

Abstract
Elevated hyaluronan expression is a hallmark of many types of cancer. Therefore, inhibition of hyaluronan biosynthesis can potentially slow the growth of tumor cells. Herein, we explore a chain termination strategy to reduce hyaluronan synthesis by tumor cells. Several analogs of glucosamine were prepared, which contained modifications at the C-3 positions. These analogs can possibly cap the nonreducing end of a growing hyaluronan chain, thus lowering the amount of hyaluronan synthesized. Upon incubation with pancreatic cancer cells, a fluorine-containing glucosamine analog was found to exhibit significant inhibitory activities of hyaluronan synthesis. Furthermore, it drastically reduced the proliferation of cancer cells.
Keywords: Biosynthesis, Chain terminator, Hyaluronan, Inhibitor, Salvage pathway
Introduction
Hyaluronan (HA), a member of the glycosaminoglycan family, is composed of repeating disaccharides of β-1,3-N-acetyl-glucosamine-β-1,4-glucuronic acid.[1] HA plays important roles in tumor development, metastasis, and drug resistance.[2–4] HA expression is elevated in a variety of tumors,[5] with the high levels of HA shown to have prognostic values for various cancers, such as non–small cell lung, bladder, gastric, and prostate cancer.[6–9] Therefore, inhibition of HA biosynthesis can be an interesting approach toward cancer treatment.
HA biosynthesis is mediated by HA synthases,[10] which use UDP-α-N-acetyl-d-glucosamine (UDP-GlcNAc) and UDP-α-d-glucuronate (UDP-GlcA) as substrates to sequentially transfer GlcNAc and GlcA to the nonreducing end of a growing HA chain. 4-Methylumbelliferone (4-MU) was found to be an HA synthesis inhibitor,[11] which functions by suppressing the expression of HA synthases.[12] 4-MU can also serve as an acceptor for UDP-GlcA, thus competing for the limited intracellular pool of UDP-GlcA and further reducing the amount of HA produced.[13,14] 4-MU can reduce proliferation, motility, invasion, and metastatic properties of various types of cancer including prostate cancer and breast cancer.[12,15–18] These observations have demonstrated the utility of HA biosynthesis inhibitors as potential antitumor agents.
Herein, we report an alternative approach to inhibit HA biosynthesis through potential early chain termination. We envision that if a modified Glc-NAc analog such as 1–3 is incorporated into an HA chain, as the C-3 position does not bear a free hydroxyl moiety, HA synthesis will be terminated, leading to a reduction of the amount of HA synthesized. The key to the success of this strategy is the incorporation of the potential chain terminators (e.g., 1–3) into the growing HA chain. The GlcNAc donor (i.e., UDP-GlcNAc) is synthesized inside the cells primarily from glucose through a series of enzymatic reactions.[19] However, there exists a salvage pathway, which utilizes exogenous GlcNAc as the source for UDP-GlcNAc.[20] The salvage pathway has a broad substrate scope and is well tolerant of GlcNAc structural modifications, which can allow the transformation of a variety of GlcNAc analogs into UDP glycosyl donors.[21] The HA synthase can potentially utilize the UDP-GlcNAc analog as a glycosyl donor and incorporate the modified GlcNAc into the HA chain. The chain termination strategy has been applied to inhibition of biosynthesis of a variety of glycans[22] including sialyl Lewis X, β-glucan, heparan sulfate, and chitin. [23–28] Herein, we report the chemical synthesis of GlcNAc analogs and investigate their abilities to inhibit HA synthesis by cancer cells. A fluorine-containing GlcNAc analog showed high inhibitory activities toward HA synthesis and cancer cell proliferation.
Results and Discussion
Our synthetic targets are the 3-OMe, 3-oxo, and 3-F[29] analogs of GlcNAc 4–6. The hydroxyl groups of these analogs are protected as acetates to reduce their hydrophilicity and increase their potential to cross cell membranes during cellular incubation. Acetate groups have been shown to significantly enhance intracellular concentrations of exogenous carbohydrates added to mammalian cells, which can then be deacetylated in situ by cytosolic esterases.[30]
In our initial route for the synthesis of these GlcNAc derivatives, we opted to use furanose oxazoline 8 as a key intermediate since it contains a free 3-OH moiety. Compound 8 was conveniently prepared in large quantities from commercially available GlcNAc 7 using anhydrous FeCl3 as a catalyst in acetone.[31] With oxazoline 8 in hand, methylation of its free 3-OH group by MeI and NaH gave compound 9 in quantitative yield. Subsequent acid-catalyzed hydrolysis of compound 9 with p-toluene sulfonic acid (PTSA) furnished the desired 3-O-methyl-N-acetyl-d-glucosamine 1, which was acetylated using acetic anhydride in pyridine to give the product 4 in 50% overall yield over the two steps (Sch. 1).
Scheme 1.

Reagents and Conditions: i) Acetone/2 eq FeCl3, reflux, 60°C, 65%; ii) 2 eq NaH, 1.5 eq Mel, DMF, 0°C to rt, 96%; iii) 0.4 eq PTSA, THF/H2O 2:1; iv) Ac2O/Pyridine, rt, 50%.
Next we explored the synthesis of the 3-oxo GlcNAc derivative 5. Swern oxidation of 8 produced the ketone 10 in 92% yield (Sch. 2a). However, acid-catalyzed deprotection of 10 in a similar manner, as in the synthesis of compound 4, failed to provide the desired product 2. One possible explanation was that compound 2 was unstable under the acidic reaction condition. Based on this consideration, we designed an alternative route to avoid the acidic condition.
Scheme 2.

Reagents and Conditions: i) 1.4 eq COCl2, 2.5 eq DMSO, 5 eq Et3N, −75°C to rt, 92%; ii) 0.4 eq PTSA, THF/H2O 2:1; iii) BnOH, HCl, 90°C, 71%, iv) 5 eq PhCH(OMe)2, 0.6 eq camphorsulfonic acid, DMF, rt, 95%, v) 1.4 eq COCl2, 2.5 eq DMSO, 5 eq Et3N, −75°C to rt, 90%; vi) H2, Pd/C, MeOH.
GlcNAc 7 was selectively benzylated at the anomeric position using concentrated HCl leading to compound 11,[28] which was followed by benzylidenation of the 4,6-hydroxyl groups generating compound 12[32] in an excellent 95% yield (Sch. 2b). Subsequent Swern oxidation of 12 produced 3-uloside derivative 13 in 90% yield. However, when subjected to the mild catalytic hydrogenation condition to remove the benzyl and benzylidene groups, compound 13 was transformed to multiple species. One of the major side products isolated was ketone 14, which was presumably formed due to a retro-Claisen reaction through nucleophilic attack of methanol on 2 (Sch. 2c). Therefore, compound 2 would most likely be unstable in a nucleophilic solvent such as water and was not pursued further.
We shifted our attention to the preparation of the 3F-GlcNAc derivative 6. The first route explored was the substitution of the free 3-OH in oxazoline 8 by a fluorine atom. However, treatment of 8 with diethylaminosulfur trifluoride (DAST) gave a complex product mixture. Exchanging 8 in this reaction with compound 12 or the corresponding epimers containing axial 3-OH did not yield any desired fluorine substitution product. This was consistent with a literature report where the yield for fluorine substitution onto C3 of a GlcNAc analog was only 10%.[29]
Kajihara and coworkers described that 6-OH of a glucosamine analog could be substituted with fluorine with a modest yield (∼38%) when phthalimide (Phth) was used as the protective group for the nitrogen.[33] To test the effect of Phth on our fluorination reaction, we started from the Phth-containing glucosamine tetraacetate 15,[34,35] which was efficiently converted to β-benzyl glycoside 16[36] using BF3.Et2O in CH2Cl2 (Sch. 3). Subsequent acetate removal with 0.3 M NaOMe in a mixture of MeOH-CH2Cl2 at −10°C to 0°C produced triol 17 in 77% yield. The low reaction temperature was important to ensure the phthalimide ring was not opened by NaOMe. The O-4 and O-6 free hydroxyl groups in 17 were protected as a benzylidene acetal by treatment with benzaldehyde dimethyl acetal in the presence of a catalytic amount of camphorsulfonic acid to yield the benzylidene glycoside 18.[36] To install the fluorine atom in the equatorial orientation, it was necessary to invert the configuration of the 3-OH group. This was accomplished through a four-step double-inversion protocol. Compound 18 was treated with Tf2O in CH2Cl2 in the presence of pyridine to convert the free hydroxyl group into a good leaving group (i.e., triflate). Displacement of the triflate by acetate via an SN2 manner yielded the 3-OAc glycoside, which was subsequently deprotected with NaOMe at −10°C to produce the corresponding allosamine glycoside 19. Fluorination of compound 19 was accomplished using DAST, resulting in the inversion of configuration at C-3 to access the fluorinated analog mixed with a side product. Upon removal of the benzylidene-protecting group, we were able to purify the desired product 20 and identify the side product as 21; 21 is presumably formed due to elimination of hydrogen fluoride followed by tautomerization and opening of the phthalimide ring. The overall yield of 20 from benzylidene glycoside 18 was 28%.
Scheme 3.

Reagents and Conditions: i) 2 eq BnOH, 3 eq BF3OEt2, CH2Cl2, 79%; ii) 0.3 M NaOMe, MeOH/CH2Cl2, −10°C; iii) PhCH(OMe)2, 0.08 eq camphorsulfonic acid, CH3CN, 77%; iv) 2 eq Tf2O, 5 eq pyridine, −20°C to 10°C; 6 eq tetrabutylammonium acetate, toluene, 60°C%; v) 6 eq DAST, CH2Cl2, −5°C to rt; vi) 6 eq AcCl, MeOH/CH2Cl2, 28% yield from 18; vii) H2N(CH2)2NH2, nBuOH, 90°C; acetic anhydride, pyridine rt, 80%; viii) H2, Pd/C, MeOH; acetic anhydride, pyridine, rt, 60%.
To remove the Phth group in 20, it was initially treated with methylamine, but this attempt resulted in multiple degradation products. In contrast, heating 20 with excess ethylene diamine in n-butanol at 90°C allowed the access to the free amine derivative, which on subsequent acetylation provided the acetamide 22 in 80% yield (Sch. 3). Pd-catalyzed hydrogenolysis of 22 followed by acetylation led to the desired product 6 with the α-anomer as the major product in 60% yield.
With compounds 4 and 6 in hand, we evaluated their abilities to inhibit HA biosynthesis in human pancreatic cancer cell line KP1-NL cells. 4-MU was used as a positive control. The amount of HA secreted into the culture medium as well as that residing on the cell surface was quantified by an ELISA-like assay using a Hyaluronan Assay Kit and reported on a per cell basis (Table 1). While compound 4 was not very effective in inhibiting HA biosynthesis, compound 6 significantly reduced the amount of HA formed, with an inhibition potency comparable to that of 4-MU.
Table 1. Amount of HA secreted into culture medium and on surface of human pancreatic cancer cell KP1-NL upon incubation with various compounds.
| Culture medium | Cell surface | ||||
|---|---|---|---|---|---|
|
|
|
||||
| Amount of HA (μg/106 cells) | % of control | Amount of HA (μg/106 cells) | % of control | ||
| DMSO | 3.02 ± 0.03 | 100 | 0.17 ± 0.02 | 100 | |
| 4-MU | 10 μM | 3.00 ± 0.05 | 99.2 | 0.17 ± 0.01 | 100.2 |
| 100 μM | 1.66 ± 0.04 | 54.9 | 0.12 ± 0.01 | 68.4 | |
| 4 | 10 μM | 2.95 ± 0.02 | 97.5 | 0.15 ± 0.02 | 89.1 |
| 100 μM | 2.65 ± 0.01 | 87.7 | 0.14 ± 0.02 | 81.8 | |
| 6 | 10 μM | 2.40 ± 0.02 | 79.6 | 0.14 ± 0.01 | 82.9 |
| 100 μM | 1.89 ± 0.06 | 62.7 | 0.11 ± 0.01 | 64.9 | |
As HA present in the pericellular matrix can create a barrier to the cell membrane, its presence can be visualized by the steric exclusion of fixed red blood cells to form a clear, halo-like area surrounding the KP1-NL cells (Fig. 3 and Fig. S1 in supporting information). Treatment of the cells with hyaluronidase, an enzyme capable of HA degradation, reduced the clear area around the cells to 48.2% (Fig. 3b; for quantification of the size of clear area see Fig. 3f). Compound 4 was much less potent, only leading to a small reduction to 93.1% of the vehicle control (Fig. 3d,f). In comparison, compound 6 (100 μM) reduced the area to 40.9% (Fig. 3e,f), which was consistent with the ELISA analysis results supporting the idea that 6 could significantly inhibit HA synthesis. 4-MU (100 μM) generated the largest change in the area under the same assay condition (Fig. 3c,f).
Figure 3.

Effect of various compounds on the formation of a pericellular HA matrix by KP1-NL cells as assessed by a particle exclusion assay. Representative pictures of KP1-NL cells following incubation with (a) DMSO control; (b) Streptomyces hyaluronidase (1.0 U/mL); (c) 4-MU (100 μM); (d) GlcNAc analog 4 (100 μM); and (e) GlcNAc analog 6 (100 μM). Fixed sheep erythrocytes (the small cells in the pictures) were then added to the cells to help visualize the pericellular matrix around the cancer cells. The pictures were taken at identical magnification (each picture represents an area of 100 μm in width). (f) Quantification of the relative sizes of areas in particle exclusion assay assessed using Image J.
Next we analyzed the effects of compounds 4 and 6 on cell proliferation using an Alamar blue assay (Fig. 4). At the concentration of 100 μM, 4-MU did not show much inhibitory activities against the pancreatic cancer cell line KP1-NL cells. 3-Methoxy analog 4 (100 μM) reduced the proliferation of the cancer cells to 60.5% of the DMSO control, while the 3-fluorine-containing analog 6 exhibited the highest antiproliferative activities, leading to a 91% reduction in cell numbers at 100 μM. The IC50 value of 6 was determined to be 30 μM.
Figure 4.
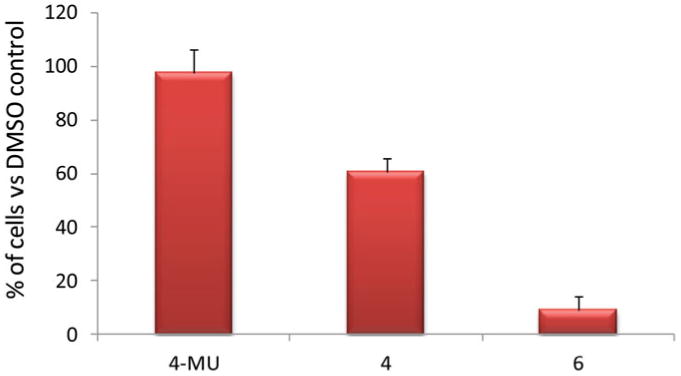
Number of KP1-NL cells upon incubation with 4-MU, compound 4, and compound 6 respectively at 100 μM for 72 hours as determined by Alamar blue assay. The number of cells treated with DMSO was set as 100% (color figure available online).
In conclusion, we have developed synthetic routes to GlcNAc analogs bearing structural modifications at C-3 positions as potential inhibitors of HA biosynthesis. While the 3-OMe compound 4 was readily prepared, the intermediate to 3-oxo analog was found to be unstable. To synthesize the 3-F-containing analog 6, fluorination of the C-3 turned out to be highly challenging. The Phth group used for protection of the nitrogen was crucial, which gave better yield for the fluorination step compared to the previous report.[29] Upon incubation of human pancreatic cell line with compounds 4 and 6, the 3-F analog 6 significantly reduced the amount of HA both in secreted form and on the cancer cell surface. The inhibitory activity of 6 was further confirmed by a particle exclusion assay and 6 exhibited significant antiproliferative activities against pancreatic cancer cells. It is possible that besides the potential for serving as a chain terminator, 3-F GlcNAc analog 6 can reduce HA biosynthesis by interfering with other cellular processes. The exact mechanism will require further investigation.[37]
Experimental Section
General Experimental Procedures
All reactions were carried out under nitrogen with anhydrous solvents in flame-dried glassware, unless otherwise noted. Chemicals used were reagent grade as supplied except where noted. Compounds were visualized by UV light (254 nm) and by staining with a yellow solution containing Ce(NH4)2(NO3)6 (0.5 g) and (NH4)6Mo7O24 4H2O (24.0 g) in 6% H2SO4 (500 mL). Flash column chromatography was performed on silica gel 60 (230–400 mesh). NMR spectra were referenced using residual CHCl3 (δ 1H NMR 7.26 ppm) and CDC13 (δ 13C NMR 77.0 ppm). Peak and coupling constant assignments are based on 1H NMR, 1H-1H gCOSY, and/or 1H-13C gHMQC and 1H-13C gHMBC experiments.
2-Methyl-(1,2-dideoxy-5,6-O-isopropylidene-α-d-glucofurano)-[2,1-d]-2-oxazoline (8)[38]
Anhydrous FeCl3 (15 g, 0.092 mol) was added to a suspension of 2-acetamido-2-deoxy-d-glucopyranose 7 (10 g, 0.068 mol) in dry acetone (200 mL), and the mixture was stirred and heated to reflux for 20 min with exclusion of moisture. The solution was cooled to 0°C, followed by addition of triethylamine (35 mL), acetone (135 mL), and a solution of sodium bicarbonate (21.3 g) in water (135 mL) with continuous stirring. Acetone, triethylamine, and some water were then removed in vacuo at <30°C. The mixture was extracted with diethyl ether (5 × 200 mL) and the combined extracts dried over MgSO4 and concentrated at rt to give compound 8 as brownish syrup (7.15 g, 65%). The 1H NMR spectrum is identical to what is reported in the literature.[38] 1H NMR (600 MHz, CDCl3) δ1.33 (s, 3H, CH3), 1.39 (s, 3H, CH3), 2.00 (s, 3H, N = COCH3), 2.92 (s, 1H, OH-3), 3.72 (dd, 1H, J3 = 3.6, 9.6 Hz, H-4), 3.97 (dd, 1H, J3 = 4.8, 8.4 Hz, H-6), 4.11 (dd, 1H, J3 = 5.4, 7.8 Hz, H-6′), 4.28−4.32 (m, 1H, H-5), 4.39 (d, 1H, J3 = 3 Hz, H-3), 4.44 (dd, 1H, J3 = 1.2, 4.8 Hz, H-2), 6.14 (d, J3 = 4.8 Hz, H-1).
2-Methyl-(1,2-di-deoxy-3-methoxy-5,6-O-isopropylidene-α-d-glucofurano)-[2,1-d]-2-oxazoline (9)
Compound 8 (0.4 g, 1.64 mmol) was dissolved in DMF (15 mL) and cooled to 0°C. NaH (0.08 g, 3.29 mmol) was added in three portions with vigorous stirring followed by dropwise addition of MeI (0.15 mL, 2.47 mmol). The reaction mixture was allowed to warm up to rt. After 3 h, DMF was evaporated in vacuo and the residue coevaporated twice with toluene. The resultant residue was dissolved in CH2Cl2 (20 mL) and washed with ice cold water and the aqueous phase extracted twice with CH2Cl2 (2 × 20 mL). The organic phases were combined, washed with brine (30 mL), and dried with Na2SO4. Upon filtration and concentration in vacuo, compound 9 was obtained in 96% yield (0.4 g). − 15.5 (c = 1, CH2Cl2); 1H NMR (600 MHz, CDCl3) δ 1.27 (s, 3H, CH3), 1.33 (s, 3H, CH3), 1.94 (s, 3H, N = CĈH3), 3.40 (s, 3H, OCH3), 3.71 (dd, 1H, J3 = 3.6, 9 Hz, H-4), 3.77 (d, 1H, J3 = 4.2 Hz, H-3), 3.91–3.94 (m, 1H, H-6), 3.97–3.99 (m, 1H, H-6′), 4.21 (m, 1H, H-5), 4.42 (dd, 1H, J3 = 1.8, 6 Hz, H-4), 6.01 (d, 1H, J3 = 6.6 Hz, H-1); 13C NMR (150 Hz, CDCl3) δ 14.3. 25.5, 26.99, 57.98, 67.0, 72.7, 74.8, 81.5, 83.7, 107.1, 109.2, 167.2. HRMS C12H20NO5 [M+ H+] calc. 258.1341 found 258.1333.
1,4,6-Tri-O-acetyl-2-acetamido-2-deoxy-3-methoxy--β-d-glucopyranoside (4)
Compound 9 (0.204 g, 0.793 mmol) was dissolved in 1:2 H2O/THF mixture (10 mL:20 mL) and p-toluenesulfonic acid (0.06 g, 0.264 mmol) was added to the reaction mixture then stirred overnight. The reaction was quenched with Et3N to a neutral pH and the solvents were evaporated in vacuo. The residue was washed several times with CH2Cl2 and the resultant white powder dried under vacuum. The residue was then dissolved in pyridine (8 mL). Ac2O (0.5 mL) was added and the reaction mixture was stirred at rt overnight. The reaction mixture was concentrated. The residue was dissolved in CH2Cl2, washed with H2O and brine, dried over Na2SO4, and purified by column chromatography (CH2Cl2/MeOH, 8:1) to give compound 4 in 50% (0.143 g) yield over two steps. + 10.5 (c = 1, CH2Cl2); 1H NMR (600 MHz, CDCl3) δ 1.99 (s, 3H, NHCOCH3), 2.07 (s, 3H, COCH3), 2.10 (s, 3H, COCH3), 2.11 (s, 3H, COCH3), 3.40 (s, 3H, OCH3), 3.75–3.84 (m, 3H, H-2, H-4, H-5), 4.09 (dd, 1H, J3 = 2.4, 10.8 Hz, H-6), 4.23 (dd, 1H, J3 = 4.5, 12Hz, H-6′), 5.02 (t, 1H, J3 = 8.4 Hz, H-3), 5.70 (d, 1H, J3 = 7.2 Hz, H-1), 5.89 (d, 1H, J3 = 6.6 Hz, CONH); 13C NMR (125 Hz, CDCl3) 20.8, 20.84, 20.9, 23.4, 53.8, 58.0, 62.0, 68.5, 72.9, 79.2, 91.97, 169.4, 169.41, 170.2, 170.7. HRMS C15H23NO9 Na[M + Na+] calc. 384.1271 found 384.1259.
Benzyl-2-acetamido-2-deoxy-α-d-glucopyranoside (11)
N-Acetyl glucosamine 7 (6.00 g, 27.1 mmol) was dissolved in benzyl alcohol (50 mL) and concentrated HCl (2.9 mL) was added. The mixture was heated to 90°C for 3 h, cooled to rt, and then poured onto 500 mL Et2O and stored overnight at −20° C. The resulting precipitate was recovered by filtration and rinsed with Et2O and hexanes to yield 17.64 g of crude material, which was purified by silica gel chromatography (8% to 15% MeOH/CH2Cl2) to provide 11 (5.98 g, 71%) as white foam. Comparison of 1H NMR with literature values confirmed the identity of compound 11.[28] 1H NMR (500 MHz, CD3OD), δ 1.95 (s, 3H, COCH3), 3.37–3.39 (m, 1H), 3.69–3.73 (m, 3H), 3.82 (d, 1H, J3 = 9.5 Hz), 3.89 (dd, 1H, J3 = 3.5, 11 Hz), 4.49 (d, 1H, J3 = 12 Hz), 4.74 (d, 1H, J3 = 11.5 Hz), 7.28–7.40 (m, 5H, aromatic).
Benzyl-2-acetamido-2-deoxy-4,6-O-benzylidene-α-d-glucopyranoside (12)
Compound 11 (0.412 g, 1.32 mmol) was dissolved in DMF (20 mL) followed by the addition of benzaldehyde dimethyl acetal (0.6 mL, 3.97 mmol) and a catalytic amount of p-toluenesulfonic acid (0.123 g, 0.53 mmol). The reaction mixture was stirred for 3 h. DMF was removed in vacuo and the resultant white residue was suspended in saturated sodium bicarbonate solution. Upon filtration, the residue was washed several times with hexanes/EtOAc/CH2Cl2 (4/1/0.5) solvent system then dried under vacuum to afford compound 12 in 95% yield (0.5 g). Comparison of 1H NMR with reported literature values confirmed the identity of compound 12.[32] 1H NMR (600 MHz, CDCl3) δ 1.82 (s, 3H, CH3), 3.59 (t, 1H, J3 = 9 Hz, H-6), 3.75 (t, 1H, J3 = 10.2 Hz, H-6′), 3.84–3.91 (m, 1H, H-5), 3.92–3.95 (m, 1H, H-4), 4.23–4.26 (m, 2H, H-2, H-3), 4.93 (d, 1H, J3 = 12 Hz, CH2Ph), 4.74 (d, 1H, J3 = 12 Hz, CH2Ph), 4.93 (d, 1H, J3 = 3.5 Hz, H-1), 5.57 (s, 1H, CHPh), 5.82 (d, 1H, J3 = 8.4 Hz, CONH), 7.33–7.51 (m, 10H, aromatic).
Benzyl-2-acetamido-2-deoxy-4,6-O-benzylidene-3-oxo-α-d-glucopyranoside (13)
To a solution of COCl2 (0.11 mL, 1.25 mmol) and DMSO (0.16 mL, 2.23 mmol) in CH2Cl2 (30 mL) cooled to −78°C and stirred for 20 min was added compound 12 (0.36 g, 0.894 mmol) dissolved in CH2Cl2 (20 mL). After 20 min, Et3N (0.63 mL, 4.47 mmol) was added and the reaction mixture allowed to warm to rt then quenched with H2O (100 mL). The aqueous layer was extracted with CH2Cl2 (50 mL). The organic layers were combined and washed with brine and dried over MgSO4. Compound 13 was obtained in 90% yield (0.32 g) by recrystallization from hexanes/EtOAc/CH2Cl2 (5/1/0.5). + 38.8 (c = 0.1, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 2.00 (s, 3H, CH3), 3.90 (t, 1H, J3 = 10.5 Hz, H-6), 4.11–4.16 (m, 1H), 4.29 (dd, 1H, J3 = 5, 10.5 Hz, H-6′), 4.37 (d, 1H, J3 = 10.5 Hz, H-4), 4.50 (d, 1H, J3 = 12 Hz, CH2Ph), 4.69 (d, 1H, J3 = 12 Hz, CH2Ph), 4.95–4.97 (m, 1H, H-2), 5.41 (d, 1H, J3 = 4.5 Hz, H-1), 5.56 (s, 1H, CHPh), 6.22 (d, 1H, J3 = 8.0 Hz, CONH), 7.25–7.49 (m, 10H, aromatic); 13C NMR (125 Hz, CDCl3) δ 23.2, 59.1, 66.6, 69.6, 70.8, 77.0, 77.2, 77.5, 82.9, 100.8, 102.2, 126.6, 128.2, 128.6, 128.9, 129.6, 136.5, 170.1, 195.2. HRMS C22H24NO6 [M + H+] calc. 398.1580 found 398.1577.
Benzyl-3,4,6-tri-O-acetyl-2-deoxy 2-phthalamido-β-d-glucopyranoside (16)
Compound 15 (2.2 g, 4.61 mmol) was dissolved in CH2Cl2 (30 mL), followed by addition of BnOH (1.1 mL, 9.22 mmol). BF3OEt2 (1.75 mL, 11.4 mmol) was added dropwise and the reaction mixture was stirred at rt for 24 h. The mixture was diluted with CH2Cl2 (20 mL) and washed with saturated NaHCO3 (3 × 20 mL). The aqueous layer was extracted with CH2Cl2 (60 mL) and the combined organic phases were dried over Na2SO4, filtered, concentrated, and purified by column purification (hexanes/EtOAc 3:2) to yield compound 16 (1.92 g, 79%). Comparison of 1H NMR with reported literature values confirmed the identity of compound 16.[36] 1H NMR (600 MHz, CDCl3) δ 1.86 (s, 3H, COCH3), 2.02 (s, 3H, COCH3), 2.13 (s, 3H, COCH3), 3.85–3.88 (m, 1H, H-5), 4.19 (dd, 1H, J3 = 2.4, 12 Hz, H-6), 4.33–4.39 (m, 2H, H-2, H-6′), 4.53 (d, 1H, J3 = 12 Hz, CHPh), 4.84 (d, 1H, J3 = 12.6 Hz, CHPh), 5.17 (t, 1H, J3 = 9.6 Hz, H-4) 5.37 (d, 1H, J3 = 8.4 Hz, H-1), 5.77 (dd, 1H, J3 = 9.0, 10.2 Hz, H-3), 7.08–7.79 (m, 9H, aromatic).
Benzyl-4,6-O-benzylidene-2-deoxy-2-phthalamido-β-d-glucopyranoside (18)
Compound 16 (1.88 g, 3.79 mmol) was dissolved in MeOH/CH2Cl2 solvent mixture (3:2, 20 mL) and cooled to −10°C followed by dropwise addition of 0.3M NaOMe in MeOH (6 mL). Upon completion of the reaction as established by TLC in 2 h, the reaction mixture was neutralized with Amberlite ion exchange resin to pH 7. The crude triol 17 was concentrated and the residue was coevaporated with toluene twice followed by drying under vacuum overnight. The residue was then suspended in CH3CN (15 mL) and PhCH(OCH3)2 (0.7 mL, 4.55 mmol) was added, followed by addition of camphorsulfonic acid (0.2 g, 0.87 mmol). The mixture was stirred for 3 h then quenched with Et3N upon completion of reaction as confirmed by TLC. Solvents were evaporated and the residue was purified by column chromatography (hexanes/EtOAc 3:1) to yield compound 18 (1.42 g, 77%). The identity of compound 18 was confirmed by comparison with reported literature values.[36] 1H NMR (600 MHz, CDCl3) 3.62–3.67 (m, 2H, H-4, H-6), 3.85–3.88 (m, 1H, H-6), 4.29–4.32 (m, 1H, H-2), 4.42 (dd, 1 H, J3 = 4.8, 10.8 Hz, H-5), 4.52 (d, 1H, J3 = 12.6 Hz, CHPh), 4.63–4.67 (m, 1H, H-3), 4.84 (d, 1H, J3 = 12 Hz, CHPh), 5.28 (d, 1H, J3 = 8.4 Hz, H-1), 5.58 (s, 1H CHPh) 7.03–7.79 (m, 14H, aromatic).
Benzyl-2-deoxy-3-fluoro-2-phthalamido-β-d-glucopyranoside (20)
To a solution of compound 18 (0.833 g, 1.71 mmol) dissolved in CH2Cl2 (30 mL), pyridine (0.7 mL, 8.54 mmol) was added and the mixture was cooled to −20°C. Tf2O (0.6 mL, 3.42 mmol) dissolved in CH2Cl2 (1 mL) was added drop-wise and the reaction mixture was allowed to warm up to rt within 1.5 h. The reaction mixture was diluted with CH2Cl2 (20 mL) and then quenched with saturated NaCHO3, washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure at ambient temperature (25–30°C). The resultant yellow syrup was dissolved in toluene, TBAOAc (3.09 g, 10.26 mmol) added, and the mixture stirred at 65°C overnight. The mixture was allowed to cool to rt then diluted with EtOAc. Solvents were evaporated and the resultant residue was purified by column chromatography (hexanes/EtOAc 3.5:1) to yield benzyl-3-O-acetyl-2-deoxy-4,6-O-benzylidene-2-phthalamido-β-d-allopyranoside. −47.0 (c = 0.5, CH2Cl2); 1H NMR (600 MHz, CDCl3) δ 2.07 (s, 3H, COCH3), 3.86–3.93 (m, 2H, H-4, H-6), 4.18–4.22 (m, 1H, H-6), 4.44–4.49 (m, 2H, H-2, H-4), 4.64 (d, 1H, J3 = 10.8 Hz, CHPh), 4.93 (d, CHPh J3 = 12 Hz, CHPh), 5.61 (s, 1H, CHPh), 5.77–5.78 (m, 1H, H-3), 6.15 (d, 1H, J3 = 8.4, H-1), 7.21–7.84 (m, 14H, aromatic). HRMS C30H27NO8Na [M + Na+] calc. 552.1634 found 552.1595. Benzyl-3-O-acetyl-2-deoxy-4,6-O-benzylidene-2-phthalamido-β-d-allopyranoside (0.6 g, 1.13 mmol) was dissolved in MeOH/CH2Cl2 (3:2, 10 mL), cooled to −10°C, and treated with 0.3M NaOMe solution in methanol (3 mL). After 1 h, the reaction was complete as confirmed by TLC and subsequently quenched with Amberlite ion exchange resin (IR 120) to pH 7. Solvents were evaporated and the resultant white solid was coevaporated with toluene (3 × 10 mL) then dried under vacuum for 2 h. The allosamine analog 19 was dissolved in CH2Cl2 (10 mL) in a 50-mL falcon tube and cooled to −5°C. This was followed by dropwise addition of DAST (0.27 mL, 6.78 mmol) and the reaction mixture was allowed to warm to rt over 3 h. The reaction mixture was cooled to −5°C followed by dropwise addition of MeOH to destroy the excess DAST, concentrated, and passed through a short silica column. The resultant mixture of compound was dissolved in MeOH/CH2Cl2 (3:2, 10 mL) followed by addition of AcCl (0.16 mL, 2.26 mmol), then stirred overnight at rt. The reaction mixture was quenched with Et3N to pH 7. Solvents were evaporated in vacuo and the resultant residue was purified by column chromatography (hexanes/EtOAc/MeOH 3:1:0.1) to acquire the desired compound 20 (0.17 g, 28% yield from 18) and elimination product 21 (0.07 g, 14.5%). 20: −64.0 (c = 0.5, CH2Cl2); 1H NMR (600 MHz, CDCl3) δ .29 (s, 1H, OH), 3.11 (s, 1H, OH), 3.52–3.54 (m, 1H, H-5), 3.89–3.99 (m, 3H, H-4, H-6, H-6′), 4.34–4.39 (m, 1H, H-2), 4.53 (d, 1H, J3 = 12 Hz, CHPh), 4.79 (d, 1H, J3 = 12 Hz, CHPh), 5.13–5.25 (m, 3H), 7.05–7.26 (m, 5H, aromatic), 7.22–7.80 (m, 4H, aromatic); 19F NMR (282 MHz, CDCl3) −194.44 (dt, 1F, J3 = 51.9, 13.8 Hz). HRMS C21H20NaO6NF [M + Na+] calc. 424.1167 found 424.1172.
Benzyl-3-deoxy-2-(methylcarbamoyl)benzoate-6-methoxy-4-oxo-β-d-glucopyranoside (21)
−51.4 (c = 0.5, CH2Cl2); 1H NMR (600 MHz, CDCl3) 2.24 (dd, 1H, J3 = 4.2, 13.2 Hz, H-3), 2.48 (t, 1H, J3 = 13.2 Hz, H-3), 3.32 (s, 3H, OCH3), 3.36 (s, 3H, OCH3), 3.73–3.74 (m, 1H, H-5), 3.88–3.96 (m, 2H, H-6, H-6′), 4.43–4.48 (m, 1H, H-2), 4.58 (d, 1H, J3 = 12 Hz, CHPh), 4.79 (d, 1H, J3 = 12 Hz, CHPh), 5.33 (d, 1H, J3 = 8.4 Hz, H-1), 7.08–7.16 (m, 5H, aromatic), 7.70–7.84 (m, 4H, aromatic); 13C NMR (150 Hz, CDCl3) 32.6, 49.4, 49.95, 50.6, 60.9, 71.2, 80.1, 98.7, 99.4, 123.3, 123.6, 127.6, 127.8, 127.9, 128.1, 128.3, 128.5, 131.4, 131.5, 131.7, 134.0, 167.6, 167.8, 204.4, 204.7. HRMS C23H29N2O7 [M + NH4+] calc. 445.1975 found 445.1994.
Benzyl-4,6-tri-O-acetyl-2-acetamido-2-deoxy-3-fluoro-β-d-glucopyranoside (22)
Compound 20 (0.34 g, 0.847 mmol) was dissolved in nbutanol (10 mL) and treated with ethylene diamine (6 mL). The reaction mixture was heated at 90°C for 23 h. Solvents were then removed under reduced pressure and the resultant residue was coevaporated with toluene (3 × 10 mL), which was followed by drying in vacuo for 3 h. The intermediate free amine was dissolved in pyridine (10 mL), followed by addition of Ac2O (6 mL), and was stirred overnight at rt. The mixture was treated with EtOH to react with excess Ac2O, concentrated, dissolved in CH2Cl2, washed with water and brine, and dried over Na2SO4, followed by purification by column chromatography (hexanes/EtOAc/CH2Cl2/MeOH, 2:1:1:0.2) to afford compound 22 (0.27 g, 80%). 1H NMR (600 MHz, CDCl3) δ 1.94 (s, 3H, NHCOCH3), 2.09 (s, 3H, COCH3), 2.10 (s, 3H, COCH3), 3.33–3.38 (m, 1H, H-2), 3.66–3.67 (m, 1H, H-5), 4.14–4.17 (m, 1H, H-6), 4.27 (dd, 1H, J3 = 5, 12.5 Hz, H-6′) 4.57 (d, 1H, J3 = 12.5 Hz, CHPh), 4.88 (d, 1H, J3 = 12 Hz, CHPh), 5.10–5.24 (m, 3H, H-1, H-3, H-4), 5.78 (d, 1H, J3 = 7 Hz, CONH); 19F NMR (282.2 MHz, CDCl3) −193.26 (dt, 1F, J3 = 53.3, 12.4 Hz). HRMS C19H25FNO7 [M + H+] calc. 398.1615 found 398.1595.
1,4,6-Tri-O-acetyl-2-acetamido-2-deoxy-3-fluoro-α-d-glucopyranoside (6)
Compound 22 (20 mg, 0.05 mmol) was dissolved in MeOH (6 mL) and treated with Pd/C (30 mg). The reaction mixture was the stirred under hydrogen for 36 h and the reaction confirmed as complete by TLC. The mixture was filtered and concentrated and the residue coevaporated with toluene (3 × 10 mL) followed by drying under vacuum. The resultant residue was dissolved in pyridine (3 mL), treated with Ac2O (0.5 mL), and stirred overnight at rt. The mixture was concentrated; washed with 1M HCl, saturated NaHCO3, and brine; and dried over Na2SO4. Column purification (hexanes/EtOAc, 1.5:1) afforded compound 6 (15 mg, 76% yield over two steps). +10.1 (c = 1, CH2Cl2); 1H NMR (600 MHz, CDCl3) δ 2.03 (s, 3H, NHCOCH3), 2.09 (s, 3H, COCH3), 2.11 (s, 3H, COCH3), 2.16 (s, 3H, COCH3), 3.93–3.96 (m, 1H, H-5), 4.06–4.09 (m, 1H, H-6), 4.22 (d, 1H, J3 = 4.2, 12.6 Hz, H-6′), 4.58–4.69 (m, 2H, H-2, H-3), 5.25–5.30 (m, 1H, H-4), 5.52 (d, 1H, J3 = 7.8 Hz, NH), 6.20 (t, 1H, J3 = 3 Hz, H-1); 13C NMR (150 Hz, CDCl3) 20.6, 20.7, 20.8, 23.2, 50.9, 50.96, 61.3, 68.1, 68.2, 69.5, 69.6, 88.97, 90.2, 90.9, 91.0, 168.4, 169.0, 170.0, 170.7; 19F NMR (282.2 MHz, CDCl3) −194.24 (dt, 1F, J3 = 50.8, 13.5 Hz,). HRMS C14H24N2O8F [M + NH4+] calc. 367.1517 found 367.1512.
Determination of HA Concentration by an ELISA-Like Assay
KP1-NL cells (passage 3) were seeded in 35-mm culture dishes (6 × 105 cells/dish) and cultured for 24 h. Cells were further cultured in fresh medium containing each compound for 48 h. The culture supernatants were collected and the cells were lysed by 1% Nonidet P-40, 140 mM NaCl, 10 mM EDTA, and 20 mM Tris-HCl (pH 7.4). HA released into the culture medium and accumulated in cell layers was quantified by an ELISA-like assay using HA-binding protein (HABP) according to the manufacturer's instructions for the Hyaluronan Assay Kit (Seikagaku Co., Tokyo, Japan). The absorbance at 490 nm (control wavelength, 630 nm) of each well was measured by a microplate spectrophotometer xMark (Bio-Rad, Tokyo, Japan). The quantity of HA was expressed per live cell number. The viability of cells was assessed by trypan blue staining.
Particle Exclusion Assay
KP1-NL cells (passage 5) plated at 2.5 × 104 cells in a 35-mm dish were cultured for 24 h and then further cultured for 48 h with or without 100 μM of the compound of interest in fresh medium containing 0.1% DMSO. An aliquot of glutaraldehyde-stabilized sheep erythrocytes (5 × 108) in 0.75 mL of PBS was then added to the culture medium. After 15 min, the culture was observed using an inverted phase-contrast microscope (Olympus IMT-2).[39] The sizes of the clear areas were quantified using ImageJ and the standard deviations of at least five areas in each image were reported.
Estimation of Cell Proliferation
Cell numbers were estimated using the Alamar Blue assay, which is based on the oxidation–reduction reaction of cells.[40] Briefly, KP1-NL cells (passage 3, 625 cells/well) were cultured overnight in 90 μL of medium in 96-well flat-bottomed microplates (Iwaki Glass Co., Chiba, Japan) in triplicate and were further cultured for 72 h with or without 0.1 mM of the test compound in freshly served medium containing 0.1% DMSO. Alamar Blue working solution (10 μL, BioSource, Camarillo, CA) was then added to each well and the plate was further incubated for 3 h at 37°C in a CO2 incubator. The fluorescence of each well was measured at ex 544 nm and em 590 nm using a Fluoroskan II (Japan Flow Laboratories, Tokyo, Japan). Reported values represented the means of replicate wells and the standard deviations for each are reported. The reaction was linear in the range of 300–3500 fluorescence units, corresponding to 300–20,000 KP1-NL cells/well.
Supplementary Material
Figure 1.
Structures of compounds 1–3 (color figure available online).
Figure 2.

Structures of compounds 4–6 (color figure available online).
Acknowledgments
We thank the National Institute of General Medical Sciences (R01 GM72667), NIH, for financial support of our work. This work was also partially supported by the Regional Innovation Strategy Support Program (City Area Type) of the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Footnotes
Supporting Information Available: Selected NMR spectra and images of the particle exclusion assay at low magnification.
References
- 1.Lapcik L, Jr, Lapcik L, De Smedt S, Demeester J, Chabrecek P. Hyaluronan: preparation, structure, properties, and applications. Chem Rev. 1998;98:2663–2684. doi: 10.1021/cr941199z. [DOI] [PubMed] [Google Scholar]
- 2.Misra S, Ghatak S, Zoltan-Jones A, Toole BP. Regulation of multidrug resistance in cancer cells by hyaluronan. J Biol Chem. 2003;278:25285–25288. doi: 10.1074/jbc.C300173200. [DOI] [PubMed] [Google Scholar]
- 3.Delpech B, Girard N, Bertrand P, Courel NM, Chauzy C, Delpech A. Hyaluronan: fundamental principles and applications in cancer. J Intern Med. 1997;242:41–48. doi: 10.1046/j.1365-2796.1997.00172.x. [DOI] [PubMed] [Google Scholar]
- 4.Knudson W. Tumor associated hyaluronan: providing an extracellular matrix that facilitates invasion. Am J Pathol. 1996;148:1721–1726. [PMC free article] [PubMed] [Google Scholar]
- 5.Franzmann EJ, Schroeder GL, Goodwin WJ, Weed DT, Fisher P, Lokeshwar VB. Expression of tumor markers hyaluronic acid and hyaluronidase (HYAL1) in head and neck tumors. Int J Cancer. 2003;106:438–445. doi: 10.1002/ijc.11252. [DOI] [PubMed] [Google Scholar]
- 6.Posey JT, Soloway MS, Ekici S, Sofer M, Civantos F, Duncan RC, Lokeshwar VB. Evaluation of the prognostic potential of hyaluronic acid and hyaluronidase (HYAL1) for prostate cancer. Cancer Res. 2003;63:2638–2644. [PubMed] [Google Scholar]
- 7.Pirinen R, Tammi R, Tammi M, Hirvikoski P, Parkkinen JJ, Johansson R, Böhm J, Hollmén S, Kosma VM. Prognostic value of hyaluronan expression in non-small-cell lung cancer: increased stromal expression indicates unfavorable outcome in patients with adenocarcinoma. Int J Cancer. 2001;95:12–17. doi: 10.1002/1097-0215(20010120)95:1<12::aid-ijc1002>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 8.Setälä LP, Tammi MI, Tammi RH, Eskelinen MJ, Lipponen PK, Ågren UM, Parkkinen J, Alhava EM, Kosma VM. Hyaluronan expression in gastric cancer cells is associated with local and nodal spread and reduced survival rate. Br J Cancer. 1999;79:1133–1138. doi: 10.1038/sj.bjc.6690180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lokeshwar VB, Öbek C, Soloway MS, Block NL. Tumor-associated hyaluronic acid: a new sensitive and specific urine marker for bladder cancer. Cancer Res. 1997;57:773–777. [PubMed] [Google Scholar]
- 10.Weigel PH, Hascall VC, Tammi M. Hyaluronan synthases. J Biol Chem. 1997;272:13997–14000. doi: 10.1074/jbc.272.22.13997. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura T, Takagaki K, Shibata S, Tanaka K, Higuchi T, Endo M. Hyaluronic-acid-deficient extracellular matrix induced by addition of 4-methylumbelliferone to the medium of cultured human skin fibroblasts. Biochem Biophys Res Commun. 1995;208:470–475. doi: 10.1006/bbrc.1995.1362. [DOI] [PubMed] [Google Scholar]
- 12.Nakazawa H, Yoshihara S, Kudo D, Morohashi H, Kakizaki I, Kon A, Takagaki K, Sasaki M. 4-Methylumbelliferone, a hyaluronan synthase suppressor, enhances the anticancer activity of gemcitabine in human pancreatic cancer cells. Cancer Chemother Pharmacol. 2006;57:165–170. doi: 10.1007/s00280-005-0016-5. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 13.Kakizaki I, Kojima K, Takagaki K, Endo M, Kannagi R, Ito M, Maruo Y, Sato H, Yasuda T, Mita S, Kimata K, Itano N. A novel mechanism for the inhibition of hyaluronan biosynthesis by 4-methylumbelliferone. J Biol Chem. 2004;279:33281–33289. doi: 10.1074/jbc.M405918200. [DOI] [PubMed] [Google Scholar]
- 14.Kultti A, Pasonen-Seppänen S, Jauhiainen M, Rilla KJ, Kärnä R, Pyöriä E, Tammi RH, Tammi MI. 4-Methylumbelliferone inhibits hyaluronan synthesis by depletion of cellular UDP-glucuronic acid and downregulation of hyaluronan synthase 2 and 3. Exp Cell Res. 2009;315:1914–1923. doi: 10.1016/j.yexcr.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 15.Kudo D, Kon A, Yoshihara S, Kakizaki I, Sasaki M, Endo M, Takagaki K. Effect of a hyaluronan synthase suppressor, 4-methylumbelliferone, on B16F-10 melanoma cell adhesion and locomotion. Biochem Biophys Res Commun. 2004;321:783–787. doi: 10.1016/j.bbrc.2004.07.041. [DOI] [PubMed] [Google Scholar]
- 16.Piccioni F, Malvicini M, Garcia MG, Rodriguez A, Atorrasagasti C, Kippes N, Buena ITP, Rizzo MM, Bayo J, Aquino J, Viola M, Passi A, Alaniz L, Mazzolini G. Antitumor effects of hyaluronic acid inhibitor 4-methylumbelliferone in an orthotopic hepatocellular carcinoma model in mice. Glycobiology. 2012;22:400–410. doi: 10.1093/glycob/cwr158. [DOI] [PubMed] [Google Scholar]
- 17.Urakawa H, Nishida Y, Wasa J, Arai E, Zhuo L, Kimata K, Kozawa E, Futamura N, Ishiguro N. Inhibition of hyaluronan synthesis in breast cancer cells by 4-methylumbelliferone suppresses tumorigenicity in vitro and metastatic lesions of bone in vivo. Int J Cancer. 2012;130:454–466. doi: 10.1002/ijc.26014. [DOI] [PubMed] [Google Scholar]
- 18.Lokeshwar VB, Lopez LE, Munoz D, Chi A, Shirodkar SP, Lokeshwar SD, Escudero DO, Dhir N, Altman N. Antitumor activity of hyaluronic acid synthesis inhibitor 4-methylumbelliferone in prostate cancer cells. Cancer Res. 2010;70:2613–2623. doi: 10.1158/0008-5472.CAN-09-3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh S, Blumenthal HJ, Davidson E, Roseman S. Glucosamine metabolism. V. Enzymatic synthesis of glucosamine 6-phosphate. J Biol Chem. 1960;235:1265–1273. [PubMed] [Google Scholar]
- 20.Boehmelt G, Wakeham A, Elia A, Sasaki T, Plyte S, Potter J, Yang YJ, Tsang E, Ruland J, Iscove NN, Dennis JW, Mak TW. Decreased UDP-GlcNAc levels abrogate proliferation control in EMeg32-deficient cells. EMBO J. 2000;19:5092–5104. doi: 10.1093/emboj/19.19.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vocadlo DJ, Hang HC, Kim EJ, Hanover JA, Bertozzi CR. A chemical approach for identifying O-GlcNAc modified proteins in cells. Proc Natl Acad Sci USA. 2003;100:9116–9121. doi: 10.1073/pnas.1632821100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gloster TM, Vocadlo DJ. Developing inhibitors of glycan processing enzymes as tools for enabling glycobiology. Nature Che mBiol. 2012;8:683–694. doi: 10.1038/nchembio.1029. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 23.Dimitroff CJ, Kupper TS, Sackstein R. Prevention of leukocyte migration to inflamed skin with a novel fluorosugar modifier of cutaneous lymphocyte-associated antigen. J Clin Invest. 2003;112:1008–1018. doi: 10.1172/JCI19220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gold H, Munneke S, Dinkelaar J, Overkleeft HS, Aerts JMG, Codée JDC, van der Marel GA. A practical synthesis of capped 4-methylumbelliferyl hyaluronan disaccharides and tetrasaccharides as potential hyaluronidase substrates. Carbohydr Res. 2011;346:1467–1478. doi: 10.1016/j.carres.2011.03.042. [DOI] [PubMed] [Google Scholar]
- 25.Danac R, Ball L, Gurr SJ, Fairbanks AJ. Synthesis of UDP-glucose derivatives modified at the 3-OH as potential chain terminators of β-glucan biosynthesis. Carbohydr Res. 2008;343:1012–1022. doi: 10.1016/j.carres.2008.01.031. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 26.Muller T, Danac R, Ball L, Gurr SJ, Fairbanks AJ. Synthesis of UDP-GlcNAc derivatives modified at OH-4 as potential chain-terminators of chitin biosynthesis. Tetrahedron Assym. 2007;18:1299–1307. [Google Scholar]
- 27.Kisilevsky R, Szarek WA, Ancsin JB, Elimova E, Marone S, Bhat S, Berkin A. Inhibition of amyloid A amyloidogenesis in vivo and in tissue culture by 4-deoxy analogues of peracetylated 2-acetamido-2-deoxy-α- and β-d-glucose. Am J Pathol. 2004;164:2127–2137. doi: 10.1016/s0002-9440(10)63771-6. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yeager AR, Finney NS. Synthesis of fluorescently labeled UDP-GlcNAc analogues and their evaluation as chitin synthase substrates. J Org Chem. 2005;70:1269–1275. doi: 10.1021/jo0483670. [DOI] [PubMed] [Google Scholar]
- 29.Sharma M, Bernacki RJ, Hillman MJ, Korytnyk W. Fluorinated carbohydrates as potential plasma membrane modifiers. Synthesis of 3-deoxy-3-fluoro derivatives of 2-acetamido-2-deoxy-d-hexopyranoses. Carbohydr Res. 1993;240:85–93. doi: 10.1016/0008-6215(93)84174-5. [DOI] [PubMed] [Google Scholar]
- 30.Dube DH, Bertozzi CR. Metabolic oligosaccharide engineering as a tool for glycobiology. Curr Opin Chem Biol. 2003;7:616–625. doi: 10.1016/j.cbpa.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 31.Cai Y, Ling CC, Bundle DR. Facile approach to 2-acetamido-2-deoxy-β-d-glucopyranosides via a furanosyl oxazoline. Org Lett. 2005;7:4021–4024. doi: 10.1021/ol051523k. [DOI] [PubMed] [Google Scholar]
- 32.Horton D, Hughes JB, Jewell JS, Philips KD, Turner WN. Anomeric equilibria in derivatives of amino sugars. Nuclear magnetic resonance studies on acetylated amino sugars and specifically deuterated analogs. J Org Chem. 1967;32:1073–1080. doi: 10.1021/jo01279a048. [DOI] [PubMed] [Google Scholar]
- 33.Kajihara Y, Kodama H, Endo T, Hashimoto H. Novel features of acceptor recognition by β-(1–4)-galactosyltransferase. Carbohydr Res. 1998;306:361–378. [Google Scholar]
- 34.Wang Z, Zhou L, El-Boubbou K, Ye XS, Huang X. Multi-component one-pot synthesis of the tumor-associated carbohydrate antigen Globo-H based on preactivation of thioglycosyl donors. J Org Chem. 2007;72:6409–6420. doi: 10.1021/jo070585g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hernandez-Torres JM, Liew ST, Achkar J, Wei A. Optimized synthesis of an orthogonally protected glucosamine. Synthesis. 2002:487–490. [Google Scholar]
- 36.Ogawa T, Nakabayashi S. Synthetic studies on cell surface glycans. Part XI. Synthesis of 3,6-di-O-acetyl-2-deoxy-2-phthalimido-4-O-(2,3,4,6-tetra-O-acetyl-β-d-galactopyranosyl)-β-d-glucopyranosyl chloride. Carbohydr Res. 1981;97:81–86. [Google Scholar]
- 37.Barthel SR, Antonopoulos A, Cedeno-Laurent F, Schaffer L, Hernandez G, Patil SA, North SJ, Dell A, Matta KL, Neelamegham S, Haslam SM, Dimitroff CJ. Peracetylated 4-fluoro-glucosamine reduces the content and repertoire of N- and O-glycans without direct incorporation. J Biol Chem. 2011;286:21717–21731. doi: 10.1074/jbc.M110.194597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mack H, Basabe JV, Brossmer R. 2-Acetamido-2-deoxy-d-gluco- and -d-manno-furanose. A simple preparation of 2-acetamido-2-deoxy-d-mannose. Carbohydr Res. 1988;175:311–316. doi: 10.1016/0008-6215(88)84154-5. [DOI] [PubMed] [Google Scholar]
- 39.Knudson W, Knudson CB. Assembly of a chondrocyte-like pericellular matrix on non-chondrogenic cells. Role of the cell surface hyaluronan receptors in the assembly of a pericellular matrix. J Cell Sci. 1991;99:227–235. doi: 10.1242/jcs.99.2.227. [DOI] [PubMed] [Google Scholar]
- 40.Ahmed SA, Gogal RMJ, Walsh JE. A new rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes: an alternative to [3H]thymidine incorporation assay. J Immunol Methods. 1994;170:211–224. doi: 10.1016/0022-1759(94)90396-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.