

Summary
The pathogenesis of heart failure with a preserved ejection fraction (HFpEF) is unclear. Myocardial fibrosis, inflammation, and cardiac hypertrophy have been suggested to contribute to the pathogenesis of HFpEF. Cardiosphere-derived cells (CDCs) are heart-derived cell products with antifibrotic and anti-inflammatory properties. This study tested whether rat CDCs were sufficient to decrease manifestations of HFpEF in hypertensive rats. Starting at 7 weeks of age, Dahl salt-sensitive rats were fed a high-salt diet for 6 to 7 weeks and randomized to receive intracoronary CDCs or placebo. Dahl rats fed normal chow served as controls. High-salt rats developed hypertension, left ventricular (LV) hypertrophy, and diastolic dysfunction, without impairment of ejection fraction. Four weeks after treatment, diastolic dysfunction resolved in CDC-treated rats but not in placebo. The improved LV relaxation was associated with lower LV end-diastolic pressure, decreased lung congestion, and enhanced survival in CDC-treated rats. Histology and echocardiography revealed no decrease in cardiac hypertrophy after CDC treatment, consistent with the finding of sustained, equally-elevated blood pressure in CDC- and placebo-treated rats. Nevertheless, CDC treatment decreased LV fibrosis and inflammatory infiltrates. Serum inflammatory cytokines were likewise decreased after CDC treatment. Whole-transcriptome analysis revealed that CDCs reversed changes in numerous transcripts associated with HFpEF, including many involved in inflammation and/or fibrosis. These studies suggest that CDCs normalized LV relaxation and LV diastolic pressure while improving survival in a rat model of HFpEF. The benefits of CDCs occurred despite persistent hypertension and cardiac hypertrophy. By selectively reversing inflammation and fibrosis, CDCs may be beneficial in the treatment of HFpEF.
Key Words: animal models, cell therapy, diastolic function, heart failure with preserved ejection fraction
Abbreviations and Acronyms: CDC, cardiosphere-derived cell; DS, Dahl salt-sensitive; E/A ratio, ratio of early to late ventricular filling velocity; HFpEF, heart failure with preserved ejection fraction; LV, left ventricular; LVEDP, left ventricular end-diastolic pressure; LVEF, left ventricular ejection fraction; MMP, matrix metalloproteinase; TIMP, tissue inhibitor of metalloproteinase
Visual Abstract
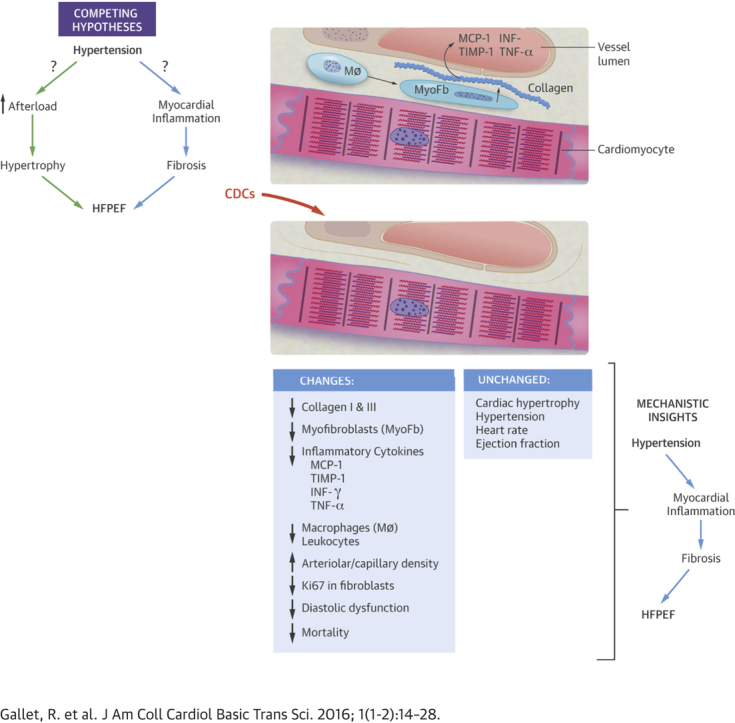
Highlights
-
•
The pathogenesis of heart failure with a preserved ejection fraction (HFpEF) is unclear.
-
•
Cardiosphere-derived cells (CDCs) are heart-derived cell products with antifibrotic and anti-inflammatory properties, which have been implicated in HFpEF.
-
•
Dahl salt-sensitive rats were fed a high-salt diet for 6 to7 weeks and randomized to receive intracoronary CDCs or placebo.
-
•
Following CDC treatment, diastolic dysfunction resolved in treated rats but not in the placebo group. Treatment with CDCs also lower LV end-diastolic pressure, decrease lung congestion, and enhance survival.
-
•
CDC treatment decreased LV fibrosis and inflammatory infiltrates, and reversed many of the transcriptomic changes associated with HFpEF, but had no effect on cardiac hypertrophy.
-
•
By selectively reversing inflammation and fibrosis, CDCs may be beneficial in the treatment of HFpEF.
Heart failure with preserved ejection fraction (HFpEF) has become a major public health concern. Its increasing prevalence now exceeds that of heart failure with reduced ejection fraction 1, 2, 3. Outcomes of patients with HFpEF are poor 4, 5, and so far, no treatment has been shown to decrease morbidity or mortality 3, 6. HFpEF is associated with various cardiovascular risk factors (especially hypertension), extracardiac comorbidities, and aging. The net result is impaired diastolic relaxation and filling of the left ventricle, increased myocardial stiffness, impaired vascular compliance, and increased diastolic pressure 7, 8. Myocardial fibrosis and inflammation have been associated with HFpEF 9, 10, 11, 12, 13, 14 and with the transition from hypertensive left ventricular (LV) hypertrophy without HFpEF to hypertensive LV hypertrophy with HFpEF (15). Cardiosphere-derived cells (CDCs) are heart cell products with antifibrotic, anti-inflammatory, and angiogenic properties 16, 17, 18, 19, 20. CDCs, which are currently in phase 2 human trials for scar reduction after myocardial infarction (5), have been shown to be beneficial in models of ischemic 17, 18, 21 and nonischemic cardiomyopathy (16). Thus, we wondered whether CDCs might have disease-modifying activity in HFpEF.
Dahl salt-sensitive (DS) rats develop hypertension, hypertrophy, and, eventually, HFpEF on a high-salt diet 22, 23, 24, 25, 26. Increased fibrosis and inflammation underlie the development of HFpEF, with resultant cachexia, pulmonary congestion, and accelerated mortality 22, 26, 27. Therefore, this model has been widely used to test new treatments for HFpEF 23, 27, 28, 29, 30, 31. Here, we tested the efficacy of CDCs in improving LV structure and function and overall outcome in DS rats with HFpEF.
Methods
An expanded “Methods” section is available in the Supplemental Appendix.
DS rats (Charles River, Wilmington, Massachusetts) were fed a 0.3% NaCl (low-salt) diet until 7 weeks of age. At that time, the diet was switched to an 8% NaCl (high-salt) diet in 54 rats by random assignment. DS rats fed the low-salt diet constituted the control group (n = 18). At 13 to 14 weeks of age, rats on the high-salt diet were randomized to receive allogeneic rat CDCs (5 × 105 resuspended in 100 μl phosphate-buffered saline) (Figure 1A) or vehicle (phosphate-buffered saline). CDCs were grown from a freshly explanted Wistar-Kyoto rat heart as previously described (19) (Figure 1B).
Figure 1.

Rat Model of HFpEF: Phenotype and Responsiveness to Treatment With Cardiosphere-Derived Cells
(A) Study design. (B) Cardiosphere-derived cell manufacturing protocol. (C) Representative images of transmitral flow by Doppler echocardiography at endpoint in control, placebo-treated, and cardiosphere-derived cell (CDC)–treated rats. (D) CDC treatment normalizes ratio of early to late ventricular filling velocity (E/A ratio) at 4 weeks, while the E/A ratio in placebo-treated rats remains depressed. Systolic function assessed by left ventricular ejection fraction (LVEF) (E,F) and by fractional area change (FAC) (G,H) is equivalent in all groups. Left ventricular end-diastolic and end-systolic volumes (I) are equivalent in all groups. (J) CDC treatment halts heart failure with preserved ejection fraction–related left atrial enlargement, while placebo does not. (Baseline and pre-treatment, n = 10 for control rats and n = 24 for placebo- and CDC-treated rats; 1 week post-treatment, n = 10 for control rats, n = 21 for placebo- and CDC-treated rats; at endpoint, n = 10 for control rats, n = 15 for placebo-treated rats, n = 18 for CDC-treated rats). *p < 0.05 versus placebo and CDC groups and †p < 0.05 versus placebo, both by analysis of variance. LAV = left atrial volume; LVV = left ventricular volume; PBS = phosphate-buffered saline; ttt = treatment.
Echocardiography was performed at baseline, before treatment, and 1 and 4 weeks after treatment to assess systolic and diastolic function. Invasive hemodynamic measurements were performed at endpoint to record systemic pressure and LV pressures and volumes. Pressure-volume loops were generated from these recordings. Rats were then euthanized and hearts were harvested for analysis. Additional follow-up (7 rats in each group, randomly chosen from the rats alive at 4 weeks) was performed for extended survival analysis (up to 6 weeks) to investigate the longer term effects of treatment.
Statistical analysis
Continuous variables are presented as mean ± SD in the text and mean ± SE in the figures. Categorical variables are expressed as absolute numbers and percentages. Normal distribution of variables was assessed using the Kolmogorov-Smirnov test. If normality was established, independent groups (n = 2) were compared using unpaired Student t test, and multiple groups were compared using 1-way analysis of variance. For variables not normally distributed, the Mann-Whitney U test was used for comparisons of 2 groups, and the Kruskal-Wallis test was used to compare multiple groups. Bonferroni correction was applied to every pairwise comparison performed after analysis of variance or the Kruskal-Wallis test. Survival analysis was performed using Kaplan-Meier analysis. A p value <0.05 was considered to indicate statistical significance.
Results
Blood pressure and cardiac hypertrophy
Table 1 shows characteristics of the high-salt and control animals at baseline and after 6 weeks of diet (13 weeks of age). As expected (25), rats fed a high-salt diet developed hypertension and cardiac hypertrophy after 6 weeks, but low-salt control rats did not. Those changes were associated with diastolic dysfunction, as shown by a decreased ratio of early (E) to late (A) ventricular filling velocity (E/A ratio) by echocardiography (1.7 ± 0.2 vs. 1.2 ± 0.2, p < 0.001), without any changes in LV volumes, LV ejection fraction (LVEF) or fractional area change (Table 1).
Table 1.
Characteristics of Rats Fed High- and Low-Salt Diets Before (Baseline) and After 6 Weeks of Diet
| Baseline |
6 Weeks |
|||||
|---|---|---|---|---|---|---|
| Low Salt | High Salt | p Value | Low Salt | High Salt | p Value | |
| SBP (mm Hg) | NA | NA | 133 ± 23 | 188 ± 17∗ | 0.001 | |
| DBP (mm Hg) | NA | NA | 92 ± 15 | 130 ± 12∗ | 0.001 | |
| LVEF (%) | 73.1 ± 4.8 | 73.0 ± 5.1 | 0.90 | 71.6 ± 3.8 | 74.4 ± 5.9 | 0.10 |
| FAC (short axis) (%) | 64.1 ± 3.0 | 64.2 ± 4.9 | 0.95 | 62.3 ± 4.6 | 63.2 ± 5.9 | 0.61 |
| AW thickness (mm) | 1.2 ± 0.1 | 1.2 ± 0.1 | 0.54 | 1.2 ± 0.1 | 1.8 ± 0.2∗ | <0.001 |
| PW thickness (mm) | 1.3 ± 0.1 | 1.3 ± 0.1 | 0.27 | 1.3 ± 0.1 | 2.0 ± 0.3∗ | <0.001 |
| LVEDV (ml) | 330 ± 46 | 329 ± 70 | 0.93 | 484 ± 112 | 510 ± 110 | 0.42 |
| LVESV (ml) | 89 ± 22 | 89 ± 28 | 0.98 | 142 ± 47 | 130 ± 50 | 0.37 |
| E/A ratio | 1.7 ± 0.2 | 1.7 ± 0.3 | 0.66 | 1.7 ± 0.2 | 1.2 ± 0.2∗ | <0.001 |
| Left atrial area (mm2) | 13.7 ± 1.9 | 14.1 ± 1.9 | 0.55 | 17.9 ± 1.7 | 21.2 ± 2.9∗ | <0.001 |
| Heart weight (g) | NA | NA | 1.42 ± 0.14 | 1.67 ± 0.10∗ | 0.03 | |
| Heart weight/body weight (mg/g) | NA | NA | 4.1 ± 0.4 | 5.2 ± 0.5∗ | 0.016 | |
Values are mean ± SD.
AW = anterior wall; DBP = diastolic blood pressure; E/A ratio = ratio of early to late ventricular filling velocity; FAC = fractional area change; LVEDV = left ventricular end-diastolic volume; LVEF = left ventricular ejection fraction; LVESV = left ventricular end-systolic volume; NA = not available; PW = posterior wall; SBP = systolic blood pressure.
p < 0.05 between high-salt and low-salt groups at 6 weeks.
Echocardiographic studies: CDCs normalize E/A ratio
Having confirmed the presence of cardiac hypertrophy and diastolic dysfunction, we randomly allocated rats to intracoronary CDC or vehicle infusion. Figure 1C shows representative images of transmitral flow at endpoint in control, placebo-treated, and CDC-treated animals. Pooled data (Figure 1D) revealed that after 6 weeks of diet but before treatment, E/A ratios were similar in the placebo and CDC groups but lower than in control rats. Likewise, left atrial size was higher in the high-salt-fed rats compared with control rats, indicating already increased LV filling pressure. After intracoronary infusion of CDCs (but not placebo), E/A ratio increased over time (Figure 1D), a change that was evident as soon as 1 week after treatment. At endpoint, E/A ratios had returned to control levels in CDC-treated animals (1.7 ± 0.2 for CDC-treated vs. 1.8 ± 0.16 for control rats, p = 0.36), whereas they remained depressed in placebo-treated animals (1.2 ± 0.3, p < 0.001 vs. CDC-treated rats and vs. control rats), indicating a likely normalization of LV relaxation with CDC treatment (an interpretation verified later by hemodynamic recordings). In addition, left atrial dimensions kept increasing in the placebo animals, while CDC treatment halted left atrial enlargement. In contrast, LVEF (measured in long-axis views) (Figures 1E and 1F), fractional area change (from short-axis views) (Figures 1G and 1H), and LV volumes (Figures 1I and 1J) were equivalent in all groups.
Hemodynamic studies: CDC treatment normalizes LV relaxation and prevents elevation of LV end-diastolic pressure
Figure 2A shows representative recordings of pressure-volume loop families at endpoint. The time constant of isovolumic LV pressure fall (tau) was prolonged in placebo-treated animals compared with CDC-treated animals (21 ± 8 s vs. 13 ± 1 s in control rats [p = 0.011] and 14 ± 4 s in CDC-treated rats [p = 0.006]) (Figure 2B) and control rats, while −dP/dt minimum was lower, indicating impaired relaxation (Figure 2C) without changes in dP/dt maximum (Figure 2D). In parallel, pressure-volume loop analyses confirmed that LV distensibility was decreased in the placebo-treated animals, as demonstrated by the steeper slope of the end-diastolic pressure-volume relationship in placebo-treated animals compared with CDC-treated and control animals (Figure 2E), again without changes in the end-systolic pressure-volume relationship (Figure 2F). LVEDP was 2-fold higher in placebo-treated than in CDC-treated and control animals (17 ± 10 mm Hg vs. 9 ± 4 mm Hg in control rats [p = 0.015] and 8 ± 3 mm Hg in CDC-treated rats [p = 0.002]) (Figure 2G). The normalization of LVEDP and tau in CDC-treated rats confirms that the increase of E/A ratio over time in this group was due to normalization of LV relaxation rather than to progression toward a pseudonormal pattern of transmitral flow (which would have been associated with increased LVEDP and tau [32]).
Figure 2.

Hemodynamic Analysis of Control (Low Salt), High Salt Placebo (HFpEF Phenotype), and High-Salt CDC-Treated Rats
(A) Representative pressure-volume (PV) loop recordings in control, placebo-treated, and cardiosphere-derived cell (CDC)–treated rats. CDCs normalize tau (B) and −dP/dt minimum (C) in rats with heart failure with preserved ejection fraction without change in dP/dt maximum (D). PV loop analysis reveals normalization of the slope of the end-diastolic PV relationship (EDPVR) (E), with no change in the end-systolic PV relationship (ESPVR) (F). Left ventricular end-diastolic pressure (LVEDP) is normal in the CDC-treated but not the placebo-treated animals (G). The differences between CDC- and placebo-treated rats are not related to changes in systolic blood pressure (SBP) (H) or diastolic blood pressure (DBP) (I) or heart rate (HR) (J) (n = 8 for control rats and n = 12 for placebo- and CDC-treated rats; for PV loop families, n = 6 for control rats, n = 7 for placebo-treated rats, and n = 8 for CDC-treated rats). *p < 0.05 versus placebo- and CDC-treated rats, †p < 0.05 versus control and CDC-treated rats, and ‡p < 0.05, all by analysis of variance.
We did not observe any differences in blood pressure or heart rate between CDC- and placebo-treated animals that could have confounded relaxation and LVEDP measurements (although blood pressure was lower in the control group, as expected) (Figures 2H to 2J). Thus, the improvements in diastolic function were not due to antihypertensive or chronotropic effects of CDCs.
CDC treatment improves survival and decreases lung congestion
Consistent with the improvement of diastolic function, we observed a dramatic improvement of survival in CDC-treated rats (Kaplan-Meier survival curves) (Figure 3A) (log-rank p = 0.027). Post-mortem lung weight and lung weight/body weight ratios were higher in placebo-treated rats, indicative of pulmonary congestion (Figure 3B). In parallel, Figure 3C shows that animals treated with CDCs resumed some physiological weight gain, while placebo rats lost weight, presumably because of cardiac cachexia (an impression that was confirmed visually).
Figure 3.

Effects of CDCs on Mortality and Heart Failure
Cardiosphere-derived cell (CDC) treatment improves survival (A; X axis shows days from treatment, or equivalent age in control rats) and pulmonary congestion (B) (lung weight [left] and lung weight/body weight [right]) in rats with heart failure with preserved ejection fraction (HFpEF). (C) At endpoint, body weight loss induced by HFpEF is partially restored by CDC treatment (n = 10 for control rats, n = 15 for placebo-treated rats, and n = 18 for CDC-treated rats). Log-rank for CDCs versus placebo. *p < 0.05 versus placebo- and CDC-treated rats, †p < 0.05 versus control and CDC-treated rats, and ‡p < 0.05 versus placebo-treated rats, all by analysis of variance. ttt = treatment.
Mechanism
Improvement of LV relaxation is not associated with quantitative changes in cardiac hypertrophy
LV hypertrophy (both macroscopic and cellular) can occur with or without diastolic dysfunction. We quantified cardiac hypertrophy using LV wall thickness by echocardiography, heart weight, and cardiomyocyte cross-sectional area. Notably, the CDC-related improvement in diastolic function was not due to a decrease in cardiac hypertrophy: wall thickness by echocardiography (Figure 4A), as well as post-mortem heart weight and cardiomyocyte cross-sectional area (Figure 4B), remained equivalent in the CDC and placebo groups. Thus, CDCs were salutary without decreasing cardiac hypertrophy.
Figure 4.

Benefit of Cardiosphere-Derived Cell Treatment Is Not Related to Decreased Cardiac Hypertrophy
Cardiac anterior wall (AW) and posterior wall (PW) thickness by echocardiography (A), heart weight and heart weight/body weight ratio (B), and cross-sectional cardiomyocyte area (CSA) (C) are equally elevated in placebo- and cardiosphere-derived cell (CDC)–treated rats relative to control. n = 10 for control rats, n = 15 for placebo-treated rats, and n = 18 for CDC-treated rats in A; n = 6, n = 11, and n = 14, respectively, in B; n = 5 in each group in C. *p < 0.05 versus placebo- and CDC-treated rats by analysis of variance.
Antifibrotic effect of CDCs
Fibrosis is increased in HFpEF 10, 12, 33. We assessed fibrosis using picrosirius red staining for total collagen and semiquantitative reverse transcriptase polymerase chain reaction to measure transcript levels for collagen 1 and 3. Figure 5A shows representative images of hearts stained with picrosirius red. Overall LV and right ventricular fibrosis was 2-fold higher in placebo- versus CDC-treated rats; fibrosis in the latter approached control values (Figures 5B and 5C). Concomitantly, collagen 1 and collagen 3 in the left ventricle (quantified by western blot) were higher in placebo-treated rats than in control or CDC-treated rats (Figure 5D). Moreover, cardiac myofibroblasts increased dramatically in placebo-treated, but not in CDC-treated, DS rats (Figure 5E). Also, transcript levels of matrix metalloproteinase (MMP)-2, MMP-7, MMP-9, and tissue inhibitor of metalloproteinase (TIMP)-1 as well as collagen 1A1 and collagen 3 were higher in the placebo-treated animals compared with the control and CDC-treated animals (which had similar levels) (Supplemental Figure 1). These increased transcript levels are suggestive of increased extracellular matrix turnover associated with HFpEF, which is normalized by CDC treatment. Because we did not measure the extent of fibrosis before treatment, we cannot distinguish between CDC-induced prevention of new fibrosis and regression of established fibrosis in the present study. However, previous work in chronic myocardial infarction models 20, 21, 34 and in humans 35, 36 has shown that CDCs (and cardiospheres) can reduce established scar.
Figure 5.

Antifibrotic Effects Underlie the Benefits of CDCs in HFpEF
(A) Representative heart sections stained with picrosirius red in control, placebo-treated, and cardiosphere-derived cell (CDC)–treated rats. Left ventricular (LV) fibrosis (B) and right ventricular (RV) fibrosis (C) quantified from such images is higher in placebo- than in CDC-treated and control rats. (D) Collagen 1A1 and collagen 3 content is higher in placebo- than in CDC-treated and control rats. (E) Immunostaining for α–smooth muscle actin (SMA) in control and placebo- and CDC-treated rats. Myofibroblast infiltration into the heart is higher in placebo- than in CDC-treated and control rats. n = 6 to 8 in each group. †p < 0.05 versus control and CDC-treated rats by analysis of variance.
Attenuation of inflammation
HFpEF is associated with increased levels of circulating cytokines and infiltration of macrophages and other inflammatory cells in the heart (9). Quantification of cytokines in the serum revealed lower levels of proinflammatory and profibrotic cytokines in CDC-treated rats compared with placebo-treated rats; the levels in CDC-treated rats were comparable with those in control rats (Figure 6A). Among those cytokines, some have been linked to the development of HFpEF (especially monocyte chemotactic protein-1, interleukin-6, and tumor necrosis factor-α) and to the accumulation of collagen (TIMP-1) 12, 37. CDC treatment was also associated with a 2-fold reduction of macrophages (CD68-positive cells) and leukocytes (CD45-positive cells) in the heart compared with placebo, approaching control levels (Figure 6B).
Figure 6.

Anti-Inflammatory Effects of CDCs in HFpEF
(A) Cardiosphere-derived cell (CDC) treatment normalizes the expression of proinflammatory and profibrotic cytokines in serum, including tumor necrosis factor (TNF)-α, interleukin (IL)-6, monocyte chemotactic protein (MCP)-1, and tissue inhibitor of metalloproteinase (TIMP)-1 (n = 4 in each group). (B) CDC treatment decreases myocardial infiltration by macrophages (CD68) and leukocytes (CD45) in the left ventricle (n = 5 in each group). *p < 0.05 versus placebo- and CDC-treated rats and †p < 0.05 versus control and CDC-treated rats, by nonparametric tests for (A) and by analysis of variance for (B). CINC = cytokine-induced neutrophil chemoattractant; CINC = cytokine-induced neutrophil chemoattractant; CNTF = ciliary neurotrophic factor; GMCSF = granulocyte-macrophage colony-stimulating factor; INF = interferon; IL = interleukin; NGF = nerve growth factor; VEGF = vascular endothelial growth factor.
Vessel density and cell proliferation
Because microvascular rarefaction has been associated with HFpEF in numerous studies 38, 39, 40, we investigated arteriolar and capillary density in the left ventricle (Figure 7A). Both vascular densities were lower in placebo-treated rats compared with control rats (Figures 7B and 7C); CDC treatment normalized arteriolar density and significantly increased capillary density compared with placebo, although capillary density did not reach the value measured in control rats. Parallel measurements of cell proliferation using Ki67 immunostaining (Figure 7D) revealed that CDCs stimulated cardiomyocyte proliferation (cells positive for both α-sarcomeric actinin and Ki67) (Figure 7E). In contrast, the number of proliferating fibroblasts (Ki67-positive, vimentin-positive cells) was greatly increased in placebo-treated (but not CDC-treated) high-salt DS rat hearts relative to low-salt control rats (Figure 7F).
Figure 7.

Microvascular Density and Cardioproliferation Enhanced by CDCs, While Fibroblast Proliferation Is Suppressed
(A) Immunostaining for von Willebrand factor (VWF) and smooth muscle actin (SMA) in control, placebo-treated, and cardiosphere-derived cell (CDC)–treated rats. CDC treatment increases arteriolar (B) and capillary (C) density in the left ventricle. (D) Immunostaining for Ki67 and α-actinin (SA) and for Ki67 and vimentin in control, placebo-treated, and CDC-treated rats. CDC treatment increased cardiomyocyte (CM) proliferation (E) and decreased the proliferation of fibroblasts (F) compared with placebo. n = 5 in each group. †p < 0.05 versus control and CDC-treated rats, both by analysis of variance; ‡p < 0.05 versus control and placebo-treated rats. DAPI = 4′,6-diamidino-2-phenylindole.
Next-generation ribonucleic acid sequencing
Next-generation sequencing was performed in the 3 groups. Supplemental Figures 2A to 2C show head-to-head pairwise comparison of gene expression in the 3 groups. The heat maps reveal that the HFpEF phenotype is associated with major global changes in gene expression, as shown by the comparison between placebo and control groups (Supplemental Figure 2A). More important, the comparison between CDC- and placebo-treated rats (Supplemental Figure 2B) reveals that CDC treatment dramatically changed gene expression. Interestingly, >300 genes whose expression was up- or downregulated in HFpEF (i.e., in high-salt placebo hearts) had their expression levels “rescued” by CDC treatment (Figure 8A). Some of these transcript changes involved genes that underlie HFpEF-related pathophysiologic features that we and others have identified (a nonexhaustive list is shown in Figure 8B). Indeed, key genes involved in fibrosis, inflammation, and macrophage signaling, or associated with the consequences of HFpEF (brain natriuretic and atrial natriuretic peptides), were upregulated in placebo hearts but returned fully or partially to control levels after CDC treatment. These profound changes in the transcriptome reveal HFpEF-related activation, and CDC-induced inhibition, of key disease-associated signaling and effector pathways (Figure 8C).
Figure 8.

Transcriptomic Analysis Reveals Profound CDC-Responsive Changes of Gene Expression in HFpEF
(A) Heat map showing the transcripts that are up- or downregulated by heart failure with preserved ejection fraction (HFpEF) and normalized partially or completely by cardiosphere-derived cell (CDC) treatment. (B) Selected genes involved in inflammation and fibrosis or associated with HFpEF that are rescued by CDC treatment (abbreviations in Supplemental Table 1). (C) Selected pathways modified by CDC treatment compared with placebo; blue, inhibited; orange, activated; white, not clear from the database (published research missing).
Discussion
The challenge of HFpEF is increasing as the population ages and comorbidities become more prevalent. The HFpEF hospitalization rate is now greater than that for heart failure with reduced LVEF (3). So far, no treatment for HFpEF has proved effective (6). Here, we have demonstrated that cell therapy by CDCs can reverse the functional abnormalities of HFpEF and improve survival in a rat model of hypertension-induced HFpEF. The CDC-induced reversal of HFpEF occurred without a reduction in either blood pressure or cardiac hypertrophy. The selective correction of functional HFpEF abnormalities creates an unprecedented opportunity for mechanistic insights. Potentially causal pathways (i.e., those that accompany the abnormalities in HFpEF) can now be distinguished from those that are merely associative. Our findings support the concept that fibrosis and inflammation are causative in HFpEF (10): reductions in those 2 pathophysiological processes underlie the resolution of HFpEF, while hypertrophy and hypertension remain unchanged.
Cardiac hypertrophy has long been thought to be the linchpin in HFpEF 8, 41, 42. However, several recent studies in animal models and humans have implicated inflammation and collagen infiltration 10, 12, 13, 14, 43, 44, 45. Hypertension and other comorbidities can favor a systemic proinflammatory state with high circulating cytokine levels, including interleukin-6, tumor necrosis factor–α, and monocyte chemotactic protein–1 9, 10, 37. Inflammation leads to activation, recruitment, and transendothelial migration of leukocytes and monocytes or macrophages into the heart. These inflammatory cells contribute to LV fibrosis by promoting the differentiation of fibroblasts into myofibroblasts 33, 46, 47. The resulting increase in LV collagen content is the main contributor to the increase in passive myocardial fiber stiffness, a major component of diastolic impairment in HFpEF (12). The observed phenotypic improvements after CDC treatment were associated with decreases in circulating inflammatory cytokines (including interleukin-6, tumor necrosis factor–α, and monocyte chemotactic protein-1) and less myocardial inflammation. In addition, myofibroblast infiltration, collagen content, and collagen production were increased in placebo-treated animals but fell markedly after CDC treatment. The parallel decrease in transcripts for MMPs and TIMPs (Supplemental Figure 1) suggests, but does not prove, increased extracellular matrix turnover in the placebo-treated animals that is normalized by CDC treatment. Increased extracellular matrix turnover in HFpEF has been described, and MMPs and TIMPs have been suggested as biomarkers for the diagnosis and prognosis of HFpEF 48, 49, 50. Taken together, these findings strengthen the hypothesis that proinflammatory and profibrotic stimuli play a major role in the development of HFpEF (10). Furthermore, our results suggest that modulating those stimuli may improve HFpEF phenotype and outcomes. The mechanism whereby CDCs modify inflammation and fibrosis clearly involves major changes in gene expression (Figure 8C). Such changes are long-lasting, as the transcriptome was analyzed 4 weeks after CDC injection (at which point injected allogeneic cells are no longer detectable) (18). Our working hypothesis posits that CDCs secrete exosomes laden with micro-ribonucleic acids and other noncoding ribonucleic acids that collectively mold the target transcriptome. Although in-depth exploration of this hypothesis is beyond the scope of this initial report, multiple lines of evidence in other models show that exosomes mediate the benefits of CDCs and modify phenotype and gene expression in recipient cells 51, 52, 53. We are intrigued by the possibility that the drastic changes in the expression of the genes involved in fibrosis and inflammation seen here may be in vivo manifestations of exosome-mediated phenotypic conversions such as those we have described in skin fibroblasts (52).
It is noteworthy that no changes in the magnitude of cardiac hypertrophy were observed after CDC treatment. Cardiac hypertrophy assessed using 3 different techniques (echocardiography, heart weight, and cardiomyocyte cross-sectional area) was present and virtually identical in both high-salt groups at endpoint but not in control rats. Here, decreased inflammation and fibrosis underlie the resolution of HFpEF, despite persistent hypertrophy and hypertension. Thus, attenuation of cardiac hypertrophy is not required to normalize diastolic function. Regarding the mechanical properties of the cardiomyocytes, we observed that CDCs enhance cardiomyocyte (but not fibroblast) proliferation. Nevertheless, the absolute number of new myocytes remains low, and cardiomyocytes remain hypertrophic after CDC treatment. Thus, the correction of altered mechanical properties is more likely related to intrinsic changes in preexisting cardiomyocytes than to a dominant effect of newly generated cardiomyocytes. Our ongoing characterization of cardiomyocytes isolated from the various groups is consistent with this prediction, but such studies are beyond the scope of this initial report.
Microvascular dysfunction and rarefaction have been reported as additional contributors to HFpEF. Several studies have shown that even in the absence of coronary artery disease, patients with HFpEF have fewer microvessels and lower coronary reserve 38, 39, 40. In addition to their anti-inflammatory and antifibrotic properties, CDCs are able to promote the growth of new vessels 18, 21. We observed increases in the numbers of arterioles and capillaries in the left ventricle after CDC treatment (Figure 7). Although the vascular findings here are limited to histologic findings, we have previously demonstrated that cardiosphere-related increases in vessels by histology were associated with augmented myocardial perfusion and coronary reserve (54).
Study limitations
HFpEF is a multifactorial disease involving aging and cardiovascular risk factors. Although DS rats reproduce most of the key features of HFpEF (including hypertension, inflammation, fibrosis, and microvascular rarefaction), some typical contributors to this disease (especially aging) are absent in this model. Also, our follow-up was relatively short. Because death and/or progression toward systolic dysfunction occurs in DS rats around 19 to 20 weeks of age 55, 56, we decided to set the endpoint before the terminal decrease in LVEF, which might otherwise have confounded the analysis of HFpEF. Therefore, we have no information (other than the observed mortality benefit at 6 weeks) regarding the potential long-term persistence of the benefit. Regarding remodeling of the extracellular matrix, we have shown directionally appropriate changes in various transcripts and proteins, but we have not performed zymography to directly evaluate MMP activity. Finally, we have yet to demonstrate the involvement of exosomes and micro-ribonucleic acids in the dramatic changes observed after CDC treatment here. For now, the proposed mechanisms remain at the level of plausibility, as bolstered by previous studies 51, 52.
Conclusions
CDCs normalized LV relaxation and improved survival in a rat model of HFpEF, without blunting hypertension or hypertrophy. Given that CDCs are already in advanced clinical testing for other indications (57), the present findings motivate clinical trials of CDCs in HFpEF.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: Intracoronary CDC administration normalizes diastolic function and improves survival in rats with HFpEF. Reversal of inflammation and fibrosis, but not attenuation of cardiac hypertrophy, underlies these functional benefits.
TRANSLATIONAL OUTLOOK: Given that CDCs are already in advanced clinical testing for other indications, the present study motivates clinical trials of CDCs in HFpEF. Also, the selective correction of functional HFpEF abnormalities observed here creates an unprecedented opportunity for mechanistic insights.
Footnotes
This study was funded by the Cedars-Sinai Board of Governors Heart Stem Cell Center. General support for the laboratory is provided by the National Institutes of Health. Dr. Gallet has received a grant from the French Society of Cardiology. Dr. Marbán owns equity in Capricor Inc. Dr. Zile has received consulting fees from Capricor Inc. as an external advisor. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose. Drs. Gallet and de Couto contributed equally to this work.
Appendix
References
- 1.Hogg K., Swedberg K., McMurray J. Heart failure with preserved left ventricular systolic function; epidemiology, clinical characteristics, and prognosis. J Am Coll Cardiol. 2004;43:317–327. doi: 10.1016/j.jacc.2003.07.046. [DOI] [PubMed] [Google Scholar]
- 2.Lam C.S., Donal E., Kraigher-Krainer E., Vasan R.S. Epidemiology and clinical course of heart failure with preserved ejection fraction. Eur J Heart Fail. 2011;13:18–28. doi: 10.1093/eurjhf/hfq121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Owan T.E., Hodge D.O., Herges R.M., Jacobsen S.J., Roger V.L., Redfield M.M. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355:251–259. doi: 10.1056/NEJMoa052256. [DOI] [PubMed] [Google Scholar]
- 4.Brouwers F.P., de Boer R.A., van der Harst P. Incidence and epidemiology of new onset heart failure with preserved vs. reduced ejection fraction in a community-based cohort: 11-year follow-up of PREVEND. Eur Heart J. 2013;34:1424–1431. doi: 10.1093/eurheartj/eht066. [DOI] [PubMed] [Google Scholar]
- 5.Tribouilloy C., Rusinaru D., Mahjoub H. Prognosis of heart failure with preserved ejection fraction: a 5 year prospective population-based study. Eur Heart J. 2008;29:339–347. doi: 10.1093/eurheartj/ehm554. [DOI] [PubMed] [Google Scholar]
- 6.McMurray J.J., Adamopoulos S., Anker S.D. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2012;33:1787–1847. doi: 10.1093/eurheartj/ehs104. [DOI] [PubMed] [Google Scholar]
- 7.Borlaug B.A., Paulus W.J. Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J. 2011;32:670–679. doi: 10.1093/eurheartj/ehq426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zile M.R., Baicu C.F., Gaasch W.H. Diastolic heart failure—abnormalities in active relaxation and passive stiffness of the left ventricle. N Engl J Med. 2004;350:1953–1959. doi: 10.1056/NEJMoa032566. [DOI] [PubMed] [Google Scholar]
- 9.Glezeva N., Baugh J.A. Role of inflammation in the pathogenesis of heart failure with preserved ejection fraction and its potential as a therapeutic target. Heart Fail Rev. 2014;19:681–694. doi: 10.1007/s10741-013-9405-8. [DOI] [PubMed] [Google Scholar]
- 10.Paulus W.J., Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263–271. doi: 10.1016/j.jacc.2013.02.092. [DOI] [PubMed] [Google Scholar]
- 11.van Empel V., Brunner-La Rocca H.P. Inflammation in HFpEF: key or circumstantial? Int J Cardiol. 2015;189:259–263. doi: 10.1016/j.ijcard.2015.04.110. [DOI] [PubMed] [Google Scholar]
- 12.Zile M.R., Baicu C.F., Ikonomidis J.S. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation. 2015;131:1247–1259. doi: 10.1161/CIRCULATIONAHA.114.013215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Westermann D., Lindner D., Kasner M. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. 2011;4:44–52. doi: 10.1161/CIRCHEARTFAILURE.109.931451. [DOI] [PubMed] [Google Scholar]
- 14.Kasner M., Westermann D., Lopez B. Diastolic tissue Doppler indexes correlate with the degree of collagen expression and cross-linking in heart failure and normal ejection fraction. J Am Coll Cardiol. 2011;57:977–985. doi: 10.1016/j.jacc.2010.10.024. [DOI] [PubMed] [Google Scholar]
- 15.Dorn G.W., II The fuzzy logic of physiological cardiac hypertrophy. Hypertension. 2007;49:962–970. doi: 10.1161/HYPERTENSIONAHA.106.079426. [DOI] [PubMed] [Google Scholar]
- 16.Aminzadeh M.A., Tseliou E., Sun B. Therapeutic efficacy of cardiosphere-derived cells in a transgenic mouse model of non-ischaemic dilated cardiomyopathy. Eur Heart J. 2015;36:751–762. doi: 10.1093/eurheartj/ehu196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Couto G., Liu W., Tseliou E. Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J Clin Invest. 2015;125:3147–3162. doi: 10.1172/JCI81321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malliaras K., Li T.S., Luthringer D. Safety and efficacy of allogeneic cell therapy in infarcted rats transplanted with mismatched cardiosphere-derived cells. Circulation. 2012;125:100–112. doi: 10.1161/CIRCULATIONAHA.111.042598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith R.R., Barile L., Cho H.C. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation. 2007;115:896–908. doi: 10.1161/CIRCULATIONAHA.106.655209. [DOI] [PubMed] [Google Scholar]
- 20.Tseliou E., Reich H., de Couto G. Cardiospheres reverse adverse remodeling in chronic rat myocardial infarction: roles of soluble endoglin and Tgf-beta signaling. Basic Res Cardiol. 2014;109:443. doi: 10.1007/s00395-014-0443-8. [DOI] [PubMed] [Google Scholar]
- 21.Malliaras K., Smith R.R., Kanazawa H. Validation of contrast-enhanced magnetic resonance imaging to monitor regenerative efficacy after cell therapy in a porcine model of convalescent myocardial infarction. Circulation. 2013;128:2764–2775. doi: 10.1161/CIRCULATIONAHA.113.002863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doi R., Masuyama T., Yamamoto K. Development of different phenotypes of hypertensive heart failure: systolic versus diastolic failure in Dahl salt-sensitive rats. J Hypertens. 2000;18:111–120. doi: 10.1097/00004872-200018010-00016. [DOI] [PubMed] [Google Scholar]
- 23.Horgan S., Watson C., Glezeva N., Baugh J. Murine models of diastolic dysfunction and heart failure with preserved ejection fraction. J Card Fail. 2014;20:984–995. doi: 10.1016/j.cardfail.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 24.Koshizuka R., Ishizu T., Kameda Y., Kawamura R., Seo Y., Aonuma K. Longitudinal strain impairment as a marker of the progression of heart failure with preserved ejection fraction in a rat model. J Am Soc Echocardiogr. 2013;26:316–323. doi: 10.1016/j.echo.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 25.Masuyama T., Yamamoto K., Sakata Y. Evolving changes in Doppler mitral flow velocity pattern in rats with hypertensive hypertrophy. J Am Coll Cardiol. 2000;36:2333–2338. doi: 10.1016/s0735-1097(00)01000-7. [DOI] [PubMed] [Google Scholar]
- 26.Ishizu T., Seo Y., Kameda Y. Left ventricular strain and transmural distribution of structural remodeling in hypertensive heart disease. Hypertension. 2014;63:500–506. doi: 10.1161/HYPERTENSIONAHA.113.02149. [DOI] [PubMed] [Google Scholar]
- 27.Bowen T.S., Rolim N.P., Fischer T. Heart failure with preserved ejection fraction induces molecular, mitochondrial, histological, and functional alterations in rat respiratory and limb skeletal muscle. Eur J Heart Fail. 2015;17:263–272. doi: 10.1002/ejhf.239. [DOI] [PubMed] [Google Scholar]
- 28.Bae S., Yalamarti B., Ke Q. Preventing progression of cardiac hypertrophy and development of heart failure by paricalcitol therapy in rats. Cardiovasc Res. 2011;91:632–639. doi: 10.1093/cvr/cvr133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim S., Yoshiyama M., Izumi Y. Effects of combination of ACE inhibitor and angiotensin receptor blocker on cardiac remodeling, cardiac function, and survival in rat heart failure. Circulation. 2001;103:148–154. doi: 10.1161/01.cir.103.1.148. [DOI] [PubMed] [Google Scholar]
- 30.Rimbaud S., Ruiz M., Piquereau J. Resveratrol improves survival, hemodynamics and energetics in a rat model of hypertension leading to heart failure. PLoS One. 2011;6:e26391. doi: 10.1371/journal.pone.0026391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yazawa H., Miyachi M., Furukawa M. Angiotensin-converting enzyme inhibition promotes coronary angiogenesis in the failing heart of Dahl salt-sensitive hypertensive rats. J Card Fail. 2011;17:1041–1050. doi: 10.1016/j.cardfail.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 32.Appleton C.P., Hatle L.K., Popp R.L. Relation of transmitral flow velocity patterns to left ventricular diastolic function: new insights from a combined hemodynamic and Doppler echocardiographic study. J Am Coll Cardiol. 1988;12:426–440. doi: 10.1016/0735-1097(88)90416-0. [DOI] [PubMed] [Google Scholar]
- 33.Kai H., Kuwahara F., Tokuda K., Imaizumi T. Diastolic dysfunction in hypertensive hearts: roles of perivascular inflammation and reactive myocardial fibrosis. Hypertens Res. 2005;28:483–490. doi: 10.1291/hypres.28.483. [DOI] [PubMed] [Google Scholar]
- 34.Tseliou E., de Couto G., Terrovitis J. Angiogenesis, cardiomyocyte proliferation and anti-fibrotic effects underlie structural preservation post-infarction by intramyocardially-injected cardiospheres. PLoS One. 2014;9:e88590. doi: 10.1371/journal.pone.0088590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Makkar R.R., Smith R.R., Cheng K. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet. 2012;379:895–904. doi: 10.1016/S0140-6736(12)60195-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malliaras K., Makkar R.R., Smith R.R. Intracoronary cardiosphere-derived cells after myocardial infarction: evidence of therapeutic regeneration in the final 1-year results of the CADUCEUS trial (Cardiosphere-Derived Autologous Stem Cells to Reverse Ventricular Dysfunction) J Am Coll Cardiol. 2014;63:110–122. doi: 10.1016/j.jacc.2013.08.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Collier P., Watson C.J., Voon V. Can emerging biomarkers of myocardial remodelling identify asymptomatic hypertensive patients at risk for diastolic dysfunction and diastolic heart failure? Eur J Heart Fail. 2011;13:1087–1095. doi: 10.1093/eurjhf/hfr079. [DOI] [PubMed] [Google Scholar]
- 38.Goligorsky M.S. Microvascular rarefaction: the decline and fall of blood vessels. Organogenesis. 2010;6:1–10. doi: 10.4161/org.6.1.10427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoenig M.R., Bianchi C., Rosenzweig A., Sellke F.W. The cardiac microvasculature in hypertension, cardiac hypertrophy and diastolic heart failure. Curr Vasc Pharmacol. 2008;6:292–300. doi: 10.2174/157016108785909779. [DOI] [PubMed] [Google Scholar]
- 40.Mohammed S.F., Hussain S., Mirzoyev S.A., Edwards W.D., Maleszewski J.J., Redfield M.M. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. 2015;131:550–559. doi: 10.1161/CIRCULATIONAHA.114.009625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shah A.M., Pfeffer M.A. The many faces of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2012;9:555–556. doi: 10.1038/nrcardio.2012.123. [DOI] [PubMed] [Google Scholar]
- 42.Maurer M.S., King D.L., El-Khoury Rumbarger L., Packer M., Burkhoff D. Left heart failure with a normal ejection fraction: identification of different pathophysiologic mechanisms. J Card Fail. 2005;11:177–187. doi: 10.1016/j.cardfail.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 43.Desai A.S. Heart failure with preserved ejection fraction: time for a new approach? J Am Coll Cardiol. 2013;62:272–274. doi: 10.1016/j.jacc.2013.03.075. [DOI] [PubMed] [Google Scholar]
- 44.Kuwahara F. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. 2002;106:130–135. doi: 10.1161/01.cir.0000020689.12472.e0. [DOI] [PubMed] [Google Scholar]
- 45.Melendez G.C., McLarty J.L., Levick S.P., Du Y., Janicki J.S., Brower G.L. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. 2010;56:225–231. doi: 10.1161/HYPERTENSIONAHA.109.148635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nicoletti A., Heudes D., Mandet C., Hinglais N., Bariety J., Michel J.B. Inflammatory cells and myocardial fibrosis: spatial and temporal distribution in renovascular hypertensive rats. Cardiovasc Res. 1996;32:1096–1107. doi: 10.1016/s0008-6363(96)00158-7. [DOI] [PubMed] [Google Scholar]
- 47.Sciarretta S., Ferrucci A., Ciavarella G.M. Markers of inflammation and fibrosis are related to cardiovascular damage in hypertensive patients with metabolic syndrome. Am J Hypertens. 2007;20:784–791. doi: 10.1016/j.amjhyper.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 48.Zouein F.A., de Castro Bras L.E., da Costa D.V., Lindsey M.L., Kurdi M., Booz G.W. Heart failure with preserved ejection fraction: emerging drug strategies. J Cardiovasc Pharmacol. 2013;62:13–21. doi: 10.1097/FJC.0b013e31829a4e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zile M.R., Desantis S.M., Baicu C.F. Plasma biomarkers that reflect determinants of matrix composition identify the presence of left ventricular hypertrophy and diastolic heart failure. Circ Heart Fail. 2011;4:246–256. doi: 10.1161/CIRCHEARTFAILURE.110.958199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zannad F., Rossignol P., Iraqi W. Extracellular matrix fibrotic markers in heart failure. Heart Fail Rev. 2010;15:319–329. doi: 10.1007/s10741-009-9143-0. [DOI] [PubMed] [Google Scholar]
- 51.Ibrahim A.G., Cheng K., Marban E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Reports. 2014;2:606–619. doi: 10.1016/j.stemcr.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tseliou E., Fouad J., Reich H. Fibroblasts rendered antifibrotic, antiapoptotic, and angiogenic by priming with cardiosphere-derived extracellular membrane vesicles. J Am Coll Cardiol. 2015;66:599–611. doi: 10.1016/j.jacc.2015.05.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Couto G., Makkar N., Marbán E. Cardiosphere-derived cell exosomes confer acute cardioprotection following ischemia-reperfusion injury through macrophage polarization. Circulation. 2015;132:A16991. [Google Scholar]
- 54.Gallet R., Tseliou E., Dawkins J. Intracoronary delivery of self-assembling heart-derived microtissues (cardiospheres) for prevention of adverse remodeling in a pig model of convalescent myocardial infarction. Circ Cardiovasc Interv. 2015;8 doi: 10.1161/CIRCINTERVENTIONS.115.002391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klotz S., Hay I., Zhang G., Maurer M., Wang J., Burkhoff D. Development of heart failure in chronic hypertensive Dahl rats: focus on heart failure with preserved ejection fraction. Hypertension. 2006;47:901–911. doi: 10.1161/01.HYP.0000215579.81408.8e. [DOI] [PubMed] [Google Scholar]
- 56.Qu P., Hamada M., Ikeda S., Hiasa G., Shigematsu Y., Hiwada K. Time-course changes in left ventricular geometry and function during the development of hypertension in Dahl salt-sensitive rats. Hypertens Res. 2000;23:613–623. doi: 10.1291/hypres.23.613. [DOI] [PubMed] [Google Scholar]
- 57.Marban E. Breakthroughs in cell therapy for heart disease: focus on cardiosphere-derived cells. Mayo Clin Proc. 2014;89:850–858. doi: 10.1016/j.mayocp.2014.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.