

Abstract
Molybdenum is the only second row transition metal essential for biological systems, which is biologically available as molybdate ion. In eukarya, bacteria and archaea, molybdenum is bound to either to a tricyclic pyranopterin, thereby forming the molybdenum cofactor (Moco), or in some bacteria to the FeS cluster based iron-molybdenum cofactor (FeMoco), which forms the active site of nitrogenase. To date more than 50 Moco-containing enzymes have been purified and biochemically or structurally characterized. The physiological role of molybdenum in these enzymes is fundamental to organisms, since the reactions include the catalysis of key steps in carbon, nitrogen and sulfur metabolism. The catalyzed reactions are in most cases oxo-transfer reactions or the hydroxylation of carbon centers. The biosynthesis of Moco has been intensively studied, in addition to its insertion into molybdoenzymes. In particular, a link between the biosynthesis and maturation of molybdoenzymes and the biosynthesis and distribution of FeS clusters has been identified in the last years: 1) The synthesis of the first intermediate in Moco biosynthesis requires an FeS-cluster containing protein, 2) The sulfurtransferase for the dithiolene group in Moco is common also for the synthesis of FeS clusters, thiamin and thiolated tRNAs, 3) the modification of the active site with a sulfur atom additionally involves a sulfurtransferase, 4) most molybdoenzymes in bacteria require FeS clusters as additional redox active cofactors. In this review we will focus on the biosynthesis of the molybdenum cofactor in bacteria, its modification and insertion into molybdoenzymes, with an emphasis to its link to FeS cluster biosynthesis and sulfur transfer.
1. An introduction into the biosynthesis of the molybdenum cofactor
In all organisms, Moco is synthesized by a conserved biosynthetic pathway that can be divided into three general steps, according to the stable biosynthetic intermediates which can be isolated (Figure 1) [1,2,3]: the synthesis of cyclic pyranopterin monophosphate (cPMP) [4], conversion of cPMP into MPT by introduction of two sulfur atoms [5], and insertion of molybdate to form Moco [6]. These reactions are highly conserved among all kingdoms of life. In prokaryotes, Moco is further modified by the attachment of GMP or CMP to the phosphate group of MPT, forming the dinucleotide variants of Moco, MPT-guanine dinucleotide (MGD) [7] and MPT-cytosine dinucleotide (MCD) [8,9] (Figure 1). The different forms of Moco are inserted into molybdoenzymes which are categorized into three families based on the ligands at the molybdenum atom (Figure 2): the xanthine oxidase (XO) family, the sulfite oxidase (SO) family and the dimethyl sulfoxide (DMSO) reductase family, which is present only in prokaryotes [2]. The chemical nature of Moco has been determined by Rajagopalan and coworkers in 1982 [10]. Here, we will focus on the biosynthesis of Moco in bacteria.
Figure 1. The biosynthesis of Moco.
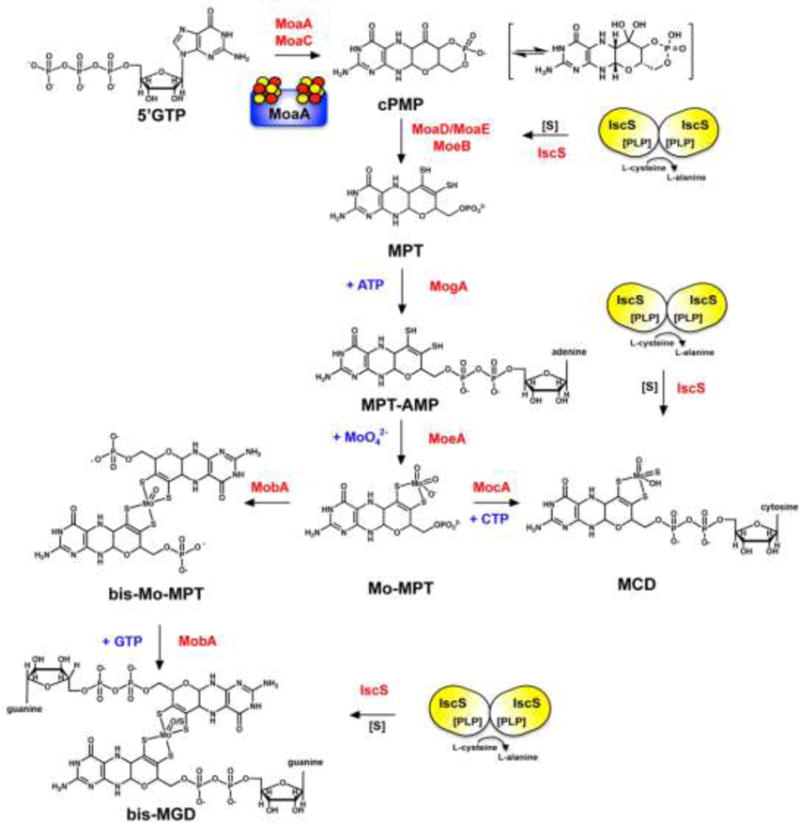
Shown is a scheme of the biosynthetic pathway for Moco biosynthesis in bacteria and the proteins involved in this pathway (which were mainly identified by studies using the E. coli proteins). Mo-MPT is formed from 5′GTP with cPMP, MPT and MPT-AMP as intermediates. The first reaction involves an FeS cluster containing protein (schematically the MoaA protein is shown containing two 4Fe4S clusters). Mo-MPT can be further modified by formation of an bis-Mo-MPT intermediate, and further addition of a GMP molecule to each MPT unit, forming the bis-MGD cofactor. Both reactions are catalyzed by the MobA protein. Bis-MGD can be further modified by the addition of a sulfido-ligand at the Mo-active site, a reaction catalyzed by the L-cysteine desulfurase IscS. Alternatively, Mo-MPT is modified by the addition of CMP to form the MCD form of the cofactor. Here, a terminal sulfur ligand is added to the molybdenum site, generating sulfurated MCD, a reaction catalyzed by the L-cysteine desulfurase IscS. The names of the proteins involved in the reactions are colored in red, nucleotides required and molybdate are colored in blue.
Figure 2. The different structures of Moco in bacteria.
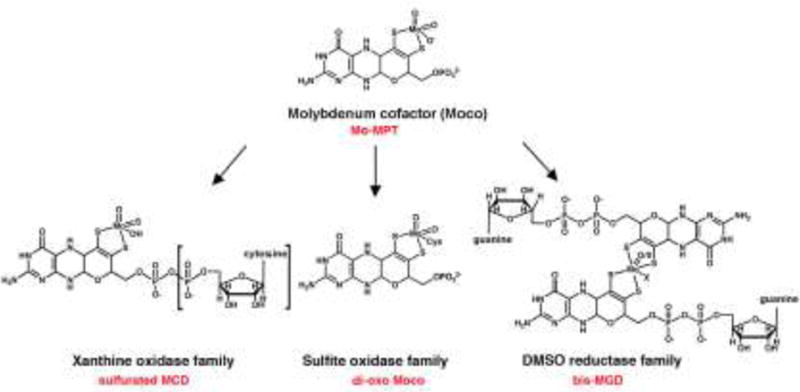
The basic form of Moco is a 5,6,7,8-tetrahydropyranopterin with a unique dithiolene group coordinating the molybdenum atom, named Mo-MPT. Mo-MPT (shown in the tri-oxo structure [28]) can be further modified and three different molybdenum-containing enzyme families exist in bacteria classified according to their coordination at the molybdenum atom: the xanthine oxidase, sulfite oxidase, and DMSO reductase families. In E. coli, the xanthine oxidase family contains the sulfurated MCD cofactor. In bacteria like R. capsulatus enzymes of the xanthine oxidase family contain the sulfurated Mo-MPT cofactor (the additional CMP is boxed). The sulfite oxidase family is characterized by a di-oxo Moco with an additional protein ligand, which usually is a cysteine. The DMSO reductase family contains two MGDs ligated to one molybdenum atom with additional ligands being an oxygen or a sulfur, and a sixth ligand X, which can be a serine, a cysteine, a selenocysteine, an aspartate or a hydroxide and/or water molecule. MCD: molybdopterin cytosine dinucleotide cofactor; MGD: molybdopterin guanine dinucleotide cofactor; MPT: molybdopterin.
The biosynthesis of Moco starts from 5′-GTP and produces the first stable intermediate of Moco, cPMP, (Figure 1) [4]. cPMP is a 6-alkyl pterin with a cyclic phosphate group at the C2′ and C4′ atoms [11]. This transformation requires two enzymes, MoaA and MoaC [4,12] (Figure 1). While the individual catalytic functions of MoaA and MoaC had long been unknown, recent studies showed that MoaA catalyzes the conversion of GTP to (8S)-3′,8-cyclo-7,8-dihydroguanosine 5′triphosphate (3′,8-cH2GTP), and MoaC catalyzes the conversion of 3′,8-cH2GTP to cPMP (Figure 3). [13]. Details of the recent updates and the details of MoaA mechanism, where 4Fe-4S clusters play central roles, will be discussed in detail below. Two forms of cPMP are described in the literature, one in the hydrate form and one in the ketone form [11,14]. The ketone form was proposed based on the early characterization studies by chemical derivatization [4,15] and an X-ray crystallography of the MPT synthase in complex with cPMP [14]. The hydrate form was based on the NMR and MS characterization of cPMP under acidic pH (<3) [11]. Recently, the hydrated form of cPMP was synthesized chemically [16]. Considering that, in general, ketones and hydrates are in equilibrium in aqueous solutions, the two models are chemically similar. Whether biosynthetic enzymes distinguish the two forms remains to be identified.
Figure 3. Reaction catalyzed by MoaA.
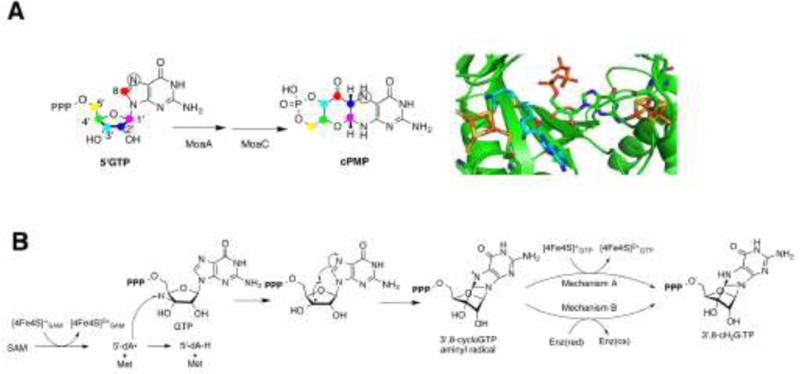
A) The conversion of 5′GTP to cPMP is catalyzed by MoaA and MoaC. The results of the isotope tracer experiments showed that all carbon atoms of the 5′GTP are found within cPMP. The C8 atom from the guanine ring is inserted between the C2′ and C3′ atoms of the ribose. The model of the active site of MoaA shows the binding of SAM (shown in light blue) (PDB ID: 1TV8) [38] and GTP (PDB ID: 2FB3) [37]. Oxygens are shown in red, nitrogens in blue, sulfurs in yellow, phosphorous in orange, and irons in brown. B) In the reaction catalyzed by MoaA, SAM is reductively cleaved to the 5′-dA• radical and L-methionine (Met) by oxidizing [4Fe-4S]SAM. The 5′-dA• then abstracts the H-3′ atom of GTP, resulting in the C3′ centered radical which attacks the C8 of the guanine to forms an aminyl radical intermediate. This intermediate is then reduced to form the 3′,8-cH2GTP intermediate. Two possible mechanisms of aminyl radical reduction are shown. In the mechanism A, [4Fe-4S]GTP cluster is the electron donor. In mechanism B, the electron is provided by an exogenous donor; an uncharacterized redox enzyme (Enz) in vivo.
In the next step of Moco biosynthesis, cPMP is converted to MPT [5,17,18,19,20]. In this reaction two sulfur atoms are inserted to the C1′ and C2′ positions of cPMP [14]. This reaction is catalyzed by MPT synthase, which forms an (αβ)2 heterodimer composed of two MoaD and two MoaE subunits [21]. The sulfur atoms required for this reaction are present at the C-terminus of MoaD in form of a thiocarboxylate group [22,23]. Studies of the reaction mechanism showed that the first sulfur is added by one MoaD molecule at the C2′ position of cPMP, resulting in a hemi-sulfurated intermediate (Fig. 4). This reaction is coupled to the hydrolysis of the cPMP cyclic phosphate group [14]. In the hemi-sulfurated intermediate, the MoaD C-terminus is covalently linked via a thioester linkage, which is hydrolyzed by a water molecule in the next reaction. After transfer of its thiocarboxylate sulfur to cPMP, the first MoaD subunit dissociates from the MPT synthase complex [14,24] and a new MoaD molecule containing a thiocarboxylate group (MoaD-SH) associates with the complex. After the first sulfur transfer, the opening of the cyclic phosphate is proposed to shift the location of the intermediate within the protein so that the C1′ position then becomes more accessible to the attack by the second MoaD-SH (Figure 4). Further, a second covalent intermediate is formed with the new MoaD-SH protein. MPT is formed by the elimination of a water molecule and hydrolysis of this MoaD-thioester intermediate.
Figure 4. The biosynthesis of MPT from cPMP.
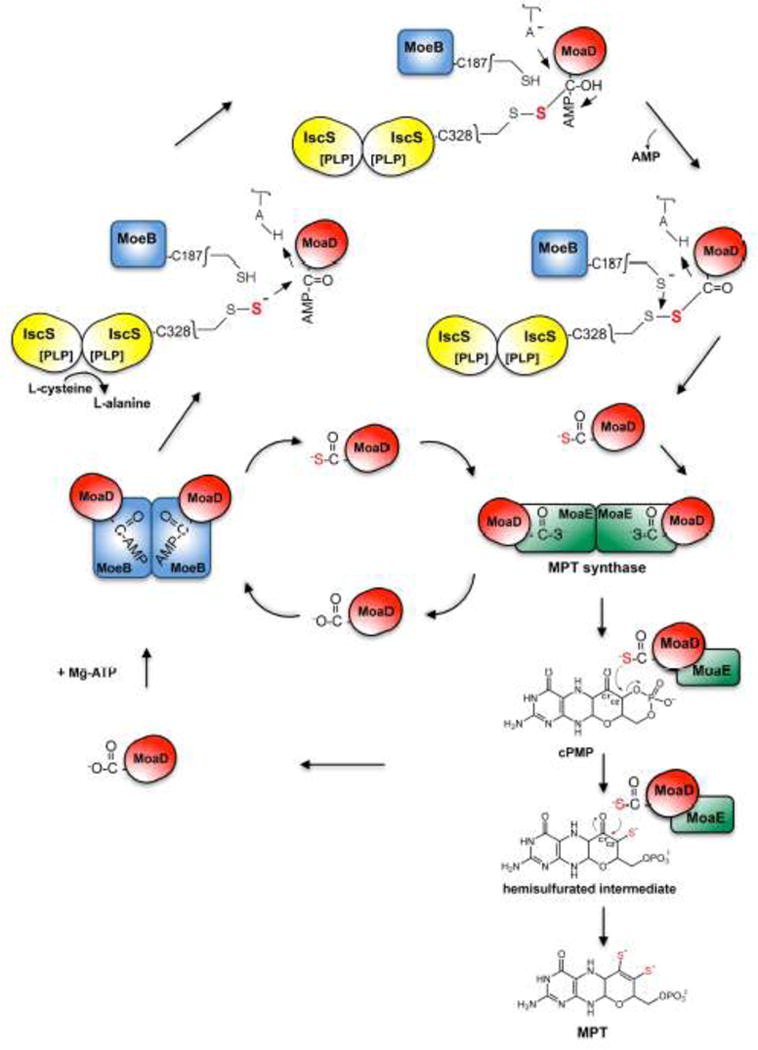
The MPT synthase tetramer is built of two MoaE and two MoaD subunits. In the MPT synthase mechanism, cPMP is bound to the MoaE subunit. The initial attack and transfer of the first thiocarboxylated MoaD-SH sulfur atom occurs at the C2′ position of cPMP, coupled to the hydrolysis of the cPMP cyclic phosphate. A new MoaD-SH thiocarboxylate attacks the C1′ of the newly formed hemisulfurated intermediate, which is converted to MPT via the elimination of a water molecule. MoaD is regenerated and a new MoaD-SH thiocarboxylate is formed on MoeB, where MoaD is first activated under ATP consumption to form an activated MoaD-acyl adenylate. MoaD-AMP is then sulfurated by a protein-bound persulfide, e.g. from Cys328 of IscS or from other sulfur transferring proteins like YnjE, TusA or SufS (not shown). Likely, an MoeB-MoaD disulfide intermediate is formed during the reaction, which is further cleaved by reductive cleavage (e.g. MoeB-Cys187). After formation of the thiocarboxylate group, MoaD-SH dissociates from the MoeB dimer and reassociates with MoaE, forming the active MPT synthase. The mechanism of MPT synthase was adapted from the one proposed in [14].
After the synthesis of MPT, molybdate is inserted to the dithiolene sulfurs and Moco is formed. The specific insertion of molybdenum into MPT is catalyzed by the joined action of the MoeA and MogA proteins (Figure 1) [25,26]. MogA was shown to form an MPT-AMP intermediate under ATP consumption [27]. This intermediate is then transferred to MoeA, which mediates molybdenum ligation at low concentrations of MoO42−[26]. The endproduct of the MoeA and MogA reaction is Mo-MPT in a tri-oxo form, the basic form of the molybdenum cofactor which can be further modified by nucleotide addition [28]. Alternatively, the Mo-MPT cofactor can be directly inserted into enzymes of the sulfite oxidase family, where Moco is coordinated by a cysteine ligand which is provided by the polypeptide chain of the protein, forming an MPT-MoVIO2 core in its oxidized state [2].
The proteins of the DMSO reductase family in bacteria contain the bis-MGD cofactor [2] (Figure 2). The synthesis of the bis-MGD was shown to occur in two step reaction which requires Mo-MPT, MobA and Mg-GTP [28]. In the first reaction, the bis-Mo-MPT intermediate is formed on MobA with Mo-MPT as substrate (Figure 1). For this reaction the ligation of molybdenum to MPT is essential [29], but no further cofactors or molecules are required. In the second reaction, two GMP moieties from GTP are added to the C4′ phosphate of bis-Mo-MPT, forming the bis-MGD cofactor [30,31]. After the attachment of two GMP molecules to the bis-Mo-MPT intermediate, the bis-MGD cofactor is formed and released from MobA. Since bis-MGD is not stable in its free form, it is immediately bound by Moco-binding chaperones which insert the cofactor specifically into target enzymes of the DMSO reductase family. The molybdenum coordination in enzymes of the DMSO reductase family is a MPT2-MoVIO/S(X) core, where the sixth ligand, X, can be a serine, a cysteine, a selenocysteine, an aspartate from the protein backbone or a hydroxide and/or water molecule.
Enzymes of the xanthine oxidase family in some bacteria like E. coli, contain the MCD form of Moco (Figure 2) [32]. MCD formation is catalyzed by MocA, a protein that shares a high amino acid sequence identities to MobA [9,33]. In contrast to MobA, MocA is specific for the pyrimidine nucleotide CTP. The overall catalytic reaction of MocA is similar to the second part of the reaction of MobA, in that it acts as a molybdopterin CTP transferase and covalently links MPT and CMP with the concomitant release of the β- and γ-phosphates of CTP as pyrophosphate [9]. However, in this reaction MCD is the endproduct and the bis-form is not formed. Instead, the MCD cofactor for all enzymes of the xanthine oxidase family is further modified and contains an equatorial sulfido ligand at its active site, which is essential for enzyme activity. Enzymes of the xanthine oxidase family contain either an MCD cofactor or an Mo-MPT cofactor (without an additional nucleotide) with an MPT-MoVIOS(OH) core in the oxidized state [32]. There is no amino acid ligand from the protein backbone to the molybdenum atom.
2. The role of FeS clusters in the conversion of 5′GTP to cPMP
The first link between the Fe-S cluster and Moco biosynthesis appears in the first step of Moco biosynthesis, a conversion of GTP into cPMP, which proceeds through a complex rearrangement reaction, where C8 of guanine is being inserted between C2′ and C3′ of ribose [12] (Fig. 3A). During this transformation, MoaA plays a key role. MoaA is a [4Fe-4S] cluster protein, and belongs to the radical S-adenosyl methionine (SAM) superfamily [34,35]. Radical SAM enzymes catalyze a common reaction, in which SAM is reductively cleaved by an electron from a [4Fe-4S] cluster to generate 5′-deoxyadenosyl radical (5′-dA•) [36]. This organic radical species is subsequently used to abstract a H-atom from the substrate to initiate radical reactions. Many of the radical SAM enzymes catalyze diverse and chemically challenging reactions [36].
MoaA harbors two 4Fe-4S clusters [37,38] (Figure 3A). One cluster ([4Fe-4S]SAM) locates at the N-terminus, and binds SAM in crystals in a manner similar to other radical SAM enzymes [38], supporting its annotation as a radical SAM enzyme. The second cluster ([4Fe-4S]GTP) was unique to MoaA and shown to bind GTP in crystals as well as in solution (Kd = 0.29 μM) [37,39]. While both of these clusters were shown to be essential for Moco biosynthesis, their functions, especially that of [4Fe-4S]GTP, remained elusive due the lack of understanding in the chemical reaction catalyzed by MoaA [35,38,40].
The understanding of the functions of the 4Fe-4S clusters requires knowledge inthe catalytic function of MoaA. However, this has been challenging, primarily due to the poor stability of the MoaA product. This technical challenge has resulted in multiple proposals for the structure of the MoaA product, which are currently under active debate [13,37,41,42]. However, despite such discussion, accumulating evidence suggest that MoaA catalyzes the conversion of GTP into 3′,8-cyclo-7,8-dihydroguanosine 5′-triphosphate (3′,8-cH2GTP). Initially, an involvement of a free radical generated at C3′ was indicated by isotope labeling experiments [13,42], where a deuterium atom at the 3′-position was shown to be transferred to 5′-deoxyadenosine, a product of reductive cleavage of SAM. More definitive evidence was provided by Hover et al., in which 3′,8-cH2GTP was isolated from the in vitro MoaA assay solution using GTP and SAM as substrates [13]. In this study, 3′,8-cH2GTP was found to be highly sensitive to oxygen, and the isolation was achieved under strict anaerobic conditions. These observations explain the absence of its isolation until this study. Importantly, the isolated 3′,8-cH2GTP was converted to cPMP by MoaC with high specificity (Km < 60 nM for MoaC) [13]. This reaction was independent of MoaA. Therefore, based on these observations, Hover et al. proposed 3′,8-cH2GTP being the product of MoaA, and the conversion of 3′,8-cH2GTP to cPMP being catalyzed by MoaC [13]. This is a sharp contrast to the previous notion of the cPMP formation, where the pyranopterin structure is formed by MoaA, and MoaC has no or little role in the rearrangement reaction. The presence of 3′,8-cH2GTP was later supported by an observation reported by Mehta et al., where a product of MoaA reaction using 2′-deoxy-GTP, a non-physiological substrate [43]. It is noteworthy that MoaA was also proposed to catalyze the conversion of GTP into pyranopterin triphosphate [41,42]. However, physiological relevance of such model remains unclear, especially when MoaC catalyzes the conversion of 3′,8-cH2GTP to cPMP with high specificity (Km < 60 nM).
The conversion of GTP into 3′,8-cH2GTP by MoaA proceeds through a radical formation at C3′ and a stoichiometric consumption of SAM (Fig. 3B) [13,42]. This reaction is initiated by the abstraction of H-3′ atom of GTP by 5′-dA•. The [4Fe-4S]SAM cluster likely serves as an electron donor for the reductive cleavage of SAM, as has been proposed for other radical SAM enzymes. The resulting C3′ centered radical attacks C8 to form the aminyl radical intermediate, which is then reduced by transfer of an electron and a proton to form 3′,8-cH2GTP.
One major unresolved question about the reaction of MoaA catalysis is the role of the [4Fe-4S]GTP cluster, for which two possibilities are conceivable. In one model (mechanism A in Fig. 3B), the [4Fe-4S]GTP cluster serves as the electron donor for the aminyl radical reduction. This model is based on the consideration of the redox potentials of related FeS clusters (−0.4 ~ −0.6 V) relative to aminyl radicals on pyrimidine and pterin molecules (+0.1 ~ +0.3 V)[13]. In a mutant MoaA (or its human homolog, MOCS1A) that lacks the [4Fe-4S]SAM cluster, the [4Fe-4S]GTP cluster was shown to take two different redox states (1+/2+), and the 1+ state of the cluster binds GTP [33, 37] The redox properties of the [4Fe-4S]GTP cluster in catalytically competent MoaA is currently unknown, and are the subjects of future investigation.
Alternatively, in the second model (mechanism B in Fig. 3B), the [4Fe-4S]GTP cluster does not have redox functions during the catalytic turnover, and serves only for structural stabilization and/or to provide a specific binding site for GTP. In this model, the electron for the aminyl radical reduction is provided exogenously. In vitro, this electron is likely provided by dithionite added in the assay solution in large excess. In vivo, this electron is likely provided by a redox enzyme. While flavodoxin is generally considered as the physiological reductant of radical SAM enzymes [44], pathway specific reducing enzymes are also known for other radical SAM enzymes [45]. Currently, the physiological reductant for MoaA is unknown, and its identification might help to distinguish the two possible roles of the [4Fe-4S]GTP cluster.
Understanding of the role of the [4Fe-4S]GTP cluster will expand our mechanistic insights into the radical SAM enzymes. MoaA is a representative member of radical SAM enzymes with more than one 4Fe-4S cluster, and catalyzes chemical reactions that require an electron transfer step. Recently, radical SAM enzymes with multiple 4Fe-4S clusters were classified into a subfamily with more than 7,000 annotated sequences [46]. The classification is based on the presence of a conserved SPASM/twitch domain that contains Cys ligands for auxiliary [4Fe-4S] clusters (the [4Fe-4S]GTP cluster in case of MoaA). All the functionally characterized members of this subfamily require an electron transfer step for the catalysis. While the mechanism of the electron transfer step is generally unknown, the involvement of an auxiliary cluster was proposed for anaerobic sulfatase maturase [47]. Further mechanistic investigation of the functions of the 4Fe-4S clusters in MoaA will promote the understanding of this emerging group of enzymes.
3. The dithiolene sulfurs of MPT link Moco biosynthesis to the general mechanism of sulfur trafficking in the cell
After the formation of cPMP by MoaA and MoaC, two sulfur atoms are inserted to the C1′ and C2′ atoms of cPMP, forming the dithiolene group of MPT. The direct sulfur donor on this reaction are two MoaD-SH subunits, which carry the sulfurs in form of a thiocarboxylate group at the C-terminal glycine of the protein. After the reaction, a new thiocarboxylate group has to be formed on MoaD for a new round of catalysis.
The regeneration of sulfur at the C-terminal glycine of MoaD is catalyzed by MoeB [48] and resembles the first step of the activation of ubiquitin for the ubiquitin-dependent protein degradation system in eukaryotes (Figure 4) [49]. In the (MoaD–MoeB)2 complex, ATP is bound and the C-terminus of MoaD is activated by the formation of an acyl-adenylate group at its terminal glycine. The hydrolysis of Mg-ATP and the release of PPi in the reaction were shown both in the X-ray structure and in biochemical assays [48,50]. In its activated form, MoaD-AMP then receives the sulfur from a sulfurtransferase and MoaD-SH is formed. The sulfur is directly transferred to MoaD in the (MoaD–MoeB)2 complex. After the reaction, MoaD-SH dissociates from the complex, additionally releasing MoeB2 and AMP, and reassociates with MoaE2 to form the active MPT synthase heterotetramer (Fig. 4) [51,52]. It was determined that in E. coli, L-cysteine serves as the origin of the MPT dithiolene sulfurs [53]. In this reaction, the L-cysteine desulfurase IscS is involved in the initial mobilization step.
L-cysteine desulfurases in general are pyridoxal 5′-phosphate (PLP)-containing enzymes that catalyze the desulfuration of L-cysteine to yield L-alanine and a protein-bound persulfide group [54]. This persulfide group can be further transferred either to sulfur-accepting proteins or to biomolecules [55]. In E. coli, three L-cysteine desulfurases are present, which are grouped into two different classes of cysteine desulfurases according to their amino acid sequence homology: IscS belongs to the class V fold-type I aminotransferase group I [56,57] and SufS and CsdA to group II [58,59]. The main difference in these two groups is the amino acid region around the catalytically active cysteine residue of these proteins [55]. The L-cysteine desulfurase IscS has been originally described to be involved in the transfer of sulfur for the formation of FeS clusters [60]. IscS is encoded by a gene that is part of a larger operon, iscRSUA-hscBA-fdx-iscX, in which the other gene products are involved in the biosynthesis of FeS clusters in E. coli [61]. For FeS cluster biosynthesis IscS interacts with IscU, which serves as a scaffold protein for FeS cluster assembly [62,63] (Figure 5). IscS is a pyridoxal 5′-phosphate-containing homodimer that decomposes L-cysteine to L-alanine and sulfane sulfur via the formation of an enzyme-bound persulfide intermediate [61]. The persulfide-sulfur is then transferred for the formation of FeS clusters in the IscS–IscU complex.
Figure 5. Sulfur transfer to sulfur-containing biomolecules involving the IscS-bound persulfide.
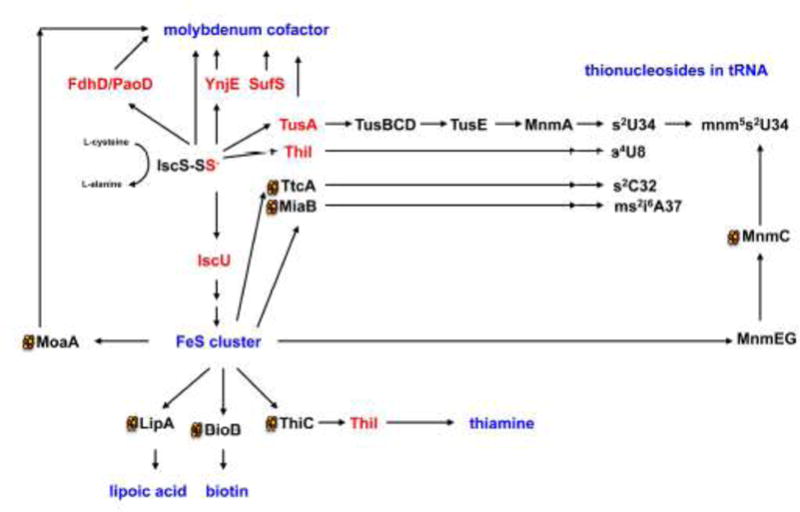
A protein-bound persulfide-group is formed on the L-cysteine desulfurase IscS from L-cysteine, releasing L-alanine. The persulfide-sulfur is further transferred to proteins like IscU, TusA, ThiI, YnjE, or FdhD (highlighted in red). IscU is the primary scaffold for the assembly of FeS clusters. FeS clusters are required for proteins involved in the pathways for the biosynthesis Moco, lipoic acid, biotin, thiamine and also for the addition of thionucleosides in tRNA. In E. coli tRNA thiolation involves the synthesis of s2C by TtcA, ms2i6A by MiaB, s4U by ThiI and a sulfur relay system involving TusA, TusBCD, TusE and MnmA for the formation of s2U in tRNA. In most cases, additional proteins are involved in the biosynthetic pathways. Persulfide-containing proteins are highlighted in red and names of the final sulfur-containing molecules are colored in blue. Proteins which containing 4Fe4S clusters are marked with a cluster.
It was shown that IscU is not the only interaction partner of IscS, since IscS interacts with a number of other proteins for delivery of sulfur including the involvement of CyaY, IscX for FeS cluster formation, TusA for the thiolation of tRNA, and ThiI for the synthesis of thiamine and the thiolation of tRNA (Figure 5) [55]. Different binding sites for some of these molecules were mapped on E. coli IscS and show overlapping binding modes [64]. This rises the question on the direction of sulfur transfer, since for each biomolecule this fist sulfur mobilization step is essential. It has been suggested that the direction of sulfurtransfer of the IscS-persulfide to a sulfur acceptor protein is mediated by the availability of the latter [64,65]. This would require a precise regulation of the direction of sulfurtransfer in the cell, a mechanism which is not completely understood so far.
Recently, a role of TusA for Moco biosynthesis was identified [65]. This implied that for the sulfurtransfer to MoaD, IscS does not directly interact with MoaD, but rather transfers the sulfur to sulfurtransferases like TusA, in a sulfur relay system, which then transfer the sulfur further to the acceptor protein. It was shown that deletion of tusA in E. coli affected the activity of molybdoenzymes under aerobic and anaerobic conditions. Characterization of the ΔtusA strain under aerobic conditions showed an overall low MPT content and an accumulation of cPMP. Under anaerobic conditions the activity of nitrate reductase was only 50% reduced, suggesting that TusA is not essential for Moco biosynthesis and can be replaced by other proteins. Under these conditions, the rhodanese-like protein YnjE or the L-cysteine desulfurase SufS are able to replace IscS and TusA in its role as sulfur donor for MoaD [65,66]. The expression of ynjE was shown to be increased under anaerobic conditions and in the absence of TusA. Additionally, SufS can replace IscS in its role in providing the sulfur under certain conditions. SufS has been mainly described to be involved in the synthesis of FeS clusters which have been oxidatively damaged. Thus, systems exist in the cell, which allow alternative sulfurtransfer pathways.
The TusA protein has been originally described to be involved in the formation of s2U of the modified wobble base 5-methylaminomethyl (mnm)5s2U in tRNA of tRNALys, tRNAGlu and tRNAGln[67] (Figure 5). This thiomodification is crucial for precise decoding of the genetic code. TusA was shown to transfer the sulfur from IscS further onto the TusBCD complex and finally via TusE to MnmA. MnmA has a P-loop in its active site that recognizes tRNA and activates the C2 position of the uracil ring at position 34 by forming an adenylate intermediate [68]. This shows that TusA is involved in sulfurtransfer for at least two pathways, Moco biosynthesis and the biosynthesis of s2U in tRNA. Therefore, it has been suggested that the role of TusA might also be to provide a balanced regulation of the availability of IscS for the various biomolecules in E. coli [65].
It has been suggested that the pleiotropic effects of a tusA deletion might be caused by changes in the FeS cluster concentration in the cell, leading to differences in the sulfur transfer to tRNA and for Moco biosynthesis [65]. Studies showed that elevated levels of TusA in E. coli decreased the level of FeS clusters (Figure 6). A decrease in FeS cluster biosynthesis resulted in an inactive MoaA protein and thus in a decreased activity of molybdoenzymes, showing that Moco biosynthesis and FeS cluster biosynthesis are directly linked. On the other side, overexpression of IscU also reduced the level of active molybdoenzymes in E. coli (Figure 6). This observation was explained by the fact that when IscU is present in high amounts, it forms a tight complex with IscS, making it unavailable for the interaction with TusA (based on overlapping binding sites), thus resulting in a lack of sulfurtransfer for the conversion of cPMP to MPT (Figure 6). Conclusively, this suggests that TusA is involved in regulating the availability of IscS for the different sulfurtransfer pathways.
Figure 6. Model for the effect of altered concentrations of TusA or IscU on Moco biosynthesis.
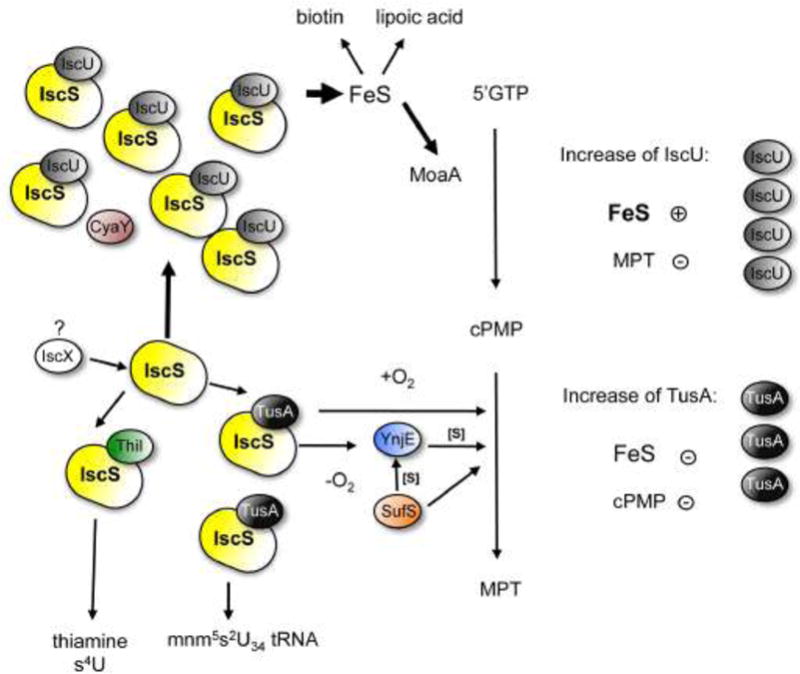
IscS interacts with IscU, TusA, and ThiI for sulfurtransfer to FeS clusters, tRNA, Moco biosynthesis and thiamine/tRNA, respectively. FeS clusters are inserted into target proteins. Among these proteins is MoaA, involved in Moco biosynthesis (conversion of 5′GTP to cPMP). TusA is involved in the sulfurtransfer for the conversion of cPMP to MPT, or in sulfurtransfer for s2U formation. Increased levels of IscU reduce the level of active molybdoenzymes in E. coli. When IscU is present in high amounts, it forms a complex with IscS, making it unavailable for the interaction with TusA, thus, resulting in a lack of sulfurtransfer for the conversion of cPMP to MPT. When the amount of TusA is increased, the levels of FeS clusters are decreased which further result in an inactive MoaA protein and thus in a decreased activity of molybdoenzymes. Detailed descriptions are given in the text.
The studies also showed that the pleiotropic effect of a tusA deletion might be caused by changes in the FeS cluster concentration in the cell leading additionally to major differences in gene regulation. Microarray analysis of a ΔtusA strain showed that the expression of genes regulated by FNR (regulator for fumarate and nitrate reductase) were increased. The same effect was observed after overexpression of FNR in E. coli under aerobiosis, which led to the induction of the isc operon due to a higher FeS cluster demand in the cell [69]. FNR directly senses the ambient oxygen concentration via the disassembly and reassembly of its [4Fe-4S] clusters [70]. A higher level of FeS clusters in the cell stabilizes holoFNR, thus stimulating the transcription of FNR regulated genes (like the genes for nitrate reductase). It is concluded that the absence of TusA changes the level of IscS which is available for FeS cluster biosynthesis [65], and thus, results in an increase in FeS clusters in the cell. As a consequence, the sulfur transfer of IscS to other biosynthetic pathways like Moco, thiamine or thiolated tRNA is reduced. Thus, the availability and amount of IscS has an effect on various pathways in the cell. Further studies are necessary to dissect the complex network of sulfur transfer in E. coli in detail.
4. Alteration of ligands at the molybdenum atom of Moco involves the L-cysteine desulfurase IscS
After the insertion of two sulfur atoms into cPMP, MPT is formed and the dithiolene sulfurs of MPT serve as backbone for molybdenum ligation. After molybdate addition, formed Mo-MPT is either inserted into enzymes of the sulfite oxidase family or is further modified by CMP addition to Mo-MPT or GMP addition to bis-Mo-MPT, forming the MCD or bis-MGD cofactor, respectively.
It was reported that the bis-MGD cofactor can be further modified by sulfuration. For proteins like E. coli formate dehydrogenase (FdhF) [71,72] and Cupriavidus necator periplasmic nitrate reductase (Nap) [73] it was suggested that at the molybdenum site, the apical ligand is rather a sulfur ligand instead of an oxygen ligand. Thus, in the oxidized state, the enzyme contains the four pterin sulfur ligands at the Mo site, a selenocysteine ligand (in case of FdhF) or a cysteine ligand (in case of NapA) and a –SH ligand [2]. The chaperone involved in sulfuration of the Moco for FdhF was shown to be the FdhD protein [71]. The protein involved in the sulfuration reaction of bis-MGD in E. coli was suggested to be the L-cysteine desulfurase IscS (Figure 7). For the E. coli FdhD protein it was reported that it acts as a sulfurtransferase between the L-cysteine desulfurase IscS and FdhF, a mechanism which is essential to yield active FdhF. Thus, the additional role of bis-MGD binding chaperones might be to bind bis-MGD for the further modification of Moco by sulfuration (Figure 7). An interaction between IscS and FdhD was demonstrated [71]. This adds another interaction partner to IscS, which receives the persulfide-sulfur formed at the active site of IscS (Figure 5). An interaction of IscS with other bis-MGD-binding chaperones has not been demonstrated to date.
Figure 7. Formation of sulfurated bis-MGD.
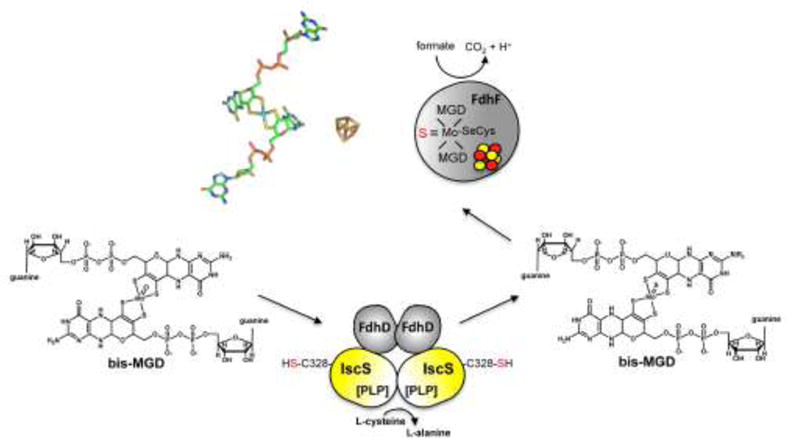
It is proposed that bis-MDG is further modified by sulfuration. In the reaction for bis-MGD sulfuration for formate dehydrogenase FdhF in E. coli, the proteins FdhD and IscS are involved. FdhD likely binfd bis-MGD and IscS transfers the sulfur to the Mo atom by exchanging an oxo-group and adding the sulfido ligand. Afterwards, sulfurated bis-MGD is inserted into FdhF, which contains additionally an [4Fe4S] cluster.
Additionally, in enzymes of the xanthine oxidase family the Mo-active site (which is either MCD or Mo-MPT) is further modified and contains an equatorial sulfido ligand which is essential for enzyme activity (Figure 2). After MCD synthesis by MocA, the cofactor is handed over to Moco binding chaperones. The best characterized chaperone for Moco sulfuration of bacterial enzymes of the xanthine oxidase family is the R. capsulatus XdhC protein. XdhC is the specific chaperone for the maturation of R. capsulatus xanthine dehydrogenase (XDH) [74]. Investigation of R. capsulatus XdhC showed that it binds the Mo-MPT form of Moco and protects it from oxidation. Additionally, XdhC interacts with the R. capsulatus L-cysteine desulfurase, NifS4, the protein that replaces the equatorial oxygen ligand of Mo-MPT by a sulfido ligand [75] (Figure 8). The sulfur for this reaction originates from L-cysteine, and a NifS4 persulfide group is formed during the course of the reaction. After the sulfuration reaction, it is believed that XdhC with its bound sulfurated Moco dissociates from NifS4 and forms a new interaction with the XdhB subunits of the R. capsulatus (αβ)2 XDH heterotetramer [75,76]. Thus, XdhC-like proteins perform a number of functions including stabilization of the newly formed Mo-MPT, interaction with an L-cysteine desulfurase to ensure Mo-MPT sulfuration and further interaction with their specific target proteins for insertion of the sulfurated Moco. Because Moco is deeply buried in the protein, it is also believed that the XdhC proteins may act additionally as chaperones to facilitate the proper folding of the target proteins after Moco insertion (Figure 8).
Figure 8. Model for the assembly of R. capsulatus XDH.
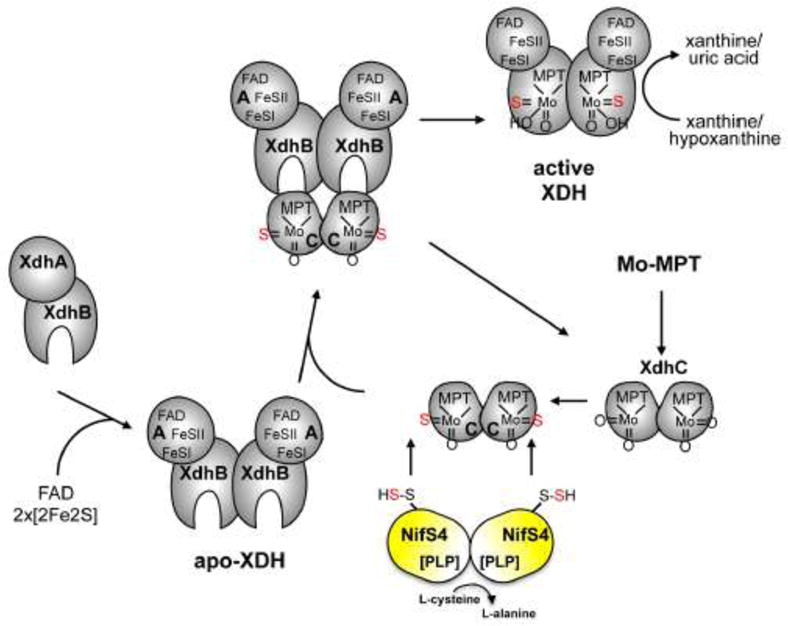
The assembly of R. capsulatus XDH involves the synthesis of the XdhA and XdhB subunits, the dimerization of both subunits, the insertion of FeSI, FeSII and FAD into the XdhA subunit, dimerization of two (αβ) dimers via the XdhB subunit to form the Moco-free apo-XDH, and finally, insertion of sulfurated Moco into of XdhC. XdhC binds Mo-MPT and the equatorial Mo=S is inserted into Moco before its incorporation into XDH by the sulfurtransferase function of the NifS4 protein. After the formation of sulfurated Mo-MPT, XdhC interacts with XDH for final Moco insertion.
Since homologues to the family of XdhC-like Moco-binding chaperones exists in E. coli, like the PaoD protein for the periplasmic aldehyde oxidoreductase PaoABC, the mechanisms for Moco sulfuration is expected to be similar for all members of this protein family. Thus, it is expected that PaoD binds MCD and facilitates its sulfuration and insertion into the PaoC subunit [74]. The specific L-cysteine desulfurase involved in the sulfuration of PaoD-bound MCD has not been identified in E. coli to date, but it is expected that IscS might perform this role in E. coli. With the interaction of the chaperones for MCD and bis-MGD binding to IscS, Moco modification by sulfuration is directly linked to the availability of IscS in the cell.
5. FeS cluster containing molybdoenzymes in bacteria
Molybdoenzymes are often composed of different subunits harboring several metal cofactors in addition to Moco, such as cytochromes, FeS cluster or FAD/FMN which are involved in intramolecular electron transfer reactions [2]. The molybdenum atom exists in various oxidation states under physiological conditions and couples oxide or proton transfer and acts as a transducers between 2 electron transfer and 1 electron transfer processes to the additional cofactors.
X-ray crystallographic studies of all known molybdoenzymes revealed that Moco is not located at the surface of the protein, but is buried deeply within the enzyme and, in some cases, is in close proximity to [FeS] clusters [77]. This suggested that Moco insertion is connected to protein folding, insertion of other prosthetic groups and subunit assembly. Most molybdoenzymes of the DMSO reductase family in bacteria are located in the periplasm and/or are tethered to the cytoplasmic membrane through c-or b-type cytochromes [2]. These membrane subunits connect the cytoplasmic or periplasmic redox reactions with electron transport to or from the respiratory quinone/quinole pool. The successful assembly of molybdoenzymes requires several steps including the synthesis of the different subunits in the cytoplasm, the incorporation of Moco and the additional prosthetic groups in addition to anchoring the complex to the membrane [78]. In case of periplasmic molybdoenzymes, the assembly and metal cofactor insertion takes place in the cytoplasm prior to the translocation of the protein complex across the inner membrane by the Tat-machinery [79]. The membrane subunits are located to the inner membrane by the Sec system with the help of the accessory protein YidC [80]. While usually the insertion of Moco has been well studied for molybdoenzymes, there is only limited knowledge about the proteins which insert the FeS cofactors into molybdoenzymes. Usually, Moco insertion occurs as the last step of molybdoenzyme maturation, after the insertion of the FeS clusters or other prosthetic groups [81]. Here, we will give an overview of well studied molybdoenzymes in E. coli and R. capsulatus which contain FeS clusters as additional redox active cofactors and will explain the roles of these cofactors for enzyme activity and electron transfer.
5.1 Nitrate reductases
E. coli contains three dissimilatory nitrate reductases which are members of the DMSO reductase family: nitrate reductase A (NarGHI), nitrate reductase Z (NarZYV) and the periplasmic nitrate reductase NapAB. All three enzymes catalyze the reduction of nitrate to nitrite, however, their expression patterns differ: at high concentrations of nitrate, only the membranous NarGHI system is synthesized [82], whereas at very low concentrations the Nap system is produced [83]. The operon encoding a third system, narZYWV, is expressed during the early stationary phase independent of the presence of nitrate [84]. While NarGHI and NarZYV are facing the cytoplasm and are linked to formate reductases in the formate:nitrate reductase complexes (Figure 9), NapAB receives the electrons from the NapCGH complex in the membrane. NapC is essential for electron transfer from both ubiquinol and menaquinol to NapAB [83]. The periplasmic nitrate reductase Nap system is found in many different organisms [85,86,87,88]. In E. coli, it is made up of five proteins: the NapAB dimer, NapA being the catalytic subunit and NapC that transfers electrons to the NapAB complex, and two other proteins, NapG a periplasmic protein and NpH an integral membrane protein, containing both iron-sulfur centers are also involved in the electron transfer pathway from the ubiquinol pool to NapC [83]. Reduction of nitrate occurs in the catalytic site of NapA containing the bis-MGD form of the cofactor (which coordinates a sulfido group and a cysteine residue) and a [4Fe4S] cluster [89]. Similar to all periplasmic molybdoenzymes, NapA is matured in the cytoplasm and its translocation depends on the Tat machinery [90]. NapB contains two b-type cytochromes. The X-ray structure of NapA from various organisms showed that the overall fold of NapA is similar to the one of FdhF [2]. NapD is the proposed molecular chaperone for the insertion of the bis-MGD cofactor into NapA [91]. Both Moco insertion and FeS cluster insertion into NapA have not been analyzed in detail so far.
Figure 9. Schematic presentation of molybdoenzymes, their localization and cofactor composition.
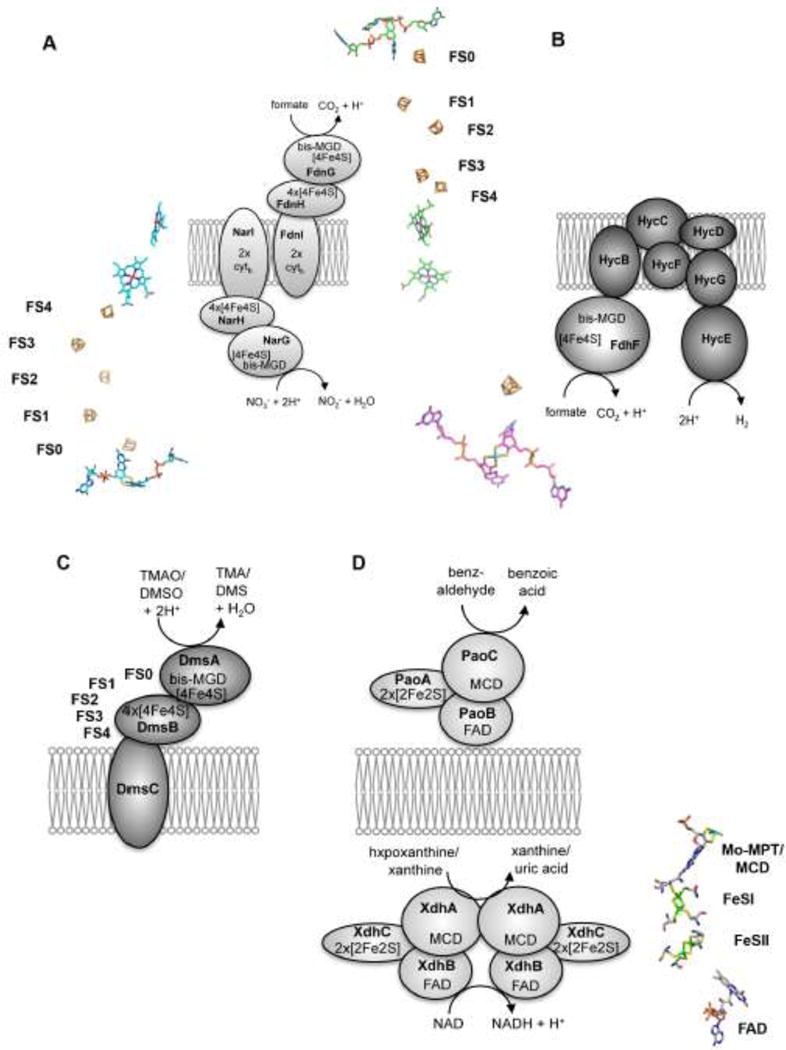
(A) Localization of the membrane associated multienzyme complex build of formate dehydrogenase N and nitrate reductase A. FdnGHI are components of the nitrate respiratory pathway, in which formate oxidation is coupled to nitrate reduction via NarGHI. The bis-MGD cofactor, FeS clusters and cytochromes are shown next to the subunits of each enzyme complex. The FeS clusters are numbers FS0- 4. (B) Formate dehydrogenase H is part of the formate:hydrogen lyase complex. The cofactors present in the hydrogenase subunits are not shown. The 4Fe4S cluster in FdhF has a similar localization to the bis-MGD cofactor as in NarG, FdnG or DmsA. (C) Shown is a schematic representation of E. coli DMSO reductase. The crystal structure of the enzyme is not solved so far. Cytochromes were not identified to be present in DmsC, but DmsC has a predicted menachinol binding site. The arrangement of the FeS clusters is similar to formate dehydrogenase, forming the FS4-FS3-FS2-FS1-FS0-bis-MGD electron transfer chain. (D) Localization of enzymes of the xanthine oxidase family. PaoABC is localized in the periplasm and detoxifies aromatic aldehydes. XdhABC is the cytoplasmic xanthine dehydrogenase using NAD+ as terminal electron acceptor. Since structures of these enzymes are not available so far, the arrangement of the cofactors of R. capsulatus xanthine dehydrogenase are shown, which have a similar arrangement as the cofactors found in PaoABC or XdhABC.
The X-ray structure of NarGHI showed that the protein forms a (αβγ)2 heterotrimer [92,93]. The NarG subunit faces the cytoplasm and binds the bis-MGD cofactor in addition to an [4Fe4S] cluster (Figure 9). The bis-MGD cofactor contains an oxo group and an aspartate from the protein backbone as additional ligands. The [4Fe4S] cluster is coordinated by 3 cysteines and one histidine, which increases the redox potential in comparison to other proteins containing the [4Fe4S] cluster in the same organization in proximity to the bis-MGD cofactor. Replacement of this histidine to serine of this so-called FS0 cluster results in loss of enzymatic activity, which shows the importance of this cluster in the electron transfer reaction [94]. NarH contains four FeS clusters in total, three [4Fe4S] clusters and one distal [3Fe4S] cluster, which is coordinated by three cysteines. The homologous subunits FdnH, FdoH or DmsB contain four [4Fe4S] in comparison. Finally, the cytoplasmically exposed NarGH subunits bind to the membrane-integral subunit NarI, which binds two b-type cytochromes [95]. The electron flow occurs from menaquinol in the direction to the hemes of NarI to the [3Fe4S] FS4 cluster and the three [4Fe4S] FS3, FS2, FS1 clusters in NarH further via FS0 in NarG to the bis-MGD cofactor (Figure 9). The specific chaperone for NarGHI assembly is the NarJ protein, the function of which has been intensively studies [30,96,97]. NarJ was shown to mediate a chaperone-assisted quality control for the assembly of NarGHI, thereby preventing membrane anchoring of the soluble and cytoplasmic NarGH complex before all maturation events have been completed [98]. It was shown that FS0 insertion must precede bis-MGD insertion into NarG [99,100], a process which was confirmed by X-ray structural analysis of specific mutants of the FS0 coordination sphere [91,94]. In the absence of NarJ, a defect in cofactor insertion into NarG was observed, while the proximal heme b of NarI is additionally absent due to loss of coordination between maturation of the NarI and NarGH components [101]. Thus, NarJ ensures complete maturation of the b-type cytochrome NarI by proper timing of membrane anchoring of the cytoplasmic NarGH complex after the concerted cofactor insertion into the latter [102].
A third nitrate reductase in E. coli is NarZYV, which is highly homologous to NarGHI in its subunit composition. The operon contains the narZYWV genes and is expressed during the early stationary phase independent of the presence of nitrate [84].
5.2 Formate dehydrogenases
E. coli harbors three different molybdenum containing formate dehydrogenases which are members of the DMSO reductase family; FdhF, FdnGHI and FdoGHI. Formate dehydrogenases catalyze the conversion of formate to CO2 by concomitant release of H+ and 2e−. Formate dehydrogenase H (FdhF) is part of the hydrogen lyase complex [103,104,105]. The FdhF subunit of formate dehydrogenase H harbors the bis-MGD cofactor in addition to a [4Fe4S] cluster, designated as FS0 [106]. FdhF contains the bis-MGD cofactor with a sulfido group and selenocysteine as additional ligands [107]. The electrons released from substrate oxidation are transferred via the [4Fe4S] cluster further to the hydrogenases Hyd3 or Hyd4, depending on the growth conditions, to produce H2 [108] (Figure 9).
E. coli contains two structurally related but differentially expressed respiratory formate dehydrogenases: formate dehydrogenase-O and formate dehydrogenase-N [109]. FdhGHI is a component of the nitrate respiratory pathway, where, under anaerobic conditions, formate oxidation is coupled to nitrate reduction (NarG) via lipid-soluble quinone [109]. FdoGHI and NarZYV are the corresponding isoenzymes that are additional present under aerobic conditions to ensure rapid adaptation during a shift from aerobiosis to anaerobiosis [110]. E. coli formate dehydrogenase N and O are organized as an (αβγ)3 trimer which faces the periplasm [111]. The γ subunit of each formate dehydrogenase harbors the bis-MGD cofactor which is coordinated by a selenocysteine and likely an additional sulfido ligand. Additionally, the subunit also contains the FS0 [4Fe4S] cluster similar to FdhF or NarG. Both FdnH and FdoH contain four [4Fe4S] clusters which are arranged as FS1-FS2-FS3-FS4 (Figure 9), an arrangement which is also present in NarH and other systems, including the [NiFe] hydrogenases and NADH dehydrogenase [2]. The FdnI and FdoI subunits are membrane proteins that bind two b-type cytochromes. While the X-ray structure of FdnGHI has been solved, the structure of FdoGHI is not available so far. Both FdnG and FdoG contain the Tat-leader peptide for the translocation to the periplasm in conjunction with the FdnH and FdoH subunits, respectively. While for bis-MGD insertion into FDHs in E. coli the FdhD protein is involved [91], the specific insertion of the FeS clusters into the α and β subunits is not completely understood so far.
In R. capsulatus, one FDH is present in the cytoplasm [112]. The physiological role of the enzyme for R. capsulatus has not been resolved to date. The enzyme is composed of a (αβγ)2 heterotrimer encoded by fdsGBA, five [Fe4S4] clusters two [Fe2S2] cluster, FMN and sulfurated bis-MGD as prosthetic groups in total. The terminal electron acceptor for formate oxidation is specifically NAD+ (NADP+ is not used), which accepts the two electrons from the FMN cofactor. Since in the absence of the chaperones FdsC and FdsD during expression, a heterotrimeric enzyme lacking Moco but containing the set of [FeS] clusters and FMN is formed, it is believed that bis-MGD is inserted into the enzyme after the insertion of the other cofactors into the heterotrimer [113]. So far, the FeS clusters have not been further characterized in R. capsulatus FDH and nothing is known about the proteins that specifically insert the FeS clusters or FMN into FdsGBA.
5.3 DMSO reductase
The DMSO reductase from E. coli is a αβγ heterotrimer that is located in the periplasm [114] (Figure 9). There is no X-ray structure available for the E. coli enzyme so far. The DmsA subunit harbors the bis-MGD cofactor and [4Fe4S] cluster, which has been designated as FS0 [115,116]. The DmsB subunit contains four [4Fe4S] clusters. DmsC is the integral transmembrane subunit with 8 transmembrane helices, which does not contain any redox-active cofactors [117]. A menaquinole-binding site has been identified on DmsC, which serves an electron donor for substrate reduction [118]. E. coli DMSO reductase has a broad substrate specificity, being able to reduce S- and N-oxides. The molybdenum active site is composed of the bis-MGD cofactor with an oxo-ligand and a serine ligand from the protein backbone. The presence of the FS0 cluster in the DmsA subunit is based on amino acid sequence homologies to NarG and FdhF [2]. Both proteins contain a [4Fe4S] cluster in proximity to the bis-MGD cofactor, which is coordinated by Cys34, Cys38, Cys42 and Cys75 in DmsA [115]. The four [4Fe4S] clusters in DmsB were designated as FS1-4 [119,120]. The reduction potentials were determined for the four clusters and on the basis of crystal structures of similar proteins, they are expected to be arranged as FS1-FS2-FS3-FS4, with the potentials of −240 mV, −330 mM, −120 mV and − 50 mV, respectively. The Mo(VI/V) and Mo(V/IV) potentials are in the range of +0 mV and − 140 mV, thus, the clusters are not arranged in thermodynamic order for electron transfer from the menaquinone via FS4, FS3, FS2, FS1, FS0 to the molybdenum center. However, it has been suggested that the clusters are in close proximity enabling rapid electron transfer during catalysis. The DmsA subunit contains the Tat-leader sequence for the transport to the periplasm and both DmsA and DmsB are cotransported after maturation in the cytoplasm [121]. The chaperone for bis-MGD insertion into DmsA was shown to be the DmsD protein [121,122]. DmsD thereby is shared by DmsABC and YnfE/F for maturation [32,123]. So far, it has not been investigated how the FeS clusters are inserted into DmsA or DmsB.
5.4 Periplasmic aldehyde oxidoreductase and xanthine dehydrogenase
Among the enzymes of the xanthine oxidase family in E. coli, only the periplasmic aldehyde oxidoreductase has been characterized in detail so far [124]. The paoABCD operon encodes for a molybdenum-containing iron–sulfur flavoprotein which is located in the periplasm (Figure 9). The 135 kDa enzyme comprises a noncovalent (αβγ) heterotrimer with a large (78.1 kDa) molybdenum cofactor (Moco)-containing PaoC subunit, a medium (33.9 kDa) FAD-containing PaoB subunit, and a small (21.0 kDa) 2x[2Fe2S]-containing PaoA subunit, which also contains the Tat-leader peptide for the localization to the periplasm [125]. PaoD is not a subunit of the mature enzyme, and the protein was shown to bind Moco which likely is inserted into PaoC after sulfuration on PaoD [126]. Analysis of the form of Moco present in PaoABC revealed the presence of the MCD cofactor [9]. Kinetic characterization of the enzyme showed that PaoABC converts a broad spectrum of aldehydes, with a preference for aromatic aldehydes [124]. The physiologic electron acceptor in this reaction is not completely clear to date. Complete growth inhibition of E. coli cells devoid of genes from the paoABC operon was observed by the addition of cinnamaldehyde to a low-pH medium. This finding showed that PaoABC might have a role in the detoxification of aromatic aldehydes for E. coli under certain growth conditions. In the absence of Moco, the enzyme was shown to be instable. Unlike other enzymes of the xanthine oxidase family, the heterotrimeric αβγ enzyme does not dimerize via its molybdenum-containing PaoC domain which was been confirmed by SAXS studies [125]. Studies about the order of cofactor insertion of MCD, FAD and the 2x[2Fe2S] clusters into PaoABC were not performed so far, however it is believed that MCD insertion is the last step of PaoABC maturation before its translocation to the periplasm [74]. Like for other molybdoenzymes, studies on the insertion of FeS clusters and the specific protein involved in this reaction were not performed on these enzymes so far.
A well studied enzyme from the xanthine oxidase family is R. capsulatus xanthine dehydrogenase [127,128]. The active form of the enzyme is a (αβ)2 herterodimer encoded by the xdhAB genes, with each of the (aβ) dimers containing two non identical [2Fe2S] clusters, FAD and the sulfurated form of Mo-MPT as catalytically acting units [129]. The oxidation of xanthine takes place at the molybdenum center and the electrons thus introduced are rapidly distributed to the other centers according to their relative redox potentials [130]. The re-oxidation of the reduced enzyme by the oxidant substrate NAD+ occurs through FAD. The two [2Fe2S] clusters (FeSI and FeSII) are indistinguishable in terms of their absorption spectra, but the midpoint redox potential of FeSII is generally more positive than that of the FeSI center (Figure 11). The insertion and assembly of R. capsulatus XDH has been studied and it was suggested that insertion of the three different redox centers is a complex process that occurs in an ordered manner (Fig. 8) [81]. The assembly involves the synthesis of the XdhA and XdhB subunits, the dimerization of both subunits, the insertion of FeSI, FeSII and FAD into the XdhA subunit, dimerization of two (αβ) dimers via the XdhB subunit and finally, insertion of sulfurated Moco by XdhC into XdhB (Fig. 8). The dimerization of the (αβ) subunits is required to stabilize a structure of the protein which makes the protein suitable for Moco insertion. The step of Moco insertion is strictly regulated, since in vivo di-oxo Moco is not inserted into R. capsulatus XDH. Thus XdhC has to perform two “quality control” steps: (i) to ensure that Moco is sulfurated by the interaction with the L-cysteine desulfurase NifS4 before insertion into XDH, and (ii) to control that XDH is correctly assembled as an (αβ)2 heteroteramer before sulfurated Moco is inserted [74,81]. Since Moco is deeply buried in the protein it is also believed that XdhC is involved in proper folding of XDH after Moco insertion. So far the proteins that insert specifically the FeS clusters or FAD into XDH were not identified in R. capsulatus.
6. Insertion of FeS clusters into molybdoenzymes
As revealed above, the link between FeS cluster biosynthesis and insertion and Moco biosynthesis for the production of active molybdoenzymes is only at the beginning to be understood. The insertion of the molybdenum cofactor into molybdoenzymes has been studied in detail. Since the majority of molybdoenzymes in E. coli are membrane associated and the majority of the enzymes are facing the periplasm, the maturation and transport of molybdoenzymes is linked to the Tat system. Here, the transport of enzymes occurs in the folded state after the insertion of the Moco and FeS clusters. The “quality” control step is performed by the molecular chaperones which bind and insert the mature form of the molybdenum cofactor and in addition, protect the Tat signal peptide. After the final folding of the enzyme, which contains the full complement of cofactors, the enzyme is directed to the Tat-translocon and is exported to the periplasm. This implies, that in addition to Moco, the FeS clusters have to be present before the translocation of the molybdoenzyme. For FeS cluster insertion, several proteins, designated as A-type carriers (ATC) are involved [131]. These proteins, which are SufA, IscA or ErpA in E. coli carry and insert the FeS clusters to designated target proteins. Recent reports showed that ErpA is essential for the formation of an active formate-nitrate reductase complex in E. coli [132]. It was shown that in an E. coli erpA mutant both FdnGHI and NarGHI were inactive. In both enzymes, the FeS cluster-containing H-subunit was missing. In addition, FdnG was not correctly translocated and still contained the Tat signal sequence. IscA was able to partially complement the erpA mutant, showing that these proteins might have overlapping roles, but ErpA seems to be the more specific enzyme for nitrate reductase and formate reductase maturation [132]. However, an aspect which was missing in this study is the overlapping effects of FeS cluster biosynthesis and insertion and Moco biosynthesis. Since MoaA contains FeS clusters, the effect of the erpA mutant might have been already at the level of MoaA, leading to a lack of Moco in the cell. Thus, active nitrate reductase of formate dehydrogenase will not be produced, and bis-MGD is not inserted into FdnG or NarG. This might explain the level of unmatured FdnG and NarG, still containing the Tatleader in the erpA mutant. Additionally, ErpA might be specific for FeS cluster insertion into the H-subunit of these enzymes. Thus, it is difficult to dissect the effects of Moco biosynthesis and FeS cluster insertion. More detailed studies are necessary to overcome the overlapping effects of both pathways.
Highlights.
-
-
the link between the biosynthesis of the molybdenum cofactor and Fes clusters
-
-
a mechanism for MoaA is proposed, where 4Fe-4S clusters play central roles
-
-
IscS transfers the sulfur for the biosynthesis of the dithiolene group in Moco
-
-
the biosynthesis of sulfur-containing cofactors influence each other by the availability of IscS
-
-
the insertion of FeS clusters into molybdoenzymes is still unknown
Acknowledgments
The authors thanks all current and former members of the research group in addition to collaboration partners who were involved in the work over the past years. Special thanks goes to K.V. Rajagopalan, the founder of the field of Moco biosynthesis for his support over the years and the helpful discussions. The work was mainly supported by continuous grants of the Deutsche Forschungsgemeinschaft (to S.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rajagopalan KV. Biosynthesis of the molybdenum cofactor. In: Neidhardt FC, editor. Escherichia coli and Salmonella Cellular and Molecular Biology. Washington, DC: ASM Press; 1996. pp. 674–679. [Google Scholar]
- 2.Hille R, Hall J, Basu P. The Mononuclear Molybdenum Enzymes. Chemical reviews. 2014 doi: 10.1021/cr400443z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leimkühler S, Wuebbens MM, Rajagopalan KV. The History of the Discovery of the Molybdenum Cofactor and Novel Aspects of its Biosynthesis in Bacteria. Coordination chemistry reviews. 2011;255:1129–1144. doi: 10.1016/j.ccr.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wuebbens MM, Rajagopalan KV. Structural characterization of a molybdopterin precursor. J Biol Chem. 1993;268:13493–13498. [PubMed] [Google Scholar]
- 5.Pitterle DM, Johnson JL, Rajagopalan KV. In vitro synthesis of molybdopterin from precursor Z using purified converting factor. Role of protein-bound sulfur in formation of the dithiolene. J Biol Chem. 1993;268:13506–13509. [PubMed] [Google Scholar]
- 6.Joshi MS, Johnson JL, Rajagopalan KV. Molybdenum cofactor biosynthesis in Escherichia coli mod and mog mutants. J Bacteriol. 1996;178:4310–4312. doi: 10.1128/jb.178.14.4310-4312.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson JL, Bastian NR, Rajagopalan KV. Molybdopterin guanine dinucleotide: a modified form of molybdopterin identified in the molybdenum cofactor of dimethyl sulfoxide reductase from Rhodobacter sphaeroides forma specialis denitrificans. Proc Natl Acad Sci U S A. 1990;87:3190–3194. doi: 10.1073/pnas.87.8.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyer O, Rajagopalan KV. Molybdopterin in carbon monoxide oxidase from carboxydotrophic bacteria. J Bacteriol. 1984;157:643–648. doi: 10.1128/jb.157.2.643-648.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neumann M, Mittelstädt G, Seduk F, Iobbi-Nivol C, Leimkühler S. MocA is a specific cytidylyltransferase involved in molybdopterin cytosine dinucleotide biosynthesis in Escherichia coli. The Journal of biological chemistry. 2009;284:21891–21898. doi: 10.1074/jbc.M109.008565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajagopalan KV, Johnson JL. The pterin molybdenum cofactors. J Biol Chem. 1992;267:10199–10202. [PubMed] [Google Scholar]
- 11.Santamaria-Araujo JA, Fischer B, Otte T, Nimtz M, Mendel RR, et al. The tetrahydropyranopterin structure of the sulfur- and metal-free molybdenum cofactor precursor. J Biol Chem. 2004;279:15994–15999. doi: 10.1074/jbc.M311815200. [DOI] [PubMed] [Google Scholar]
- 12.Wuebbens MM, Rajagopalan KV. Investigation of the early steps of molybdopterin biosynthesis in Escherichia coli through the use of in vivo labeling studies. J Biol Chem. 1995;270:1082–1087. doi: 10.1074/jbc.270.3.1082. [DOI] [PubMed] [Google Scholar]
- 13.Hover BM, Loksztejn A, Ribeiro AA, Yokoyama K. Identification of a cyclic nucleotide as a cryptic intermediate in molybdenum cofactor biosynthesis. Journal of the American Chemical Society. 2013;135:7019–7032. doi: 10.1021/ja401781t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daniels JN, Wuebbens MM, Rajagopalan KV, Schindelin H. Crystal Structure of a Molybdopterin Synthase-Precursor Z Complex: Insight into Its Sulfur Transfer Mechanism and Its Role in Molybdenum Cofactor Deficiency(,) Biochemistry. 2008;47:615–626. doi: 10.1021/bi701734g. [DOI] [PubMed] [Google Scholar]
- 15.Johnson JL, Wuebbens MM, Rajagopalan KV. The structure of a molybdopterin precursor. Characterization of a stable, oxidized derivative. J Biol Chem. 1989;264:13440–13447. [PubMed] [Google Scholar]
- 16.Clinch K, Watt DK, Dixon RA, Baars SM, Gainsford GJ, et al. Synthesis of cyclic pyranopterin monophosphate, a biosynthetic intermediate in the molybdenum cofactor pathway. Journal of medicinal chemistry. 2013;56:1730–1738. doi: 10.1021/jm301855r. [DOI] [PubMed] [Google Scholar]
- 17.Pitterle DM, Johnson JL, Rajagopalan KV. Molybdopterin formation by converting factor of E. coli chlA1. FASEB J. 1990;4:A1957. [Google Scholar]
- 18.Pitterle DM, Rajagopalan KV. Two proteins encoded at the chlA locus constitute the converting factor of Escherichia coli chlA1. J Bacteriol. 1989;171:3373–3378. doi: 10.1128/jb.171.6.3373-3378.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pitterle DM, Rajagopalan KV. Purification and characterization of the converting factor from E. coli chlA1. FASEB J. 1991;5:A468. [Google Scholar]
- 20.Pitterle DM, Rajagopalan KV. The biosynthesis of molybdopterin in Escherichia coli. Purification and characterization of the converting factor. J Biol Chem. 1993;268:13499–13505. [PubMed] [Google Scholar]
- 21.Rudolph MJ, Wuebbens MM, Rajagopalan KV, Schindelin H. Crystal structure of molybdopterin synthase and its evolutionary relationship to ubiquitin activation. Nat Struct Biol. 2001;8:42–46. doi: 10.1038/83034. [DOI] [PubMed] [Google Scholar]
- 22.Gutzke G, Fischer B, Mendel RR, Schwarz G. Thiocarboxylation of molybdopterin synthase provides evidence for the mechanism of dithiolene formation in metal-binding pterins. J Biol Chem. 2001;276:36268–36274. doi: 10.1074/jbc.M105321200. [DOI] [PubMed] [Google Scholar]
- 23.Leimkühler S, Freuer A, Araujo JA, Rajagopalan KV, Mendel RR. Mechanistic studies of human molybdopterin synthase reaction and characterization of mutants identified in group B patients of molybdenum cofactor deficiency. The Journal of biological chemistry. 2003;278:26127–26134. doi: 10.1074/jbc.M303092200. [DOI] [PubMed] [Google Scholar]
- 24.Wuebbens MM, Rajagopalan KV. Mechanistic and mutational studies of Escherichia coli molybdopterin synthase clarify the final step of molybdopterin biosynthesis. J Biol Chem. 2003;278:14523–14532. doi: 10.1074/jbc.M300453200. [DOI] [PubMed] [Google Scholar]
- 25.Nichols J, Rajagopalan KV. Escherichia coli MoeA and MogA. Function in metal incorporation step of molybdenum cofactor biosynthesis. J Biol Chem. 2002;277:24995–25000. doi: 10.1074/jbc.M203238200. [DOI] [PubMed] [Google Scholar]
- 26.Nichols JD, Rajagopalan KV. In vitro molybdenum ligation to molybdopterin using purified components. J Biol Chem. 2005;280:7817–7822. doi: 10.1074/jbc.M413783200. [DOI] [PubMed] [Google Scholar]
- 27.Kuper J, Llamas A, Hecht HJ, Mendel RR, Schwarz G. Structure of the molybdopterin-bound Cnx1G domain links molybdenum and copper metabolism. Nature. 2004;430:803–806. doi: 10.1038/nature02681. [DOI] [PubMed] [Google Scholar]
- 28.Reschke S, Sigfridsson KG, Kaufmann P, Leidel N, Horn S, et al. Identification of a Bis-molybdopterin Intermediate in Molybdenum Cofactor Biosynthesis in Escherichia coli. The Journal of biological chemistry. 2013;288:29736–29745. doi: 10.1074/jbc.M113.497453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Temple CA, Rajagopalan KV. Mechanism of assembly of the Bis(Molybdopterin guanine Dinucleotide)Molybdenum cofactor in Rhodobacter sphaeroides dimethyl sulfoxide reductase. J Biol Chem. 2000;275:40202–40210. doi: 10.1074/jbc.M007407200. [DOI] [PubMed] [Google Scholar]
- 30.Palmer T, Santini C-L, Iobbi-Nivol C, Eaves DJ, Boxer DH, et al. Involvement of the narJ and mob gene products in the biosynthesis of the molybdoenzyme nitrate reductase in Escherichia coli. Mol Microbiol. 1996;20:875–884. doi: 10.1111/j.1365-2958.1996.tb02525.x. [DOI] [PubMed] [Google Scholar]
- 31.Lake MW, Temple CA, Rajagopalan KV, Schindelin H. The crystal structure of the Escherichia coli MobA protein provides insight into molybdopterin guanine dinucleotide biosynthesis. J Biol Chem. 2000;275:40211–40217. doi: 10.1074/jbc.M007406200. [DOI] [PubMed] [Google Scholar]
- 32.Iobbi-Nivol C, Leimkühler S. Molybdenum enzymes, their maturation and molybdenum cofactor biosynthesis in Escherichia coli. Biochimica et biophysica acta. 2013;1827:1086–1101. doi: 10.1016/j.bbabio.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 33.Neumann M, Seduk F, Iobbi-Nivol C, Leimkühler S. Molybdopterin dinucleotide biosynthesis in Escherichia coli: identification of amino acid residues of molybdopterin dinucleotide transferases that determine specificity for binding of guanine or cytosine nucleotides. The Journal of biological chemistry. 2011;286:1400–1408. doi: 10.1074/jbc.M110.155671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hänzelmann P, Hernandez HL, Menzel C, Garcia-Serres R, Huynh BH, et al. Characterization of MOCS1A, an oxygen-sensitive iron-sulfur protein involved in human molybdenum cofactor biosynthesis. The Journal of biological chemistry. 2004;279:34721–34732. doi: 10.1074/jbc.M313398200. [DOI] [PubMed] [Google Scholar]
- 36.Frey PA, Hegeman AD, Ruzicka FJ. The Radical SAM Superfamily. Critical reviews in biochemistry and molecular biology. 2008;43:63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- 37.Hänzelmann P, Schindelin H. Binding of 5′-GTP to the C-terminal FeS cluster of the radical S-adenosylmethionine enzyme MoaA provides insights into its mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:6829–6834. doi: 10.1073/pnas.0510711103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hänzelmann P, Schindelin H. Crystal structure of the S-adenosylmethionine-dependent enzyme MoaA and its implications for molybdenum cofactor deficiency in humans. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:12870–12875. doi: 10.1073/pnas.0404624101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lees NS, Hänzelmann P, Hernandez HL, Subramanian S, Schindelin H, et al. ENDOR spectroscopy shows that guanine N1 binds to [4Fe-4S] cluster II of the S-adenosylmethionine-dependent enzyme MoaA: mechanistic implications. Journal of the American Chemical Society. 2009;131:9184–9185. doi: 10.1021/ja903978u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hänzelmann P, Schwarz G, Mendel RR. Functionality of alternative splice forms of the first enzymes involved in human molybdenum cofactor biosynthesis. The Journal of biological chemistry. 2002;277:18303–18312. doi: 10.1074/jbc.M200947200. [DOI] [PubMed] [Google Scholar]
- 41.Mehta AP, Abdelwahed SH, Xu H, Begley TP. Molybdopterin Biosynthesis: Trapping of Intermediates for the MoaA-Catalyzed Reaction Using 2′-DeoxyGTP and 2′-ChloroGTP as Substrate Analogues. Journal of the American Chemical Society. 2014 doi: 10.1021/ja502663k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mehta AP, Hanes JW, Abdelwahed SH, Hilmey DG, Hänzelmann P, et al. Catalysis of a New Ribose Carbon-Insertion Reaction by the Molybdenum Cofactor Biosynthetic Enzyme MoaA. Biochemistry. 2013;52:1134–1136. doi: 10.1021/bi3016026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mehta AP, Abdelwahed SH, Begley TP. Molybdopterin biosynthesis: trapping an unusual purine ribose adduct in the MoaA-catalyzed reaction. Journal of the American Chemical Society. 2013;135:10883–10885. doi: 10.1021/ja4041048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frey PA, Magnusson OT. S-Adenosylmethionine: a wolf in sheep’s clothing, or a rich man’s adenosylcobalamin? Chemical reviews. 2003;103:2129–2148. doi: 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- 45.Dong M, Su X, Dzikovski B, Dando EE, Zhu X, et al. Dph3 is an electron donor for Dph1-Dph2 in the first step of eukaryotic diphthamide biosynthesis. Journal of the American Chemical Society. 2014;136:1754–1757. doi: 10.1021/ja4118957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goldman PJ, Grove TL, Sites LA, McLaughlin MI, Booker SJ, et al. X-ray structure of an AdoMet radical activase reveals an anaerobic solution for formylglycine posttranslational modification. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:8519–8524. doi: 10.1073/pnas.1302417110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grove TL, Ahlum JH, Qin RM, Lanz ND, Radle MI, et al. Further Characterization of Cys-Type and Ser-Type Anaerobic Sulfatase Maturating Enzymes Suggests a Commonality in the Mechanism of Catalysis. Biochemistry. 2013;52:2874–2887. doi: 10.1021/bi400136u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leimkühler S, Wuebbens MM, Rajagopalan KV. Characterization of Escherichia coli MoeB and its involvement in the activation of molybdopterin synthase for the biosynthesis of the molybdenum cofactor. The Journal of biological chemistry. 2001;276:34695–34701. doi: 10.1074/jbc.M102787200. [DOI] [PubMed] [Google Scholar]
- 49.Schindelin H. Evolutionary Origin of the Activation Step During Ubiquitin-dependent Protein Degradation. In: Mayer RJ, Ciechanover A, Rechsteiner M, editors. Protein Degradation: Ubiquitin and the Chemistry of Life. Weinheim: WILEY-VCH; 2005. pp. 21–43. [Google Scholar]
- 50.Lake MW, Wuebbens MM, Rajagopalan KV, Schindelin H. Mechanism of ubiquitin activation revealed by the structure of a bacterial MoeB-MoaD complex. Nature. 2001;414:325–329. doi: 10.1038/35104586. [DOI] [PubMed] [Google Scholar]
- 51.Schmitz J, Wuebbens MM, Rajagopalan KV, Leimkühler S. Role of the C-Terminal Gly-Gly Motif of Escherichia Coli MoaD, a Molybdenum Cofactor Biosynthesis Protein with a Ubiquitin Fold. Biochemistry. 2007;46:909–916. doi: 10.1021/bi062011w. [DOI] [PubMed] [Google Scholar]
- 52.Tong Y, Wuebbens MM, Rajagopalan KV, Fitzgerald MC. Thermodynamic analysis of subunit interactions in Escherichia coli molybdopterin synthase. Biochemistry. 2005;44:2595–2601. doi: 10.1021/bi047762h. [DOI] [PubMed] [Google Scholar]
- 53.Leimkühler S, Rajagopalan KV. An Escherichia coli NifS-like sulfurtransferase is required for the transfer of cysteine sulfur in the in vitro synthesis of molybdopterin from precursor Z. J Biol Chem. 2001;276:22024–22031. doi: 10.1074/jbc.M102072200. [DOI] [PubMed] [Google Scholar]
- 54.Zheng L, White RH, Cash VL, Dean DR. Mechanism for the desulfurization of L-cysteine catalyzed by the nifS gene product. Biochemistry. 1994;33:4714–4720. doi: 10.1021/bi00181a031. [DOI] [PubMed] [Google Scholar]
- 55.Hidese R, Mihara H, Esaki N. Bacterial cysteine desulfurases: versatile key players in biosynthetic pathways of sulfur-containing biofactors. Applied microbiology and biotechnology. 2011;91:47–61. doi: 10.1007/s00253-011-3336-x. [DOI] [PubMed] [Google Scholar]
- 56.Nakamura M, Saeki K, Takahashi Y. Hyperproduction of recombinant ferredoxins in escherichia coli by coexpression of the ORF1-ORF2-iscS-iscU-iscA-hscB-hs cA-fdx-ORF3 gene cluster. J Biochem (Tokyo) 1999;126:10–18. doi: 10.1093/oxfordjournals.jbchem.a022409. [DOI] [PubMed] [Google Scholar]
- 57.Cupp-Vickery JR, Urbina H, Vickery LE. Crystal structure of IscS, a cysteine desulfurase from Escherichia coli. Journal of molecular biology. 2003;330:1049–1059. doi: 10.1016/s0022-2836(03)00690-9. [DOI] [PubMed] [Google Scholar]
- 58.Mihara H, Kurihara T, Yoshimura T, Soda K, Esaki N. Cysteine sulfinate desulfinase, a NIFS-like protein of Escherichia coli with selenocysteine lyase and cysteine desulfurase activities. Gene cloning, purification, and characterization of a novel pyridoxal enzyme. J Biol Chem. 1997;272:22417–22424. doi: 10.1074/jbc.272.36.22417. [DOI] [PubMed] [Google Scholar]
- 59.Mihara H, Maeda M, Fujii T, Kurihara T, Hata Y, et al. A nifS-like gene, csdB, encodes an Escherichia coli counterpart of mammalian selenocysteine lyase. Gene cloning, purification, characterization and preliminary x-ray crystallographic studies. J Biol Chem. 1999;274:14768–14772. doi: 10.1074/jbc.274.21.14768. [DOI] [PubMed] [Google Scholar]
- 60.Yuvaniyama P, Agar JN, Cash VL, Johnson MK, Dean DR. NifS-directed assembly of a transient [2Fe-2S] cluster within the NifU protein. Proc Natl Acad Sci U S A. 2000;97:599–604. doi: 10.1073/pnas.97.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zheng L, Cash VL, Flint DH, Dean DR. Assembly of iron-sulfur clusters. Identification of an iscSUA-hscBA-fdx gene cluster from Azotobacter vinelandii. J Biol Chem. 1998;273:13264–13272. doi: 10.1074/jbc.273.21.13264. [DOI] [PubMed] [Google Scholar]
- 62.Marinoni EN, de Oliveira JS, Nicolet Y, Raulfs EC, Amara P, et al. (IscS-IscU)2 complex structures provide insights into Fe2S2 biogenesis and transfer. Angewandte Chemie. 2012;51:5439–5442. doi: 10.1002/anie.201201708. [DOI] [PubMed] [Google Scholar]
- 63.Yamanaka Y, Zeppieri L, Nicolet Y, Marinoni EN, de Oliveira JS, et al. Crystal structure and functional studies of an unusual L-cysteine desulfurase from Archaeoglobus fulgidus. Dalton transactions. 2013;42:3092–3099. doi: 10.1039/c2dt32101g. [DOI] [PubMed] [Google Scholar]
- 64.Shi R, Proteau A, Villarroya M, Moukadiri I, Zhang L, et al. Structural basis for Fe-S cluster assembly and tRNA thiolation mediated by IscS protein-protein interactions. PLoS biology. 2010;8:e1000354. doi: 10.1371/journal.pbio.1000354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dahl JU, Radon C, Bühning M, Nimtz M, Leichert LI, et al. The Sulfur Carrier Protein TusA Has a Pleiotropic Role in Escherichia coli That Also Affects Molybdenum Cofactor Biosynthesis. The Journal of biological chemistry. 2013;288:5426–5442. doi: 10.1074/jbc.M112.431569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dahl JU, Urban A, Bolte A, Sriyabhaya P, Donahue JL, et al. The identification of a novel protein involved in molybdenum cofactor biosynthesis in Escherichia coli. The Journal of biological chemistry. 2011;286:35801–35812. doi: 10.1074/jbc.M111.282368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ikeuchi Y, Shigi N, Kato J, Nishimura A, Suzuki T. Mechanistic insights into sulfur relay by multiple sulfur mediators involved in thiouridine biosynthesis at tRNA wobble positions. Molecular cell. 2006;21:97–108. doi: 10.1016/j.molcel.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 68.Numata T, Ikeuchi Y, Fukai S, Suzuki T, Nureki O. Snapshots of tRNA sulphuration via an adenylated intermediate. Nature. 2006;442:419–424. doi: 10.1038/nature04896. [DOI] [PubMed] [Google Scholar]
- 69.Giel JL, Nesbit AD, Mettert EL, Fleischhacker AS, Wanta BT, et al. Regulation of iron-sulphur cluster homeostasis through transcriptional control of the Isc pathway by [2Fe-2S]-IscR in Escherichia coli. Molecular microbiology. 2013;87:478–492. doi: 10.1111/mmi.12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Unden G, Schirawski J. The oxygen-responsive transcriptional regulator FNR of Escherichia coli: the search for signals and reactions. Mol Microbiol. 1997;25:205–210. doi: 10.1046/j.1365-2958.1997.4731841.x. [DOI] [PubMed] [Google Scholar]
- 71.Thome R, Gust A, Toci R, Mendel R, Bittner F, et al. A sulfurtransferase is essential for activity of formate dehydrogenases in Escherichia coli. The Journal of biological chemistry. 2012;287:4671–4678. doi: 10.1074/jbc.M111.327122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Raaijmakers HC, Romao MJ. Formate-reduced E. coli formate dehydrogenase H: The reinterpretation of the crystal structure suggests a new reaction mechanism. Journal of biological inorganic chemistry: JBIC: a publication of the Society of Biological Inorganic Chemistry. 2006;11:849–854. doi: 10.1007/s00775-006-0129-2. [DOI] [PubMed] [Google Scholar]
- 73.Coelho C, Gonzalez PJ, Moura JG, Moura I, Trincao J, et al. The crystal structure of Cupriavidus necator nitrate reductase in oxidized and partially reduced states. Journal of molecular biology. 2011;408:932–948. doi: 10.1016/j.jmb.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 74.Neumann M, Leimkühler S. The role of system-specific molecular chaperones in the maturation of molybdoenzymes in bacteria. Biochemistry research international. 2011;2011:850924. doi: 10.1155/2011/850924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Neumann M, Stöcklein W, Walburger A, Magalon A, Leimkühler S. Identification of a Rhodobacter capsulatus L-cysteine desulfurase that sulfurates the molybdenum cofactor when bound to XdhC and before its insertion into xanthine dehydrogenase. Biochemistry. 2007;46:9586–9595. doi: 10.1021/bi700630p. [DOI] [PubMed] [Google Scholar]
- 76.Neumann M, Stöcklein W, Leimkühler S. Transfer of the Molybdenum Cofactor Synthesized by Rhodobacter capsulatus MoeA to XdhC and MobA. J Biol Chem. 2007;282:28493–28500. doi: 10.1074/jbc.M704020200. [DOI] [PubMed] [Google Scholar]
- 77.Kisker C, Schindelin H, Rees DC. Molybdenum-cofactor-containing enzymes: structure and mechanism. Ann Rev Biochem. 1997;66:233–267. doi: 10.1146/annurev.biochem.66.1.233. [DOI] [PubMed] [Google Scholar]
- 78.Genest O, Mejean V, Iobbi-Nivol C. Multiple roles of TorD-like chaperones in the biogenesis of molybdoenzymes. FEMS microbiology letters. 2009;297:1–9. doi: 10.1111/j.1574-6968.2009.01660.x. [DOI] [PubMed] [Google Scholar]
- 79.Jack RL, Buchanan G, Dubini A, Hatzixanthis K, Palmer T, et al. Coordinating assembly and export of complex bacterial proteins. Embo J. 2004;23:3962–3972. doi: 10.1038/sj.emboj.7600409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schulze RJ, Komar J, Botte M, Allen WJ, Whitehouse S, et al. Membrane protein insertion and proton-motive-force-dependent secretion through the bacterial holo-translocon SecYEG-SecDF-YajC-YidC. Proceedings of the National Academy of Sciences of the United States of America. 2014 doi: 10.1073/pnas.1315901111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schumann S, Saggu M, Möller N, Anker SD, Lendzian F, et al. The mechanism of assembly and cofactor insertion into Rhodobacter capsulatus xanthine dehydrogenase. The Journal of biological chemistry. 2008;283:16602–16611. doi: 10.1074/jbc.M709894200. [DOI] [PubMed] [Google Scholar]
- 82.Stewart V. Nitrate respiration in relation to facultative metabolism in enterobacteria. Microbiol Rev. 1988;52:190–232. doi: 10.1128/mr.52.2.190-232.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brondijk TH, Nilavongse A, Filenko N, Richardson DJ, Cole JA. NapGH components of the periplasmic nitrate reductase of Escherichia coli K-12: location, topology and physiological roles in quinol oxidation and redox balancing. The Biochemical journal. 2004;379:47–55. doi: 10.1042/BJ20031115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chang L, Wei LI, Audia JP, Morton RA, Schellhorn HE. Expression of the Escherichia coli NRZ nitrate reductase is highly growth phase dependent and is controlled by RpoS, the alternative vegetative sigma factor. Mol Microbiol. 1999;34:756–766. doi: 10.1046/j.1365-2958.1999.01637.x. [DOI] [PubMed] [Google Scholar]
- 85.Grove J, Tanapongpipat S, Thomas G, Griffiths L, Crooke H, et al. Escherichia coli K-12 genes essential for the synthesis of c-type cytochromes and a third nitrate reductase located in the periplasm. Molecular microbiology. 1996;19:467–481. doi: 10.1046/j.1365-2958.1996.383914.x. [DOI] [PubMed] [Google Scholar]
- 86.Potter L, Angove H, Richardson D, Cole J. Nitrate reduction in the periplasm of gram-negative bacteria. Advances in microbial physiology. 2001;45:51–112. doi: 10.1016/s0065-2911(01)45002-8. [DOI] [PubMed] [Google Scholar]
- 87.Maillard J, Spronk CA, Buchanan G, Lyall V, Richardson DJ, et al. Structural diversity in twin-arginine signal peptide-binding proteins. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:15641–15646. doi: 10.1073/pnas.0703967104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Richardson DJ, Berks BC, Russell DA, Spiro S, Taylor CJ. Functional, biochemical and genetic diversity of prokaryotic nitrate reductases. Cellular and molecular life sciences: CMLS. 2001;58:165–178. doi: 10.1007/PL00000845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jepson BJ, Marietou A, Mohan S, Cole JA, Butler CS, et al. Evolution of the soluble nitrate reductase: defining the monomeric periplasmic nitrate reductase subgroup. Biochemical Society transactions. 2006;34:122–126. doi: 10.1042/BST0340122. [DOI] [PubMed] [Google Scholar]
- 90.Thomas G, Potter L, Cole JA. The periplasmic nitrate reductase from Escherichia coli: a heterodimeric molybdoprotein with a double-arginine signal sequence and an unusual leader peptide cleavage site. FEMS microbiology letters. 1999;174:167–171. doi: 10.1111/j.1574-6968.1999.tb13564.x. [DOI] [PubMed] [Google Scholar]
- 91.Magalon A, Fedor JG, Walburger A, Weiner JA. Molybdenum enzymes in bacteria and their maturation. Coordination chemistry reviews. 2011;255:1159–1178. [Google Scholar]
- 92.Bertero MG, Rothery RA, Boroumand N, Palak M, Blasco F, et al. Structural and biochemical characterization of a quinol binding site of Escherichia coli nitrate reductase A. The Journal of biological chemistry. 2005;280:14836–14843. doi: 10.1074/jbc.M410457200. [DOI] [PubMed] [Google Scholar]
- 93.Bertero MG, Rothery RA, Palak M, Hou C, Lim D, et al. Insights into the respiratory electron transfer pathway from the structure of nitrate reductase A. Nature structural biology. 2003;10:681–687. doi: 10.1038/nsb969. [DOI] [PubMed] [Google Scholar]
- 94.Rothery RA, Bertero MG, Spreter T, Bouromand N, Strynadka NC, et al. Protein crystallography reveals a role for the FS0 cluster of Escherichia coli nitrate reductase A (NarGHI) in enzyme maturation. The Journal of biological chemistry. 2010;285:8801–8807. doi: 10.1074/jbc.M109.066027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bertero MG, Rothery RA, Palak M, Hou C, Lim D, et al. Insights into the respiratory electron transfer pathway from the structure of nitrate reductase A. Nat Struct Biol. 2003;10:681–687. doi: 10.1038/nsb969. [DOI] [PubMed] [Google Scholar]
- 96.Blasco F, Pommier J, Augier V, Chippaux M, Giordano G. Involvement of the narJ or narW gene product in the formation of active nitrate reductase in Escherichia coli. Molecular microbiology. 1992;6:221–230. doi: 10.1111/j.1365-2958.1992.tb02003.x. [DOI] [PubMed] [Google Scholar]
- 97.Blasco F, Dos Santos JP, Magalon A, Frixon C, Guigliarelli B, et al. NarJ is a specific chaperone required for molybdenum cofactor assembly in nitrate reductase A of Escherichia coli. Mol Microbiol. 1998;28:435–447. doi: 10.1046/j.1365-2958.1998.00795.x. [DOI] [PubMed] [Google Scholar]
- 98.Zakian S, Lafitte D, Vergnes A, Pimentel C, Sebban-Kreuzer C, et al. Basis of recognition between the NarJ chaperone and the N-terminus of the NarG subunit from Escherichia coli nitrate reductase. The FEBS journal. 2010;277:1886–1895. doi: 10.1111/j.1742-4658.2010.07611.x. [DOI] [PubMed] [Google Scholar]
- 99.Magalon A, Asso M, Guigliarelli B, Rothery RA, Bertrand P, et al. Molybdenum cofactor properties and [Fe-S] cluster coordination in Escherichia coli nitrate reductase A: investigation by site-directed mutagenesis of the conserved his-50 residue in the NarG subunit. Biochemistry. 1998;37:7363–7370. doi: 10.1021/bi972858f. [DOI] [PubMed] [Google Scholar]
- 100.Rothery RA, Magalon A, Giordano G, Guigliarelli B, Blasco F, et al. The molybdenum cofactor of Escherichia coli nitrate reductase A (NarGHI). Effect of a mobAB mutation and interactions with [Fe-S] clusters. The Journal of biological chemistry. 1998;273:7462–7469. doi: 10.1074/jbc.273.13.7462. [DOI] [PubMed] [Google Scholar]
- 101.Lanciano P, Vergnes A, Grimaldi S, Guigliarelli B, Magalon A. Biogenesis of a respiratory complex is orchestrated by a single accessory protein. The Journal of biological chemistry. 2007;282:17468–17474. doi: 10.1074/jbc.M700994200. [DOI] [PubMed] [Google Scholar]
- 102.Grimaldi S, Schoepp-Cothenet B, Ceccaldi P, Guigliarelli B, Magalon A. The prokaryotic Mo/W-bisPGD enzymes family: a catalytic workhorse in bioenergetic. Biochimica et biophysica acta. 2013;1827:1048–1085. doi: 10.1016/j.bbabio.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 103.Axley MJ, Bock A, Stadtman TC. Catalytic properties of an Escherichia coli formate dehydrogenase mutant in which sulfur replaces selenium. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:8450–8454. doi: 10.1073/pnas.88.19.8450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Axley MJ, Grahame DA. Kinetics for formate dehydrogenase of Escherichia coli formate-hydrogenlyase. The Journal of biological chemistry. 1991;266:13731–13736. [PubMed] [Google Scholar]
- 105.Axley MJ, Grahame DA, Stadtman TC. Escherichia coli formate-hydrogen lyase. Purification and properties of the selenium-dependent formate dehydrogenase component. J Biol Chem. 1990;265:18213–18218. [PubMed] [Google Scholar]
- 106.Boyington JC, Gladyshev VN, Khangulov SV, Stadtman TC, Sun PD. Crystal structure of formate dehydrogenase H: catalysis involving Mo, molybdopterin, selenocysteine, and an Fe4S4 cluster. Science. 1997;275:1305–1308. doi: 10.1126/science.275.5304.1305. [DOI] [PubMed] [Google Scholar]
- 107.Khangulov SV, Gladyshev VN, Dismukes GC, Stadtman TC. Selenium-containing formate dehydrogenase H from Escherichia coli: a molybdopterin enzyme that catalyzes formate oxidation without oxygen transfer. Biochemistry. 1998;37:3518–3528. doi: 10.1021/bi972177k. [DOI] [PubMed] [Google Scholar]
- 108.Zhang Y, Rump S, Gladyshev VN. Comparative Genomics and Evolution of Molybdenum Utilization. Coordination chemistry reviews. 2011;255:1206–1217. doi: 10.1016/j.ccr.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jormakka M, Byrne B, Iwata S. Formate dehydrogenase–a versatile enzyme in changing environments. Curr Opin Struct Biol. 2003;13:418–423. doi: 10.1016/s0959-440x(03)00098-8. [DOI] [PubMed] [Google Scholar]
- 110.Abaibou H, Pommier J, Benoit S, Giordano G, Mandrand-Berthelot MA. Expression and characterization of the Escherichia coli fdo locus and a possible physiological role for aerobic formate dehydrogenase. J Bacteriol. 1995;177:7141–7149. doi: 10.1128/jb.177.24.7141-7149.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jormakka M, Tornroth S, Abramson J, Byrne B, Iwata S. Purification and crystallization of the respiratory complex formate dehydrogenase-N from Escherichia coli. Acta crystallographica Section D, Biological crystallography. 2002;58:160–162. doi: 10.1107/s0907444901017723. [DOI] [PubMed] [Google Scholar]
- 112.Hartmann T, Leimkühler S. The oxygen-tolerant and NAD(+) -dependent formate dehydrogenase from Rhodobacter capsulatus is able to catalyze the reduction of CO2 to formate. The FEBS journal. 2013;280:6083–6096. doi: 10.1111/febs.12528. [DOI] [PubMed] [Google Scholar]
- 113.Böhmer N, Hartmann T, Leimkühler S. The chaperone FdsC for Rhodobacter capsulatus formate dehydrogenase binds the bis-molybdopterin guanine dinucleotide cofactor. FEBS letters. 2014;588:531–7. doi: 10.1016/j.febslet.2013.12.033. [DOI] [PubMed] [Google Scholar]
- 114.Bilous PT, Weiner JH. Molecular cloning and expression of the Escherichia coli dimethyl sulfoxide reductase operon. J Bacteriol. 1988;170:1511–1518. doi: 10.1128/jb.170.4.1511-1518.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Trieber CA, Rothery RA, Weiner JH. Consequences of removal of a molybdenum ligand (DmsA-Ser-176) of Escherichia coli dimethyl sulfoxide reductase. J Biol Chem. 1996;271:27339–27345. doi: 10.1074/jbc.271.44.27339. [DOI] [PubMed] [Google Scholar]
- 116.Cheng VW, Rothery RA, Bertero MG, Strynadka NC, Weiner JH. Investigation of the environment surrounding iron-sulfur cluster 4 of Escherichia coli dimethylsulfoxide reductase. Biochemistry. 2005;44:8068–8077. doi: 10.1021/bi050362p. [DOI] [PubMed] [Google Scholar]
- 117.Ujiiye T, Yamamoto I, Nakama H, Okubo A, Yamazaki S, et al. Nucleotide sequence of the genes, encoding the pentaheme cytochrome (dmsC) and the transmembrane protein (dmsB), involved in dimethyl sulfoxide respiration from Rhodobacter sphaeroides f. sp. denitrificans. Biochim Biophys Acta. 1996;1277:1–5. doi: 10.1016/s0005-2728(96)00101-6. [DOI] [PubMed] [Google Scholar]
- 118.Rothery RA, Weiner JH. Topological characterization of Escherichia coli DMSO reductase by electron paramagnetic resonance spectroscopy of an engineered [3Fe-4S] cluster. Biochemistry. 1993;32:5855–5861. doi: 10.1021/bi00073a019. [DOI] [PubMed] [Google Scholar]
- 119.Cammack R, Weiner JH. Electron paramagnetic resonance spectroscopic characterization of dimethyl sulfoxide reductase of Escherichia coli. Biochemistry. 1990;29:8410–8416. doi: 10.1021/bi00488a030. [DOI] [PubMed] [Google Scholar]
- 120.Rothery RA, Workun GJ, Weiner JH. The prokaryotic complex iron-sulfur molybdoenzyme family. Biochimica et biophysica acta. 2008;1778:1897–1929. doi: 10.1016/j.bbamem.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 121.Chan CS, Winstone TM, Chang L, Stevens CM, Workentine ML, et al. Identification of residues in DmsD for twin-arginine leader peptide binding, defined through random and bioinformatics-directed mutagenesis. Biochemistry. 2008;47:2749–2759. doi: 10.1021/bi702138a. [DOI] [PubMed] [Google Scholar]
- 122.Ray N, Oates J, Turner RJ, Robinson C. DmsD is required for the biogenesis of DMSO reductase in Escherichia coli but not for the interaction of the DmsA signal peptide with the Tat apparatus. FEBS letters. 2003;534:156–160. doi: 10.1016/s0014-5793(02)03839-5. [DOI] [PubMed] [Google Scholar]
- 123.Guymer D, Maillard J, Sargent F. A genetic analysis of in vivo selenate reduction by Salmonella enterica serovar Typhimurium LT2 and Escherichia coli K12. Archives of microbiology. 2009;191:519–528. doi: 10.1007/s00203-009-0478-7. [DOI] [PubMed] [Google Scholar]
- 124.Neumann M, Mittelstädt G, Iobbi-Nivol C, Saggu M, Lendzian F, et al. A periplasmic aldehyde oxidoreductase represents the first molybdopterin cytosine dinucleotide cofactor containing molybdo-flavoenzyme from Escherichia coli. The FEBS journal. 2009;276:2762–2774. doi: 10.1111/j.1742-4658.2009.07000.x. [DOI] [PubMed] [Google Scholar]
- 125.Otrelo-Cardoso AR, da Silva Correia MA, Schwuchow V, Svergun DI, Romao MJ, et al. Structural data on the periplasmic aldehyde oxidoreductase PaoABC from Escherichia coli: SAXS and preliminary X-ray crystallography analysis. International journal of molecular sciences. 2014;15:2223–2236. doi: 10.3390/ijms15022223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Otrelo-Cardoso AR, Schwuchow V, Rodrigues D, Cabrita EJ, Leimkühler S, et al. Biochemical, stabilization and crystallization studies on a molecular chaperone (PaoD) involved in the maturation of molybdoenzymes. PloS one. 2014;9:e87295. doi: 10.1371/journal.pone.0087295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Leimkühler S, Kern M, Solomon PS, McEwan AG, Schwarz G, et al. Xanthine dehydrogenase from the phototrophic purple bacterium Rhodobacter capsulatus is more similar to its eukaryotic counterparts than to prokaryotic molybdenum enzymes. Molecular microbiology. 1998;27:853–869. doi: 10.1046/j.1365-2958.1998.00733.x. [DOI] [PubMed] [Google Scholar]
- 128.Leimkühler S, Hodson R, George GN, Rajagopalan KV. Recombinant Rhodobacter capsulatus xanthine dehydrogenase, a useful model system for the characterization of protein variants leading to xanthinuria I in humans. The Journal of biological chemistry. 2003;278:20802–20811. doi: 10.1074/jbc.M303091200. [DOI] [PubMed] [Google Scholar]
- 129.Truglio JJ, Theis K, Leimkühler S, Rappa R, Rajagopalan KV, et al. Crystal Structures of the Active and Alloxanthine-Inhibited Forms of Xanthine Dehydrogenase from Rhodobacter capsulatus. Structure (Camb) 2002;10:115–125. doi: 10.1016/s0969-2126(01)00697-9. [DOI] [PubMed] [Google Scholar]
- 130.Aguey-Zinsou KF, Bernhardt P, Leimkühler S. Protein film voltammetry of Rhodobacter capsulatus xanthine dehydrogenase. J Am Chem Soc. 2003;125:15352–15358. doi: 10.1021/ja037940e. [DOI] [PubMed] [Google Scholar]
- 131.Vinella D, Brochier-Armanet C, Loiseau L, Talla E, Barras F. Iron-sulfur (Fe/S) protein biogenesis: phylogenomic and genetic studies of A-type carriers. PLoS genetics. 2009;5:e1000497. doi: 10.1371/journal.pgen.1000497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pinske C, Sawers RG. A-type carrier protein ErpA is essential for formation of an active formate-nitrate respiratory pathway in Escherichia coli K-12. Journal of bacteriology. 2012;194:346–353. doi: 10.1128/JB.06024-11. [DOI] [PMC free article] [PubMed] [Google Scholar]