

Abstract
Purpose
While the existence of CSC in ACC has been proposed, lack of assays for their propagation and uncertainty about molecular markers prevented their characterization. Our objective was to isolate CSC from ACC and provide insight into signaling pathways that support their propagation.
Experimental design
To isolate CSC from ACC and characterize them, we used ROCK inhibitor-supplemented cell culture, immunomagnetic cell sorting, and in vitro/in vivo assays for CSC viability and tumorigenicity.
Results
We identified in ACC CD133-positive CSC that expressed NOTCH1 and SOX10, formed spheroids, and initiated tumors in nude mice. CD133+ ACC cells produced activated NOTCH1 (N1ICD) and generated CD133− cells that expressed JAG1 as well as neural differentiation factors NR2F1, NR2F2, and p27Kip1. Knockdowns of NOTCH1, SOX10, and their common effector FABP7 had negative effects on each other, inhibited spheroidogenesis, and induced cell death pointing at their essential roles in CSC maintenance. Downstream effects of FABP7 knockdown included suppression of a broad spectrum of genes involved in proliferation, ribosome biogenesis, and metabolism. Among proliferation-linked NOTCH1/FABP7 targets we identified SKP2 and its substrate p27Kip1. A γ-secretase inhibitor, DAPT, selectively depleted CD133+ cells, suppressed N1ICD and SKP2, induced p27Kip1, inhibited ACC growth in vivo, and sensitized CD133+ cells to radiation.
Conclusions
These results establish in the majority of ACC the presence of a previously uncharacterized population of CD133+ cells with neural stem properties, which are driven by SOX10, NOTCH1, and FABP7. Sensitivity of these cells to Notch inhibition and their dependence on SKP2 offer new opportunities for targeted ACC therapies.
Introduction
Adenoid cystic carcinoma (ACC) accounts for nearly one quarter of malignant neoplasms of the salivary gland, and is a slow-growing, yet unpredictable tumor with a propensity for insidious local spread, perineural invasion, and distant pulmonary and brain metastases. Three major histology types are distinguished in morphologically heterogeneous ACC tumors: tubular, cribriform, and solid. Although variation in clinical presentation of these types has been reported, all three patterns may be neuroinvasive and show extremely high rates of recurrence. Treatment options for ACC are currently limited to surgery with or without radiation, consistent with limited insight into its molecular drivers (1). ACC has a high recurrence rate and dismal survival in part because of its intrinsic resistance to radio- and chemotherapies (2). Existence of cancer stem cells (CSC) in ACC has been suggested (3), but their molecular identity remained elusive due to difficulties with ACC culture. Development of targeted therapies for ACC has been further complicated by lack of reliable in vitro models, as there are currently no ACC cell lines available from centralized resources, and six previously created and shared cell lines were proven to be grossly contaminated or misidentified (4).
Recently, we used primary tumor specimens and patient-derived mouse xenografts (PDX) (5) to characterize genes differentially expressed in ACC compared to other head and neck cancers. These subcutaneous PDX models recapitulate basic ACC features, such as histologic appearance of the original tumor, characteristic t(6;9) translocations, and gene expression patterns (5, 6). While drawbacks of PDX models include relatively high maintenance costs and lack of interactions with the immune system, their ability to at least partially preserve tumor cell heterogeneity including CSC holds a potential to advance our knowledge of cancer biology and perform feasible pre-clinical studies (7-10). Our analysis of clinical and PDX data revealed neuronal genes and stem cell markers intrinsic to ACC, suggesting aberrant activation of a transcriptional program that controls neural stem cells (NSC). This hypothesis was supported by the association of ACC with activation of SOX10, a major transcriptional regulator and molecular marker of normal and malignant cells that originate from the neural crest (11, 12). Similar to ACC, SOX10 gene signatures were also established in basal-like breast carcinoma, melanoma, neuroblastoma, and glioma (13).
Here, we adopted a ROCK inhibitor-based approach that supports propagation of stem cells (14, 15) to produce sustainable ACC cell cultures that maintain cell lineage identity. Using this new approach, we characterized in ACC a previously unknown population of tumorigenic CD133+ cells that expressed SOX10, NOTCH1, activated intracellular NOTCH1 domain (N1ICD), and canonical NOTCH1 targets including SKP2, an E3 ubiquitin ligase that targets p27Kip1 for degradation and stimulates proliferation of CSC (16, 17). On the other hand, CD133- cells expressed JAG1 (a Notch ligand), p27Kip1 (a key cell cycle regulator), and neural differentiation genes NR2F1 and NR2F2. As Notch signaling is linked to cell proliferation and radiation resistance (18, 19) and can be pharmaceutically blocked (20), we investigated whether NOTCH1 inhibition in cultured ACC cells depletes CD133+ cells and sensitizes them to irradiation. Overall, we have identified in ACC a population of stem-like cells and delineated principal signaling pathways that may be used in the near future for ACC treatment.
Materials and Methods
PDX and primary tumor specimen
Patient-derived xenograft (PDX) models of ACC were created and validated as described in (5, 6). One clinical ACC specimen was collected from the Smilow Cancer Center at Yale New Haven Hospital (HIC# 1206010419).
Tissue processing
5-10 mg of fresh or cryopreserved (90% FBS and 10% DMSO) tumor tissue were rinsed once with PBS, 70% EtOH, 100X Anti-Anti (GIBCO), twice with PBS containing 1:500 ceftazidime, and minced. Digestion was performed at 37°C for 1-2 h with occasional agitation in 3 mL of DMEM media (10% FBS, 1x Pen/Strep, 1x L-Glutamine) supplemented with 1 mL of Dispase (BD Biosciences, San Jose, CA), 30-150 μL hyaluronidase (Sigma, St. Louis, MO), and 30-150 μL collagenase (Roche, Indianapolis, IN). Digested tissue was collected at 1,500 rpm for 3 min., rinsed with PBS, re-centrifuged, transferred into 3 mL of F+Y media (15), and filtered using a 100 μm cell strainer. Tumor cells were cultured in a CO2 incubator with irradiated 3T3-J2 cells or conditioned media derived from these cells (see below).
Cell culture
3T3-J2 feeder cells were grown as described (15). To create conditioned media, irradiated 3T3-J2 cells were incubated in a T-150 flask supplemented with 30 mL of DMEM media for 4 days. Media was filter-sterilized and then mixed in a 1:4 ratio with F+Y media.
Real-time RT-PCR
RNA was isolated from frozen cell pellets using RNeasy kit (Qiagen, Valencia, CA). cDNA was generated using Bio-Rad iScript Reverse Transcriptase kit. Real-time PCR was performed using iQ SYBR Green Supermix and CFX96 real-time detection system (Bio-Rad, Hercules, CA).
Western blot analysis
We used the following antibodies: SOX10 (Abcam, ab155279), NOTCH1 (Cell Signaling, #3608), FABP7 (Cell Signaling, #13347), cleaved NOTCH1 (Cell Signaling, #4147), p27Kip1 (Cell Signaling, #2552), SKP2 (Cell Signaling, #4313), β-actin (Santa Cruz, sc-47778), and GAPDH (Santa Cruz, sc-25778). Pre-cast gels, Rapid Transfer Turbo-blot System, and gel imaging software were from Bio-Rad.
Microsatellite analysis
For cell line authentication, we used Short Tandem Repeat (STR) analysis recommended by ATCC https://www.atcc.org/∼/media/PDFs/Technical%20Bulletins/tb08.ashx. Using Promega GenePrint 10 STR analysis PCR kit with fluorescent tagging, PCR products were analyzed on Applied Biosystems 3730xL DNA Analyzer at the Yale Keck Facility and data processed using GeneMapper 3.7 (Applied Biosystems) software. The results were compared to STR databases (http://www.cstl.nist.gov/strbase/, http://www.dsmz.de/services/services-human-and-animal-cell-lines/online-str-analysis.html).
Flow cytometry
Cells were collected and incubated with CD133-PE antibody (Miltenyi) for 10 minutes. Fluorescence-activated cell sorting (FACS) was performed to determine percentage of CD133+ cells using a FACS Caliber BD machine. Data analysis performed using Flowing Software version 2.5.0.
Cell sorting
For separation of CD133+ and CD133- cells from cultured cells and grafted tissue we used CD133 Tumor Tissue MicroBead Kit from Miltenyi (San Diego, CA) according to the manufacturer's protocol.
Tumorigenicity assays
All mouse experiments were performed in accordance with NIH national guidelines and approved by Yale IACUC. Athymic NCr-nu/nu mice were purchased from NCI-Frederick (Frederick, MD) and used for subcutaneous injections in the flanks with a specified number of viable tumor cells.
Spheroidogenesis and spheroid viability assays
For spheroid studies we used tools and approaches previously described (21). Briefly, spheroid formation and disintegration were studied in 6-well plates with or without DAPT (Eli Lilly, GSI-IX) from Selleckchem, Houston, TX, or DMSO control. At 24h intervals, 3 separate representative digital images per well at 4X magnification were used to quantify spheroid number.
Immunofluorescence staining
Cells were plated onto glass slides at a density of 25,000-30,000, incubated for 24 h, washed twice with PBS, and fixed with 3-4% paraformaldehyde at RT for 30 min. Cell were permeabilized with 0.15% Triton X-100 in PBS for 5 minutes, washed twice, and blocked with 3% BSA for 30 minutes. Incubation with primary antibodies diluted in 3% BSA was done at 40C overnight with subsequent washing once with PBS and once with 3% BSA. The following antibodies were used: SOX10 (Abcam, ab155279), FABP7 (D8N3N), NOTCH1 (D1E11), and SKP2 (#4313) from Cell Signaling, JAG1 (C-20) and NR2F1 (sc-74561) from Santa Cruz, NR2F2 (H7147) from Parsons Proteomics, and AlexaFluor secondary antibodies from Life Tech.
Immunohistochemical staining
The following antibodies were used: cleaved NOTCH1 (Val1744) antibodies from Cell Signaling (Danvers, MA), SOX10 antibodies from Cell Marque (Rocklin, CA), and FABP7 antibodies from Abcam (Ab32423, Cambridge, MA).
Proliferation assays
Cells were cultured in 96-well black clear bottom plates and treated with DAPT versus control (DMSO). After cells were grown for 24-96 h, viable cells were assayed using the Cell Titer Glo system (Promega).
Cell cycle analysis
Cells were fixed in 70% cold ethanol and stained with propidium iodide. A FACS Caliber BD machine was used and data analysis performed using Flowing Software version 2.5.0.
Cell death assay
Cells were incubated for 10 minutes with CD133-FITC antibody (Miltenyi) as well as Annexin V antibody and 7-AAD DNA stain (Annexin V:PE apoptosis detection kit I, BD Biosciences). A FACS Caliber BD platform was used and data analysis was performed using Flowing Software version 2.5.0.
Radiosensitivity assay
Cells plated in T-25 flasks were irradiated using Mark I Cesium-137 irradiator, incubated for 48 h in the presence or absence of DAPT, and stained with CD133-PE antibody. FACS was performed to determine percentage of CD133+ cells. Statistical differences in tumor volume were determined using a two-tailed t-test.
DAPT studies on ACC xenografts
In vivo efficacy of DAPT was evaluated in an Accx11 xenograft model generated via subcutaneous injection of 106 cultured Accx11 cells. Tumor cell-injected mice were randomized into control (vehicle-injected) and experimental (DAPT in corn oil) groups 15 days after cell injection. DAPT was given via i.p. injection at 20 mg/kg, following a 3/4 injection schedule (3 days of treatment/4 days rest) for 35 days. To assess DAPT toxicity, animals were observed daily and weighed weekly using a digital scale. To assess DAPT efficacy, tumor dimensions were measured weekly by a digital caliper and data including individual and mean estimated tumor volumes (mean TV±s.e.m.) recorded for each group; tumor volume was calculated using the formula TV=width2 × length × 0.52. Statistical differences in tumor volume were determined using a two-tailed t-test.
Results
Generation and validation of primary cell cultures from ACC tissue
ACC cells do not normally grow in regular cell culture media (our unpublished observations). To overcome scarcity of clinical material and develop a robust assay for ACC culture, we used PDX models that have been validated for ACC-intrinsic markers and histology (5, 6). Optimization of a recently published conditional reprogramming cell culture (CRC) protocol (15) allowed us to produce intermediate and long-term cell cultures from 5 ACC xenografts and one primary ACC tumor (Fig. 1A and B). To confirm authenticity of newly created cell cultures, we used STR DNA profiling (22) recommended by ATCC to compare microsatellite patterns of cultured cells with parental PDX tumors (Suppl. Table 1).
Figure 1. Clinical, cytological, and molecular properties of ACC cell cultures.
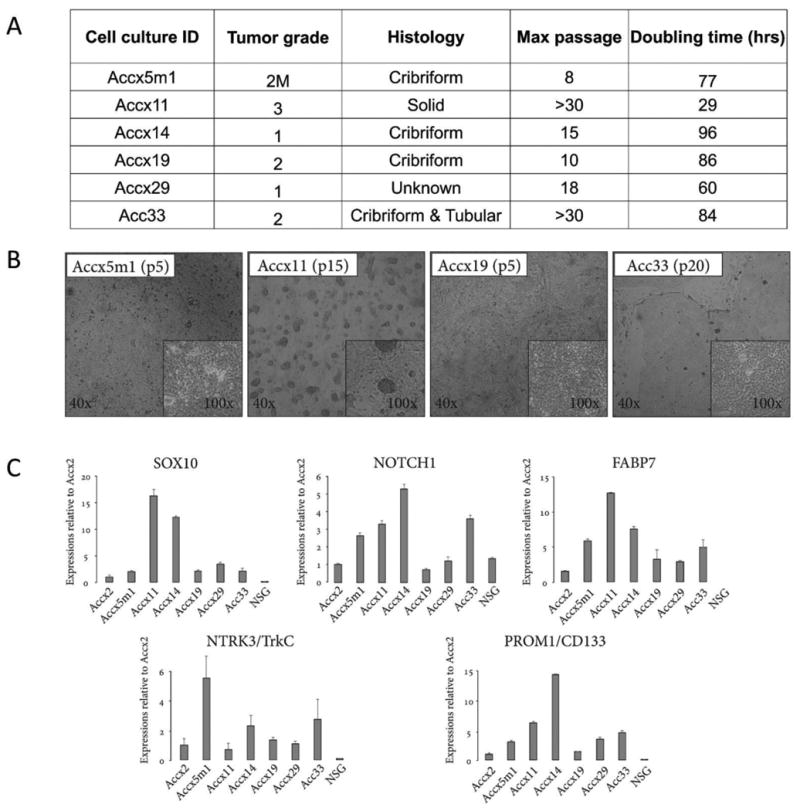
(A) ACC cell cultures produced from PDXs (Accx) and one clinical specimen (Acc33). M, metastases. (B) Low and high magnification brightfield images of cultured ACC cells at indicated passages for Accx5m1, Accx11, Accx19, and Acc33. (C) Real-time PCR quantification (qRT-PCR) of gene expression for SOX10, NOTCH1, FABP7, NTRK3/TrkC, and PROM1/CD133 in cultured ACC cells compared to cultured cells isolated from normal salivary gland (NSG). In all qRT-PCR experiments, expression is normalized to β-actin and error bars show standard errors representative of at least two independent experiments.
We also used ACC markers that we recently introduced to validate stem cell identity of cultured cells. In previous studies, we linked TrkC/NTRK3, SOX10, NOTCH1, and FABP7 expression in clinical specimens to an ACC-intrinsic gene signature that contained ∼200 genes (6, 13). Reassuringly, all ACC cell cultures expressed all four neural stem genes (Fig. 1C) recapitulating clinical ACC specimens and PDX used for cell culture (Suppl. Fig. 1). Cultures of normal salivary gland cells, however, showed no FABP7 expression and barely detectable levels of SOX10 and NTRK3. Thus, the ROCK inhibitor and cell feeder-based protocol is an effective solution to the ACC cell culture problem that may significantly advance translational ACC research.
Isolation and characterization of previously unrecognized CD133+/SOX10+/NOTCH1+ ACC cells
Expression of SOX10, a key neural stem cell marker, in clinical ACC specimens and cancers that originate from neural crest (6, 13) suggested existence in ACC of SOX10+ cells with neural stem properties. To validate this hypothesis, we profiled ACC for expression of cell surface markers that could be used for CSC isolation and noticed that CD133/PROM1, a CSC cell surface marker (23), is expressed in nearly all clinical ACC specimens and PDX (Suppl. Fig. 1). While CD133 expression was recently reported in ACC (24), there have been no published attempts to isolate and characterize CD133+ ACC cells. Interestingly, CD133 was expressed in all ACC cell cultures that we generated but not in normal salivary epithelial cells (Fig. 1C) suggesting that it may be used as a tool for CSC isolation.
To produce sufficient amounts of CD133-expressing ACC cells, a robustly proliferating and spheroid-forming culture, Accx11 (Fig. 1A and B), was used. Accx11 was derived from a xenograft of grade 3 ACC that are distinguished with solid histology and poor prognosis (25). In Accx11, 2-15% of cells expressed CD133 according to FACS analysis (Suppl. Fig. 2). Magnetic activated cell sorting (MACS) was then performed and purified CD133+ and CD133- fractions characterized (Fig. 2). This separation produced more than 50-fold enrichment of CD133 expression (Fig. 2A). Noteworthy, CD133+ cells expressed at least 25-fold higher levels of SOX10, NOTCH1, and FABP7 as compared to CD133- cells suggesting that expression of these genes co-segregates with CD133 (Fig. 2B and Suppl. Table 2).
Figure 2. Isolation and characterization of CD133+ and CD133- cells in Accx11 culture.
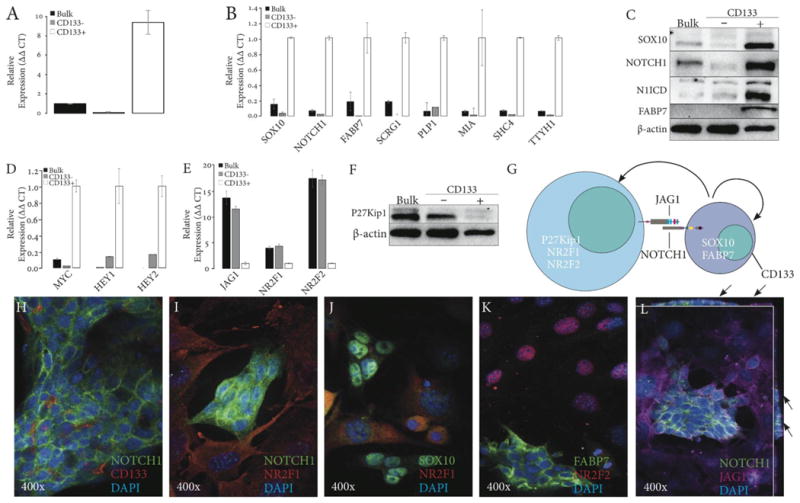
(A) qRT-PCR of CD133 expression in Accx11 cell fractions following magnetic-activated cell sorting (MACS). (B) Expression analysis by qRT-PCR of ACC-associated (SOX10 & FABP7) and NSC-associated (NOTCH1, SCRG1, PLP1, MIA, SHC4, & TTYH1) genes in MACS-sorted Accx11 cell fractions. (C) Immunoblot analysis shows selective expression of SOX10, NOTCH1, activated intracellular domain of Notch-1 (N1ICD), and FABP7 in CD133+ cells. β-actin serves as a loading control. (D) Expression analysis by qRT-PCR of Notch targets MYC, HEY1, and HEY2 in MACS-sorted Accx11 cell fractions. (E) Expression analysis by qRT-PCR of JAG1 and neural differentiation markers NR2F1 and NR2F2 in MACS-sorted Accx11 cell fractions. (F) Immunoblot analysis of CD133 fractions demonstrates CD133- cell-specific p27Kip1 expression. β-actin serves as a loading control. (G) Molecular determinants of stem-like CD133+ cells and more differentiated CD133- cells. (H-L) Immunofluorescence (IF) staining demonstrates great selectivity of CD133+ and CD133- cell markers and confirms spheroid-forming property of CD133+ cells. Z-stack images (L) show NOTCH1 and JAG1 co-localization at sites of spheroidal and non-spheroidal cell contacts (arrows).
After separation, cultured CD133+ cells generated both spheroid-forming and tightly adherent cells. Cultured CD133- Accx11, however, remained adherent and were not able to produce CD133+ cells or spheroids (data not shown). All these data suggested that CD133+, but not CD133- cells, express key stem cell markers and are capable of asymmetric division recreating both cell populations. To further explore stem properties of CD133+ cells, expression of SCRG1, PLP1, MIA, SHC4, and TTYH1 previously linked to the SOX10 neural gene signature in ACC (13) was investigated. Remarkably, without exception these genes were more highly expressed in CD133+ ACC cells compared to bulk or CD133- cells (Fig. 2B and Suppl. Table 2). In light of the association of these genes with NSC cancers (26-29), their selective expression in CD133+ ACC provided more support into possible ACC origin from NSC-like cells.
Activation of Notch is a common feature of NSC and CSC (30). Immunoblotting revealed high expression of NOTCH1 and N1ICD in CD133+, but not in CD133- or unsorted cells and confirmed high expression levels of SOX10 and FABP7 in CD133+ cells (Fig. 2C and Suppl. Table 2). In line with Notch activation, its canonical targets HEY1, HEY2, and MYC were predominantly expressed in CD133+ cells (Fig. 2D and Suppl. Table 2).
To determine if other ACC cultures harbored similar cell populations, we examined Accx19 cells (Fig. 1A, B, and C). In agreement with lower expression levels of SOX10, NOTCH1, and FABP7, the percentage of CD133+ cells in Accx19 determined by FACS analysis was lower than in Accx11 comprising <8% of total cells (data not shown). MACS separation of Accx19 cells produced 2.5-3-fold enrichment of CD133 expression in CD133+ cells compared to CD133- cells. In CD133+ Accx19 cells, expression of NOTCH1, SOX10, and FABP7 was similarly enriched (Suppl. Fig. 3A). Overall, these data supported the existence in ACC of a previously unknown population of SOX10-positive CD133+ cells with activated NOTCH1.
CD133− ACC cells express neuronal differentiation genes NR2F1 and NR2F2, p27Kip1, and a Notch ligand, JAG1
Differentiation states of CD133-sorted cells were analyzed via two orphan nuclear receptors, NR2F1 and NR2F2, required for specification of NSC (31). This comparison revealed markedly increased expression of both genes in CD133- cells (Fig. 2E and Suppl. Table 2) suggesting that these cells are more advanced toward differentiation.
Jagged-1 (JAG1), a canonical Notch ligand, is an essential component of Notch signaling, and in many cancers it is expressed not in stem cells but in supporting niches (32, 33). Interestingly, JAG1 expression was markedly higher (∼11-fold) in CD133- cells (Fig. 2E and Suppl. Table 2). Enhanced expression of NR2F2 and JAG1 was also observed in CD133- cells isolated from Accx19 cultures (Suppl. Fig. 3B).
p27Kip1 is a critical effector of neural differentiation, and in NSC NOTCH1 blocks p27Kip1 expression (34, 35). Given that NOTCH1 is preferentially activated in CD133+ cells and that CD133- cells express markers of neural differentiation, we measured p27Kip1 expression in both fractions. Immunoblotting of unsorted and CD133- cells revealed significantly higher p27Kip1 levels compared to the CD133+ population (Fig. 2F).
Based on selective expression of NOTCH1 and its targets in CD133+ cells and JAG1 and NR2F1/2 in CD133- cells, we suggested that these cell subpopulations may communicate via NOTCH1/JAG1 interaction (Fig. 2G) and explored this hypothesis using immunofluorescent staining. In agreement with RT-PCR and immunoblot data (Fig. 2B and C), membrane CD133 and NOTCH1 staining was detected in cells associated with spheroids, but not in the adherent cell population (Fig. 2H and I). Similarly, SOX10 and FABP7 stained cells in spheroids, while differentiation markers NR2F1 or NR2F2 were seen in cells surrounding spheroids (Fig. 2I, J, and K). Remarkably, NOTCH1-expressing cells within spheroids were closely surrounded by adherent cells expressing membrane-localized Jagged-1 (Fig. 2L). Z stack images revealed that Jagged-1 and NOTCH1 were co-localized on the surface of the spheroid (Fig. 2L, see arrows) supporting the proposed NOTCH1/JAG1 communication and signal-sending role of JAG1-producing CD133- cells.
CD133+ and CD133- populations identified in grafted ACC tissue
To rule out possible cell culture artifacts, the existence of CD133+ and CD133- cell populations was validated on PDX tissue derived from 5 ACC patients. To this end, PDX tumors were dissociated into cells, and CD133+ and CD133- populations immediately separated. CD133+ cells comprised from 21 to 65% of viable cells derived from xenografts (Suppl. Fig. 4A), and real-time PCR revealed that these cells expressed higher levels of SOX10, NOTCH1, and FABP7, with a few exceptions (Suppl. Fig. 4B-E). On the other hand, CD133- cells universally expressed higher levels of JAG1, while expression of NR2F1 was more variable (Suppl. Fig. 4F and G). These results mirrored our findings in cultured cells derived from ACCX11 and ACCX19 (Fig. 2 and Suppl. Fig. 3, respectively). Collectively, these data confirmed the existence of two cell phenotypes, SOX10+/NOTCH1+/FABP7+/CD133+ and JAG1+/CD133- , in the majority of ACC tested.
Tumorigenicity of ACC cultures and dependency on CD133 status
The ability of cultured ACC cells to initiate tumors was investigated via subcutaneous injections of 106 cells into flanks of nude mice. Among ACC cultures tested to date, only Accx11 cells were tumorigenic in nude mice creating tumors with solid histology and an intervening stromal component similar to the parental patient tumor and PDX (Fig.3A). Relative expression of CD133, SOX10, NOTCH1, and FABP7 was similar in the original PDX and in the tumor produced from injection of cultured cells (Fig. 3B and C).
Figure 3. Tumorigenic properties of bulk and CD133-fractionated Accx11 cells.
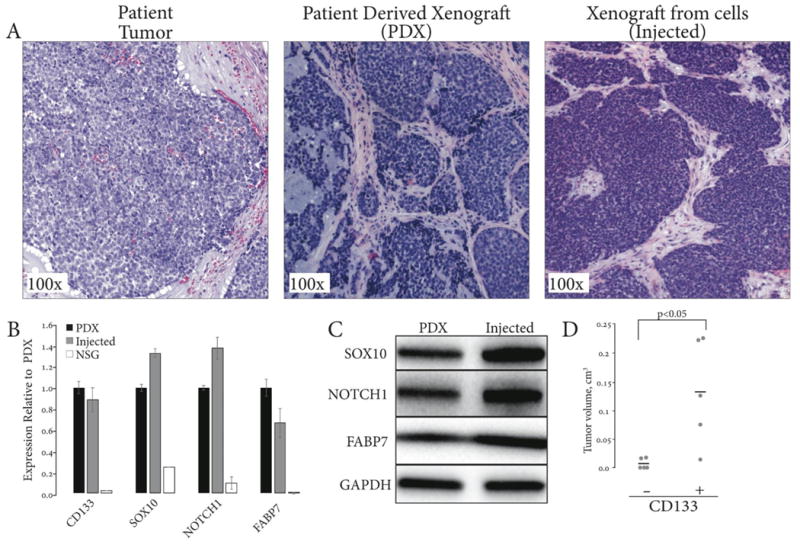
(A) Cultured bulk Accx11 subcutaneously injected in athymic nude mice produce tumors with histology similar to patient tumor and parental xenograft (H&E staining). (B) CD133, SOX10, NOTCH1, and FABP7 expression confirms ACC identity of PDX and tumors generated from injection of bulk Accx11 cells. (C) Immunoblot analysis confirms similar expression of SOX10, NOTCH1, and FABP7 in PDX and tumor formed from injected bulk Accx11 cells. GAPDH serves as a loading control. (D) Dot plot of tumor volumes 90 days after injection of 105 CD133- or CD133+ MACS-sorted Accx11 cells.
In various cancers, CD133 expression defines populations of CSC with enhanced tumor-initiating properties (36). To investigate CSC properties of CD133-expressing ACC cells, bulk Accx11 cells were sorted as above and 104 cells injected subcutaneously into right (CD133+) and left (CD133-) flanks of three nude mice. After 22 weeks, tumors were observed in 3 of 3 sites injected with CD133+ cells, but in none of 3 sites injected with CD133- cells. At 32 weeks, a single tumor formed at a site injected with CD133- cells. Histologic evaluation of the parental ACCX11 tumor, tumors derived from CD133+ cell injections, and the single tumor from a CD133- cell injection revealed similar histologic appearance (Suppl. Fig. 5A and data not shown). Expression analyses of CD133, SOX10, NOTCH1, and FABP7 by qRT-PCR and immunoblotting revealed expression of all markers in tumors derived from both CD133+ and CD133- cells (Suppl. Fig. 5B and C) suggesting that small amounts of CD133+ are trapped in the CD133- fraction or that injected CD133- cells can convert to CD133+ cells.
As experience with CD133 cell separation increased, we found that single round of separation using CD133 antibody conjugated to beads left detectable traces of CD133+ cells in the CD133- population reflecting technical limitations of this procedure. Also, separation decreased viability of CD133+ cells more than CD133- cells, possibly due to the damage inflicted by elution of column-bound cells (data not shown). To more adequately compare tumorigenicity of CD133+ and CD133- cells, a second round of magnetic separation was performed on the unbound cells to isolate a more pure CD133- population. Following this double separation, 105 viable cells of each population were injected into the flanks of nude mice. Tumors began to form at the sites of CD133+ injections as early as 6.5 weeks after injection. By 13 weeks post-injection, 4 of 5 CD133+ injection sites had distinct tumors while none of 5 CD133- injections showed signs of tumor formation (Fig. 3D). With a marked delay, 2 of 5 injection sites of CD133- cell injections formed small tumors with the earliest tumor developing at 19 weeks. At 27.5 weeks, all of 5 CD133+ injection sites formed tumors, while no additional tumors grew in CD133- injection sites. Overall, these experiments demonstrated enhanced tumorigenicity of CD133+ cells compared to CD133- cells.
Interdependence between NOTCH1, SOX10, and their common effector FABP7
Essential individual roles for SOX10 and NOTCH1 in NSC maintenance are well established, and loss of Notch signaling decreases SOX10 expression in neural progenitors (37, 38). FABP7 has recently been described as critical for proliferation and prevention of differentiation in neural progenitors (39). Given that SOX10, NOTCH1, and FABP7 are each implicated in neural stemness, their interdependence in ACC cells was explored. Following individual depletions, expression of all three genes was measured by qRT-PCR and immunoblotting. Knockdown of SOX10 markedly decreased NOTCH1 expression and, similarly, depletion of NOTCH1 was associated with decreased SOX10 both at the mRNA and protein levels (Fig. 4A and B) suggesting interdependence and functional cooperation. As expected, since FABP7 is a common NOTCH1/SOX10 target (40, 41), depletion of either NOTCH1 or SOX10 markedly suppressed FABP7 mRNA and protein levels. Depletion of FABP7 had less suppressive effects on NOTCH1 and SOX10 mRNA than did either NOTCH1 or SOX10 knockdowns. At the protein level, FABP7 had no obvious effect on NOTCH1 expression while modestly decreasing SOX10 levels (Fig. 4A and B).
Figure 4. Interrelated stimulatory effects of SOX10, NOTCH1, and FABP7 on ACC cells.
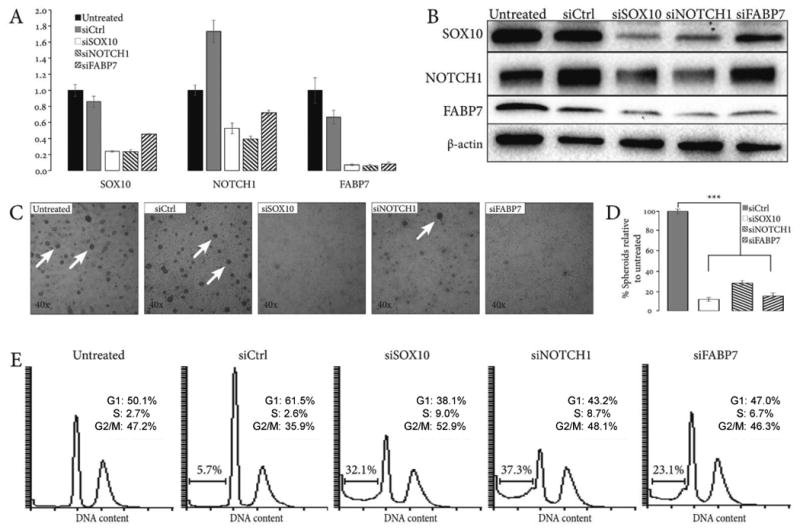
(A) qRT-PCR analysis of SOX10, NOTCH1, and FABP7 expression following siRNA knockdowns reveals their regulatory interdependence. (B) Validation of SOX10, NOTCH1, and FABP7 interdependence by Western blot. β-actin serves as a loading control. (C) Depletion of SOX10, NOTCH1, and FABP7 by siRNA suppresses spheroidogenesis of Accx11 cells at 72 h. Arrows: brightfield images of spheroids. (D) Quantification of suppressive SOX10, NOTCH1, and FABP7 knockdown effects on spheroids compared to untreated Accx11 cells. Error bars represent standard errors and are representative of at least two independent experiments. In each case, siRNA knockdown resulted in a significant decrease in spheroid formation (p<0.001, Student's t-test). (E) Suppression of SOX10, NOTCH1, and FABP7 by siRNA increases sub-G1 cell population indicative of cell death. Histograms represent cell counts based on propidium iodide (PI) staining of Accx11 cells. Percentages of live cells in different cell cycle phases are shown.
Individual roles of NOTCH1 and SOX10 in CSC and NSC survival are well established, but their co-operation via FABP7 has not yet been studied. To begin exploring the role of FABP7 in adenoid cystic carcinoma signaling, it was depleted in Accx11 cells followed by gene expression profiling. Analyses revealed that 1158 genes were downregulated (Suppl. Table 4, 2-fold threshold), and KEGG pathway analysis (http://bioinfo.vanderbilt.edu/webgestalt/) implicated these genes in cell cycle regulation, ribosome biogenesis, cell metabolism, and signaling pathways involved in these processes (Suppl. Table 4 and data not shown). Among these genes, 11 belonged to the Notch signaling network and 26 genes overlapped with the ACC SOX10 gene signature that we previously characterized (13). This observation further supported the feedback signaling from FABP7 to NOTCH1 and SOX10. In addition, SKP2, a major NOTCH1 effector involved in the regulation of cell cycle and proliferation (42), was among 31 cell cycle progression and 11 NOTCH1-regulated genes downregulated by FABP7 knockdown (Suppl. Table 4). In summary, these data suggested that NOTCH1, SOX10, and FABP7 may be co-regulated in ACC, and that FABP7 may serve as a pro-survival NOTCH1 and SOX10 effector.
SOX10, NOTCH1, and FABP7 stimulate spheroidogenesis of Accx11 cells
NSC and CSC have the propensity to form spheroids in culture (43). Using spheroid formation as a surrogate of stem cell survival and proliferation, functional consequences of SOX10, NOTCH1, and FABP7 knockdowns were determined. Individual depletion of these genes significantly suppressed spheroid formation in Accx11 cells (Fig. 4C and D). These data suggested that SOX10, NOTCH1, and FABP7 are vital for spheroid formation, which could be an effect on CSC survival and growth. Flow cytometry analyses of Accx11 were performed to assess the effects of SOX10, NOTCH1, and FABP7 depletion on cell cycle progression and cell survival. A marked (∼4-6-fold) increase in the subG1 populations, decrease in G1, and increase in S phases were observed following NOTCH1, SOX10, or FABP7 depletion (Fig. 4E). Altogether, these results suggested that NOTCH1, SOX10, and FABP7 are important for survival of ACC cells, cell cycle progression, and maintenance of stem-like phenotype.
NOTCH1 depletion and γ-secretase inhibition decrease the proportion of CD133+, suppress spheroidogenesis, inhibit tumor growth in nude mice, and sensitize CD133+ to radiation
Inhibitors of SOX10 and FABP7 are not available for clinical use at this time, but Notch is targeted through γ-secretase inhibitors (GSI) that have advanced to clinical trials (44). The suppressive effects on spheroid formation and cell viability seen with NOTCH1 loss (Fig. 4C-E) suggested that NOTCH1 is required for survival of CD133+ ACC cells. To test this hypothesis, NOTCH1 was depleted in bulk Accx11 cells and the percentage of CD133+ cells measured. NOTCH1 depletion resulted in a ∼2.5-fold decrease in the proportion of CD133+ cells (Fig. 5A). Bulk Accx11 cells treated with DAPT (a GSI developed by Eli Lilly) showed suppression of spheroid formation in a concentration-dependent manner (Fig. 5B and C). To determine if γ-secretase activity is also required for maintenance of pre-formed spheroids, Accx11 cells were grown to confluency to allow spheroid formation as shown in Fig. 1B. Over a 9-day time course, a dose-dependent loss of pre-formed spheroids was observed in DAPT-treated Accx11 cells compared to controls (Fig. 5D); however, the effect of γ-secretase inhibition on maintenance of preformed spheroids was not as dramatic as its effect on spheroidogenesis.
Figure 5. Inhibition of Notch signaling selectively depletes CD133+ cells.
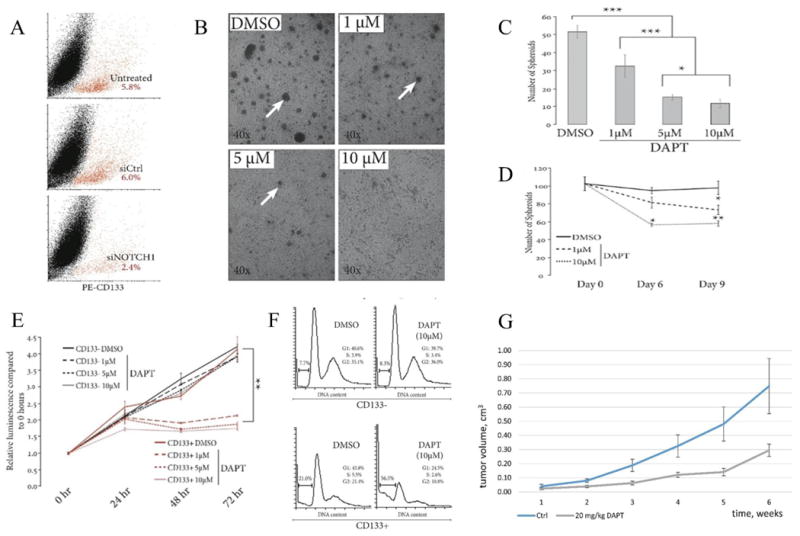
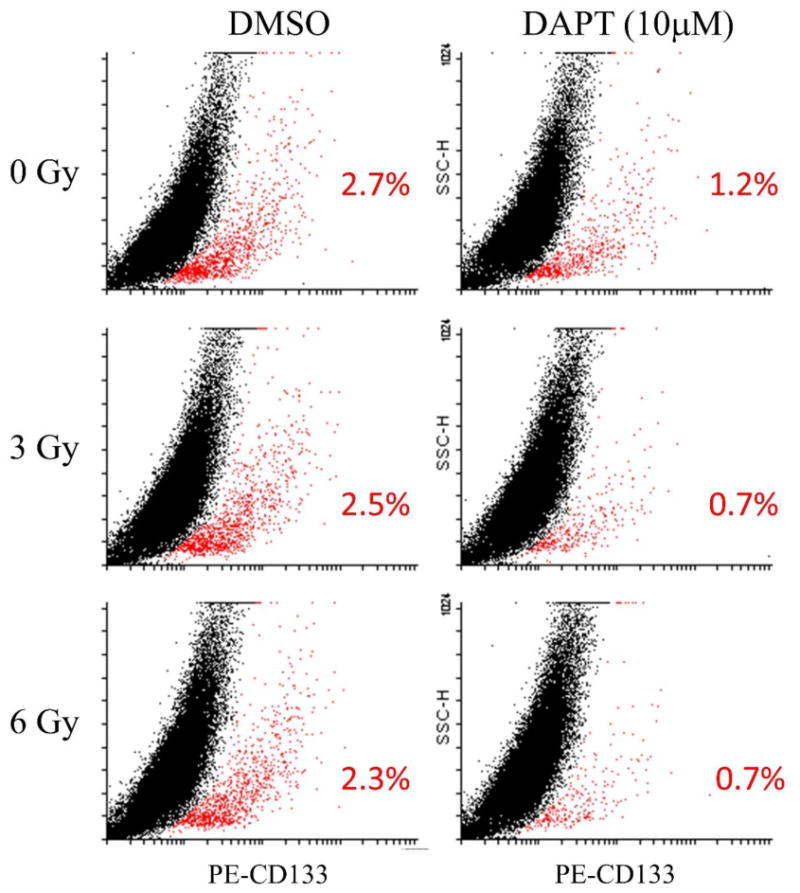
(A) Suppression of NOTCH1 signaling by siRNA decreases the CD133+ cell fraction (red) as demonstrated by FACS analysis using PE-conjugated CD133 antibody. (B) Inhibition of Notch signaling with γ-secretase inhibitor DAPT demonstrates dose-dependent suppressive effects on spheroid formation (arrows) in Accx11 culture at 48 h. (C) Quantification of DAPT effects on spheroid formation compared to control (DMSO-treated) Accx11 cells, p<0.05 (*), p<0.01 (**), p<0.001 (***), Student's t-test. (D) Breakdown of pre-formed spheroids induced by DAPT as shown by quantification of remaining spheroids following DAPT treatment versus control (DMSO-treated) Accx11 cells, p<0.05 (*), p<0.01 (**). (E) Inhibitory DAPT effect on cell proliferation is highly CD133+-selective and dose-dependent. At 72 h, there is a statistically significant decrease (p<0.01) in luminescence (Cell Titer Glo assay), p<0.01 (**). (F) DAPT selectively induces cell death in the CD133+ population as demonstrated by increase in sub-G1 cell population. Histograms represent cell counts based on PI-staining of separated Accx11 cells (G) Suppressive effect of DAPT on tumor growth in a nude mouse model subcutaneously injected with Accx11. Starting at week2, p<0.01. (H) Combination of DAPT with radiation is more detrimental to CD133+ cells (red) than DAPT or radiation alone. Representatives of 3 replicates are shown (p<0.05 in all comparisons except 3 Gy vs 6 Gy, radiation only).
Effects of NOTCH1 inhibition on spheroids in Accx11 coupled with analyses showing that spheroids are enriched in CD133+ cells suggested that Notch may be critical for maintenance and survival of CSC. To validate DAPT selectivity towards CD133+ cells, ACC cells were CD133-sorted and treated with DAPT for 72 h. DAPT treatment suppressed proliferation of CD133+ cells with differences noted at all doses (1, 5, and 10uM), while proliferation of CD133- cells was not affected (Fig. 5E). FACS analysis confirmed selective DAPT effect demonstrating a ∼2.5-fold increase in sub-G1 population in CD133+ cells without obvious effects on the G1, S, and G2/M ratios. Neither sub-G1 nor cell cycle progression was altered in CD133- cells (Fig. 5F). Annexin and propidium iodide co-staining confirmed that DAPT treatment selectively killed CD133+ cells, but did not reveal a pattern typical of apoptosis (Suppl. Fig. 6). Finally, to perform pre-clinical assessment of DAPT as a single agent, we used nude mice subcutaneously injected with Accx11 cells. In this study, DAPT had a statistically significant tumor-suppressive effect starting at week 2 (Fig. 5G) but exhibited no obvious toxicity. Overall, these in vitro and in vivo data suggest that GSI effects as a monotherapy should be evaluated in clinical trials with ACC patients.
Because CSC have been implicated in radiation resistance in many tumor types (36), the effect of targeting CD133+ cells through Notch inhibition in combination with radiation was determined by treating cells with DAPT for 24 h and then exposing them to increasing doses of radiation. Radiation alone had minimal effect on the amount of CD133+ cells: even 6 Gy of radiation decreased this fraction only moderately, from 2.7 to 2.3 %, which translates into a ∼15% depletion (Fig. 5H). However, DAPT as a single agent showed a two-fold (∼56%) decrease in the proportion of CD133+ cells. Remarkably, when a single 3Gy or 6Gy dose of radiation was added to DAPT treatment, the combined effect resulted in an almost 2-fold enhancement of CD133+ cell depletion as compared with DAPT alone (compare 0.7% with 1.2% in Fig. 5H, p<0.05). The enhanced loss of CD133+ ACC cells following radiation in the presence of DAPT suggested that combination of radiation with GSI therapy may be worth exploring in this radiation-resistant tumor.
NOTCH1 depletion down-regulates SKP2, an E3 ubiquitin ligase that controls p27Kip1
Activated NOTCH1 reduces p27Kip1 levels by stimulating expression of the p27Kip1 E3 ubiquitin-ligase SKP2 (45). Identification of SKP2 downstream from a NOTCH1 effector, FABP7 (Suppl. Table 3), suggested that SKP2 and p27Kip1 are engaged in the pro-survival NOTCH1 signaling in stem-like ACC cells. Indeed, we observed decreased expression of p27Kip1 in CD133+ Accx11 cells, where NOTCH1 was activated (Fig. 2C and F). Assessment of SKP2 expression by qRT-PCR and immunoblotting showed up-regulation of SKP2 in CD133+ compared to CD133- cells (Fig. 6A and B). Immunofluorescent staining of Accx11 cultures revealed that SKP2 was detected primarily in NR2F1-negative spheroidal cells, and was localized to the cytoplasm, where it has been associated with oncogenic activity (46) (Fig. 6C). Since SKP2 is a transcriptional NOTCH1 target, inhibition of NOTCH1 activity is expected to suppress SKP2 expression and, in turn, increase p27Kip1 protein stability and levels. In agreement with this expectation, siRNA-mediated NOTCH1 knockdown dramatically decreased SKP2 mRNA levels in Accx11 cells, as did DAPT treatment in a dose-dependent manner (Fig. 6D and E). To determine if Notch effects on SKP2 and p27Kip1 are seen predominantly in CD133+ cells, bulk Accx11 cells, CD133+, and CD133- populations were treated with DAPT for 48 hours, and then cell lysates immunoblotted. As expected, CD133+ cells expressed higher levels of N1ICD than bulk or CD133- cells, and DAPT effectively inhibited NOTCH1 activation as marked by decreased N1ICD in these cells (Fig. 6F). SKP2 was detected at very low levels in bulk and CD133- Accx11 cells (long exposure, data not shown), but was readily detected in CD133+ cells, whereas p27Kip1 was detected in bulk cells and in CD133- cells at much higher levels than in CD133+ cells. Treatment with DAPT did not markedly alter expression of p27Kip1 in bulk or CD133- cells, but dramatically enhanced its expression in CD133+ cells (Fig. 6F). Together, these data suggested that NOTCH1 inhibition up-regulates p27Kip1 selectively in CD133+ cells.
Figure 6. CD133+-cell-selective and NOTCH1-dependent SKP2 expression in ACC cells.
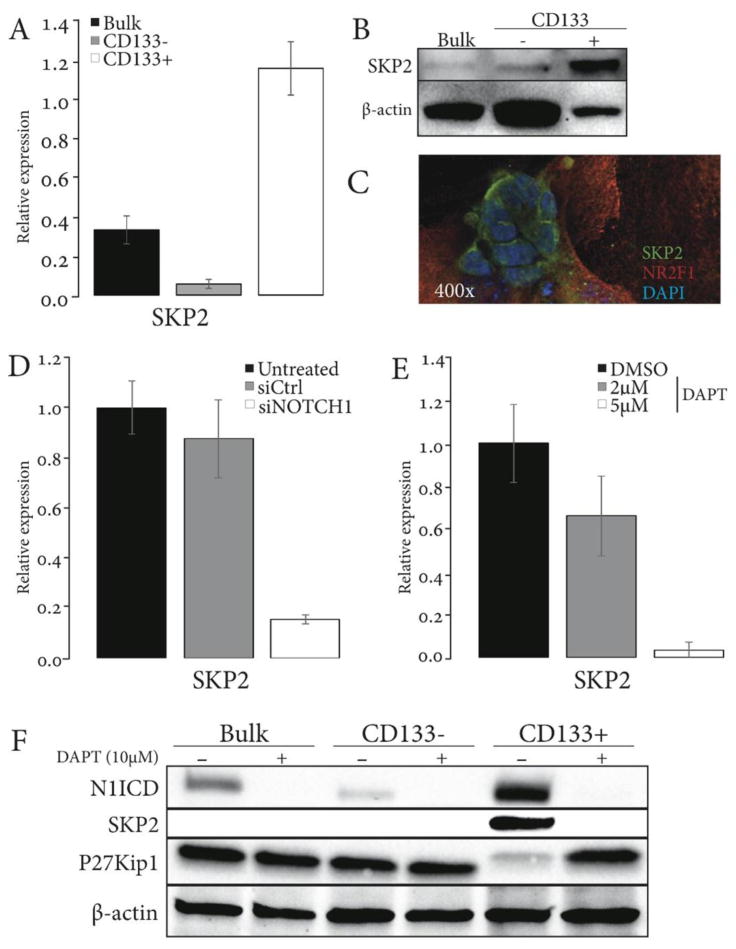
(A-C) SKP2 is selectively expressed in CD133+ Accx11 cells as demonstrated by qRT-PCR (A), immunoblot (B), and shows cytoplasmic localization in spheroid-forming CD133+ cells as demonstrated by IF (C). β-actin serves as a control for (A) and (B). (D-E) Inhibition of Notch signaling by siRNA (D) and DAPT (E) decreases expression of SKP2 in bulk Accx11 cells (qRT-PCR). (F) DAPT blocks expression of activated NOTCH1 (N1ICD) and its target SKP2 in CD133+ ACC cells and up-regulates p27Kip1, a SKP2 substrate. β-actin serves as a loading control.
Discussion
CSC isolated from various malignancies possess many features of embryonic or tissue stem cells and can be targeted via highly conserved Notch, Hedgehog (HH), and Wnt pathways activated in these cells (47). While previous studies confirmed the existence of CSC in ACC (3), difficulties with ACC culture and lack of reliable stem cell markers prevented their characterization. Our recent studies provided a set of novel ACC markers, such as NOTCH1, SOX10, FABP7, and other genes that control propagation and differentiation of NSC (6, 13), suggesting that isolation of CSC from ACC can be achieved. Here, we developed and implemented a novel technology and an additional set of stem cell markers for isolation, propagation, reliable validation, and characterization of these cells. For the first time, ACC cells with CSC properties were isolated directly from tumor tissue or propagated and purified from primary ACC cultures. Using these new approaches, a population of SOX10+/NOTCH1+/FABP7+ ACC cells was identified that also expressed CD133, a cell surface marker used for CSC isolation. As we demonstrated, ACC cells that express CD133 have basic characteristics of CSC including the ability to generate both CD133+ and CD133- populations, form spheroids in culture, and initiate tumors in nude mice.
Gene knockdown studies highlighted the essential roles and potential functional cooperation between NOTCH1 and SOX10 in maintenance of CD133+ ACC cells. An important insight into this cooperation, which has not been previously appreciated, was provided by the co-expression of FABP7 that relied on both NOTCH1 and SOX10, and the role of FABP7 in regulating expression of genes critical for cell survival. FABP7 has been recently described as a novel diagnostic and prognostic ACC marker (48) and was also implicated in glioblastoma, melanoma, and basal-like breast cancer; however, its downstream targets and molecular mechanisms of activation have not been studied. Our data associate FABP7 with the expression of SKP2 and multiple other genes involved in survival, proliferation, and metabolic pathways pointing at a novel signaling pathway that stimulates CSC in ACC.
In this study, we demonstrated that SOX10 is one of the most consistent markers of CD133+ stem-like ACC cells. Expression of SOX10 is also seen in other cancers suggesting that they may contain similar stem-like cells. We and others have reported SOX10 expression in basal-like breast carcinomas, a histologically and clinically distinct subtype of breast cancer that lacks targeted therapy and expresses NOTCH1 and FABP7 (13, 49). It would be interesting in lieu of possible therapeutic implications to determine if basal-like breast carcinoma is SOX10- and NOTCH1-dependent similar to ACC and neural crest-derived cancers such as melanoma, neuroblastoma, and glioblastoma.
ACC is a radioresistant tumor, and currently there are no curative treatment options for patients with unresectable disease. Targeting CSC via Notch has been linked with sensitization of glioblastoma to radiation (18). Thus, Notch-targeting therapies offer a new opportunity for ACC, and while there are no available drugs to inhibit SOX10 or FABP7, agents targeting Notch are currently in clinical trials (30). Here, we demonstrate that activated NOTCH1 stimulates proliferation and survival of ACC cells with neural stem properties. In line with this scenario, NOTCH1 targeting selectively inhibited proliferation of CD133+ ACC cells and depleted them triggering cell death. Notch inhibitors have strong antineoplastic activity in numerous preclinical models when combined with DNA damaging therapies (47), and here we show that DAPT is efficacious in pre-clinical ACC studies and can be also used as sensitizer of CD133+ ACC cells to γ-irradiation. Overall, these experiments provide preliminary data that stimulate interest to using Notch inhibitors for ACC therapy either as a single agent or in combination with radiation.
Mutations in the NOTCH1 gene are detected in 5% of patients (50), and a pattern of activating NOTCH1 mutations in ACC is beginning to emerge (51, 52). The low incidence of these mutations, however, suggest the existence in this cancer of other mechanisms that trigger Notch activation and supports CD133+ cells in ACC. Intrinsically high NOTCH1 expression in ACC is an important pre-requisite for its activation achieved via ligand-binding. Hence, association of NOTCH1 in ACC with JAG1, a NOTCH1 ligand produced by CD133- cells, provides a potential mechanism for oncogenic NOTCH1 activation and aggressive tumor growth. Indeed, up-regulation of JAG1 has been previously linked to NOTCH1 activation and poor survival in breast, pancreatic, and ovarian cancers (32).
In search of additional actionable targets associated with NOTCH1 signaling in ACC, p27Kip1 and its ubiquitin ligase SKP2 were identified. Expression of SKP2 and p27Kip1 inversely correlated in CD133+ and CD133- cells, and inhibition of NOTCH1 markedly decreased SKP2 levels in CD133+ cells and stimulated p27Kip1. In addition to its central roles in the regulation of proliferation, apoptosis, and senescence, which are mediated at least partially through degradation of p27Kip1 (53), SKP2 is a tumor survival factor during energy stress (54). Multifaceted SKP2 activities at a crossroad of many oncogenic signaling axes suggest that SKP2 may be an additional therapeutic target in ACC.
Overall, characterization of neural stem-like cells in ACC and delineating signaling events essential for their maintenance provide a new concept for ACC research and treatment that focuses on cancer cells with neural stem properties. Notch inhibitors that show promise in neuroblastoma and brain tumors (30) may be used as therapy for ACC in combination with Trk inhibition that we previously tested on PDX (6) and cytotoxic modalities. Future combination strategies for ACC may also include SKP2 inhibitors that are currently in development (16).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Translational Relevance.
Adenoid cystic carcinoma (ACC) is a slow-growing but highly lethal neuroinvasive tumor, which is prone to recurrence and metastases. ACC is an orphan tumor, which remains one of the least studied cancers with therapy limited to surgery and radiation in the absence of molecular targets and validated in vitro models. Cancer stem cells (CSC) are an attractive therapeutic target and understanding their role in ACC may advance therapy. In this study, we isolated from ACC a previously unrecognized population of neural stem-like cells and identified ACC-intrinsic CSC markers. Novel aspects of this research link CSC propagation in ACC to SOX10 and NOTCH1 activation producing a proof of concept for pharmaceutical targeting of these cells and sensitizing them to radiation.
Acknowledgments
Grant Support. This study was supported by funds from the Adenoid Cystic Carcinoma Research Foundation and by grants 5R21DE023228 (WGY) and 5R21DE022641 (SVI) from the National Institute of Dental and Craniofacial Research. This work was also supported in part by funds from the Department of Surgery, Yale School of Medicine, by an endowment to the Barry Baker Laboratory for Head and Neck Oncology, by Laura and Isaac Perlmutter Cancer Center Support Grant NIH/NCI P30CA016087, and the National Institutes of Health S10 Grants NIH/ORIP S10OD01058 and S10OD018338.
Footnotes
C. Moskaluk is a consultant/advisory board member for Novartis. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Dodd RL, Slevin NJ. Salivary gland adenoid cystic carcinoma: a review of chemotherapy and molecular therapies. Oral oncology. 2006;42(8):759–69. doi: 10.1016/j.oraloncology.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Jensen AD, Poulakis M, Nikoghosyan AV, Chaudhri N, Uhl M, Munter MW, et al. Re-irradiation of adenoid cystic carcinoma: Analysis and evaluation of outcome in 52 consecutive patients treated with raster-scanned carbon ion therapy. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 2015;114(2):182–8. doi: 10.1016/j.radonc.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 3.Adams A, Warner K, Nor JE. Salivary gland cancer stem cells. Oral oncology. 2013;49(9):845–53. doi: 10.1016/j.oraloncology.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Phuchareon J, Ohta Y, Woo JM, Eisele DW, Tetsu O. Genetic profiling reveals cross-contamination and misidentification of 6 adenoid cystic carcinoma cell lines: ACC2, ACC3, ACCM, ACCNS, ACCS and CAC2. PloS one. 2009;4(6):e6040. doi: 10.1371/journal.pone.0006040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moskaluk CA, Baras AS, Mancuso SA, Fan H, Davidson RJ, Dirks DC, et al. Development and characterization of xenograft model systems for adenoid cystic carcinoma. Laboratory investigation; a journal of technical methods and pathology. 2011;91(10):1480–90. doi: 10.1038/labinvest.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ivanov SV, Panaccione A, Brown B, Guo Y, Moskaluk CA, Wick MJ, et al. TrkC signaling is activated in adenoid cystic carcinoma and requires NT-3 to stimulate invasive behavior. Oncogene. 2013;32(32):3698–710. doi: 10.1038/onc.2012.377. [DOI] [PubMed] [Google Scholar]
- 7.Cassidy JW, Caldas C, Bruna A. Maintaining Tumor Heterogeneity in Patient-Derived Tumor Xenografts. Cancer research. 2015;75(15):2963–8. doi: 10.1158/0008-5472.CAN-15-0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gach PC, Attayek PJ, Herrera G, Yeh JJ, Allbritton NL. Isolation and in vitro culture of rare cancer stem cells from patient-derived xenografts of pancreatic ductal adenocarcinoma. Anal Chem. 2013;85(15):7271–8. doi: 10.1021/ac401165s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hasmim M, Bruno S, Azzi S, Gallerne C, Michel JG, Chiabotto G, et al. Isolation and characterization of renal cancer stem cells from patient-derived xenografts. Oncotarget. 2015 doi: 10.18632/oncotarget.6266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moro M, Bertolini G, Tortoreto M, Pastorino U, Sozzi G, Roz L. Patient-derived xenografts of non small cell lung cancer: resurgence of an old model for investigation of modern concepts of tailored therapy and cancer stem cells. J Biomed Biotechnol. 2012;2012:568567. doi: 10.1155/2012/568567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKeown SJ, Lee VM, Bronner-Fraser M, Newgreen DF, Farlie PG. Sox10 overexpression induces neural crest-like cells from all dorsoventral levels of the neural tube but inhibits differentiation. Developmental dynamics : an official publication of the American Association of Anatomists. 2005;233(2):430–44. doi: 10.1002/dvdy.20341. [DOI] [PubMed] [Google Scholar]
- 12.Shakhova O, Zingg D, Schaefer SM, Hari L, Civenni G, Blunschi J, et al. Sox10 promotes the formation and maintenance of giant congenital naevi and melanoma. Nature cell biology. 2012;14(8):882–90. doi: 10.1038/ncb2535. [DOI] [PubMed] [Google Scholar]
- 13.Ivanov SV, Panaccione A, Nonaka D, Prasad ML, Boyd KL, Brown B, et al. Diagnostic SOX10 gene signatures in salivary adenoid cystic and breast basal-like carcinomas. British journal of cancer. 2013;109(2):444–51. doi: 10.1038/bjc.2013.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nature biotechnology. 2007;25(6):681–6. doi: 10.1038/nbt1310. [DOI] [PubMed] [Google Scholar]
- 15.Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. The American journal of pathology. 2012;180(2):599–607. doi: 10.1016/j.ajpath.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hao Z, Huang S. E3 ubiquitin ligase Skp2 as an attractive target in cancer therapy. Frontiers in bioscience. 2015;20:474–90. doi: 10.2741/4320. [DOI] [PubMed] [Google Scholar]
- 17.Chan CH, Morrow JK, Li CF, Gao Y, Jin G, Moten A, et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell. 2013;154(3):556–68. doi: 10.1016/j.cell.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Wakeman TP, Lathia JD, Hjelmeland AB, Wang XF, White RR, et al. Notch promotes radioresistance of glioma stem cells. Stem cells. 2010;28(1):17–28. doi: 10.1002/stem.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiba S. Notch signaling in stem cell systems. Stem cells. 2006;24(11):2437–47. doi: 10.1634/stemcells.2005-0661. [DOI] [PubMed] [Google Scholar]
- 20.Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling--are we there yet? Nature reviews Drug discovery. 2014;13(5):357–78. doi: 10.1038/nrd4252. [DOI] [PubMed] [Google Scholar]
- 21.Friedrich J, Seidel C, Ebner R, Kunz-Schughart LA. Spheroid-based drug screen: considerations and practical approach. Nat Protoc. 2009;4(3):309–24. doi: 10.1038/nprot.2008.226. [DOI] [PubMed] [Google Scholar]
- 22.Reid Y, Storts D, Riss T, Minor L. Authentication of Human Cell Lines by STR DNA Profiling Analysis. In: Sittampalam GS, Coussens NP, Nelson H, Arkin M, Auld D, Austin C, et al., editors. Assay Guidance Manual. Bethesda (MD): 2004. [Google Scholar]
- 23.Grosse-Gehling P, Fargeas CA, Dittfeld C, Garbe Y, Alison MR, Corbeil D, et al. CD133 as a biomarker for putative cancer stem cells in solid tumours: limitations, problems and challenges. The Journal of pathology. 2013;229(3):355–78. doi: 10.1002/path.4086. [DOI] [PubMed] [Google Scholar]
- 24.Karbanova J, Laco J, Marzesco AM, Janich P, Vobornikova M, Mokry J, et al. Human prominin-1 (CD133) is detected in both neoplastic and non-neoplastic salivary gland diseases and released into saliva in a ubiquitinated form. PloS one. 2014;9(6):e98927. doi: 10.1371/journal.pone.0098927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lima RA, Tavares MR, Dias FL, Kligerman J, Nascimento MF, Barbosa MM, et al. Clinical prognostic factors in malignant parotid gland tumors. Otolaryngology--head and neck surgery : official journal of American Academy of Otolaryngology-Head and Neck Surgery. 2005;133(5):702–8. doi: 10.1016/j.otohns.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 26.Archer TC, Pomeroy SL. A developmental program drives aggressive embryonal brain tumors. Nature genetics. 2014;46(1):2–3. doi: 10.1038/ng.2857. [DOI] [PubMed] [Google Scholar]
- 27.Chen Z, Liu C, Patel AJ, Liao CP, Wang Y, Le LQ. Cells of origin in the embryonic nerve roots for NF1-associated plexiform neurofibroma. Cancer cell. 2014;26(5):695–706. doi: 10.1016/j.ccell.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fagiani E, Giardina G, Luzi L, Cesaroni M, Quarto M, Capra M, et al. RaLP, a new member of the Src homology and collagen family, regulates cell migration and tumor growth of metastatic melanomas. Cancer research. 2007;67(7):3064–73. doi: 10.1158/0008-5472.CAN-06-2301. [DOI] [PubMed] [Google Scholar]
- 29.Hau P, Apfel R, Wiese P, Tschertner I, Blesch A, Bogdahn U. Melanoma-inhibiting activity (MIA/CD-RAP) is expressed in a variety of malignant tumors of mainly neuroectodermal origin. Anticancer research. 2002;22(2A):577–83. [PubMed] [Google Scholar]
- 30.Teodorczyk M, Schmidt MH. Notching on Cancer's Door: Notch Signaling in Brain Tumors. Frontiers in oncology. 2014;4:341. doi: 10.3389/fonc.2014.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naka H, Nakamura S, Shimazaki T, Okano H. Requirement for COUP-TFI and II in the temporal specification of neural stem cells in CNS development. Nature neuroscience. 2008;11(9):1014–23. doi: 10.1038/nn.2168. [DOI] [PubMed] [Google Scholar]
- 32.Li D, Masiero M, Banham AH, Harris AL. The notch ligand JAGGED1 as a target for anti-tumor therapy. Frontiers in oncology. 2014;4:254. doi: 10.3389/fonc.2014.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xing F, Kobayashi A, Okuda H, Watabe M, Pai SK, Pandey PR, et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO molecular medicine. 2013;5(3):384–96. doi: 10.1002/emmm.201201623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haupt S, Borghese L, Brustle O, Edenhofer F. Non-genetic modulation of Notch activity by artificial delivery of Notch intracellular domain into neural stem cells. Stem cell reviews. 2012;8(3):672–84. doi: 10.1007/s12015-011-9335-6. [DOI] [PubMed] [Google Scholar]
- 35.Qiu J, Takagi Y, Harada J, Topalkara K, Wang Y, Sims JR, et al. p27Kip1 constrains proliferation of neural progenitor cells in adult brain under homeostatic and ischemic conditions. Stem cells. 2009;27(4):920–7. doi: 10.1002/stem.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clevers H. The cancer stem cell: premises, promises and challenges. Nature medicine. 2011;17(3):313–9. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- 37.Kim J, Lo L, Dormand E, Anderson DJ. SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron. 2003;38(1):17–31. doi: 10.1016/s0896-6273(03)00163-6. [DOI] [PubMed] [Google Scholar]
- 38.Okamura Y, Saga Y. Notch signaling is required for the maintenance of enteric neural crest progenitors. Development. 2008;135(21):3555–65. doi: 10.1242/dev.022319. [DOI] [PubMed] [Google Scholar]
- 39.Matsumata M, Sakayori N, Maekawa M, Owada Y, Yoshikawa T, Osumi N. The effects of Fabp7 and Fabp5 on postnatal hippocampal neurogenesis in the mouse. Stem cells. 2012;30(7):1532–43. doi: 10.1002/stem.1124. [DOI] [PubMed] [Google Scholar]
- 40.Jacob C, Lotscher P, Engler S, Baggiolini A, Varum Tavares S, Brugger V, et al. HDAC1 and HDAC2 control the specification of neural crest cells into peripheral glia. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34(17):6112–22. doi: 10.1523/JNEUROSCI.5212-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anthony TE, Mason HA, Gridley T, Fishell G, Heintz N. Brain lipid-binding protein is a direct target of Notch signaling in radial glial cells. Genes & development. 2005;19(9):1028–33. doi: 10.1101/gad.1302105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sarmento LM, Huang H, Limon A, Gordon W, Fernandes J, Tavares MJ, et al. Notch1 modulates timing of G1-S progression by inducing SKP2 transcription and p27 Kip1 degradation. The Journal of experimental medicine. 2005;202(1):157–68. doi: 10.1084/jem.20050559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tirino V, Desiderio V, Paino F, De Rosa A, Papaccio F, La Noce M, et al. Cancer stem cells in solid tumors: an overview and new approaches for their isolation and characterization. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2013;27(1):13–24. doi: 10.1096/fj.12-218222. [DOI] [PubMed] [Google Scholar]
- 44.Capaccione KM, Pine SR. The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis. 2013;34(7):1420–30. doi: 10.1093/carcin/bgt127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dohda T, Maljukova A, Liu L, Heyman M, Grander D, Brodin D, et al. Notch signaling induces SKP2 expression and promotes reduction of p27Kip1 in T-cell acute lymphoblastic leukemia cell lines. Experimental cell research. 2007;313(14):3141–52. doi: 10.1016/j.yexcr.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 46.Gao D, Inuzuka H, Tseng A, Chin RY, Toker A, Wei W. Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nature cell biology. 2009;11(4):397–408. doi: 10.1038/ncb1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nature reviews Clinical oncology. 2015 doi: 10.1038/nrclinonc.2015.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Phuchareon J, Overdevest JB, McCormick F, Eisele DW, van Zante A, Tetsu O. Fatty Acid binding protein 7 is a molecular marker in adenoid cystic carcinoma of the salivary glands: implications for clinical significance. Translational oncology. 2014;7(6):780–7. doi: 10.1016/j.tranon.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cimino-Mathews A, Subhawong AP, Elwood H, Warzecha HN, Sharma R, Park BH, et al. Neural crest transcription factor Sox10 is preferentially expressed in triple-negative and metaplastic breast carcinomas. Human pathology. 2013;44(6):959–65. doi: 10.1016/j.humpath.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ho AS, Kannan K, Roy DM, Morris LG, Ganly I, Katabi N, et al. The mutational landscape of adenoid cystic carcinoma. Nature genetics. 2013;45(7):791–8. doi: 10.1038/ng.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stoeck A, Lejnine S, Truong A, Pan L, Wang H, Zang C, et al. Discovery of biomarkers predictive of GSI response in triple-negative breast cancer and adenoid cystic carcinoma. Cancer discovery. 2014;4(10):1154–67. doi: 10.1158/2159-8290.CD-13-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stephens PJ, Davies HR, Mitani Y, Van Loo P, Shlien A, Tarpey PS, et al. Whole exome sequencing of adenoid cystic carcinoma. The Journal of clinical investigation. 2013;123(7):2965–8. doi: 10.1172/JCI67201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chan CH, Morrow JK, Zhang S, Lin HK. Skp2: a dream target in the coming age of cancer therapy. Cell cycle. 2014;13(5):679–80. doi: 10.4161/cc.27853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chan CH, Li CF, Yang WL, Gao Y, Lee SW, Feng Z, et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell. 2012;149(5):1098–111. doi: 10.1016/j.cell.2012.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.