

Summary
The relationship between chromosomal instability and integration of the human papillomavirus (HPV) DNA into the host genome is not well understood. We found that chromosomal instability increases with HPV integration and diagnosis. However, chromosomal instability could occur without HPV integration.
Chromosomal copy number alterations (CNAs) and human papillomavirus (HPV) DNA integration into the host genome are more frequent in invasive cervical cancers compared to precancers. However, the relationship between CNAs and viral integration is not well understood. We analyzed chromosomal CNAs and HPV DNA integration in 17 biopsies from women diagnosed with cervical intraepithelial neoplasia grade 3 (CIN3) and 21 biopsies from women diagnosed with invasive cervical carcinoma. All samples were HPV16-positive. HPV DNA integration was evaluated by sequencing of chimeric transcripts or hybrid capture reads. Chromosomal copy number was measured with the aCGH 1×1M (Agilent Technologies, Santa Clara, CA). A genomic instability index (GII) was defined as the fraction of the genome with CNAs. The Wilcoxon rank-sum test was used to compare CIN3 and cancer samples. Unsupervised clustering based on CNAs identified two groups corresponding to CIN3 and cancer. Most differential CNAs were found in chromosomes 3 and 8. HPV DNA was present in episomal form in 15 samples and integrated in 23 samples. The mean GII was 0.12 and 0.21 for CIN3 and cancer, respectively (P = 0.039). The GII was significantly higher in integrated samples (mean GII in episomal samples: 0.12; and integrated samples: 0.20; P = 0.02), but not within CIN3 or cancer. Integration sites were more frequently observed in amplified regions than expected by chance (P = 0.008). Our findings demonstrate that GII increases with HPV integration and at the transition from CIN3 to cancer. However, chromosomal instability can occur in the absence of integration, suggesting that it may facilitate integration.
Introduction
Persistent genital infection with carcinogenic human papillomaviruses (HPV) is the necessary cause of cervical cancer (1). Carcinogenic HPV infections of the cervix are usually transient, with approximately half clearing in 12 months (2). However, about 5% of infections persist for an extended period of time (3), which increases the risk of developing into cervical precancers (i.e. cervical intraepithelial neoplasia or CIN) that may progress to invasive cervical cancer, typically over many years. The underlying mechanisms of progression from initial infection to precancer and from precancer to cancer are not well understood. A better understanding of the natural history of cervical cancer on the molecular level could help to define biomarkers that distinguish the women who are at high risk of developing invasive cancer and women who could avoid unnecessary medical interventions.
Over 40 different HPV types have been found to infect the genital tract, with 12 of them being classified as group 1 carcinogens (4). It is estimated that approximately 60% of all cervical cancers are caused by HPV16 (5). Upon infection, the HPV genome enters the host cell nucleus. Viral oncoproteins E6 and E7 can affect many cellular processes, including cell cycle, apoptosis and maintenance of chromosomal stability. The high degree of genomic instability reported in cervical cancers is thought to be the consequence of the interaction of viral E6 and E7 with host tumor suppressor gene products, p53 and pRb, respectively (6–8). Several recurrent chromosomal gains and losses have been observed in cervical cancers. A recent meta-analysis of chromosomal gains and losses in cervical CIN and cancer reported that gains in 3q were present in both cancer and precancerous lesions, while losses of 3p and gains of 5p were uncommon in precancers, but more common in cancers (9). However, the majority of the reported studies used technologies restricted to measuring large chromosomal aberrations. Moreover, very few studies have measured chromosomal aberrations in both precancers and cancers, which allow evaluation of chromosomal aberrations at the two disease stages with similar experimental conditions, technologies and analytical techniques to determine copy number changes. Array comparative genomic hybridization (aCGH) substantially increases the resolution to detect chromosomal aberrations that can be measured compared to traditional CGH methods performed on chromosomal spreads.
Once the HPV genome is incorporated in the host cells, it replicates in a circular, episomal state as part of the normal viral life cycle. However, as the disease progresses from infection to precancer and cancer, HPV DNA is found more commonly integrated into the genome of the host cell (10–12). In addition, integrated viral transcripts enhance the transforming capacity compared to episomal transcripts possibly due to the longer half-life of the integrated transcripts (13) potentially conferring neoplastic growth advantage (14). It has been hypothesized that host genes can be affected by integration of HPV genome sequences, contributing to the clonal selection and the progression of the disease (15,16).
The relationship between the host genomic structural variation and HPV integration is not well understood. Specifically, it is not clear whether chromosomal aberrations precede and thereby facilitate HPV integration, or whether HPV integration triggers more extensive chromosomal changes. While host genomic instability and HPV genome integration are more common in invasive cancers as compared to CIN lesions (17), it is not known whether genomic instability differs between HPV integrated and non-integrated genomes.
In a well-characterized epidemiologic study of cervical cancer and its precursors, we analyzed the degree of chromosomal changes and HPV integration in cervical precancers and invasive cancers, and we evaluated whether host genome amplifications co-locate with integrations sites in precancers and cancers.
Materials and methods
Study population
Subjects included in this analysis were selected from the Study to Understand Cervical Cancer Early Endpoints and Determinants (SUCCEED), a cross-sectional epidemiologic study conducted at the Dysplasia Clinic at the University of Oklahoma Health Sciences Center (OUHSC). The study design and methodology has been described previously (18,19). Briefly, women with an abnormal Pap smear diagnosis or a biopsy diagnosis of CIN who were scheduled for diagnostic colposcopy were enrolled between November 2003 and September 2007. Exclusion criteria to participate in the study included women who were less than 18 years of age, pregnant at the time of their visit, previously treated with chemotherapy or radiation for any cancer or scheduled for colposcopy. All participants completed an in-person interview with a standardized questionnaire to obtain demographic and important information on known HPV risk factors. Written consent was obtained from all participants enrolled in the study and the study was approved by the Institutional Review Boards at OUHSC and the National Cancer Institute.
A physician conducted the colposcopic examination according to routine practice. Before the biopsy or loop electrosurgical excision procedure, cervical samples were obtained with a Papette broom (Wallach Surgical, Orange, CT) and rinsed directly into PreservCyt solution (Hologic, Marlborough, MA) (20). The cytology specimen was used for ThinPrep (Hologic, Marlborough, MA) cytology and for HPV genotype determinations using the Linear Array (LA) HPV Genotyping System (Roche Molecular Diagnostics, Branchburg, NJ). Biopsy specimens were obtained from any colposcopy suspected of having cervical CIN or cancer lesions and were placed in separate prelabeled vials containing 10% buffered formalin (21) for histological evaluation. In addition, adjacent biopsies from suspected lesions were snap frozen for research purposes. Details of DNA isolation and HPV genotyping procedure used in SUCCEED have been described elsewhere (19,22).
Analytical population
A total of 3013 women enrolled in the SUCCEED study, of which 2294 (76.1%) provided cervical samples. Of these women, 936 (40.8%) tested positive for HPV16. The diagnosis for 285 (30.4%) of these women was CIN3, the histologic lesions recognized as precancer, and invasive squamous cell cervical cancer for 128 (13.7%). We randomly selected 38 women for our analysis among the 412 HPV16-positive women with CIN3 and invasive cancer diagnoses for which other profiling data were available, stratified by disease status. Of the 38 women, 21 were diagnosed with invasive squamous cell cervical cancer and 17 with CIN3.
Array copy number assay
Laser capture microdissection was used on the snap frozen colposcopic biopsies to select epithelial cells from the lesions and minimize the contamination with other cell types. Captured tissues were resuspended in TE pH8.0 with 0.5%SDS, followed by 30–60min consecutive incubations with RNAseA and proteinaseK at 37°C. DNA was extracted using two phenol/chloroform extractions and ethanol precipitation in the presence of linear acrylamide. Total DNA yields ranged from 15 to 1100ng of which 200ng or otherwise half of the DNA preparation was used for whole genome amplification (WGA) by Phi29 polymerase using Qiagen’s RepliG kit. Approximately 5–10 µg of the amplified DNA was used to measure DNA copy number (double deletions, losses, neutral, gains and amplifications) using the Agilent SurePrint G3 Human CGH 1×1M (Agilent Technologies, Santa Clara, CA) at Oxford Gene Technology (OTG, Oxfordshire, UK) based on the GRCh36/hg18 build. Hybridization and data acquisition were according to the Agilent’s standard protocol including the use of unamplified male reference DNA.
A pilot study was run to compare the aCGH results for DNA from five samples subject to prior WGA or without prior WGA. The mean Pearson’s correlation coefficient of estimated segment means between unamplified and WGA samples was 0.88 indicating good performance of the aCGH after WGA. Following this pilot, all DNA samples were subjected to WGA using DNA from new biopsies prior to aCGH analysis. One sample was run in duplicate for quality control.
HPV genomic integration assays
HPV integrations assays were specific for HPV16. DNA for the integration assays was extracted from the same biopsies as those used for aCGH analyses.
Integration sites were detected using the Amplification of Papillomavirus Oncogene Transcripts (APOT) assay (10). This assay exploits the structural differences between the 3′ ends of the various viral oncogenes transcripts. Briefly, RNA transcripts derived from integrated HPV16 E6 and E7 oncogenes usually incorporate viral genomic sequences at their 5′ ends and human genomic sequences at their 3′ ends, following the polyadenylation (poly-A) site. In contrast, transcripts derived from episomal HPV16 E6-E7 are commonly spliced at the E1- to the E4-splice acceptor site and terminate at the viral poly-A site (23). HPV16 oncogenes transcripts (episomal and integrated) were amplified using E7-specific and poly-A tail primers. Integrated-derived transcripts were differentiated from episome-derived transcripts using Southern blot hybridization analysis with HPV16 E7- and E4-specific oligonucleotides.
Some samples (n = 5) where the APOT assay failed to detect a fusion transcript between the HPV and the human genomes were subjected to a ligation-mediated PCR assay for the detection of integrated papillomavirus sequences (DIPS) (24). This assay uses restriction enzyme digestions, the ligation of an enzyme-specific double stranded adapter, an initial PCR using HPV16-specific primers for linear amplification and a second PCR using an HPV16-specific primer set and an adaptor-specific primer (24). The amplified DNA sequence includes the fusion between the viral and the human genomic DNA.
Additionally, 30 samples were subjected to a HPV16 specific capture sequencing assay, a high-throughput and cost-effective sequencing assay. Briefly, custom Ion AmpliSeq libraries were prepared to capture the HPV16 genome. Libraries were hybridized with HPV16 probes and washed to remove un-captured fragments. Captured-fragments were amplified using PCR and sequenced using Ion Torrent Sequencing technology following the manufacturer’s recommended protocols (25). Reads were aligned to the human genome and the HPV16 genome. Reads that perfectly aligned to either of the genomes were excluded so only chimeric reads (partially aligned to the human genome and partially aligned to the HPV16 genome) were further analyzed to determine the exact position of the putative integration event. Those with 100 or more supporting reads were considered integration sites.
Genomic integration sites were determined from analysis of amplified HPV integration sequences using the Basic Local Alignment Search Tool (BLAST, http://blast.ncbi.nlm.nih.gov/Blast.cgi) for the GRCh36/hg18 build.
Statistical analysis
Analyses were performed using the R software (version 3.1.1) and restricted to the 22 autosomal chromosomes.
Background correction and normalization of the aCGH raw signal intensities were done using the Bioconductor limma package with the minimum method for background correction and the print-tip loess normalization, which takes into account signal intensity and spatial position on the array (26). Diagnostic plots were used to assure that signal biases were not present in the data after normalization. Binary logarithm ratios (log2 ratios) of the two intensity channels were then computed. For the subject with duplicate runs, the log2 ratios of the two runs were averaged for analysis. The log2 ratios were subjected to the circular binary segmentation algorithm (27) by means of the R package ParDNAcopy, which is a parallel version of the Bioconductor package DNAcopy. Copy number (double deletions, losses, neutral, gains and amplifications) were determined using the CGHcall Bioconductor package (28). We restricted our analyses to aberrations of sizes of 40kb or larger, to avoid including outliers due to technical artifacts as a consequence of the GC content or the aCGH array (29,30). In addition, this size is well-below the median aberration size observed in most tumors using recent copy number arrays (31). However, the results including all aberrations did not qualitatively differ from the results after excluding them (data not shown).
Unsupervised hierarchical clustering analysis of the log2 ratio data, using the Euclidean metric and the complete linkage method, was performed to determine whether aberrations differ between CIN3 and invasive cancer samples. In order to quantify the level of genomic instability in each sample, we defined a genomic instability index (GII) as the fraction of the genome that was altered (32). The Wilcoxon rank-sum test was used to compare the distribution of the GII across different groups. To evaluate the relationship between GII and age, we downloaded level 3 copy number data (i.e., segmented; levels 1 and 2 were protected) from 223 women diagnosed with cervical squamous cell carcinoma with available blood copy number and not missing age from the TCGA database. TCGA data was generated from Affymetrix SNP array 6.0. Calls were computed as described above.
We used data from the ENCyclopedia Of DNA Elements (ENCODE) (33) to describe the genomic characteristics in 50Kb windows around each HPV integration site. Information regarding fragile sites was obtained from a previous publication (34).
Finally, we explored whether integration sites for CIN3 and cervical cancers co-localized with chromosomal gains. To that end, the observed distribution for the minimal distance between the integration site and chromosomal gains was computed for each subject with HPV DNA integrated. Then, for each of these subjects, we randomly permuted 10 000 integrations sites across the genome (only loci in the genome covered by the aCGH). The expected distribution was computed as the minimal distance between these random integration sites and chromosomal gains. The observed and expected distributions were compared using distance thresholds and the Fisher’s exact test.
Results
Our analysis included 38 women, 17 of them with a diagnosis of cervical CIN3 and 21 with cervical carcinoma. Characteristics of the women are shown in Table 1. Compared to women with CIN3 diagnosis, women with cervical cancer tended to be older, more likely to have less education and less likely to be current smokers. Three women with cervical cancer and one with CIN3 diagnosis were former smokers. Smoking information was missing for four cancer cases. The age at first sexual contact was similar for women in both groups. The average number of years from first sexual contact to CIN3 diagnosis was 12.2 (SD = 9.8) versus 25.5 (SD = 8.5) to cancer.
Table 1.
Characteristics of women in analytic population
| Characteristicsa | CIN3 (N = 17) | Cervical cancer (N = 21) |
|---|---|---|
| Age (years), mean (SD) | 29.4 (9.1) | 45.0 (11.1) |
| Age (years), n (%) | ||
| <25 | 7 (41.2) | — |
| 25–34 | 6 (35.3) | 5 (23.8) |
| 35–44 | 2 (11.8) | 7 (33.3) |
| 45–54 | 2 (11.8) | 3 (14.3) |
| ≥55 | — | 6 (28.6) |
| Education, n (%) | ||
| High/vocational school or less | 6 (37.5) | 12 (70.6) |
| Some college or more | 10 (62.5) | 5 (29.4) |
| Number of sexual partners, n (%) | ||
| ≤5 | 7 (43.8) | 8 (50.0) |
| >5 | 9 (56.2) | 8 (50.0) |
| Age at first sexual contact (years), mean (SD) | 16.2 (3.7) | 16.5 (2.3) |
| Age at first sexual contact (years), n (%) | ||
| <15 | 3 (17.6) | 4 (23.5) |
| 15–18 | 9 (52.9) | 7 (41.2) |
| ≥18 | 5 (29.4) | 6 (35.3) |
| Smoking, n (%) | ||
| Never or former | 6 (35.3) | 11 (64.7) |
| Current | 11 (64.7) | 6 (35.3) |
| HPV DNA integration statusb, n (%) | ||
| Episomal | 9 (53.9) | 6 (28.6) |
| Integrated | 8 (47.1) | 15 (71.4) |
Numbers may not add to total due to missing values. SD, Standard deviation.
aAt the baseline questionnaire.
bHPV DNA integration assays was performed in 16 women with CIN3 diagnosis (17 samples) and 20 women with cancer diagnosis (21 samples). One woman with CIN3 had two samples (one integrated and one not integrated) and one women with cancer also had two samples (non-integrated). DNA from 2 samples in two different women diagnosed with CIN3 and 6 samples in six different women diagnosed with cancer did not amplified correctly and the integration assays could not be not run.
There were 23 samples with integrated HPV DNA and 15 samples with episomal HPV DNA. Of the 23 samples, 15 (65.2%) were observed in cancer samples. The genomic characterization for the 27 HPV DNA integrated events in the human genome is shown in Table 2. As expected, integration sites occurred at various sites across the genome. Four samples, one in 15q23, two in 15q24.2, and another in 15q25, had integration events ~14Mb apart. Characterization of all integration sites indicated that these occurred in transcriptionally active regions, as suggested by the number of proximal genes, the percent of CpG islands and the number of peaks of open chromatin. Integration of HPV DNA also occurred in genomic regions with an abundance of repeat elements. Five integration sites ware within 50kb of a fragile site.
Table 2.
Genomic characterization of HPV DNA integration locations
| ID | Pathology | Cytoband | Nucleotide positiona | Assay to determine integration locus | Genesb | Fragile sitesc | % CpG islandsb | Repeat element class (number of events)b | Number of recombination hotspotsb | Number of peaks of open chromatind | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| FAIRE | DNaseI | ||||||||||
| Subject 1 | CIN3 | 1p36.22 | 9 819 841 | Sequencing | CLSTN1, CTNNBIP1 |
FRA1A | 19.2 | LINE (18), SINE (73), LTR (5), other (15) | 0 | 5 | 3 |
| Subject 1 | CIN3 | 3p11.2 | 88 525 362 | Sequencing | — | — | 0 | LINE (19), SINE (14), LTR (15), other (26) | 0 | 0 | 0 |
| Subject 1 | CIN3 | 9q33.3 | 126 313 825 | Sequencing | NR5A1, NR6A1 |
— | 17.2 | LINE (26), SINE (31), LTR (9), other (17) | 0 | 2 | 2 |
| Subject 1 | CIN3 | 10q24.2 | 99 428 623 | Sequencing | PI4K2A, AVPI1 |
— | 19.6 | LINE (30), SINE (35), LTR (42), other (28) | 0 | 7 | 15 |
| Subject 5 | CIN3 | 15q24.2 | 74 295 689 | APOT | ETFA | — | 20.1 | LINE (16), SINE (61), LTR (11), other (5) | 0 | 4 | 8 |
| Subject 6 | CIN3 | 1q32.2 | 207 609 939 | Sequencing | — | — | 0 | LINE (21), SINE (34), LTR (12), other (22) | 0 | 3 | 5 |
| Subject 8 | CIN3 | 3q28 | 191 025 132 | Sequencing | TP63, MIR944 | — | 0 | LINE (26), SINE (25), LTR (7), other (25) | 0 | 1 | 7 |
| Subject 9 | CIN3 | 15q24.2 | 74 297 043 | Sequencing | ETFA | 0 | LINE (16), SINE (60), LTR (13), other (6) | 0 | 4 | 8 | |
| Subject 12 | CIN3 | 4q21.23 | 85 671 277 | Sequencing | - | — | 0 | LINE (22), SINE (33), LTR (13), other (15) | 1 | 1 | 2 |
| Subject 15 | CIN3 | 22q11.23 | 22 060 494 | APOT | CES5AP1, ZDHHC8P1 | — | 17.3 | LINE (10), SINE (31), LTR (9), other (33) | 0 | 1 | 2 |
| Subject 16 | CIN3 | 20q13.13 | 48 435 994 | APOT | - | — | 0 | LINE (44), SINE (84), LTR (7), other (24) | 0 | 4 | 12 |
| Subject 18 | Cancer | 7q11.23 | 74 757 074 | Sequencing | PMS2P5, SPDYE8P | FRA7J | 0 | LINE (40), SINE (65), LTR (8), other (27) | 0 | 0 | 0 |
| Subject 18 | Cancer | 15q23 | 70 231 187 | Sequencing | MY09A, SENP8, GRAMD2 | — | 0 | LINE (31), SINE (54), LTR (9), other (8) | 0 | 3 | 2 |
| Subject 20 | Cancer | 3p26.2 | 4 804 305 | Sequencing | ITPR1 | — | 0 | LINE (22), SINE (14), LTR (25), other (8) | 0 | 3 | 1 |
| Subject 21 | Cancer | 13q22.1 | 72 885 297 | APOT | - | — | 0 | LINE (40), SINE (36), LTR (5), other (8) | 0 | 3 | 10 |
| Subject 23 | Cancer | 14q32.2 | 98 775 862 | Sequencing | BCL11B | — | 16.7 | LINE (32), SINE (17), LTR (5), other (11) | 0 | 0 | 0 |
| Subject 24 | Cancer | 8q22.1 | 96 153 569 | Sequencing | NDUFAF6, MIR3150A, MIR3150B | FRA8B | 20.5 | LINE (26), SINE (24), LTR (33), other (23) | 0 | 10 | 8 |
| Subject 25 | Cancer | 15q25.3 | 83 735 261 | Sequencing | AKAP13 | — | 23.2 | LINE (40), SINE (57), LTR (6), other (13) | 0 | 6 | 13 |
| Subject 26 | Cancer | 21q22.3 | 42 078 171 | Sequencing | RIPK4, PRDM15 | — | 16.6 | LINE (19), SINE (35), LTR (11), other (16) | 0 | 7 | 8 |
| Subject 27 | Cancer | 6p25.2 | 2 847 972 | Sequencing | SERPINB9 | — | 18.9 | LINE (9), SINE (18), LTR (3), other (45) | 0 | 3 | 4 |
| Subject 29 | Cancer | 8q24.21 | 128 844 999 | Sequencing | MYC | — | 13.6 | LINE (50), SINE (39), LTR (24), other (35) | 0 | 6 | 14 |
| Subject 30 | Cancer | 9p24.2 | 3 364 982 | Sequencing | RFX3 | — | 0 | LINE (30), SINE (30), LTR (27), other (24) | 0 | 0 | 1 |
| Subject 31 | Cancer | 10p14 | 8 648 393 | Sequencing | — | — | 0 | LINE (24), SINE (47), LTR (2), other (23) | 1 | 0 | 0 |
| Subject 33 | Cancer | 1p34.2 | 43 770 932 | Sequencing | PTPRF | - | 17.7 | LINE (35), SINE (33), LTR (10), other (15) | 0 | 2 | 7 |
| Subject 35 | Cancer | 9q34.3 | 138 625 713 | APOT | — | — | 12.6 | LINE (19), SINE (34), LTR (3), other (10) | 0 | 7 | 7 |
| Subject 37 | Cancer | 19q13.2 | 45 789 652 | Sequencing | SPTBN4, SHKBP1, LTBP4 | FRA19A | 18.1 | LINE (26), SINE (46), LTR (13), other (11) | 0 | 16 | 21 |
| Subject 38 | Cancer | 11q13.1 | 66 165 789 | Sequencing | RBM14, RBM4, RBM4B | FRA11H | 18.4 | LINE (38), SINE (36), LTR (11), other (14) | 0 | 2 | 6 |
aNucleotide position within corresponding chromosome based on reference genome NCBI Build 36/hg18.
bWithin a window of 50Kb around the integration site. Information from the ENCODE database.
cWithin a window of 50Kb around the integration site.
dWithin a window of 50Kb around the integration site. Information from the ENCODE database. Based on the HeLa cell line. For different open chromatin assays see Ref. (35).
Cancer samples had a higher frequency of chromosomal aberrations as compared to the CIN3 samples (Figure 1). There were, on average, 36.3 CNAs per sample, with and average size of 12.4Mb. Cancers tended to have larger size of copy number alterations (CNAs) (average size 17.5Mb) compared to CIN3 (average size 7.6Mb). Chromosome 3 had the most distinct differences between CIN3 and cancer samples with respect to gains and losses. Approximately 40% of the cancer samples had losses in 3p and over 50% of the cancer samples had gains in 3q. In contrast, losses in 3p were present in only 10% of the CIN3 samples and gains in 3q in approximately 25% of the samples. The region hosting oncogene MLF1 in 3q25.32 had the most gains in cancer samples (over 60%) and the greatest difference between CIN3 and cancers (40%). Approximately 33% difference in gains between cancer and CIN3 (over 60 versus 30%, respectively) were observed in the 3q28 locus where LPP, a gene related to cell–cell adhesion, cell motility and tumor growth, is located. With respect to 3p, over 42% of cancer samples had a loss in tumor suppressor gene FOXP1 in 3p14.1 compared to only 18% of CIN3. Chromosome 8q had gains in over 25% of the cancer samples. The most commonly altered region in 8q was the region surrounding and including oncogene MYC and genes POU5F1 and POU5F1P1 which play a key role in stem cell pluripotency, with gains in approximately 33% of the cancer samples. Chromosome 8q did not have noticeable CNAs in CIN3 samples.
Figure 1.
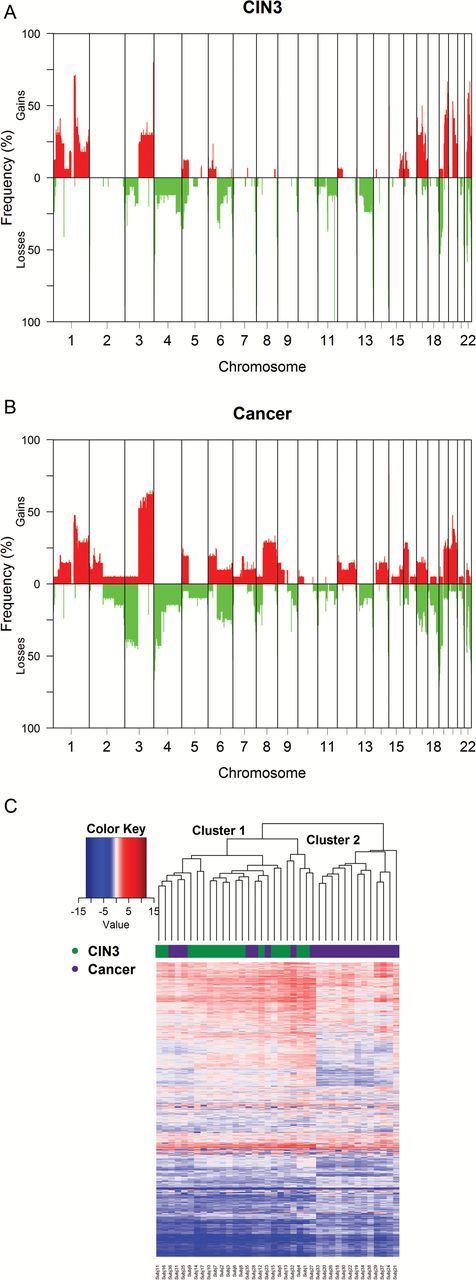
Frequencies of losses and gains for CIN3 (A) and cancers (B). Frequencies compute using one sample per subject. (C) Unsupervised hierarchical clustering analysis of the log2 ratio data for the 8000 most variables probes as measured by the median absolute deviation (MAD). The Euclidean metric and the complete linkage method were employed for the clustering. Subjects 1–17 were diagnosed with CIN3 and subjects 18–38 were diagnosed with cancer.
Unsupervised hierarchical clustering analysis of the copy number for the 21 cancer samples and 17 CIN3 samples resulted in good separation of the two diagnoses (Figure 1C). Cluster 2 only contained cancer samples while all CIN3 and eight cancer samples were in cluster 1. Accordingly, the mean age of women in cluster 1 was 33.3 years old vs. 47.2 years old in cluster 2 (P = 0.001). Women with cancer in cluster 1 were slightly younger (mean 41.5 years) than women in cluster 2 (mean 47.2 years), but the difference was not statistically different (P = 0.14).
The extent of genomic instability in each sample was quantified using the GII (Figure 2A). The majority of cancer samples had higher GII than CIN3 samples. Specifically, the mean GII for CIN3 samples was 0.12 while the mean GII for cancer samples was 0.21 (Wilcoxon rank-sum test P = 0.039). Integration samples had greater GII values (mean GII in episomal samples: 0.12; mean GII in integrated samples: 0.20; Wilcoxon rank-sum test P = 0.02).We explored whether the GII differed by HPV DNA integration status within a diagnosis (Figure 2B). Among samples with CIN3, the mean GII in episomal HPV DNA samples was 0.09 while in integrated HPV DNA was 0.15 (Wilcoxon rank-sum test P = 0.24). Similarly among samples with cancer, the mean GII in episomal HPV DNA samples was 0.17 while in integrated HPV DNA samples was 0.22 (Wilcoxon rank-sum test P = 0.30). The GII was not significantly correlated with age in CIN3 patients (age range: 19–51 years old; Spearman’s correlation coefficient: 0.03; t-test P = 0.88) or in cancer patients (age range: 29–65 years old; Spearman’s correlation coefficient: 0.09; t-test test P = 0.73; Supplementary Figure 1, available at Carcinogenesis Online). To further understand the effect of age on the GII, we looked at copy number in blood samples from 223 women diagnosed with cervical squamous cell carcinoma in TCGA and observed a very small increase of GII with age (Supplementary Figure 2, available at Carcinogenesis Online), which cannot explain the difference in GII observed in CIN3 and cancer.The GII was not associated with education (P = 0.81) or number of sexual partners (P = 0.52). GII was inversely associated with current smoking (P = 0.02), but after adjusting for diagnosis, it was no longer significant (P = 0.064).
Figure 2.

Genomic instability index (GII) for CIN3 and cancer samples. (A) Heatmap of copy number alterations of 250 000 probes that were randomly selected for illustration purposes. Red represents gains and blue represents losses. Columns are samples and rows are positions of the probes along the 22 autosomes. The top column track indicates the diagnosis of the sample (CIN3 versus cancer) and the bottom column track provides the GII score. Samples were ordered according to their GII value. (B) GII values for each subject according to the diagnosis and integration status. Dots indicate individual values and lines are mean values.
Finally, we explored whether integration sites colocate with chromosomal gains. Six integration siteswere directly at locations of chromosomal gains. Thirty percent of observed integration sites were within 200kb of chromosomal gains compared to the expected 11% (Fisher’s Exact test P = 0.008).
Discussion
In this analysis, we evaluated DNA CNAs in 38 CIN3 and cervical cancer micro-dissected biopsies and tested the relationship between genomic instability and integration events in precancers and cancers. Unsupervised hierarchical clustering analysis identified two distinct genomic subgroups, which were largely characterized by CIN3 and cancer diagnosis. Chromosomes 3 and 8 were particularly important in distinguishing CIN3 from invasive cancer. Our findings are in agreement with previous studies using conventional CGH, which reported consistent high-level gains of 3q and losses of 3p in cancer but to a lesser extent in CIN3. In fact, in a recent meta-analysis, amplification of 3q was observed in 64 out of 240 samples (27%) (9), which is close to our estimate of 30%. As is Bierkens et al., we also observed losses in chromosomes 2, 3p, 4, 7 and 17 to be more common in cancers than CIN3 (36). The most common chromosomal gains and losses included oncogenes and tumor suppressor genes, respectively.
Similar to previous findings (11,14), integration sites were mapped to different loci across the genome. We found an integration event in 8q24.21, near MYC, and another in 13q22.1 which have been previously found to be integration hot-spots (14,37,38). Many others were in cytobands that have been previously observed to have integration sites (14,39). In general, integration sites were close to transcription regions and repeat elements and in gene rich regions. Therefore, it is possible that HPV integration may disrupt or interfere with transcription of important pathways that promote tumor growth. For instance, four distinct samples had integration sites within 14MB in the 15q arm possibly targeting some oncogenic pathways, such as the RAS oncogenic pathway via RASGRF1 in 15q25. The integration site region 8q22.1 is a gene rich region, which includes cancer related genes CCNE2, TP53INP1 and RAD54B.
To summarize the extent of genomic alterations in each sample, we defined an instability index as the proportion of genome that was altered. We observed that the proportion of the altered genomes was significantly higher in cancer samples than in CIN3 samples. Up to now, measures of genomic instability and HPV DNA integration have not been compared in cervical precancers and cancers. Moreover, a relationship between integration and copy number has not been formally evaluated. Integrated samples had higher genomic instability, although we did not observe significant relationships between GII and HPV integration status within a diagnosis. However, in general, cancers had higher genomic instability than CIN3, regardless whether integration had occurred in CIN3. Previous studies have suggested that HPV DNA integration may lead to chromosomal aberrations based on to the co-localization of integration sites with chromosomal gains (40,41). However, co-location does not imply a temporal relationship. In addition, those studies only included tumors or tumor derived cell lines, and the latter may not represent the natural history of cervical cancer. In contrast to the previous study, a clinical study of 85 samples encompassing pre-cancerous and cancerous biopsies observed that most of samples with integrated HPV were aneuploid, concluding that aneuploidy is likely to precede integration of HPV genomes (17). Similar to previous reports (40,41), we also observed that there was a tendency of integration sites in CIN3 and cancers samples to co-locate with chromosomal gains. This suggests that CNAs and HPV integration are not independent of each other. However, an alternative hypothesis supported by our data is that chromosomal aberrations are already present in precancer lesions due to the effects of episomal oncoproteins E6 and E7 on DNA replication and abnormal centrosome duplication (42). This initial chromosomal instability may provide an environment where HPV DNA is more likely to be integrated into the host genome. In turn, HPV integration may further enhance the activity of E6 and E7 promoting cellular outgrowth, and eventually leading to invasive cervical cancer by selecting cell clones with integrated HPV DNA and strong oncoprotein activity.
Our study has several strengths. It is the first study to measure CNAs and HPV DNA integration in cancer and CIN3 specimens from the same population. This allowed us to evaluate differences in cervical cancer and its immediate precursor. However, due to the cross-sectional nature of this study, inferences regarding temporal relationships cannot be made. This is a ubiquitous limitation of cervical cancer studies in screening populations, since cervical precancer is treated and cannot ethically be followed for progression to cancer. Our study used micro-dissected fresh frozen specimens, which are of superior quality to formalin-fixed paraffin embedded samples and better reflect the true pathological state of the tissue by limiting contamination with adjacent normal epithelial or stromal cells. In this study, we demonstrated the feasibility of measuring chromosomal abnormalities using array CGH and HPV integration in meticulously microdissected cervical lesions. This was in part related to the use of the APOT HPV DNA integrations, which require smaller amounts of nucleic acids compared to current whole genome next-generation sequencing techniques. While large amounts of DNA can be obtained from some tumor samples, it is unlikely that small pre-cancer lesions found in a screening population can provide enough DNA yields, especially since micro-dissection is required for intraepithelial lesions. However, APOT is a laborious assay which makes it challenging to evaluate large samples sizes. We have also used a newly developed HPV capture sequencing assay, which allows to sequencing only genomic fragments with HPV present. This assay is more sensitive than APOT and additional integration sites were found. A further strength of our study was the restriction to HPV16-positive cases, eliminating biologic differences related to HPV genotypes. Previous studies included different HPV types in their analysis without carefully examining whether type-dependent differences in biology could have influenced their findings (40,41). A limitation of our study was the need to perform WGA to yield enough DNA for aCGH from micro-dissected material. Although WGA is relatively uniform across the genome, there are regions which are known to have higher than average CG content and are consistently under-amplified (43). This can introduce artifacts and confound the analysis of aCGH data (44). However, with the use of frozen, microdissected samples likely produced high quality of the genomic DNA used and the help of algorithms to reduce such artifacts, we observed excellent correlations between aCGH results from unamplified and WGA DNA.
In summary, we found that chromosomal aberrations can robustly distinguish CIN3 and cancers. The proportion of genomic alterations was greater in cancer compared to CIN3, and increased with integration status. However, chromosomal instability can occur in the absence of integration, which may suggest that some level of instability may be necessary to facilitate integration.
Supplementary material
Supplementary Figures 1 and 2 can be found at http://carcin.oxfordjournals.org/
Funding
Intramural Research Program of the National Cancer Institute, NIH grant P01 CA022443, and by funds from the Morgridge Institute for Research. P. A. is an investigator of the Howard Hughes Medical Institute.
Conflict of Interest Statement: None declared.
Supplementary Material
Glossary
Abbreviations
- aCGH
array comparative genomic hybridization
- CIN
cervical intraepithelial neoplasia
- CNA
copy number alteration
- HPV
human papillomavirus
References
- 1. Schiffman M., et al. (2007) Human papillomavirus and cervical cancer. Lancet, 370, 890–907. [DOI] [PubMed] [Google Scholar]
- 2. Rodríguez A.C., et al. ; Proyecto Epidemiológico Guanacaste Group. (2008) Rapid clearance of human papillomavirus and implications for clinical focus on persistent infections. J. Natl. Cancer Inst., 100, 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rodríguez A.C., et al. (2010) Longitudinal study of human papillomavirus persistence and cervical intraepithelial neoplasia grade 2/3: critical role of duration of infection. J. Natl. Cancer Inst., 102, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bouvard V., et al. ; WHO International Agency for Research on Cancer Monograph Working Group. (2009) A review of human carcinogens–Part B: biological agents. Lancet. Oncol., 10, 321–322. [DOI] [PubMed] [Google Scholar]
- 5. de Sanjose S., et al. ; Retrospective International Survey and HPV Time Trends Study Group. (2010) Human papillomavirus genotype attribution in invasive cervical cancer: a retrospective cross-sectional worldwide study. Lancet. Oncol., 11, 1048–1056. [DOI] [PubMed] [Google Scholar]
- 6. Mantovani F., et al. (2001) The human papillomavirus E6 protein and its contribution to malignant progression. Oncogene, 20, 7874–7887. [DOI] [PubMed] [Google Scholar]
- 7. Münger K., et al. (2001) Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene, 20, 7888–7898. [DOI] [PubMed] [Google Scholar]
- 8. Korzeniewski N., et al. (2011) Genomic instability and cancer: lessons learned from human papillomaviruses. Cancer Lett., 305, 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thomas L.K., et al. (2014) Chromosomal gains and losses in human papillomavirus-associated neoplasia of the lower genital tract - a systematic review and meta-analysis. Eur. J. Cancer, 50, 85–98. [DOI] [PubMed] [Google Scholar]
- 10. Klaes R., et al. (1999) Detection of high-risk cervical intraepithelial neoplasia and cervical cancer by amplification of transcripts derived from integrated papillomavirus oncogenes. Cancer Res., 59, 6132–6136. [PubMed] [Google Scholar]
- 11. Wentzensen N., et al. (2002) Characterization of viral-cellular fusion transcripts in a large series of HPV16 and 18 positive anogenital lesions. Oncogene, 21, 419–426. [DOI] [PubMed] [Google Scholar]
- 12. Vinokurova S., et al. (2008) Type-dependent integration frequency of human papillomavirus genomes in cervical lesions. Cancer Res., 68, 307–313. [DOI] [PubMed] [Google Scholar]
- 13. Jeon S., et al. (1995) Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis. Proc. Natl. Acad. Sci. USA, 92, 1654–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wentzensen N., et al. (2004) Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Res., 64, 3878–3884. [DOI] [PubMed] [Google Scholar]
- 15. Reuter S., et al. (1998) APM-1, a novel human gene, identified by aberrant co-transcription with papillomavirus oncogenes in a cervical carcinoma cell line, encodes a BTB/POZ-zinc finger protein with growth inhibitory activity. EMBO J., 17, 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schmitz M., et al. (2012) Loss of gene function as a consequence of human papillomavirus DNA integration. Int. J. Cancer, 131, E593–E602. [DOI] [PubMed] [Google Scholar]
- 17. Melsheimer P., et al. (2004) DNA aneuploidy and integration of human papillomavirus type 16 e6/e7 oncogenes in intraepithelial neoplasia and invasive squamous cell carcinoma of the cervix uteri. Clin. Cancer Res., 10, 3059–3063. [DOI] [PubMed] [Google Scholar]
- 18. Wang S.S., et al. (2009) Human papillomavirus cofactors by disease progression and human papillomavirus types in the study to understand cervical cancer early endpoints and determinants. Cancer Epidemiol. Biomarkers Prev., 18, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wentzensen N., et al. (2009) Multiple human papillomavirus genotype infections in cervical cancer progression in the study to understand cervical cancer early endpoints and determinants. Int. J. Cancer, 125, 2151–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schiffman M., et al. (2000) ASCUS-LSIL Triage Study. Acta Cytologica, 44, 726–742. [DOI] [PubMed] [Google Scholar]
- 21. Wang S.S., et al. (2006) Cervical tissue collection methods for RNA preservation: comparison of snap-frozen, ethanol-fixed, and RNAlater-fixation. Diagn. Mol. Pathol., 15, 144–148. [DOI] [PubMed] [Google Scholar]
- 22. Dunn S.T., et al. (2007) DNA extraction: an understudied and important aspect of HPV genotyping using PCR-based methods. J. Virol. Methods, 143, 45–54. [DOI] [PubMed] [Google Scholar]
- 23. Sherman L., et al. (1992) Expression and splicing patterns of human papillomavirus type-16 mRNAs in pre-cancerous lesions and carcinomas of the cervix, in human keratinocytes immortalized by HPV 16, and in cell lines established from cervical cancers. Int. J. Cancer, 50, 356–364. [DOI] [PubMed] [Google Scholar]
- 24. Luft F., et al. (2001) Detection of integrated papillomavirus sequences by ligation-mediated PCR (DIPS-PCR) and molecular characterization in cervical cancer cells. Int. J. Cancer, 92, 9–17. [PubMed] [Google Scholar]
- 25. Cullen M., et al. (2015) Deep sequencing of HPV16 genomes: A new high-throughput tool for exploring the carcinogenicity and natural history of HPV16 infection. Papillomavirus Res., 1, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smyth G.K., et al. (2003) Normalization of cDNA microarray data. Methods, 31, 265–273. [DOI] [PubMed] [Google Scholar]
- 27. Venkatraman E.S., et al. (2007) A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics, 23, 657–663. [DOI] [PubMed] [Google Scholar]
- 28. van de Wiel M.A., et al. (2007) CGHcall: calling aberrations for array CGH tumor profiles. Bioinformatics, 23, 892–4. [DOI] [PubMed] [Google Scholar]
- 29. Marioni J.C., et al. (2007) Breaking the waves: improved detection of copy number variation from microarray-based comparative genomic hybridization. Genome Biol., 8, R228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Halper-Stromberg E., et al. (2011) Performance assessment of copy number microarray platforms using a spike-in experiment. Bioinformatics, 27, 1052–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zack T.I., et al. (2013) Pan-cancer patterns of somatic copy number alteration. Nat. Genet., 45, 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Curtis C., et al. (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature, 486, 346–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. The ENCODE Project Consortium (2004) The ENCODE (ENCyclopedia Of DNA Elements) Project. Science, 306, 636–640. [DOI] [PubMed] [Google Scholar]
- 34. Lukusa T., et al. (2008) Human chromosome fragility. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms, 1779, 3–16. [DOI] [PubMed] [Google Scholar]
- 35. Song L., et al. (2011) Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Research, 21, 1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bierkens M., et al. (2012) Chromosomal profiles of high-grade cervical intraepithelial neoplasia relate to duration of preceding high-risk human papillomavirus infection. Int. J. Cancer, 131, E579–E585. [DOI] [PubMed] [Google Scholar]
- 37. Kraus I., et al. (2008) The majority of viral-cellular fusion transcripts in cervical carcinomas cotranscribe cellular sequences of known or predicted genes. Cancer Res., 68, 2514–2522. [DOI] [PubMed] [Google Scholar]
- 38. Schmitz M., et al. (2012) Non-random integration of the HPV genome in cervical cancer. PLoS One, 7, e39632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xu B., et al. (2013) Multiplex identification of human papillomavirus 16 DNA integration sites in cervical carcinomas. PLoS One, 8, e66693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Peter M., et al. (2010) Frequent genomic structural alterations at HPV insertion sites in cervical carcinoma. J. Pathol., 221, 320–330. [DOI] [PubMed] [Google Scholar]
- 41. Ojesina A.I., et al. (2014) Landscape of genomic alterations in cervical carcinomas. Nature, 506, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Duensing S., et al. (2004) Cyclin-dependent kinase inhibitor indirubin-3'-oxime selectively inhibits human papillomavirus type 16 E7-induced numerical centrosome anomalies. Oncogene, 23, 8206–8215. [DOI] [PubMed] [Google Scholar]
- 43. Han T., et al. (2012) Characterization of whole genome amplified (WGA) DNA for use in genotyping assay development. BMC Genomics, 13, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Przybytkowski E., et al. (2011) The use of ultra-dense array CGH analysis for the discovery of micro-copy number alterations and gene fusions in the cancer genome. BMC Med. Genomics, 4, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.