 60 % had imprinting effects. In an earlier study, 54 % of 602 genes expressed in human kidney or liver tissues were shown to have strong parent-of-origin effects caused by preferential expression, with some of them not located in known imprinted genomic regions [99]. Therefore, based on these studies, we decided to increase the proportion of imprinted QTL in our simulation over the 1 % mentioned earlier.
Moreover, for the approximately 200 identified imprinted genes, a vast majority are growth- and/or development-related. This was shown when the famous “parent-offspring conflict hypothesis” was proposed [100–102] to explain the evolution of imprinting. Although Lush often stated the view that all complex traits are possibly affected by all genes at various degrees [103, 104], it is unlikely that all tens of thousands of genes in the mammalian genome affect a trait jointly [105]. Since there is no consensus on how many genes affect specific complex traits, tens to several hundreds might be a reasonable guess. Hence, within the hundreds of genes that control a single trait, say, fetal growth, it is possible that a considerable proportion is subject to imprinting. In addition, imprinting is a major cause of parent-of-origin effects, but not the only one [106]. Therefore, when imprinting was considered as the only cause to simplify the source of parent-of-origin effects in the simulation, we set the proportion of imprinted QTL up to 60 %.
Other sources of information than DNA polymorphisms
Incorporating parent-of-origin effects into a prediction model may be helpful if it accounts for a considerable proportion of the total variance. In practice, additive variance is the major contributor to phenotypic variability for most complex traits [107]. Along with the overfitting problems associated with the POE model, the preceding implies that the POE model may bring only a minimal advantage in most cases. Therefore, it might be helpful to consider other sources of information in whole genome prediction to incorporate parent-of-origin effects. Since epigenetics, a main cause of parent-of-origin effects, is the study of heritable variation that does not involve a change of DNA sequence [108–110], our prediction model may fail under many situations because only variation at the DNA level (e.g., SNPs) is used as input. Hence, incorporating epigenetic information in addition to SNPs might be useful [111], as it has already been successfully used to identify disease-related genomic regions through epigenome-wide association studies (EWAS) [112–114].
Including epigenetic information in whole-genome prediction has been previously investigated and seems promising (e.g., [115, 116]). However, it can also be challenging. One aspect is the amount of information one needs to deal with. Consider DNA methylation as an example: it is the addition of a methyl group to either the 5-position carbon atom of the cytosine pyrimidine ring, or to the 6-position nitrogen atom of the adenine purine ring, with the latter observed mainly in mitochondrial DNA of flowering plants [117]. Two important features of DNA methylation are: (1) it is tissue and developmental-stage specific; (2) it is reversible, since the added methyl group can be removed from the methylated DNA. Due to this second feature, methylation status is unstable compared to DNA polymorphisms and, for a certain cytosine locus, it may shift between methylated and unmethylated states. Thus, although modern technologies are able to convert the unstable methylation information into stable sequence information via bisulfite treatment (e.g., [118, 119]), the methylation profile is for a specific time in a specific sample of cells. The term “methylome” is thus abused: in many studies, it actually refers to a “snap shot” of the entire methylome at a certain time point from a certain tissue given the first feature of DNA methylation. Compared to DNA sequence information which is size-invariant (unless a somatic mutation occurs) throughout an individual’s life time, the size of the methylome is highly variable and can be extremely large. Along with other epigenetic mechanisms such as histone modification, the size of the human epigenome is potentially enormous. For example, the diploid human epigenome contains more than cytosines (of which are found in CpG dinucleotides, the major target of mammalian DNA methylation) and more than histone tails (the target of histone modification) that can all potentially vary [112]. It has been estimated that the human epigenome could be thousands of times larger than the genome [120]! Given this magnitude, choosing appropriate epigenetic information from a suitable tissue is crucial, and powerful and reliable analytical tools must be developed to ensure an appropriate use of the information.
Apart from the size of the epigenome, epigenetic mechanisms are affected by environmental effects. For instance, the methyl group added to a DNA molecule must come from a methyl group donor. One major source is the diet [121], so different diets can result in different methylation profiles that lead to different phenotypes. Several cases demonstrate the impact of nutrition on epigenetics. In mice, the coat color of genetically identical individuals showed variation when their mothers were fed with different diets during pregnancy [122, 123]. In honey bees, almost all female individuals in a colony are (almost, if not exactly) genetically identical. However, the royalactin found in the royal jelly turns one (and only one) individual into a queen and the others remain as workers [124]. In livestock, maternal diet during pregnancy can alter the DNA methylation of the fetus and, hence, result in changes in gene expression [125]. This evidence indicates that environmental variation brings extra difficulties to the already complicated epigenetic analysis.
Furthermore, epigenome profiling is very expensive. In the case of methylation, due to the massive number of CpG sites within the mammalian genome, high-resolution methylation profiles are very costly. Although reduced representation bisulfite sequencing (RRBS, [126]) can reduce the profiling costs by selecting a small proportion of representative CpG sites from certain regions (e.g., gene promoter regions) of the genome, methylation profiling of a large cohort (e.g., thousands of individuals in a WGP study) is still expensive, especially when multiple “snapshots” of the methylome need to be considered.
In short, epigenetic polymorphisms could contribute to genetic studies and open a door to a better understanding of biological systems. However, many challenges need to be resolved before this information can be efficiently used to advantage.
Conclusions
We propose a model that is capable of incorporating parent-of-origin effects into whole-genome prediction using pedigree and DNA information. Our study on real and simulated data suggested that the POE model could be useful when parent-of-origin effects contributed a large proportion to the genetic variation, which was in agreement with a recent study that incorporated parent-of-origin effects in whole-genome prediction under a GBLUP framework [62]. In addition, our results draw attention to a possible overfitting problem when considering parent-of-origin effects in WGP with a Bayesian implementation, and indicate that one should be careful when using a POE model for prediction if the true signal attributed to parent-of-origin effects is weak in practice.
Owing to the discovery of more imprinted genes and of parent-of-origin-effects-affected complex traits, obtaining predictions that take parent-of-origin effects into account seems attractive. However, our simulation indicated that it did not always work well unless parent-of-origin effects contributed to the complex trait substantially. Hence, assessing the contribution of parent-of-origin effects to the total genetic variance (e.g., [127]) prior to model training might be helpful, as well as considering other sources of information than that from DNA polymorphisms (e.g., epigenetic variation) in evaluating parent-of-origin effects. Because many technical challenges need to be faced at the current stage of knowledge, future studies need to explore more effective prediction machines for parent-of-origin-effects-affected complex traits in animals, plants, and humans.
Authors' contributions
YH conceived the study, performed data analysis, and drafted the manuscript; GJMR and DG advised data analysis and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors are grateful to Co-Editors Dr. Hayes and Dr. Dekkers and two anonymous reviewers for their valuable comments on our manuscript. The authors also thank Monsanto Fellowship in Plant Breeding and Graduate Student Support Funding from the Department of Animal Sciences, University of Wisconsin-Madison to YH, Wisconsin Agriculture Experiment Station Hatch grant (142-PRJ28RT) from USDA to GJMR, and Wisconsin Agriculture Experiment Station Hatch grant (142-PRJ63CV) from USDA to DG.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Yaodong Hu, Email: yhu32@wisc.edu.
Guilherme J. M. Rosa, Email: grosa@wisc.edu
Daniel Gianola, Email: gianola@ansci.wisc.edu.
References
- 1.Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–5. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- 2.Delaval K, Feil R. Epigenetic regulation of mammalian genomic imprinting. Curr Opin Genet Dev. 2004;14:188–95. doi: 10.1016/j.gde.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 3.McEwen KR, Ferguson-Smith AC. Genomic imprinting—a model for roles of histone modifications in epigenetic control. In: Ferguson-Smith AC, Greally JM, Martienssen RA, editors. Epigenomics. Netherlands: Springer; 2009. pp. 235–258. [Google Scholar]
- 4.Hall JG. Genomic imprinting: review and relevance to human diseases. Am J Hum Genet. 1990;46:857–73. [PMC free article] [PubMed] [Google Scholar]
- 5.Solter D. Relevance of genomic imprinting to human diseases. Curr Opin Biotechnol. 1992;3:632–6. doi: 10.1016/0958-1669(92)90007-6. [DOI] [PubMed] [Google Scholar]
- 6.Falls JG, Pulford DJ, Wylie AA, Jirtle RL. Genomic imprinting: implications for human disease. Am J Pathol. 1999;154:635–47. doi: 10.1016/S0002-9440(10)65309-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clayton-Smith J. Genomic imprinting as a cause of disease. BMJ. 2003;327:1121–2. doi: 10.1136/bmj.327.7424.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Úbeda F, Wilkins JF. Imprinted genes and human disease: an evolutionary perspective. In: Wilkins JF, editor. Genomic imprinting. Vol. 626 of Advances in experimental medicine and biology. Austin, TX: Springer, New York & Landes Bioscience; 2008. p. 101–15. [PubMed]
- 9.Meijers-Heijboer EJ, Sandkuijl LA, Brunner HG, Smeets HJ, Hoogeboom AJ, Deelen WH, et al. Linkage analysis with chromosome 15q11-13 markers shows genomic imprinting in familial Angelman syndrome. J Med Genet. 1992;29:853–7. doi: 10.1136/jmg.29.12.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nicholls RD, Saitoh S, Horsthemke B. Imprinting in Prader–Willi and Angelman syndromes. Trends Genet. 1998;14:194–200. doi: 10.1016/S0168-9525(98)01432-2. [DOI] [PubMed] [Google Scholar]
- 11.Wolf JB, Hager R, Cheverud JM. Genomic imprinting effects on complex traits: a phenotype-based perspective. Epigenetics. 2008;3:295–9. doi: 10.4161/epi.3.6.7257. [DOI] [PubMed] [Google Scholar]
- 12.Kilpinen H, Dermitzakis ET. Genetic and epigenetic contribution to complex traits. Hum Mol Genet. 2012;21:R24–8. doi: 10.1093/hmg/dds383. [DOI] [PubMed] [Google Scholar]
- 13.Lawson HA, Cheverud JM, Wolf JB. Genomic imprinting and parent-of-origin effects on complex traits. Nat Rev Genet. 2013;14:609–17. doi: 10.1038/nrg3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knott SA, Marklund L, Haley CS, Andersson K, Davies W, Ellegren H, et al. Multiple marker mapping of quantitative trait loci in a cross between outbred wild boar and large white pigs. Genetics. 1998;149:1069–80. doi: 10.1093/genetics/149.2.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeon JT, Carlborg O, Tornsten A, Giuffra E, Amarger V, Chardon P, et al. A paternally expressed QTL affecting skeletal and cardiac muscle mass in pigs maps to the IGF2 locus. Nat Genet. 1999;21:157–8. doi: 10.1038/5938. [DOI] [PubMed] [Google Scholar]
- 16.Nezer C, Moreau L, Brouwers B, Coppieters W, Detilleux J, Hanset R, et al. An imprinted QTL with major effect on muscle mass and fat deposition maps to the IGF2 locus in pigs. Nat Genet. 1999;21:155–6. doi: 10.1038/5935. [DOI] [PubMed] [Google Scholar]
- 17.de Koning DJ, Bovenhuis H, van Arendonk JA. On the detection of imprinted quantitative trait loci in experimental crosses of outbred species. Genetics. 2002;161:931–8. doi: 10.1093/genetics/161.2.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stella A, Stalder KJ, Saxton AM, Boettcher PJ. Estimation of variances for gametic effects on litter size in Yorkshire and Landrace swine. J Anim Sci. 2003;81:2171–8. doi: 10.2527/2003.8192171x. [DOI] [PubMed] [Google Scholar]
- 19.Lee HK, Lee SS, Kim TH, Jeon GJ, Jung HW, Shin YS, et al. Detection of imprinted quantitative trait loci (QTL) for growth traits in pigs. Asian Aust J Anim Sci. 2003;16:1087–1092. doi: 10.5713/ajas.2003.1087. [DOI] [Google Scholar]
- 20.Thomsen H, Lee HK, Rothschild MF, Malek M, Dekkers JCM. Characterization of quantitative trait loci for growth and meat quality in a cross between commercial breeds of swine. J Anim Sci. 2004;82:2213–28. doi: 10.2527/2004.8282213x. [DOI] [PubMed] [Google Scholar]
- 21.Kim EH, Choi BH, Kim KS, Lee CK, Cho BW, Kim TH, et al. Detection of mendelian and parent-of-origin quantitative trait loci in a cross between korean native pig and landrace. I. Growth and body composition traits. Asian-Aust. J Anim Sci. 2007;20:669–76. [Google Scholar]
- 22.Engellandt TH, Tier B. Genetic variances due to imprinted genes in cattle. J Anim Breed Genet. 2002;119:154–65. doi: 10.1046/j.1439-0388.2002.00323.x. [DOI] [Google Scholar]
- 23.Essl A, Voith K. Genomic imprinting effects on dairy- and fitness-related traits in cattle. J Anim Breed Genet. 2002;119:182–9. doi: 10.1046/j.1439-0388.2002.00334.x. [DOI] [Google Scholar]
- 24.Meyer K, Tier B. Estimates of variances due to parent of origin effects for weights of Australian beef cattle. Anim Prod Sci. 2012;52:215–24. doi: 10.1071/AN11195. [DOI] [Google Scholar]
- 25.Lewis A, Redrup L. Genetic imprinting: conflict at the Callipyge locus. Curr Biol. 2005;15:R291–4. doi: 10.1016/j.cub.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 26.Cui Y, Lu Q, Cheverud JM, Littell RC, Wu R. Model for mapping imprinted quantitative trait loci in an inbred design. Genomics. 2006;87:543–51. doi: 10.1016/j.ygeno.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 27.Cui Y, Cheverud JM, Wu R. A statistical model for dissecting genomic imprinting through genetic mapping. Genetica. 2007;130:227–39. doi: 10.1007/s10709-006-9101-x. [DOI] [PubMed] [Google Scholar]
- 28.Liu T, Todhunter RJ, Wu S, Hou W, Mateescu R, Zhang Z, et al. A random model for mapping imprinted quantitative trait loci in a structured pedigree: an implication for mapping canine hip dysplasia. Genomics. 2007;90:276–84. doi: 10.1016/j.ygeno.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 29.Holl JW, Cassady JP, Pomp D, Johnson RK. A genome scan for quantitative trait loci and imprinted regions affecting reproduction in pigs. J Anim Sci. 2004;82:3421–9. doi: 10.2527/2004.82123421x. [DOI] [PubMed] [Google Scholar]
- 30.Wolf JB, Cheverud JM, Roseman C, Hager R. Genome-wide analysis reveals a complex pattern of genomic imprinting in mice. PLoS Genet. 2008;4:e1000091. doi: 10.1371/journal.pgen.1000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheverud JM, Hager R, Roseman C, Fawcett G, Wang B, Wolf JB. Genomic imprinting effects on adult body composition in mice. Proc Natl Acad Sci USA. 2008;105:4253–8. doi: 10.1073/pnas.0706562105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imumorin IG, Kim EH, Lee YM, De Koning DJ, van Arendonk JA, De Donato M, et al. Genome scan for parent-of-origin QTL effects on Bovine growth and carcass traits. Front Genet. 2011;2:44. doi: 10.3389/fgene.2011.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kärst S, Vahdati AR, Brockmann GA, Hager R. Genomic imprinting and genetic effects on muscle traits in mice. BMC Genomics. 2012;13:408. doi: 10.1186/1471-2164-13-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coster A, Madsen O, Heuven HC, Dibbits B, Groenen MA, van Arendonk JA, et al. The imprinted gene DIO3 is a candidate gene for litter size in pigs. PLoS One. 2012;7:e31825. doi: 10.1371/journal.pone.0031825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ribaut JM, Hoisington DA. Marker-assisted selection: new tools and strategies. Trends Plant Sci. 1998;3:236–9. doi: 10.1016/S1360-1385(98)01240-0. [DOI] [Google Scholar]
- 36.Guimarães EP, Ruane J, Scherf BD, Sonnio A, Dargie JD, editors. Marker-assisted selection: current status and future perspectives in crops, livestock, forestry and fish. Food and Agriculture Organization of the United Nations; 2007.
- 37.Young ND. A cautiously optimistic vision for marker-assisted breeding. Mol Breed. 1999;5:505–510. doi: 10.1023/A:1009684409326. [DOI] [Google Scholar]
- 38.Dekkers JC. Commercial application of marker- and gene-assisted selection in livestock: strategies and lessons. J Anim Sci. 2004;82(e–Suppl.):313–328. doi: 10.2527/2004.8213_supplE313x. [DOI] [PubMed] [Google Scholar]
- 39.Henderson CR. Applications of linear models in animal breeding. Guelph: University of Guelph; 1984. [Google Scholar]
- 40.Mrode RA. Linear models for the prediction of animal breeding values. 3. Wallingford: CAB International; 2014. [Google Scholar]
- 41.Meuwissen TH, Hayes BJ, Goddard ME. Prediction of total genetic value using genome-wide dense marker maps. Genetics. 2001;157:1819–29. doi: 10.1093/genetics/157.4.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goddard M. Genomic selection: prediction of accuracy and maximisation of long term response. Genetica. 2009;136:245–57. doi: 10.1007/s10709-008-9308-0. [DOI] [PubMed] [Google Scholar]
- 43.de los Campos G, Hickey JM, Pong-Wong R, Daetwyler HD, Calus MP. Whole-genome regression and prediction methods applied to plant and animal breeding. Genetics. 2013;193:327–345. doi: 10.1534/genetics.112.143313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schaeffer LR. Strategy for applying genome-wide selection in dairy cattle. J Anim Breed Genet. 2006;123:218–23. doi: 10.1111/j.1439-0388.2006.00595.x. [DOI] [PubMed] [Google Scholar]
- 45.Jonas E, de Koning DJ. Does genomic selection have a future in plant breeding? Trends Biotechnol. 2013;31:497–504. doi: 10.1016/j.tibtech.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 46.Nakaya A, Isobe SN. Will genomic selection be a practical method for plant breeding? Ann Bot. 2012;110:1303–16. doi: 10.1093/aob/mcs109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gianola D, de los Campos G, Hill WG, Manfredi E, Fernando R. Additive genetic variability and the Bayesian alphabet. Genetics. 2009;183:347–363. doi: 10.1534/genetics.109.103952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gianola D. Priors in whole-genome regression: the Bayesian alphabet returns. Genetics. 2013;194:573–96. doi: 10.1534/genetics.113.151753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.VanRaden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–23. doi: 10.3168/jds.2007-0980. [DOI] [PubMed] [Google Scholar]
- 50.Legarra A, Aguilar I, Misztal I. A relationship matrix including full pedigree and genomic information. J Dairy Sci. 2009;92:4656–63. doi: 10.3168/jds.2009-2061. [DOI] [PubMed] [Google Scholar]
- 51.Gianola D, Fernando RL, Stella A. Genomic-assisted prediction of genetic value with semiparametric procedures. Genetics. 2006;173:1761–76. doi: 10.1534/genetics.105.049510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gianola D, de los Campos G. Inferring genetic values for quantitative traits non-parametrically. Genet Res (Camb) 2008;90:525–540. doi: 10.1017/S0016672308009890. [DOI] [PubMed] [Google Scholar]
- 53.de los Campos G, Gianola D, Rosa GJ. Reproducing kernel Hilbert spaces regression: a general framework for genetic evaluation. J Anim Sci. 2009;87:1883–1887. doi: 10.2527/jas.2008-1259. [DOI] [PubMed] [Google Scholar]
- 54.Morota G, Gianola D. Kernel-based whole-genome prediction of complex traits: a review. Front Genet. 2014;5:363. doi: 10.3389/fgene.2014.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gianola D, Okut H, Weigel KA, Rosa GJ. Predicting complex quantitative traits with Bayesian neural networks: a case study with Jersey cows and wheat. BMC Genet. 2011;12:87. doi: 10.1186/1471-2156-12-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.González-Camacho JM, de Los Campos G, Pérez P, Gianola D, Cairns JE, Mahuku G, et al. Genome-enabled prediction of genetic values using radial basis function neural networks. Theor Appl Genet. 2012;125:759–71. doi: 10.1007/s00122-012-1868-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pérez-Rodríguez P, Gianola D, González-Camacho JM, Crossa J, Manès Y, Dreisigacker S. Comparison between linear and non-parametric regression models for genome-enabled prediction in wheat. G3 (Bethesda) 2012;2:1595–1605. doi: 10.1534/g3.112.003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mott R, Yuan W, Kaisaki P, Gan X, Cleak J, Edwards A, et al. The architecture of parent-of-origin effects in mice. Cell. 2014;156:332–42. doi: 10.1016/j.cell.2013.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spencer HG. The correlation between relatives on the supposition of genomic imprinting. Genetics. 2002;161:411–7. doi: 10.1093/genetics/161.1.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shete S, Amos CI. Testing for genetic linkage in families by a variance-components approach in the presence of genomic imprinting. Am J Hum Genet. 2002;70:751–7. doi: 10.1086/338931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu Y, Rosa GJ, Gianola D. A GWAS assessment of the contribution of genomic imprinting to the variation of body mass index in mice. BMC Genomics. 2015;16:576. doi: 10.1186/s12864-015-1721-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nishio M, Satoh M. Genomic best linear unbiased prediction method including imprinting effects for genomic evaluation. Genet Sel Evol. 2015;47:32. doi: 10.1186/s12711-015-0091-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hager R, Cheverud JM, Wolf JB. Maternal effects as the cause of parent-of-origin effects that mimic genomic imprinting. Genetics. 2008;178:1755–62. doi: 10.1534/genetics.107.080697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tuiskula-Haavisto M, de Koning DJ, Honkatukia M, Schulman NF, Maki-Tanila A, Vilkki J. Quantitative trait loci with parent-of-origin effects in chicken. Genet Res. 2004;84:57–66. doi: 10.1017/S0016672304006950. [DOI] [PubMed] [Google Scholar]
- 65.Tuiskula-Haavisto M, Vilkki J. Parent-of-origin specific QTL—a possibility towards understanding reciprocal effects in chicken and the origin of imprinting. Cytogenet Genome Res. 2007;117:305–312. doi: 10.1159/000103192. [DOI] [PubMed] [Google Scholar]
- 66.O’Neill MJ, Ingram RS, Vrana PB, Tilghman SM. Allelic expression of IGF2 in marsupials and birds. Dev Genes Evol. 2000;210:18–20. doi: 10.1007/PL00008182. [DOI] [PubMed] [Google Scholar]
- 67.Nolan CM, Killian JK, Petitte JN, Jirtle RL. Imprint status of M6P/IGF2R and IGF2 in chickens. Dev Genes Evol. 2001;211:179–83. doi: 10.1007/s004270000132. [DOI] [PubMed] [Google Scholar]
- 68.Frésard L, Morisson M, Brun JM, Collin A, Pain B, Minvielle F, et al. Epigenetics and phenotypic variability: some interesting insights from birds. Genet Sel Evol. 2013;45:16. doi: 10.1186/1297-9686-45-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de los Campos G, Naya H, Gianola D, Crossa J, Legarra A, Manfredi E, et al. Predicting quantitative traits with regression models for dense molecular markers and pedigree. Genetics. 2009;182:375–385. doi: 10.1534/genetics.109.101501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Erbe M, Pimentel ECG, Sharifi AR, Simianer H. Assessment of cross-validation strategies for genomic prediction in cattle. In: Proceedings of the 9th World Congress on Genetics Applied to Livestock Production. Leipzig, Germany; 2010.
- 71.Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2:21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- 72.Gibson JP, Kennedy BW, Schaeffer LR, Southwood OI. Gametic models for estimation of autosomally inherited genetic effects that are expressed only when received from either a male or female parent. J Dairy Sci. 1988;71 (Suppl. 1):143 (Abstr.).
- 73.Schaeffer LR, Kennedy BW, Gibson JP. The inverse of the gametic relationship matrix. J Dairy Sci. 1989;72:1266–1272. doi: 10.3168/jds.S0022-0302(89)79231-6. [DOI] [Google Scholar]
- 74.Dong C, Li WD, Geller F, Lei L, Li D, Gorlova OY, et al. Possible genomic imprinting of three human obesity-related genetic loci. Am J Hum Genet. 2005;76:427–37. doi: 10.1086/428438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rance KA, Fustin JM, Dalgleish G, Hambly C, Bunger L, Speakman JR. A paternally imprinted QTL for mature body mass on mouse chromosome 8. Mamm Genome. 2005;16:567–77. doi: 10.1007/s00335-005-0012-4. [DOI] [PubMed] [Google Scholar]
- 76.Gorlova OY, Amos CI, Wang NW, Shete S, Turner ST, Boerwinkle E. Genetic linkage and imprinting effects on body mass index in children and young adults. Eur J Hum Genet. 2003;11:425–32. doi: 10.1038/sj.ejhg.5200979. [DOI] [PubMed] [Google Scholar]
- 77.Legarra A, Robert-Granie C, Manfredi E, Elsen JM. Performance of genomic selection in mice. Genetics. 2008;180:611–8. doi: 10.1534/genetics.108.088575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Valdar W, Solberg LC, Gauguier D, Burnett S, Klenerman P, Cookson WO, et al. Genome-wide genetic association of complex traits in heterogeneous stock mice. Nat Genet. 2006;38:879–87. doi: 10.1038/ng1840. [DOI] [PubMed] [Google Scholar]
- 79.Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84:210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Browning BL. BEAGLE 3.3.2 User’s manual; 2011. http://faculty.washington.edu/browning/beagle/beagle.html.
- 81.Sargolzaei M, Schenkel FS. QMSim: a large-scale genome simulator for livestock. Bioinformatics. 2009;25:680–1. doi: 10.1093/bioinformatics/btp045. [DOI] [PubMed] [Google Scholar]
- 82.Barlow DP, Bartolomei MS. Genomic imprinting in mammals. Cold Spring Harb Perspect Biol. 2014;6:a018382. doi: 10.1101/cshperspect.a018382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Khatib H. Is it genomic imprinting or preferential expression? Bioessays. 2007;29:1022–8. doi: 10.1002/bies.20637. [DOI] [PubMed] [Google Scholar]
- 84.Abramowitz LK, Bartolomei MS. An in vitro ES cell imprinting model shows that imprinted expression of the Igf2r gene arises from an allele-specific expression bias. Development. 2009;136:437–48. doi: 10.1242/dev.032060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Barlow DP. Genomic imprinting: a mammalian epigenetic discovery model. Annu Rev Genet. 2011;45:379–403. doi: 10.1146/annurev-genet-110410-132459. [DOI] [PubMed] [Google Scholar]
- 86.Morcos L, Ge B, Koka V, Lam KC, Pokholok DK, Gunderson KL, et al. Genome-wide assessment of imprinted expression in human cells. Genome Biol. 2011;12:R25. doi: 10.1186/gb-2011-12-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wimmer V, Albrecht T, Auinger HJ, Schoen CC. Synbreed: a framework for the analysis of genomic prediction data using R. Bioinformatics. 2012;28:2086–2087. doi: 10.1093/bioinformatics/bts335. [DOI] [PubMed] [Google Scholar]
- 88.Pérez P, de los Campos G. Genome-wide regression and prediction with the BGLR statistical package. Genetics. 2014;198:483–495. doi: 10.1534/genetics.114.164442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.de los Campos G, Pérez Rodríguez P. BGLR: Bayesian generalized linear regression; 2014. R package version 1.0.3. http://CRAN.R-project.org/package=BGLR.
- 90.Park T, Casella G. The Bayesian Lasso. J Am Stat Assoc. 2008;103:681–686. doi: 10.1198/016214508000000337. [DOI] [Google Scholar]
- 91.Falconer DS, Mackay TFC. Introduction to quantitative genetics. 4. Englewood Cliffs: Prentice Hall; 1996. [Google Scholar]
- 92.Lynch M, Walsh B. Genetics and analysis of quantitative traits. Sunderland: Sinauer Associates; 1998. [Google Scholar]
- 93.Hastie T, Tibshirani R, Friedman J. The elements of statistical learning: data mining, inference and prediction. 2. Berlin: Springer; 2009. [Google Scholar]
- 94.Tier B, Meyer K. Analysing quantitative parent-of-origin effects with examples from ultrasonic measures of body composition in Australian beef cattle. J Anim Breed Genet. 2012;129:359–68. doi: 10.1111/j.1439-0388.2012.00996.x. [DOI] [PubMed] [Google Scholar]
- 95.Luedi PP, Hartemink AJ, Jirtle RL. Genome-wide prediction of imprinted murine genes. Genome Res. 2005;15:875–84. doi: 10.1101/gr.3303505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brideau CM, Eilertson KE, Hagarman JA, Bustamante CD, Soloway PD. Successful computational prediction of novel imprinted genes from epigenomic features. Mol Cell Biol. 2010;30:3357–70. doi: 10.1128/MCB.01355-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kelsey G, Bartolomei MS. Imprinted genes ... and the number is? PLoS Genet. 2012;8:e1002601. doi: 10.1371/journal.pgen.1002601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sha K. A mechanistic view of genomic imprinting. Annu Rev Genomics Hum Genet. 2008;9:197–216. doi: 10.1146/annurev.genom.122007.110031. [DOI] [PubMed] [Google Scholar]
- 99.Lo HS, Wang Z, Hu Y, Yang HH, Gere S, Buetow KH, et al. Allelic variation in gene expression is common in the human genome. Genome Res. 2003;13:1855–62. doi: 10.1101/gr.885403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Haig D, Westoby M. Parent-specific gene expression and the triploid endosperm. Am Nat. 1989;134:147–55. doi: 10.1086/284971. [DOI] [Google Scholar]
- 101.Haig D, Westoby M. Genomic imprinting in endosperm: its effect on seed development in crosses between species, and between different ploidies of the same species, and its implications for the evolution of apomixis. Philos Trans R Soc Lond B Biol Sci. 1991;333:1–13. doi: 10.1098/rstb.1991.0057. [DOI] [Google Scholar]
- 102.Moore T, Haig D. Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet. 1991;7:45–9. doi: 10.1016/0168-9525(91)90230-N. [DOI] [PubMed] [Google Scholar]
- 103.Lush JL. Animal breeding plans. 3. Ames: Iowa State College Press; 1945. [Google Scholar]
- 104.Lush JL. The genetics of populations (Mimeo) Ames: Iowa State Unviersity; 1948. [Google Scholar]
- 105.Gianola D, Rosa GJM. One hundred years of statistical developments in animal breeding. Annu Rev Anim Biosci. 2015;3:19–56. doi: 10.1146/annurev-animal-022114-110733. [DOI] [PubMed] [Google Scholar]
- 106.Guilmatre A, Sharp AJ. Parent of origin effects. Clin Genet. 2012;81:201–9. doi: 10.1111/j.1399-0004.2011.01790.x. [DOI] [PubMed] [Google Scholar]
- 107.Hill WG, Goddard ME, Visscher PM. Data and theory point to mainly additive genetic variance for complex traits. PLoS Genet. 2008;4:e1000008. doi: 10.1371/journal.pgen.1000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Riggs AD, Martienssen RA, Russo VEA. Introduction. In: Russo VEA, Martienssen RA, Riggs AD, editors. Epigenetic mechanisms of gene regulation. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1996. pp. 1–4. [Google Scholar]
- 109.Riggs AD, Porter TN. Overview of epigenetic mechanisms. In: Russo VEA, Martienssen RA, Riggs AD, editors. Epigenetic mechanisms of gene regulation. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1996. pp. 29–45. [Google Scholar]
- 110.Naumova AK, Greenwood CMT, editors. Epigenetics and complex traits. New York: Springer; 2013. [Google Scholar]
- 111.González-Recio O. Epigenetics: a new challenge in the post-genomic era of livestock. Front Genet. 2012;2:106. doi: 10.3389/fgene.2011.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011;12:529–41. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bell CG. Epigenome-wide association studies: potential insights into human disease. In: Naumova AK, Greenwood CMT, editors. Epigenetic and complex traits. New York: Springer; 2013. pp. 287–317. [Google Scholar]
- 114.Flanagan JM. Epigenome-wide association studies (EWAS): past, present, and future. In: Verma M, editor. Cancer epigenetics: risk assessment, diagnosis, treatment, and prognosis. Methods in molecular biology. Clifton: Humana Press; 2015. pp. 51–63. [DOI] [PubMed] [Google Scholar]
- 115.Vazquez AI, Wiener HW, Shrestha S, Tiwari H, de los Campos G. Integration of multi-layer omic data for prediction of disease risk in humans. In: Proceedings of the World Congress on genetics applied to livestock production. Vancouver, Canada; 2014.
- 116.Hu Y, Morota G, Rosa GJ, Gianola D. Prediction of plant height in Arabidopsis thaliana using DNA methylation data. Genetics. 2015;201:779–93. doi: 10.1534/genetics.115.177204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vanyushin BF. DNA methylation in plants. Curr Top Microbiol Immunol. 2006;301:67–122. doi: 10.1007/3-540-31390-7_4. [DOI] [PubMed] [Google Scholar]
- 118.Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89:1827–31. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bock C. Analysing and interpreting DNA methylation data. Nat Rev Genet. 2012;13:705–19. doi: 10.1038/nrg3273. [DOI] [PubMed] [Google Scholar]
- 120.Zhang Y, Jeltsch A. The application of next generation sequencing in DNA methylation analysis. Genes. 2010;1:85–101. doi: 10.3390/genes1010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Niculescu MD, Zeisel SH. Diet, methyl donors and DNA methylation: interactions between dietary folate, methionine and choline. J Nutr. 2002;132(Suppl. 8):2333S–5S. doi: 10.1093/jn/132.8.2333S. [DOI] [PubMed] [Google Scholar]
- 122.Morgan HD, Sutherland HG, Martin DI, Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nat Genet. 1999;23:314–318. doi: 10.1038/15490. [DOI] [PubMed] [Google Scholar]
- 123.Dolinoy DC, Weidman JR, Waterland RA, Jirtle RL. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ Health Perspect. 2006;114:567–572. doi: 10.1289/ehp.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kamakura M. Royalactin induces queen differentiation in honeybees. Nature. 2011;473:478–83. doi: 10.1038/nature10093. [DOI] [PubMed] [Google Scholar]
- 125.Lan X, Cretney EC, Kropp J, Khateeb K, Berg MA, Penagaricano F, et al. Maternal diet during pregnancy induces gene expression and DNA methylation changes in fetal tissues in sheep. Front Genet. 2013;4:49. doi: 10.3389/fgene.2013.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868–77. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lopes MS, Bastiaansen JW, Janss L, Knol EF, Bovenhuis H. Estimation of additive, dominance, and imprinting genetic variance using genomic data. G3 (Bethesda) 2015;5:2629–2637. doi: 10.1534/g3.115.019513. [DOI] [PMC free article] [PubMed] [Google Scholar]