

Abstract
Autosomal dominant omodysplasia is a rare skeletal dysplasia characterized by short humeri, radial head dislocation, short first metacarpals, facial dysmorphism and genitourinary anomalies. We performed next-generation whole-exome sequencing and comparative analysis of a proband with omodysplasia, her unaffected parents and her affected daughter. We identified a de novo mutation in FRIZZLED2 (FZD2) in the proband and her daughter that was not found in unaffected family members. The FZD2 mutation (c.1644G>A) changes a tryptophan residue at amino acid 548 to a premature stop (p.Trp548*). This altered protein is still produced in vitro, but we show reduced ability of this mutant form of FZD2 to interact with its downstream target DISHEVELLED. Furthermore, expressing the mutant form of FZD2 in vitro is not able to facilitate the cellular response to canonical Wnt signaling like wild-type FZD2. We therefore conclude that the FRIZZLED2 mutation is a de novo, novel cause for autosomal dominant omodysplasia.
Introduction
Autosomal dominant (AD) omodysplasia is a rare skeletal dysplasia characterized by short humeri, dislocated radial head, short first metacarpals, facial dysmorphism with round face, frontal bossing, short nose and long philtrum and variable degrees of genitourinary anomalies. To date, only eight cases have been reported in the literature (1–4), and no reports have identified causative gene mutations. Autosomal recessive (AR) omodysplasia, which differs clinically from the AD form, has also been reported. AR omodysplasia is associated with micromelia, severe short stature and developmental delays (5). AR omodysplasia is caused by mutations in glypican 6 [GPC6; (6)], which is thought to impair WNT5a activity (7,8). Here, we report a patient with omodysplasia and her similarly affected daughter. Using next-generation exome sequencing, we identify a single causal mutation in the affected patients not present in other family members. Taken together, our sequence analysis, along with our molecular studies, strongly implicates a Wnt-dependent signaling mechanism for AD omodysplasia.
Results
Patient 1 (proband, Table 1) was born with multiple anomalies, including bilateral cleft lip and cleft palate, short upper extremities, dysmorphic facial features and hypoplastic labia and clitoris. She was the product of a full-term vaginal delivery to a 31-year-old G3P2011 woman and her unrelated 33-year-old husband. The proband's maternal great grandfather was born with cleft lip. Birth weight was 3416 g (80th centile), length 48.3 cm (30th centile) and head circumference 35 cm (70th centile). On her initial evaluation in the Genetics Clinic, she was also noted to have frontal bossing, a flat broad nasal bridge, hypertelorism (interpupillary distance of 45 mm, 97th percentile for age), left preauricular sinus and glabellar hemangiomas, extending from forehead to nasal tip. There was rhizomelic shortening of the upper extremities. Chest and spine X rays, as well as renal ultrasound, were normal. Chromosome analysis showed a normal female karyotype of 46,XX. At the age of 10 years, her height was 132 cm (10th centile), span 103.8 cm, upper-to-lower segment ratio 0.88, head circumference 52.5 cm (50th centile) and interpupillary distance of 5.2 cm (50th centile, Fig. 1A). At 11 years, scoliosis X rays showed a 10 degree dextroscoliosis from T12 to L4 and no vertebral anomalies. During follow-up at 25 years of age (Fig. 1B), she reported a pelvic ultrasound was done to evaluate dyspareunia and a didelphic uterus was identified. Her first pregnancy resulted in the delivery of a male at 22 weeks who expired. Her subsequent pregnancy resulted in the delivery of a daughter (Patient 2) at 28 weeks of gestation (Fig. 1C).
Table 1.
Comparison of AD omodysplasia features
| Clinical features | Patient 1/proband | Patient 2 | Maroteaux patient 1 | Maroteaux Patient 2 | Maroteaux Patient 3 | Venditti Patient 1 | Venditti Patient 2 | Gordon patient | Olney Patient 1 | Olney Patient 2 |
|---|---|---|---|---|---|---|---|---|---|---|
| Gender | F | F | M | F | F | F | M | F | M | F |
| Age | 25 years | 6 years | Newborn | 27 years | 10 years | 12 years | 1 month | 48 year | 23 years | 18 months |
| Stature/length percentile | 10 | <5 | 5 | 10 | 10 | 25 | 25 | <5 | <5 | <5 |
| Skeletal features | ||||||||||
| Short humeri | + | + | + | + | + | + | + | + | + | + |
| Radial dislocation, limitation of movement | + | + | + | + | + | + | + | + | + | + |
| Short ulnae | NA | + | − | + | + | − | ND | + | − | − |
| Short first metacarpal | NA | + | + | ND | + | + | + | + | + | + |
| Femoral anomalies, proximal | NA | + | − | − | + | − | − | + | − | − |
| Short fibulae | NA | + | − | − | − | − | − | + | − | − |
| Vertebral anomalies | − | + | − | + | ND | − | − | − | − | − |
| Craniofacial features | ||||||||||
| Round face | + | + | + | ND | + | + | ND | − | ND | ND |
| Frontal bossing, prominent forehead | + | + | + | + | + | + | + | − | ND | ND |
| Small nose with broad tip | − | + | + | + | + | + | + | − | ND | ND |
| Long philtrum | + | + | − | − | + | + | + | + | ND | ND |
| Flat nasal bridge | + | + | + | ND | + | + | + | − | ND | ND |
| Cleft lip and cleft palate | + | − | − | − | − | − | − | − | − | − |
| Genitourinary features | ||||||||||
| Hypoplastic genitalia/other genital anomalies | + | − | + | ND | − | + | + | + | ND | ND |
| Uterine anomalies | + | NA | NR | ND | ND | ND | NR | + | NR | ND |
Most common features are highlighted in gray.
NA, not assessed; ND, not described; NR, not relevant.
Figure 1.

Phenotyping of proband and pedigree of patients with AD omodysplasia. Proband at 10 (A) and 25 years of age (B) presenting with skeletal dysplasia and craniofacial phenotypes. The proband's parents and her daughter, represented in the pedigree in (C), provided specimens for exome sequencing.
Patient 2 was the 1130-g product of a twenty-eight 3/7-week vaginal delivery to Patient 1 who was then a 19-year-old G2P0010 woman. The pregnancy was uncomplicated except for maternal use of cigarettes. Patient 2 had respiratory distress syndrome requiring assisted ventilation for 24 h followed by CPAP. The neonatal period was also complicated by grade I intraventricular hemorrhage and feeding difficulties with gastroesophageal reflux. Skeletal X rays showed hypoplasia of T11 vertebral body and bilateral dislocation of the radius with short humeri. At the age of 3 months, she had a round face, mildly dysplastic ears with overfolded helices and apparent hypertelorism (Fig. 2A). The nose was short with a flat nasal bridge, there was a long philtrum and mild micrognathia and the palate was intact. Genitalia appeared normal for a female infant. Follow-up at 6 years revealed that she was healthy with no major medical problems except for recurrent otitis media. Intellectual development was normal, but she was receiving speech therapy in school. Height was 101.2 cm (<second centile), weight 15.6 kg (second centile), head circumference 49 cm (fifth centile), span was 78.3 cm and upper-to-lower segment ratio was 0.99 (Fig. 2B). Her face was round; nose was short with broad nasal base. There was mild tenting of the upper lip. The ears were posteriorly rotated. The inner canthal distance was 2.9 cm (65th centile), interpupillary distance 5.1 cm (60th centile) and outer canthal distance 8.5 cm (80th centile). The musculoskeletal examination showed primarily rhizomelic shortening of the upper extremities and a mild shortening of the forearms with limited forearm supination/pronation. There was mild fifth finger clinodactyly with no brachydactyly. The neurologic examination was normal. A skeletal X-ray survey (Fig. 2C–E) showed a dysplastic T11 with anterior wedging and retrolisthesis in relation to T10 and T12. The humeri were short, and poorly formed radial heads were dislocated bilaterally. The capitella were dysplastic and the ulnas were shortened. The first metacarpals were short. There was widening of the femoral necks and mild lateral uncovering of the femoral heads bilaterally. The fibulas were shortened with hypoplastic proximal ossification centers. There was no additional family history of short stature or limb anomalies.
Figure 2.

Patient 2 with AD omodysplasia. Patient 2 at 3 months (A) and 6 years (B). Skeletal phenotypes include abnormalities of the vertebrae (C), humerus (D) and hands (E).
In order to determine the genetic cause of the dysplasia seen in these patients, we performed next-generation exome sequencing on Patients 1 and 2 as well as the parents of Patient 1. Sheared genomic DNA was enriched with NimbleGen EZ Exome V2 kit, and the exome library was sequenced using Illumina's Hi Seq 2000. Alignment and variant detection was performed using the Broad Institute's web-based Genome Analysis Tookit [GATK, (9)]. Quality control and data filtering were performed on VCF files in Golden Helix's SNP and Variation Suite. Non-synonymous coding variants were compared with control databases, including dbSNP, Human Gene Mutation Database, NHLBI's ESP6500 exome data (10), the 1000 Genomes Project (11) and an internal Cincinnati Children's Hospital Medical Center (CCHMC) control cohort. We reasoned the likely mutation was a de novo mutation in Patient 1 with AD transmission to Patient 2. Our analysis therefore targeted mutations present only in the affected individuals that were predicted to be deleterious and not present in any of the databases we searched (Table 2). We identified only one such deleterious mutation shared by the proband and affected daughter that was not present in the proband's unaffected parents. A single base pair change (c.1644G>A) within the open reading frame of the gene FRIZZLED2 (FZD2) was identified (Fig. 3A). This nonsense mutation changes a tryptophan residue at amino acid 548 to a premature stop (p.Trp548*, Fig. 3B). This variant was not found in any control databases. The identified causal p.TRP548* mutation in Patients 1 and 2 is predicted to result in a truncation of the FRIZZLED2 protein at the periphery of the canonical DISHEVELLED interaction domain, which may have significant consequences for canonical signaling (Fig. 3C). In canonical Wnt signaling, Frizzled binding to Dishevelled requires a conserved motif, KTxxxW, in the intracellular portion of the Frizzled receptor (13–15). The tryptophan residue (W) in the KTxxxW motif is the site of the nonsense mutation in our patients (Fig. 3B). Recent work has shown that the Frizzled–Dishevelled interaction is modulated by a discontinuous motif in Frizzled, which is spread across three intracellular loops of the Frizzled receptor (16). The p.TRP548* mutation would prevent translation of some of the residues in this model of Dishevelled binding as well.
Table 2.
Exome sequencing analysis
| Familial variant analysis | Number of variantsa |
|---|---|
| Total variants | 96 485 |
| Quality control | 81 617 |
| De novo mutations | 8 |
| Coding variants | 5 |
| European American variants with MAF < 0.01 (NHLBI ESP6500 exome data) | 4 |
| Variants with MAF < 0.01 [1000-genome project (12)] | 3 |
| Variants with MAF < 0.01 (internal database) | 1 |
aThe number of variants identified after exome sequencing and each step of subsequent bioinformatics analysis is indicated. The total analysis indicated only a single mutation that was coding, deleterious and not seen in any control databases.
Figure 3.

Mutations in FRIZZLED2 cause AD omodysplasia. (A) Sanger sequencing confirms findings from next-generation sequencing of heterozygous FZD2 mutations in the proband and daughter, but not the proband's parents. (B) FZD2 mutation creates a premature stop. (C) This is predicted to create an FZD2 protein lacking the majority of the intracellular cytoplasmic tail partially responsible for Dishevelled binding (gray).
In support of our finding that omodysplasia is due to a FZD2 mutation, multiple members of the Wnt pathway are expressed during both limb and craniofacial development in multiple model systems. Previous studies have reported that Fzd2 is broadly expressed throughout the developing head and limbs in various model systems (17–21). We confirmed these findings by performing in situ hybridization for Fzd2 in an avian model system. Fzd2 was expressed throughout the developing head (Fig. 4A and B) and within the proximal limb mesenchyme that contributes to the developing skeletal elements (Fig. 4C). Furthermore, FZD2 protein was detected in both the developing head and limb (Fig. 4D). Taken together, our data confirm that of many others and show that the temporal and spatial expression of both Fzd2 message and FZD2 protein is consistent with the phenotype of omodyplasia. These findings support our hypothesis that mutations in FZD2 could be causal for omodyplasia.
Figure 4.
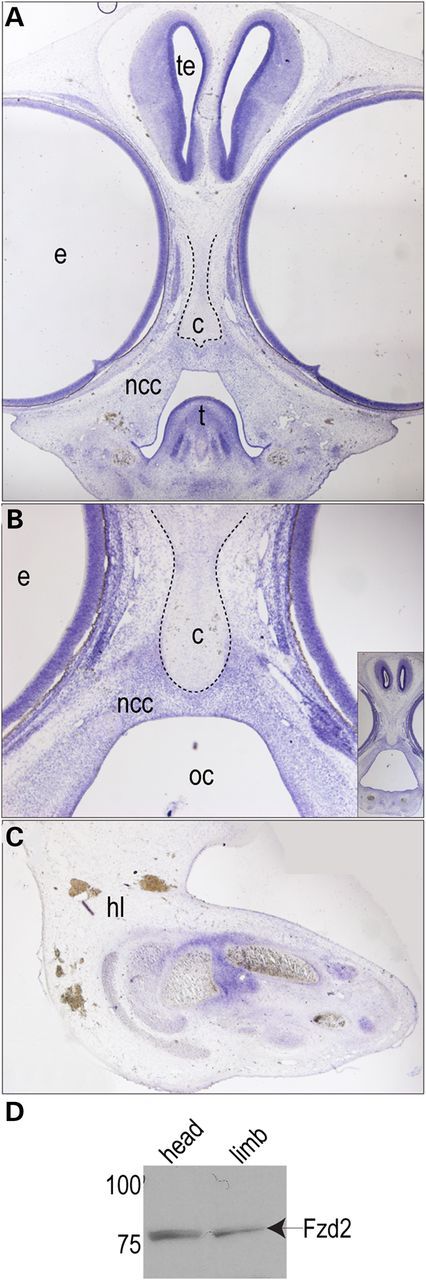
Fzd2 expression in developing craniofacial and limb tissues. RNA in situ hybridization indicates the expression of Fzd2 in HH stage 30 chicken embryos in the developing face (A and B) and limbs (C). Image in B is higher magnification view of image shown in the inset. Digoxigenin-labeled riboprobes were used according to published protocols. (D) Western immunoblotting for FZD2 protein confirms expression in these tissues from HH stage 25 embryos. c-cartilage, e-eye, hl-hindlimb, ncc-neural crest cells, oc-oral cavity, te-telencephalon and t-tongue.
To function as Wnt receptors, Fzd proteins must be properly presented on the cell surface membrane. To determine whether mislocalization of FZD2p.TRP548* was the molecular mechanism associated with AD omodysplasia, we cloned GFP-fusion protein constructs with both wild-type Fzd2 (Fzd2WT-GFP) and Fzd2 with the p.TRP548* mutation (Fzd2p.TRP548*-GFP). There were no observed differences in the subcellular localization of the Fzd2WT-GFP as compared with the Fzd2p.TRP548*-GFP in both HEK293T cells (Fig. 5A and B) and NIH3T3 cells (Fig. 5C and D). Thus, our data suggest that the p.TRP548* mutation does not affect the stability or subcellular localization of FZD2.
Figure 5.

The p.W458X mutation does not alter protein levels or subcellular localization. Expression of either Fzd2WT-GFP (A and C) or Fzd2p.TRP548*-GFP (B and D) in HEK293T (A and B) or NIH3T3 (C and D) cells does not significantly differ in abundance or subcellular localization. Scale bar is equal to 20 μm.
A key component of Wnt signal transduction is the binding of Dishvelled (Dvl) proteins to the intracellular portion of the Fzd receptor upon Wnt ligand binding. As the Fzd2 p.TRP548* mutation is predicted to result in loss of some of the Dvl-binding domain, we tested the ability of Fzd2p.TRP548*-GFP-mutant protein to recruit Dvl to the plasma upon Wnt activation. A Dvl2-FLAG fusion protein was co-expressed in both HEK293T cells and NIH3T3 cells (data not shown) along with either Fzd2WT-GFP or Fzd2p.TRP548*-GFP-mutant proteins. Prior to Wnt treatment, the Dvl2 protein was largely found in discrete puncta throughout the cell, not co-localizing with the Fzd2-GFP-fusion proteins (Fig. 6A–D and I–L). Upon stimulating cells expressing Fzd2WT-GFP and Dvl2-FLAG with Wnt media, the punctate expression of Dvl2-FLAG was lost and a significant portion of the Fzd2WT-GFP and Dvl2-FLAG proteins were co-localized (Fig. 6E–H). We next examined cells expressing Fzd2p.TRP548*-GFP-mutant protein and Dvl2-FLAG. In the absence of Wnt ligand, Dvl2-FLAG was also expressed in discrete puncta throughout the cell, not co-localizing with the Fzd2p.TRP548*-GFP-mutant protein (Fig. 6I–L). In the presence of Wnt however, we saw a significant reduction in the recruitment and persistence of the intercellular puncta, as compared with the changes we noted with Fzd2WT-GFP (Fig. 6M–P). We quantified these results via measuring both the proportion of cells in each experimental category (no, some or significant (co-localization, Fig. 6Q), as well as determining an average co-localization value for each cell in each treatment (Fig. 6R) and found that a significant co-localization was identified only in the Wnt-treated Fzd2WT-GFP cells (Fig. 6Q and R). Very little localization was detected in non-treated Fzd2WT-GFP cells or in Fzd2p.TRP548*-GFP cells regardless of Wnt treatment (Fig. 6Q and R). In fact, there was not a significant co-localization or loss of Dvl-positive puncta in any Wnt-treated cells expressing Fzd2p.TRP548*-GFP.
Figure 6.

Dvl2–Fzd2 interactions are perturbed in cells expressing truncated FZD2. Co-expression of Dvl2-FLAG and either Fzd2WT-GFP (A–H) or Fzd2p.TRP548*-GFP (I–P) in unstimulated HEK293T cells resulted in accumulations of Dvl2-FLAG puncta throughout the cell. Wnt treatment in Fzd2WT-GFP cells (E–H) resulted in increased colocalization of the Fzd and Dvl after immunocytochemistry and dissipation of most Dvl2-positive puncta, indicating successful recruitment to the plasma membrane. Treatment of Fzd2p.TRP548*-GFP-mutant cells, however, did not show a robust recruitment of Dvl2-FLAG (M–P). This was quantified as proportion of cells with ‘no’, ‘some’ or ‘significant’ colocalization and loss of puncta (Q) and as an average colocalization value for each cell (R). Number of double-transfected cells analyzed for each condition is indicated in R. Scale bar is equal to 10 μm. **P = 0.001 versus untreated cells.
We hypothesized that the expression of a prematurely truncated FZD2 protein lacking a portion of the intracellular domain would have a negative effect upon Wnt signaling. In order to assess Wnt-signaling activity, we utilized an in vitro signaling system. SuperTOPFLASH (STF) cells stably express a Wnt luciferase reporter controlled by a series of TCF-LEF binding sites known to transduce canonical Wnt signaling and serve as a robust in vitro model (22). Expressing Fzd2WT-GFP in these STF cells resulted in an almost 3-fold increase in Wnt signaling. In striking contrast, expressing Fzd2p.TRP548*-GFP in STF cells did not result in any notable increase in Wnt activity over that of background levels (Fig. 7). These data are consistent with a model wherein the FZD22p.TRP548*-mutant protein lacking a portion of the intracellular domain is significantly less efficient in transducing WNT signaling than the wild-type FZD2. Given our data and the genetic pattern of inheritance, we suggest that the patients that carry the p.TRP548* are haploinsufficient for FZD2 in skeletal development. Alternatively, the p.TRP548* mutation could also be functioning as a ‘dominant negative’ where the expression of the mutant protein is sufficient to reduce Wnt activity, likely by properly binding Wnt ligand at the surface but not propagating the signal. Either of these hypothesized molecular mechanisms would be consistent with the phenotypes arising in humans with heterozygous, de novo FZD2 mutations.
Figure 7.

Expression of truncated FZD2 attenuates canonical Wnt-signaling. STF Wnt reporter cells were transfected with control plasmids or Fzd2p.TRP548*-GFP (mut-Fzd2) and treated with Wnt-conditioned media. Expression of increasing amounts of Fzd2p.TRP548*-GFP reduced the cellular response to Wnt ligand as measured by luciferase activity. Wnt luciferase activity was normalized to the treated, non-transfected cells.**P = 0.002 versus control transfection.
Discussion
The advent of next-generation whole-exome sequencing has opened up vast new opportunities to identify novel, as well as rare and difficult-to-diagnose, single-gene disorders (23). This in turn has led to identification of new disorders and recognition of previously unrecognized phenotypes from known genes and developmental pathways. This is not only important for an understanding of genetic syndromes and disorders, but also the ability to attach specific genes and gene mutations with a specific phenotype gives us greater insight into these developmental pathways. We report two patients, a mother and daughter with AD omodysplasia, both of whom have a c.1644G>A mutation in the FZD2 gene, not previously associated with human disease. The availability of Patient 1’s unaffected biologic parents greatly facilitated the comparative analysis for exomes. This gene codes for the FRIZZLED2 protein that acts as a Wnt receptor. We show that Fzd2 is expressed in the developing face and skeleton in multiple model systems (Fig. 4). We further show that the truncated form of the protein mimicking the human mutation is stable (Fig. 5), expressed in vitro (Fig. 5), does not interact normally with Dvl (Fig. 6) and does not properly transduce canonical Wnt signaling (Fig. 7). Regarding our Dvl-interaction experimental system, we do note that in some systems the expression of Fzd2 alone is sufficient to recruit Dvl to the plasma membrane. In our system (Fig. 6), the addition of Wnt significantly alters the localization of Dvl (i.e. recruits out of the cytoplasmic punctate accumulations to more significantly appear to co-localize with Fzd2). This may be due to the differences in cell types used as compared with other systems (XENOPUS animal caps (14,24) and DROSOPHILA cells (25) as compared with mammalian cells in vitro) or the relative levels of the expression of experimental components in our assay.
There is significant evidence in the literature supporting our conclusion that FZD2 is a causal gene for AD omodysplasia. WNT5A is a known ligand for FZD2 and is mutated in patients with AD Robinow Syndrome, which is characterized by mesomelic shortening, short stature, hypertelorism and mandibular hypoplasia (26). Similarly, multiple perturbations of Wnt signaling negatively impact skeletal development in animal models. Truncations in various members of the Fzd1,2,7 subfamily lead to a delay or block in chondrocyte maturation and shortening of skeletal elements (27,28). Directly relevant to the phenotype and model we present here, retroviral expression of chicken Fzd1 and Fzd7 with deletions of intracellular C-terminal domains (24 amino acids) resulted in shortened and bent humeri, shortened ulna and bent radii (27). This deletion almost perfectly mimics the predicted effect of the mutation in our human patients [also see (29)]. Thus, there is ample precedent for disruptions in Wnt signaling leading to skeletal dysplasia similar to those seen in AD omodysplasia.
There is similarly robust evidence that Wnt signaling is crucial for craniofacial development. WNT3 mutations are found in a human syndrome that includes cleft palate among its phenotypes (30). Furthermore, the disruption of Wnt signaling via delivery of a Wnt antagonist produces similar midfacial malformations and clefting (31). The phenotypes we note in the AD omodysplaisa patients of frontal bossing, small nose, broad tip, long philtrum and flat nasal bridge do not necessarily translate as readily to mouse models making it more difficult to evaluate those phenotypes in animal models. However, Fzds and Wnts are often implicated in the more easily appreciated cleft palate phenotype. At least eleven Wnt ligands are expressed in the mouse during stages important for craniofacial development (19) and multiple mutants have craniofacial phenotypes, including Frizzled receptors (32–34).
Of note in Patient 1 was also the presence of bilateral cleft lip with cleft palate. It was noted that Patient 1’s great grandfather was born with cleft lip, but there was no additional family history of cleft lip with/without cleft palate. The cleft lip and cleft palate are not likely part of the omodysplasia phenotype. To identify candidate mutations that may explain why Patient 1 has these craniofacial phenotypes whereas Patient 2 does not, we reexamined the exomes, focusing on genes in Wnt signaling, which had variants in Patient 1 but not her daughter. We found seven variants in five WNT genes for which the genotype of Patient 1 differed from her daughter (Table 3). Patient 1 was homozygous for variants in WNT11 and WNT2B for which the daughter was heterozygous. The proband also had five additional heterozygous variants in WNT16, WNT3A and WNT9A that were absent in her daughter. None of Patient 1’s additional variants in WNT genes are predicted to have an extremely high consensus impact in the bioinformatics analysis. Wnt11 and Wnt2b are expressed in the mouse pharyngeal arches (35,36), SNPs in WNT3A have been associated with nonsyndromic cleft lip and palate (37,38) and Wnt9a is required for pharyngeal arch development in the zebrafish (39). While several of the variants are known minor alleles, in the presence of a FZD2 mutation, one or more of these could potentially explain the presence of the clefting phenotypes in the proband but not the daughter (34).
Table 3.
Wnt-signaling mutations in patients identified by exome sequencing
| Key | Reference allele | Alternate allele | dbSNP ID | dbSNP MAF | Gene | Consensus impact | Proband | Proband's daughter |
|---|---|---|---|---|---|---|---|---|
| 11_75902539_G_WNT11 | A | G | rs11236648 | 0.239 | WNT11 | MODIFIER | Homozygous variant | Heterozygous |
| 7_120969769_A_WNT16 | G | A | rs2908004 | 0.4799 | WNT16 | MODERATE | Heterozygous | No variant |
| 7_120979089_T_WNT16 | C | T | rs2707466 | 0.4716 | WNT16 | MODERATE | Heterozygous | No variant |
| 1_113063125_G_WNT2B | A | G | rs910697 | – | WNT2B | LOW | Homozygous variant | Heterozygous |
| 1_228238333_T_WNT3A | C | T | – | WNT3A | MODIFIER | Heterozygous | No variant | |
| 1_228112121_G_WNT9A | A | G | rs4653889 | 0.4863 | WNT9A | MODIFIER | Heterozygous | No variant |
| 1_228113163_G_WNT9A | A | G | rs3795768 | 0.3246 | WNT9A | LOW | Heterozygous | No variant |
In addition to canonical, beta-catenin-mediated signaling, Wnts and Fzds also activate a non-canonical/planar cell polarity (PCP) pathway that allows individual cells to sense their relative position within a field. Thus, the phenotypes we see could be due to alterations in both canonical/beta-catenin-mediated and/or non-canonical/PCP-mediated Wnt signaling. Human FZD2 and chicken Fzd1/Fzd7 have been shown to activate canonical beta-catenin Wnt signaling (28,40). Our in vitro experiments show that the expression of FZD2P.TRP548* has little activity in a canonical Wnt activity assay (Fig. 6). However, several experimental results also indicate that PCP signaling may be disrupted. Fzd2 is concentrated to the leading edge of migrating cells in vitro (41). Fzd9-mutant mice show defects in matrix mineralization but do not show defects in canonical Wnt signaling (42). In the Fzd1−/−;Fzd2−/− mutants with the cleft palate phenotype, microarray analysis showed little differences in the transcriptional profile as compared with wild-type (19). Significant changes in target genes would be predicted if nuclear beta-catenin was regulating the activity of multiple target genes in the palate. The Fzd1−/−;Fzd2−/−-mutant embryos also have a wider roof of the mouth, suggestive of a convergent extension phenotype.
Some of the most definitive experiments to correlate Fzd activities with either canonical signaling or PCP were performed by Yu and colleagues (34). In canonical Wnt-signaling assays, Fzd2 was much more active in conjunction with Wnt3 and Wnt3a than Wnt5a (canonical and non-canonical ligands, respectively). In parallel experiments to test non-canonical Wnt signaling through co-assembly with Celsr1 at the membrane surface, Fzd1,Fzd2,Fzd7 showed little colocalization suggesting limited PCP pathway activity. Taken together, these results do not allow us to make a definitive determination of which arm of the Wnt-signaling pathway is most affected in FZD2-dependent AD omdysplasia. It is most likely that both mechanisms of Wnt signaling are affected to some degree but our data show a clear decrease in canonical Wnt signaling as a result of this mutation.
Materials and Methods
Subjects
Informed consent was obtained according to CCHMC institutional review board protocol # 2012-0203. Following consent, blood was collected on the parents, residual DNA on the affected individuals and spit on the unaffected proband.
Genotyping
Library generation, exome enrichment, sequencing, alignment and variant detection were performed in the CCHMC Genetic Variation and Gene Discovery Core Facility (Cincinnati, OH, USA). Briefly, sheared genomic DNA was enriched with NimbleGen EZ Exome V2 kit (Madison, WI, USA). The exome library was sequenced using Illumina's Hi Seq 2000 (San Diego, CA, USA). Alignment and variant detection was performed using the Broad Institute's web-based GATK.
Variant filtering and pathogenicity assessment
Quality control and data filtering were performed on VCF files in Golden Helix's SNP and Variation Suite (Bozeman, MT, USA). Non-synonymous coding variants were compared with three control databases, including NHLBI's ESP6500 exome data, the 1000 genomes project and an internal CCHMC control cohort. Remaining variants were subject to AR analysis with emphasis on homozygous recessive variants found in the region of homozygosity identified by SNP microarray. The identified variant was compared with known disease genes in the OMIM and Human Gene Mutation (HGMD) databases and to reported variants in dbSNP and the Exome Variant Server. The FZD2 mutation was confirmed with Sanger Sequencing (primers: F—GCGTCTTCTCCGTGCTCTAC; R—TACAAACCTTGTGGGGGTGT).
In situ hybridization
Patterns of gene expression in the face and limb of embryos were analyzed via sectioned in situ hybridization using digoxigenin-labeled riboprobes as described on the Gallus Expression in situ hybridization Analysis site (GEISHA (43,44). Probe for FZD2 was designed according to sequences listed on GEISHA.
Western blot analysis
Facial prominences and developing limbs were dissected from HH 25 chicken embryos. Tissue was rinsed with cold chick salt (127 mm NaCl) twice and sonicated in RIPA buffer (50 mm Tris, 150 mm NaCl, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate and 1% Triton X-100) containing protease and phosphatase inhibitors (Thermo Scientific, #87786, Pittsburgh, PA, USA). Protein concentration was measured by BCA protein assay (Thermo Scientific, #23227, Pittsburgh), and 30 μg protein was applied to a 6% SDS–PAGE gel. Rabbit polyclonal anti-Fzd2 antibody (LSBio, LS-C166295, Seattle, WA, USA).
Fusion protein expression and immunocytochemistry
The pCD-rFrizzled2-myc expression vector was commercially obtained (Addgene plasmid 16812, R. Moon donation), and the Trp548Stop mutation was recapitulated using the Quik Change II Site Directed Mutagenesis kit (Stratagene). Both constructs were subcloned into the V6 CE.GFPLT CS2+ vector (Addgene plasmid 17097, R. Moon donation) to generate GFP expression constructs for both the wild-type Frizzled2 and the Trp548Stop mutation. GFP-fusion proteins were visualized directly without further antibody amplification. The Dvl2-FLAG construct was commercially obtained (Addgene plasmid 24802, J. Wrana donation) and visualized with an rabbit anti-FLAG polyclonal antibody (Abcam) and a Alexa-Flour594 goat anti-rabbit antibody (Life technologies) using 4% paraformaldehyde fixation and standard methods. GFP expression and/or Dvl-FLAG plasmids were transfected into both HEK293T (human) and NIH3T3 (mouse) cells using the Lipofectamine 3000 transfection reagent following manufacturer's instructions.
Wnt-conditioned media
WNT3A media was collected from cells stably expressing Wnt3a. DMEM/FBS was collected in two batches after 4 and 3 days of incubation. These collections were mixed, stored at 4°C and added directly to cells to induce Wnt signaling (so-called treated or stimulated).
Dvl colocalization experiments
Fzd2 and Dvl2 constructs were expressed and imaged as described earlier. Similar to previous reports (15,16,25), 24 h after transfection, cells were either fixed and stained (untreated) or treated with Wnt-conditioned media for 24 h then fixed and stained. Images were collected using a Zeiss ApoTome microscope. After collecting images, Dvl2/Fzd2 colocalization was determined by three blinded observers. Each cell was determined to have no colocalization (and assigned a value of 0), some colocalization (a value of 1) or significant colocalization (2). Data were analyzed for concordance among observers, and cells were then binned (Fig. 6Q). Additionally, an average colocalization value (0.0–2.0) was determined for each cell from the multiple observers. This average ‘Dvl2/Fzd2 colocalization’ value was compared among treatments (Fig. 6R). Statistical significance was determined with a Student's t-test (Microsoft Excel).
Wnt-signaling assay
Assay was performed largely as in previous studies (22,34). STF cells carrying a Wnt-responsive luciferase assay were transfected in a six-well plate with pBSK and/or V6.CE.GFPLT plasmids as controls, Fzd2-GFP or Fzd2p.TRP548-GFP in equal concentrations (60 ng) using Lipofectamine 3000 following manufacturer's protocols. Twenty-four hours after transfection, cells were transferred to 96-well plates at 40 000 cells/well. Six-to-eight hours after 96-well plating, cells were stimulated with Wnt3a-conditioned media or control media, and Wnt activity was measured after 24 h in vitro using the Dual-Glo luciferase System (Promega) and manufacturer's protocols. Wnt luciferase activity was normalized to the untransfected, Wnt-treated cells.
Supplementary Material
Funding
This study was supported by the Cincinnati Children's Research Foundation Research and Innovation Pilot Award (S.A.B. and R.W.S.). I.G. is a student in the Magistère de Génétique Graduate Program at Université Paris Diderot, Sorbonne Paris Cité.
Supplementary Material
Acknowledgements
We thank the family members for agreeing to be part of this study and members of the Stottmann laboratory for analysis shown in Figure 6.
Conflict of Interest statement. None declared.
References
- 1.Maroteaux P., Sauvegrain J., Chrispin A., Farriaux J.P. (1989) Omodysplasia. Am. J. Med. Genet., 32, 371–375. [DOI] [PubMed] [Google Scholar]
- 2.Venditti C.P., Farmer J., Russell K.L., Friedrich C.A., Alter C., Canning D., Whitaker L., Mennuti M.T., Driscoll D.A., Zackai E.H. (2002) Omodysplasia: an affected mother and son. Am. J. Med. Genet., 111, 169–177. [DOI] [PubMed] [Google Scholar]
- 3.Gordon B.L., Champaigne N.L., Rogers R.C., Frias J.L., Leroy J.G. (2014) Long-term observation of a patient with dominant omodysplasia. Am. J. Med. Genet. Part A, 164A, 1234–1238. [DOI] [PubMed] [Google Scholar]
- 4.Olney A., Thomas J., Anderson R., Murphy M., Hastrom J. (1990) Autosomal dominant Robinow syndrome wth rhizomelic brachymelia. Proc. Greenwood Genet. Ctr., 9, 72–73. [Google Scholar]
- 5.Borochowitz Z., Sabo E., Misselevitch I., Boss J.H. (1998) Autosomal-recessive omodysplasia: prenatal diagnosis and histomorphometric assessment of the physeal plates of the long bones. Am. J. Med. Genet., 76, 238–244. [DOI] [PubMed] [Google Scholar]
- 6.Campos-Xavier A.B., Martinet D., Bateman J., Belluoccio D., Rowley L., Tan T.Y., Baxova A., Gustavson K.H., Borochowitz Z.U., Innes A.M., et al. (2009) Mutations in the heparan-sulfate proteoglycan glypican 6 (GPC6) impair endochondral ossification and cause recessive omodysplasia. Am. J. Hum. Genet., 84, 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yiu G.K., Kaunisto A., Chin Y.R., Toker A. (2011) NFAT promotes carcinoma invasive migration through glypican-6. Biochem. J., 440, 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dwivedi P.P., Lam N., Powell B.C. (2013) Boning up on glypicans—opportunities for new insights into bone biology. Cell Biochem. Funct., 31, 91–114. [DOI] [PubMed] [Google Scholar]
- 9.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., et al. (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fu W., O'Connor T.D., Jun G., Kang H.M., Abecasis G., Leal S.M., Gabriel S., Rieder M.J., Altshuler D., Shendure J., et al. (2013) Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature, 493, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Genomes Project C., Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A. (2010) A map of human genome variation from population-scale sequencing. Nature, 467, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.International HapMap C., Altshuler D.M., Gibbs R.A., Peltonen L., Altshuler D.M., Gibbs R.A., Peltonen L., Dermitzakis E., Schaffner S.F., Yu F., et al. (2010) Integrating common and rare genetic variation in diverse human populations. Nature, 467, 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Djiane A., Yogev S., Mlodzik M. (2005) The apical determinants aPKC and dPatj regulate Frizzled-dependent planar cell polarity in the Drosophila eye. Cell, 121, 621–631. [DOI] [PubMed] [Google Scholar]
- 14.Umbhauer M., Djiane A., Goisset C., Penzo-Mendez A., Riou J.F., Boucaut J.C., Shi D.L. (2000) The C-terminal cytoplasmic Lys-thr-X-X-X-Trp motif in frizzled receptors mediates Wnt/beta-catenin signalling. EMBO J., 19, 4944–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu J., Klein T.J., Mlodzik M. (2004) Subcellular localization of frizzled receptors, mediated by their cytoplasmic tails, regulates signaling pathway specificity. PLoS Biol., 2, E158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tauriello D.V., Jordens I., Kirchner K., Slootstra J.W., Kruitwagen T., Bouwman B.A., Noutsou M., Rudiger S.G., Schwamborn K., Schambony A., et al. (2012) Wnt/beta-catenin signaling requires interaction of the Dishevelled DEP domain and C terminus with a discontinuous motif in Frizzled. Proc. Natl Acad. Sci. USA, 109, E812–E820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sisson B.E., Topczewski J. (2009) Expression of five frizzleds during zebrafish craniofacial development. Gene Express. Patterns, 9, 520–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geetha-Loganathan P., Nimmagadda S., Antoni L., Fu K., Whiting C.J., Francis-West P., Richman J.M. (2009) Expression of WNT signalling pathway genes during chicken craniofacial development. Develop. Dyn., 238, 1150–1165. [DOI] [PubMed] [Google Scholar]
- 19.Yu H., Smallwood P.M., Wang Y., Vidaltamayo R., Reed R., Nathans J. (2010) Frizzled 1 and frizzled 2 genes function in palate, ventricular septum and neural tube closure: general implications for tissue fusion processes. Development, 137, 3707–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nohno T., Kawakami Y., Wada N., Komaguchi C., Nishimatsu S. (1999) Differential expression of the frizzled family involved in Wnt signaling during chick limb development. Cell. Mol. Biol., 45, 653–659. [PubMed] [Google Scholar]
- 21.Visel A., Carson J., Oldekamp J., Warnecke M., Jakubcakova V., Zhou X., Shaw C.A., Alvarez-Bolado G., Eichele G. (2007) Regulatory pathway analysis by high-throughput in situ hybridization. PLoS Genet., 3, 1867–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Q., Wang Y., Dabdoub A., Smallwood P.M., Williams J., Woods C., Kelley M.W., Jiang L., Tasman W., Zhang K., et al. (2004) Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell, 116, 883–895. [DOI] [PubMed] [Google Scholar]
- 23.Yang Y., Muzny D.M., Reid J.G., Bainbridge M.N., Willis A., Ward P.A., Braxton A., Beuten J., Xia F., Niu Z., et al. (2013) Clinical whole-exome sequencing for the diagnosis of mendelian disorders. New Engl. J. Med., 369, 1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Axelrod J.D., Miller J.R., Shulman J.M., Moon R.T., Perrimon N. (1998) Differential recruitment of Dishevelled provides signaling specificity in the planar cell polarity and Wingless signaling pathways. Genes Develop., 12, 2610–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simons M., Gault W.J., Gotthardt D., Rohatgi R., Klein T.J., Shao Y., Lee H.J., Wu A.L., Fang Y., Satlin L.M., et al. (2009) Electrochemical cues regulate assembly of the Frizzled/Dishevelled complex at the plasma membrane during planar epithelial polarization. Nat. Cell Biol., 11, 286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Person A.D., Beiraghi S., Sieben C.M., Hermanson S., Neumann A.N., Robu M.E., Schleiffarth J.R., Billington C.J., Jr., van Bokhoven H., Hoogeboom J.M., et al. (2010) WNT5A mutations in patients with autosomal dominant Robinow syndrome. Develop. Dyn., 239, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartmann C., Tabin C.J. (2000) Dual roles of Wnt signaling during chondrogenesis in the chicken limb. Development, 127, 3141–3159. [DOI] [PubMed] [Google Scholar]
- 28.Li Y., Dudley A.T. (2009) Noncanonical frizzled signaling regulates cell polarity of growth plate chondrocytes. Development, 136, 1083–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nasevicius A., Hyatt T., Kim H., Guttman J., Walsh E., Sumanas S., Wang Y., Ekker S.C. (1998) Evidence for a frizzled-mediated wnt pathway required for zebrafish dorsal mesoderm formation. Development, 125, 4283–4292. [DOI] [PubMed] [Google Scholar]
- 30.Niemann S., Zhao C., Pascu F., Stahl U., Aulepp U., Niswander L., Weber J.L., Muller U. (2004) Homozygous WNT3 mutation causes tetra-amelia in a large consanguineous family. Am. J. Hum. Genet., 74, 558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brugmann S.A., Goodnough L.H., Gregorieff A., Leucht P., ten Berge D., Fuerer C., Clevers H., Nusse R., Helms J.A. (2007) Wnt signaling mediates regional specification in the vertebrate face. Development, 134, 3283–3295. [DOI] [PubMed] [Google Scholar]
- 32.He F., Xiong W., Yu X., Espinoza-Lewis R., Liu C., Gu S., Nishita M., Suzuki K., Yamada G., Minami Y., et al. (2008) Wnt5a regulates directional cell migration and cell proliferation via Ror2-mediated noncanonical pathway in mammalian palate development. Development, 135, 3871–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin Y.R., Han X.H., Taketo M.M., Yoon J.K. (2012) Wnt9b-dependent FGF signaling is crucial for outgrowth of the nasal and maxillary processes during upper jaw and lip development. Development, 139, 1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu H., Ye X., Guo N., Nathans J. (2012) Frizzled 2 and frizzled 7 function redundantly in convergent extension and closure of the ventricular septum and palate: evidence for a network of interacting genes. Development, 139, 4383–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kispert A., Vainio S., Shen L., Rowitch D.H., McMahon A.P. (1996) Proteoglycans are required for maintenance of Wnt-11 expression in the ureter tips. Development, 122, 3627–3637. [DOI] [PubMed] [Google Scholar]
- 36.Summerhurst K., Stark M., Sharpe J., Davidson D., Murphy P. (2008) 3D representation of Wnt and Frizzled gene expression patterns in the mouse embryo at embryonic day 11.5 (Ts19). Gene Expr. Patterns, 8, 331–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiquet B.T., Blanton S.H., Burt A., Ma D., Stal S., Mulliken J.B., Hecht J.T. (2008) Variation in WNT genes is associated with non-syndromic cleft lip with or without cleft palate. Hum. Mol. Genet., 17, 2212–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao T., Yang L., Li P.Q., Wu H., Xie H.B., Shen X., Xie X.D. (2011) Association of Wnt3A gene variants with non-syndromic cleft lip with or without cleft palate in Chinese population. Arch. Oral Biol., 56, 73–78. [DOI] [PubMed] [Google Scholar]
- 39.Curtin E., Hickey G., Kamel G., Davidson A.J., Liao E.C. (2011) Zebrafish wnt9a is expressed in pharyngeal ectoderm and is required for palate and lower jaw development. Mech. Dev., 128, 104–115. [DOI] [PubMed] [Google Scholar]
- 40.Verkaar F., van Rosmalen J.W., Smits J.F., Blankesteijn W.M., Zaman G.J. (2009) Stably overexpressed human Frizzled-2 signals through the beta-catenin pathway and does not activate Ca2+-mobilization in Human Embryonic Kidney 293 cells. Cell. Signal., 21, 22–33. [DOI] [PubMed] [Google Scholar]
- 41.Matsumoto S., Fumoto K., Okamoto T., Kaibuchi K., Kikuchi A. (2010) Binding of APC and dishevelled mediates Wnt5a-regulated focal adhesion dynamics in migrating cells. EMBO J., 29, 1192–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Albers J., Schulze J., Beil F.T., Gebauer M., Baranowsky A., Keller J., Marshall R.P., Wintges K., Friedrich F.W., Priemel M., et al. (2011) Control of bone formation by the serpentine receptor Frizzled-9. J. Cell Biol., 192, 1057–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bell G.W., Yatskievych T.A., Antin P.B. (2004) GEISHA, a whole-mount in situ hybridization gene expression screen in chicken embryos. Develop. Dyn., 229, 677–687. [DOI] [PubMed] [Google Scholar]
- 44.Darnell D.K., Kaur S., Stanislaw S., Davey S., Konieczka J.H., Yatskievych T.A., Antin P.B. (2007) GEISHA: an in situ hybridization gene expression resource for the chicken embryo. Cytogenet. Genome Res., 117, 30–35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.