

Abstract
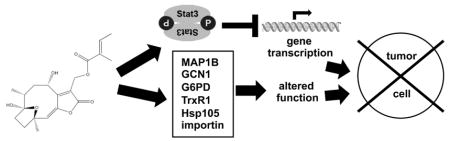
We report that hirsutinolides series, 6, 7, 10, 11, 20 and 22 and the semi-synthetic analogs, 30, 31, 33 and 36 inhibit constitutively-active Signal transducer and activator of transcription (Stat)3 and malignant glioma phenotype. A position 13 lipophilic ester group is required for activity. Molecular modeling and NMR structural analyses reveal direct hirsutinolide:Stat3 binding. One-hour treatment of cells with 6 and 22 also upregulated importin subunit α-2 levels and repressed translational activator GCN1, microtubule-associated protein (MAP)1B, thioredoxin reductase (TrxR)1 cytoplasmic isoform 3, glucose-6-phosphate 1-dehydrogenase isoform a, Hsp105, vimentin, and tumor necrosis factor α-induced protein (TNAP)2 expression. Active hirsutinolides inhibited anchorage-dependent and 3D-spheroid growth, survival, and migration of human glioma lines and glioma patients’ tumor-derived xenograft cells harboring constitutively-active Stat3. Oral gavage delivery of 6 or 22 inhibited human glioma tumor growth in subcutaneous mouse xenografts. The inhibition of Stat3 signaling represents part of the hirsutinolide-mediated mechanisms to induce antitumor effects.
Keywords: Natural products, hirsutinolides, Stat3, glioma, xenografts, antitumor effects, GCN1, Vimentin, importin α-2, microtubule-associated protein 1B, thioredoxin reductase 1 cytoplasmic isoform 3, glucose-6-phosphate 1-dehydrogenase isoform a, heat shock protein105, tumor necrosis factor α-induced protein 2
Graphical Abstract
Introduction
Human glioma is an aggressive and fatal malignant brain tumor that lacks effective treatments. Notable molecular aberrations that support malignant glioma include constitutively-active Stat3 1. The Stat family of cytoplasmic transcription factors is normally activated transiently via tyrosine (Tyr) phosphorylation by Janus kinases (JAKs), growth factor receptor kinases, or Src. Phosphorylation leads to Stat:Stat dimerization and nuclear translocation, and Stat-dependent gene regulation. Stat activation controls cell growth and proliferation, survival, and other physiological responses to growth factors and cytokines 2. By contrast, constitutively-active Stat3 dysregulates gene expression to support uncontrolled cell growth and survival, angiogenesis, and metastasis, thereby promoting tumorigenesis and tumor progression 3–5. Moreover, Stat3 suppresses tumor immune surveillance 4, 6, maintains a cross-talk with nuclear factor-kappa (NF-κ)B 7, and modulates mitochondrial biochemical processes 8 that also promote malignant development. Accordingly, Stat3 has become the focus of many drug discovery programs to develop novel anticancer therapeutics 5. In many proof-of-concept studies, the inhibition of constitutively-active Stat3 by small-molecule Stat3:Stat3 dimerization disruptors and other inhibitors of JAK-Stat3 signaling suppresses tumor cell growth in vitro and tumor growth in vivo 5, 9–21.
We made the discovery from the screening of an in-house natural product library that the hirsutinolide series of compounds, 6, 7, 10, 11, 20 and 22 inhibit Stat3 signaling. The semi-synthetic derivatives, 30, 31, 33, 38, 39, 40, 35, 37 and 36 are novel, and they as well as 6, 7, 10, 20 and 22 inhibited constitutively-active Stat3 at 1–5 μM. A lipophilic ester functionality at position 13 is vital for activity. Structurally suitable hirsutinolides directly disrupted cell-free Stat3 DNA-binding activity in vitro, presumably by binding to Stat3, as suggested by nuclear magnetic resonance (NMR) and molecular modeling studies. Moreover, global proteomic and immunoblotting analyses of whole-cell lysates from glioma cells treated for one hour with 6 or 22 showed altered expression of Hsp105, vimentin, TNAP2, TrxR1 cytoplasmic isoform 3, G6PD isoform a, MAP1B, and importin subunit α-2 proteins, suggesting the modulation of these proteins contributes to the antitumor responses to the hirsutinolides. Compounds 6, 10 and 22 suppressed proliferation, colony formation, and migration in vitro and in 3D spheroid culture of glioma cells harboring aberrantly-active Stat3. Furthermore, 6 and 22 inhibited tumor growth in vivo of subcutaneous human glioma xenografts, which is associated with the downregulation of pStat3 levels in residual tumor tissues from treated mice.
Results
Hirsutinolides and semi-synthetic analogs preferentially inhibit Stat3 activity outside and inside of tumor cells
A panel of hirsutinolides (Fig. 1A, B) were evaluated at 5 μM for inhibitory activities against Stat3 DNA-binding activity in vitro, as measured by electrophoretic mobility shift assay (EMSA) 22, 23. Nuclear extracts from v-Src-transformed mouse fibroblasts (NIH3T3/v-Src) containing activated Stat3:Stat3 dimers 24, 25 were first incubated with the hirsutinolides (5 μM) for 30 min at room temperature prior to incubating with the radiolabeled high-affinity sis-inducible element (hSIE) probe that binds Stat1 and Stat3 and performing EMSA analysis in vitro 9, 22, 23, 26. Stat3:Stat3/DNA-binding activity (complexation) was diminished by 6, 7, 10, 11, 20 and 22, while 8, 9, 14, 18, 19, 21, and 28 only weakly inhibited Stat3 activity (Fig 2A). To further investigate the specificity of the hirsutinolides, nuclear extract preparation from EGF-stimulated mouse fibroblasts (NIH3T3/hEGFR) that contains active Stat1, Stat3 and Stat5 were preincubated with 5 μM hirsutinolides for 30 min at room temperature prior to incubation with the radiolabled mammary gland factor element (MGFe) probe that binds Stat1 and Stat5 or the hSIE probe and performing EMSA analysis. Results show the active hirsutinolides, 6, 7, 10, 11, 20 or 22 preferentially inhibit Stat3:Stat3/DNA-binding activity [Fig. 2B(i), upper band; residual activity as percent of control (C) for each band is represented in the lower panel] ahead of inhibition of Stat1:Stat3 activity [Fig. 2B(i), middle band], compared to weak effects against Stat1:Stat1 activity [Fig. 2B(i) and (ii), lower band) or Stat5:Stat5 activity [Fig. 2B(ii), upper band]. Results together show that active hirsutinolides, 6, 7, 10, 11, 20 or 22 directly interfere with Stat3:Stat3 DNA-binding activity and further that they preferentially inhibit Stat3 over Stat1 or Stat5 activity.
Figure 1. Chemical structures of hirsutinolide natural products and semi-synthetic derivatives.

(A and B) Hirsutinolide natural products; and (C) newly semi-synthesized analogs of 22.
Figure 2. Hirsutinolide natural products inhibit Stat3 activation.

(A and B) Nuclear extracts containing activated (A) Stat3 from NIH3T3/v-Src fibroblasts or (B) Stat1, Stat3 and Stat5 from EGF-stimulated NIH3T3/hEGFR were pre-incubated with the designated hirsutinolides for 30 min at room temperature prior to incubating with the radiolabeled hSIE probe that binds Stat1 and Stat3 or MGFe probe that binds Stat1 and Stat5 and performing EMSA analysis; bands corresponding to STAT:DNA complexes in gel were quantified using ImageQuant and represented as percent of control (B (i), lower panel); (C and D) Stat3 DNA-binding activity with EMSA analysis using the hSIE probe that binds Stat3 of nuclear extracts prepared from U251MG or U373MG cells treated with the designated hirsutinolides at (C) 5 μM for 0–24 h or (D) 0–5 μM for 1.5 h; (E–G) Immunoblotting analysis of whole-cell lysates prepared from (E and F) U251MG cells treated with the designated hirsutinolides at (E) 5 μM for 0–24 h or (F) 0–5 μM for 2 h, or (G) human glioma patients-derived xenograft cells (i) G6-G102, untreated, or (ii) G22, treated with 5 μM for 0–24 h 6, 10, or 22 and probing for pStat3, Stat3 or GAPDH. Positions of proteins or DNA-bound STATs in gel are labeled; control lane (c, 0) represents whole-cell lysates or nuclear extracts prepared from 0.025% DMSO-treated cells or nuclear extracts pre-treated with 0.025% DMSO. Bands corresponding to Stat3:DNA complexes were scanned and quantified using ImageJ, plotted against concentration of agent from which IC50 values were derived. Data are representative of 2–3 independent determinations. *Position of supershifted complex.
Given the inhibitory effects of 6, 7, 10, 11, 20 or 22 against Stat3 DNA-binding activity in the cell-free EMSA analysis, we investigated the ability of the hirsutinolides to inhibit Stat3 activation in tumor cells. U251MG and U373MG human glioma cells harboring constitutively-active Stat3 were treated with 6, 7, 10, 11, 20 and 22 or the inactive hirsutinolides, 8, 9, 14, 18, 19, 21 and 28 as control and nuclear extracts were prepared and subjected to Stat3 DNA-binding assay/EMSA analysis using hSIE probe or whole-cell lysates were prepared and subjected to immunoblotting analysis. Results showed treatment of tumor cells with 6, 7, 10, 20 and 22 strongly inhibited Stat3 DNA-binding activity (Fig. 2C, D) and pStat3 induction (Fig. 2E, F) in a time-dependent manner, with inhibition occurring as early as 0.5 h and sustaining up to 6 h (Fig. 2C, E). Stat3 activity seemed to recover at 6–24 h (Fig. 2C, E). Moreover, we observed a dose-dependent inhibition of pStat3 (Fig. 2F) and Stat3 DNA-binding activity, with IC50 of 2.2–4.1 μM (Fig. 2D). Supershift studies with anti-Stat3 C20X antibody confirm Stat3/DNA complex (Fig. 2C, upper panel, last lane). We were interested to determine the effects of the active compounds on Stat3 activation in human glioma patient-derived mouse xenograft (pdx) cells. Consistent with published reports 27, 28, cultured pdx cells from different patients (G6-G102) showed different pStat3 levels, with moderate-to-high levels detected in 14 out of 23 samples [Fig. 2G(i)]. Phospho-Stat3 in the model G22 cells was strongly inhibited early at 3 h upon treatment with 5 μM 6 or 22, compared to moderate inhibition by 10 [Fig. 2G(ii)], while at later, 24 h time point, it was restored, although these cells lose viability (Fig. S5C). Surprisingly, similar treatment of glioma cells with the active agent, 11 (5 μM) had a weak effect on both Stat3 DNA-binding activity (Fig. 2C, 11) or pStat3 (Fig 2E, 11), despite its strong inhibitory effect on Stat3 DNA-binding activity in the cell-free EMSA analysis [Fig. 2A(ii), 11]. This descrepancy is likely due to poor membrane permeability of 11 posed by the presence of two hydroxyl functional groups. As expected, similar treatments of tumor cells with the inactive hirsutinolides, 9, 8, 14, 18, 19, 21, and 28 had little impact on pStat3 (Fig. S1A), consistent with their weak effect on Stat3 DNA-binding activity in the cell-free EMSA analysis [Fig. 2A(ii)].
We sought to identify the structural elements that contribute to the inhibitory activity of the hirsutinolides against Stat3 and pursued an extended structure-activity relationship analysis. Due to the Stat3-inhibitory activity of 22, a focused series of its semi-synthesized analogs were derived (Fig. 1C) using 14, 8 or 17 as starting materials to allow us to explore the importance of the position 13. Semisynthetic analogs were evaluated in the cell-free EMSA analysis at 5 μM for inhibitory activities against Stat3 DNA-binding activity in vitro 22, 23. Nuclear extracts from NIH3T3/v-Src containing activated Stat3:Stat3 dimers 24, 25 were first incubated with the semi-synthetic analogs (5 μM) for 30 min at room temperature prior to incubating with the radiolabeled hSIE probe that binds Stat1 and Stat3 and performing EMSA analysis in vitro 9, 22, 23, 26. Stat3:Stat3/DNA-binding activity (complexation) was diminished by 30, 31, 33, 32, 38, 39, 34, 35, 37 and 36, while 29 and 40, had moderate inhibitory effects (Fig. S2A). Therefore, the majority of the semi-synthetic analogs retained the activity of 22. We used 31 as the representative compound to examine the specificity of the analogs. Pre-incubation of nuclear extracts from EGF-stimulated NIH3T3/hEGFR that contain activated Stat1 and Stat3 with 5 μM 31 for 30 min at room temperature prior to incubation with the radiolabeled hSIE probe and performing EMSA analysis shows a stronger inhibition of Stat3:Stat3 activity over that of Stat1:Stat3, compared to the moderate effect on Stat1:Stat1 activity (Fig. S2B). Therefore, this analog retains the specificity of compound 22.
We next treated U251MG cells with 5 μM semi-synthetic analogs and prepared nuclear extracts for Stat3 DNA-binding assay with EMSA analysis or prepared whole-cell lysates for immunoblotting analysis. Results showed treatment with 30, 31, 33, 38, 39 and 36 suppressed Stat3 DNA-binding activity (Fig. S2C) and pStat3 (Fig. S2D) in a dose-dependent manner, with IC50 of 0.5–2.5 μM (Fig. S2C), indicating the analogs retained or enhanced their activity compared to the parent compound, 22. Phospho-Stat3 levels similarly recovered at 6–24 h [Fig. S2D(i)]. By contrast, treatment of U251MG cells with the weakly-active 29 had little effect on pStat3 induction (Fig. S2E), consistent with its lower activity against Stat3 DNA-binding activity in the cell-free EMSA analysis (Fig. S2A, 29). Surprisingly, treatment of U251MG cells with 32 had moderate effect on pStat3 (Fig. S2E), despite its strong inhibitory activity against Stat3 DNA-binding activity in the cell-free EMSA analysis (Fig. S2A, 32). Similar to 11, the two hydroxyl moieties may have detrimental effect on the cell membrane permeability of 32.
Structure-activity relationship (SAR) analysis
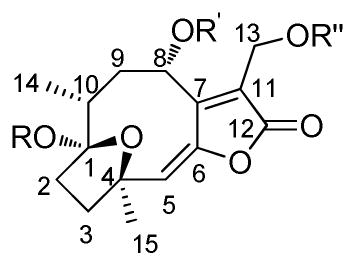
As seen for 14, the hirsutinolide scaffold alone is insufficient for Stat3-inhibitory activity. The lipophilic side chains at positions 8 and 13 promote activity (compare 14 to 6, 7, 10, 20 and 22; Table 1). The position 13 occupation by a lipophilic group is required and sufficient for activity (compare 8, 14, 15, 18, 19 and 28 to 6, 7, 10, 20 and 22), while position 8 occupation alone is insufficient (compare 8 to 6, or 19 to 20). The evaluation of the semisynthetic analogs revealed that the majority of them showed improved or retained inhibitory activity as 22 (Fig. 1C). A bulkier/longer carbon chain group at position 13 enhances intracellular activity (compare 22 to 11, and 30, 31, 33, 34, 35, 37 and 29 to 29 and 32), while a smaller group (e.g., acetoxy) confers Stat3-inhibitory activity outside of cells [see 11, Fig. 2A(ii), 29, Fig. S2A]. Moreover, a methoxy group at position 1 diminishes activity (compare 9 to 7, and 10 to 6). The observation that 11, 32, and to some extent, 29, with smaller position 13 side chains (e.g., acetoxy, 2-methylbutanoate, or 3-methyl-2-butenoate) alone directly inhibited Stat3:Stat3/DNA complexation in the cell-free EMSA analysis (Fig. 2A, 11, and Fig. S2A, 29, 32], suggests that their low intracellular activities may reflect a poor cell membrane permeability caused by the two hydroxyl groups at positions 1 and 8. The membrane permeability impact of the hydroxyl groups may be offset by the bulkier, more lipophilic group at position 13, as observed for 22.
Table 1.
Compounds, and their side chains, inhibitory activities against U251MG, MDA-MB-231, NIH3T3 cell growth, and Stat3-inhibitory activity inside and outside of cells.
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cpd | R | R′ | R″ | Inhibition of cell growth | Inhibition against Stat3 signaling | |||||
| U251MG | MDA-MB-231 | SF295 | NIH 3T3 | pYStat3 (cellular) | DNA binding activity (cellular) | Stat3/DNA complexation (cell-free) | ||||
| 6 | H |
|
|
+++ | +++ | ++ | +++ | +++ | +++ | ++ |
| 7 | H |
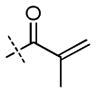
|
|
++ | + | +/− | + | ++ | nd | ++ |
| 8 | H |
|
H | +/− | +/− | +/− | − | − | nd | − |
| 9 | CH3 |
|
|
+/− | − | − | − | − | − | − |
| 10 | CH3 |
|
|
++ | + | − | +/− | ++ | +++ | + |
| 11 | H | H |
|
+/− | +/− | − | − | − | + | +++ |
| 14 | H | H | H | − | − | +/− | − | − | nd | − |
| 15 | CH3 | H | H | +/− | +/− | − | − | nd | nd | nd |
| 17 | CH3 |
|
H | +/− | − | +/− | − | nd | nd | nd |
| 18 | H |
|
H | +/− | − | − | − | − | nd | − |
| 19 | H |
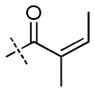
|
H | +/− | − | − | − | − | nd | − |
| 20 | H |
|
|
++ | +/− | +/− | +/− | ++ | nd | +++ |
| 21 | H |
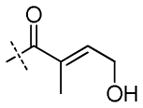
|
H | − | − | − | − | − | nd | − |
| 22 | H | H |
|
++ | +/− | +/− | +/− | +++ | +++ | +++ |
| 28 | CH3 |
|
H | +/− | − | +/− | − | − | nd | − |
Bold, most active agents; nd, not determined; +++ strongly active; ++ very active; + active; +/− moderately active or less active; − no activity; activities are measured at 5 μM; cpd, compound
Active hirsutinolides have little effects on Stat1, Stat5, JAK2, Akt, Erk1/2, SOCS or protein tyrosine phosphatases induction and block orthovanadate-induced pStat3
To determine the possibility of widespread effects on signaling proteins, cells were treated with compounds and whole-cell lysates prepared for immunoblotting analysis. Pre-treatment of NIH3T3/hEGFR fibroblasts with 5 μM 6, 10, 20 or 22 had no effect on EGF-stimulated pStat1, pStat5 or pErk1/2MAPK levels, compared to effects on pStat3 (Fig. 3A), or no effects on pS-Akt, pErk1/2MAPK, suppressors of cytokine signaling (SOCS)3, or protein tyrosine phosphatase (PTP)1B levels in U251MG glioma cells (Fig. 3B). Furthermore, treatment of cells with 10–300 μM sodium orthovanadate (PTP inhibitor) together with 5 μM 6, 10 or 22 had minimal impact on hirsutinolide-mediated pStat3 inhibition, while the hirsutinolides suppressed the robust pStat3 induction by orthovanadate (Fig. 3C, lanes 2 vs. 1, 4 vs. 3, 6 vs. 5, 8 vs. 7, and 10 vs. 9). Therefore, our data here together suggest inside cells, the active hirsutinolides have little effects on Stat1, Stat5, Erk1/2, Akt, SOCS or protein tyrosine phosphatase 1B induction, except for an apparent moderate increase in pJAK2 by compound 6, while disrupting Stat3 induction.
Figure 3. Hirsutinolides do not modulate Stat1, Stat5, JAK2, Akt, Erk1/2, SOCS3, or PTP induction.

(A–C) Immunoblotting analysis of whole-cell lysates of equal total protein prepared from (A) NIH3T3/hEGFR fibroblasts pre-treated with or without 5 μM of 6, 10, 20 or 22 for 3 h prior to stimulation with 1 μg/ml EGF for 12 min, (B) U251MG cells treated with 5 μM 6, 10 or 22 for 0–24 h, or (C) U251MG cells treated with 5 μM 6, 10 or 22 for 3 h in the absence or presence of 10–300 μM Na3VO4 and probing for pStat3, Stat3, pStat1, Stat1, pStat5, Stat5, pErk1/2, Erk1/2, pS-Akt, Akt, pJAK2, JAK2, SOCS3, PTP1B, β-actin or GAPDH. Positions of proteins in gel are labeled; control lanes (0, −) represent whole-cell lysates prepared from 0.025% DMSO-treated cells. Data are representative of 2–4 independent determinations.
Computational modeling predicts active hirsutinolides directly bind to Stat3
An unbiased computational modeling analysis of Stat3:hirsutinolide binding (MOE Site Finder, Molecular Operating Environment version 2013, Chemical Computing Group, Montreal, QC) mapped thirty-six potential binding sites within Stat3 (Stat3β/DNA complex, Protein Data Bank ID: 1BG1). Correlation was modest, but the DNA binding domain (DBD) provided the best correlation to the inhibitory against Stat3:Stat3/DNA complexation in Figure 2A out of the potential other thirty-six virtual binding sites studied (Table 2, 33 and 31Fig. 4A). In this context, key hydrophobic interactions between the lipophilic side chains and Stat3 Val375 and Leu378, with or without H-bonding with Arg379, Ser381, and/or Gly380 were evident for the most active compounds. For example, the potent inhibitors, overlap well in their respective position 13 lipophilic side chain interactions with Val375, while 31 has an additional hydrophobic interaction with Leu378 and H-bonding with Arg379 and Ser381 (Fig. 4A, upper two panels). The similarly potent 22, with a truncated position 13 lipophilic side chain, has hydrophobic interaction with Leu378 (Fig. 4A, 22). The potent compound 6, which has the position 13 side chain truncated, engages in hydrophobic interactions between its position 8 appendage and Leu378, and between its position 14 methyl group and Val375, and H-bonding with Arg379 and Gly380 (Fig. 4A, 6). By contrast, the less-active 29 (side-chain is saturated) makes several H-bonds and no hydrophobic interactions with Val375 and Leu378 (Fig. 4A, 29).
Table 2.
MOE docking scores of isolated natural hirsutinolides and semi-synthetic analogs.
| Compound | Dock score (~kcal/mol) |
|---|---|
| 36 | 25.6 |
| 33 | 25.4 |
| 40 | 25.0 |
| 31-Z | 24.0 |
| 35 | 23.7 |
| 38 | 22.9 |
| 31-E | 22.7 |
| 37 | 22.4 |
| 30 | 21.6 |
| 6 | 20.9 |
| 21 | 20.4 |
| 20 | 20.2 |
| 32 | 20.1 |
| 22 | 20.0 |
| 10 | 19.9 |
| 39 | 19.7 |
| 29 | 18.9 |
| 28 | 18.3 |
| 9 | 18.1 |
| 19 | 17.9 |
| 7 | 17.9 |
| 17 | 17.5 |
| 11 | 16.7 |
| 8 | 16.4 |
| 18 | 16.3 |
| 15 | 16.1 |
| 14 | 13.8 |
Figure 4. NMR analysis and molecular modeling of Stat3:compound interactions and the effect of over-expression of Stat3 DNA-binding domain on hirsutinolide activity.


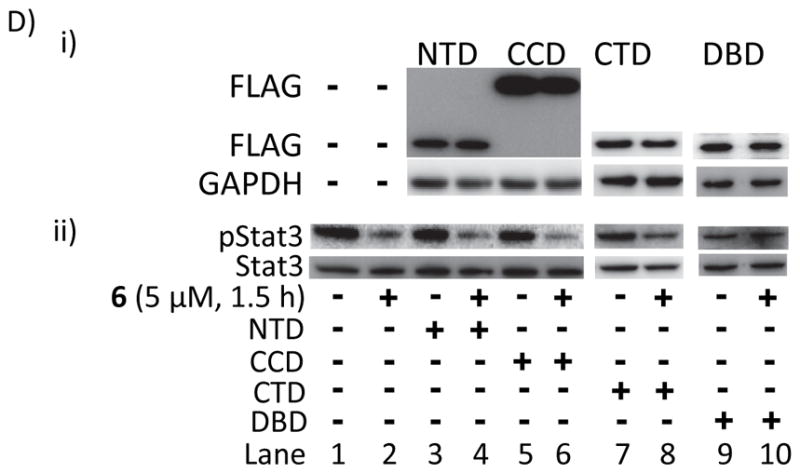
(A) Molecular modeling and docking of 33, 31, 29, 22, 6 and 7 into the Stat3 DNA-binding domain pocket showing the interactions with key amino acid residues; (B and C) Overlay of the 1H-13C HMQC spectra of (B) wild-type Stat3, free (black) or bound to 100 μM (red) or 200 μM (green) 6 and 10, and (C) wild-type Stat3, free (red) or bound to 100 or 200 μM (blue) 7, 9, 11 or 22 and showing residues with significant changes in either resonance line-widths or NMR chemical shifts that are indicated by arrowheads; and (D) immunoblots of (i) FLAG or GAPDH, or (ii) pStat3 or Stat3 in whole-cell lysates from U251MG cells transiently-transfected with Stat3 FLAG-tagged N-terminal (NTD), Coiled coil (CCD), C-terminal (CTD) or DNA-binding (DBD) domain and treated with 5 μM 6 for 1.5 h. Positions of proteins in gel are labeled; control lane (−) represents whole-cell lysates prepared from cells treated with 0.025% DMSO. Data are representative of 2 independent determinations.
Figure 6. Antitumor effects against human glioma tumor xenografts in vivo.

(A) Mice bearing U251MG subcutaneous tumor xenografts were administered 6 or 22 via oral gavage, 2 mg/kg or vehicle (1% DMSO) every other day for 33 days. Tumor sizes, measured every 3 days, were converted to tumor volumes and plotted against days of treatment; and (B) immunoblots of pStat3, Stat3 or GAPDH in tumor tissue lysates prepared from control and treated mice. Positions of proteins in gel are labeled; control lanes (c) represent tissue lysates prepared from mice treated with 1% DMSO. Values, mean ± S.D., n=6. * - <0.05.
Notwithstanding, correlations are absent for docking scores below −20 kcal/mol (Table 2). Notably, 11, has a docking score of −16.7 kcal/mol, but potently inhibits Stat3:Stat3/DNA complexation [Fig. 2A(ii)]. However, modeling shows that it interacts with Lys383 via its position 13 acetyl group, while its hemi-acetal hydroxyl group also makes a strong H-bonding with Arg423 (data not shown). Furthermore, 7, which is active, and its inactive 1-methoxy analog, 9, both share similar docking scores of −17.9 kcal/mol and −18.1 kcal/mol, respectively. However, 7 maintains stronger lipophilic interaction with Val375 and Leu378, and both of its hydroxyl group and its furan oxygen atoms at C1 make two H-bonding with Arg379 (Fig. 4A, 7), while 9 is devoid of any H-bonding (data not shown).
Nuclear Magnetic Resonance (NMR) analysis confirms direct Stat3:hirsutinolide binding
NMR studies of the 68 kDa Stat3 construct (127–711), selectively labeled at the methyl groups of 37 Val, 59 Leu, and 35 Ile residues 29–31, showed well-dispersed two-dimensional 1H-13C heteronuclear multiple quantum correlation (HMQC) spectra (Fig. 4B, C). The addition of 100 or 200 μM 6, 7, 10, 11 or 22 to Stat3 in solution caused significant chemical shift changes (CSP) in a dose-dependent manner (Fig. 4B, C, green arrowheads; a few of the amino acids with CSP are shown), indicating direct Stat3 binding. Compound:Stat3 interactions have a slow dissociation rate constant, as shown by the slow exchange rate between the free and bound state observed in the NMR spectra, consistent with their high affinities (KD < 1 μM). 6, 7, 11 and 22 that more strongly inhibited Stat3:Stat3/DNA complexation in vitro [Fig. 2A(i) and (ii)], induced the most CSP, compared to 10 or 9 (Fig. 4B, C, green arrowheads), and there are similarities. Notably, 6 and 10 (different only in the 1-methoxy group), both caused the disappearance of the same set of resonances in the Leu/Val region of the spectrum, indicating binding to the same region. However, new and different resonances emerge, indicative of significantly different binding modes and that the 1-methoxy group is sufficient to cause differences. We note that the 1-methoxy group diminishes Stat3-inhibitory activity of 10, compared to 6 [Fig. 2A(i)]. Also, significant changes in peaks are noted for 22, compared to 6 or 10, in the top left-to-middle section, while additional changes are noted for 6 in the bottom left, compared to 10 or 22.
To investigate the possibility that interactions could be non-specific, we compared the NMR spectra of Stat3:compound interactions to the spectra of Stat3 in solution with the highly reactive alkylator, N-ethylmaleimide (NEM) acquired under the identical condition as that used to study the compounds. The most likely non-specific event to induce CSP is the alkylation of the Cys residues that do not form disulfide bonds in the DBD, one of which is at the DB interface. Overlay of NMR spectra of 20 μM Stat3 with and without 100 μM NEM showed significant CSP for several residues (Fig. S3A, black arrowheads), which are indicative of alkylation. Of these changes, only one or two were similarly observed for Stat3 in solution with 6, 7, 10, 11, 22, or with the inactive compound 9 (Fig. 4B, C, black arrowheads). It is relevant that NEM, which induces strong Stat3 alkylation, inhibits Stat3:Stat3/DNA complexation in a dose-dependent manner (Fig. S3B) due to its strong reactivity 32, while 9 that induced only two of the NEM-associated CSP and also caused additional CSP (Fig. 4C, 9, black and green arrowheads) had no effect on Stat3:Stat3/DNA complexation [Fig. 2A(ii)]. These results together suggest that unlike NEM, the moderate alkylation promoted by the hirsutinolides is insufficient to induce significant changes that inhibit Stat3 activity and function. To support this, we investigated the impact of cysteine, with a free sulfhydryl group, on the hirsutinolides’ ability to inhibit Stat3:Stat3/DNA complexation by performing the cell-free DNA complexation assay to measure the effects of 22 in the presence of 50 μM Cys. Despite the addition of Cys, 22 strongly inhibited Stat3:Stat3/DNA complexation (Fig. S3C, lane 4 vs 1), while Cys alone has no effect. Thus, a free sulfhydryl (-SH) group could not blunt the activity of 22 and rescue the Stat3:Stat3/DNA complexation. Therefore, the alkylation of free sulfhydryl groups by the hirsutinolides alone is insufficient to account for the inhibitory effects of 6, 7, 10, 11 or 22 against Stat3 signaling.
To extend the molecular modeling analysis and determine whether Val375 and Leu378 in the DBD are involved in interaction with the compounds, these two residues were mutated to Ala (Stat3V375A and Stat3L378A). The resonances of Val375 and Leu378 overlap with that of other residues and thus could not be identified from the overlay of the wild type Stat3 (red) spectrum with that of V375A (green) and L378A (blue) (Fig. S3D). While we could not determine whether compound binding induced CSP on Stat3V375A and Stat3L378A, due to resonance degeneracy, the mutation of V375A and L378A caused similar CSP (Fig. S3D, green arrowheads) as that induced by the binding of 6, 7, 10, 11, and 22 to the wild-type Stat3 (Fig. 4B, C, green arrowheads), consistent with the deduction that the compounds bind to the DBD in a region including or close to Val375 and Leu378.
Over-expression of Stat3 DNA-binding domain rescues phospho-TyrStat3 from the inhibitory effects of hirsutinolides
To validate the computational docking data and the presumed binding to the Stat3 DBD, U251MG cells were transiently-transfected with Stat3 domain constructs (FLAG-Stat3 N-terminus (NTD), Coiled Coil (CCD), DBD or C-terminus (CTD) domain), treated with 5 μM 6 for 1.5 h, and whole-cell lysates prepared and immunoprobed. The over-expression of DBD, but not NTD, CCD or CTD [Fig. 4D(i)] rescued pStat3 levels from the inhibitory effect of 6 [Fig. 4D(ii)], indicating the DBD is likely a target for the active hirsutinolides.
Michael acceptor reactions could not explain the effects of hirsutinolides on Stat3 activity
We next investigated the potential reactivity of the α,β-unsaturated carbonyl groups as Michael acceptor sites 33 by treating 9, 10, 22, and the exo-methylene-γ-lactone costunolide as control with cysteamine in DMSO-d6 and performing 1H NMR analysis (Fig. S4). The spectrum of 9 in DMSO-d6 (downfield region) shows signals for the exo-methylene at H-3′a (δH 5.73 brs) and H-3′b (δH 6.19 brs), olefinic H-5 (δH 6.18 s), and the oxymethylene protons H-13 (δH 4.90 d), which all disappeared upon cysteamine addition (Fig. S4A, blue vs. red), indicative of Michael acceptor reactions. However, compared to others, 9 poorly inhibited Stat3 DNA-binding activity [Fig. 2A(ii)], despite that it mediates Michael acceptor reactions. For 10, the signals for the olefinic H-3′ (δH 6.96 q, J = 7.5 Hz) were only shifted slightly upfield (δH 6.82), while H-5 (δH 6.16, s) signals disappeared, and those of the oxymethylene H-13 protons shifted (δH 4.90→5.30) (Fig. S4B, blue vs. red) on cysteamine addition, suggesting Michael acceptor reactions have occurred. The addition of cysteamine to 22 had a similar outcome in that the signals for the olefinic H-3′ (δH 6.77 q) had a minor shift [6.77 (q, J = 7.6 Hz)], while those of H-5 (δH 6.12 s) disappeared, and the oxymethylene protons H-13 signals shifted (δH 5.02→4.05) (Fig. S4C, blue vs. red). As reported, the spectrum of costunolide treated with cysteamine shows the loss of the olefinic signals at δH 6.06 and 5.68 ppm, respectively, for H-13a and H-13b due to the formation of diastereomeric Michael adduct mixtures 1a and 1b (Fig. S4D) 33. Results here combined with the NMR data show the hirsutinolides undergo Michael acceptor reactions that weakly alkylate Stat3. However, these events are insufficient for Stat3 inhibition, given that the inactive 8, 9, 14, 18, 20, 19 and 21 similarly contain the olefinic H-5 and the exo-methylene H-3′a and H-3′b protons and do not inhibit Stat3 activity.
Active hirsutinolides suppress tumor cell phenotype in vitro and down regulate c-Myc, Bcl-2, Mcl-1, and Bcl-xL expression
We investigated the hirsutinolides effects against cell proliferation using CyQuant assay. Compounds induced differential activities against the human glioma, U251MG and SF295 and the breast cancer, MDA-MB-231 cells that harbor aberrantly-active Stat3, and normal NIH3T3 fibroblasts and breast cancer, MCF7 cells that do not (Fig. S5A, B, Table 1). 7, 10, 20 and 22 were strongly active against U251MG cells (IC50 values of 2.6, 3.4, 3.3 and 2.6 μM, respectively) [Fig. S5A(i), B, Table 1], and reasonably active against other tumor cells (MDA-MB-231, U373MG, SF295 and MCF-7), with IC50 ranges of 5.2–9.1, 6.6–8.2, 7.4–18.3 and 4.8–9.6 μM, respectively [Fig. S5A(i), B, Table 1]. Activity against normal NIH3T3 cells was also detectable, with IC50 of 7.1, 7.9, 11.8, and 6.2 μM for 7, 10, 20 and 22, respectively [Fig. S5A(i), B, Table 1]. 6 was active and non-specific against U251MG, MDA-MB-231, U373MG, SF295, MCF-7 and NIH3T3 cells, with IC50 of 1.7, 1.8, 3.5, 7.5, 10.7 and 2.0 μM, respectively [Fig. S5A(i), B, Table 1]. The most active compounds, 6, 10 and 22 at 5 μM also showed 45–60% inhibitory activity against the growth of human glioma patient-derived xenograft (pdx) cells, G10 and G22, and 25–35% inhibitory against G12 cell growth in vitro, compared to 90% inhibition of U251MG cell line (Fig. S5C). Furthermore, trypan blue exclusion with phase-contrast microscopy for viable cell numbers showed at 1 and 2.5 μM, 22 induced a preferential inhibition of growth of v-Src-transformed fibroblasts (NIH3T3/v-Src) that harbor aberrantly-active Stat3 over that of normal NIHT3T3 fibroblasts that do not (Fig. S5D).
Compounds 9 and 11 [Fig. S5A(ii)], and 8, 14, 15, 18, 19, 21 and 28 [Fig. S5A(iii)] evaluated at 5 μM are moderately or less active against the growth of the same cell lines in vitro (Table 1). For control, the structurally similar compounds, costunolide and parthenolide, which did not inhibit Stat3 activity in tumor cells [Fig. S5E(ii)] are poorly-active or non-specific in activity [Fig. S5E(i)]. The semi-synthetic analogs, 30, 31, 33, 38, 39, 40, 34, 35, 37 and 36 (5 μM) retained or improved the inhibitory potency against U251MG cell growth [Fig. S5F(i) and G]. The IC50 values for 30, 31, 33 and 36 are 1.0–1.2 μM, compared to the IC50 >5 μM against NIH3T3 cells (Fig. S5G). Compounds 39, 40, 34 and 37 also strongly inhibited MDA-MB-231 cell growth, while 38 lost cell-type specificity, compared to 39 and inhibited NIH3T3 growth [Fig. S5F(i)]. By contrast, and 29 and 32, which poorly inhibited Stat3 activation in tumor cells (Fig. S2E), are weakly-active or inactive against U251MG cell growth in vitro [Fig. S5F(ii), (iii)], despite inhibiting Stat3:Stat3/DNA complexation in the cell-free EMSA analysis (Fig. S2A).
We reasoned that 29, 32, together with 11, with less bulkier position 13 side chains have low intracellular activities likely due to poor cell membrane permeability caused by the two hydroxyl groups at positions 1 and 8. On the other hand, a bulkier/longer carbon chain group at position 13 enhances cellular activity (compare 22 to 11, and 30, 31, 33, 34, 35, 37 and 36 to 29 and 32). In addition, 30, 31, 33 and 36 with the lipophilic and bulky groups, such as the geranyl group at the 13 position showed good specificity. Our data for 7, 8, 38, 39 and 40 also suggest a group at position 8 may have a detrimental effect on specificity. Moreover, a methoxy group at position 1 diminishes activity (compare 9 to 7, and 10 to 6), while apparently improving tumor cell-specificity [compare 10 to 6, Fig. S5A(i); 39 to 38, Fig. S5F(i)].
In clonogenic assay 26, one-time treatment with 1–5 μM 22, 6 or 10 of single-cell cultures of human glioma U251MG and SF295 cells, or breast cancer MDA-MB-231 that harbor aberrantly-active Stat3 and breast cancer MCF-7 or normal NIH3T3 fibroblasts that do not induced dose-dependent reductions in the colony numbers (Fig. 5A). We observe differential sensitivities of U251MG, MDA-MB-231, MCF7 and SF295 in the descending order of preference (Fig. 5A). Compared to 6 and 22, 10 showed weaker activity (Fig. 5, 10). The effects on NIH3T3 colony numbers were moderate or none at 2.5 μM or less for 6 and 22 and 5 μM or less for 10 (Fig. 5A). Moreover, treatment of U251MG cells with 5 μM 6 or 22 for 3–24 h significantly impaired cell proliferation measured by BrdU incorporation at 24 h (Fig. 5B). Further, 19-h treatment with 5 μM 6, 10 or 22 slowed migration into the denuded area in scratch assay (Fig. 5C), with no significant loss of cell viability (Fig. S5H).
Figure 5. Hirsutinolides inhibit in vitro cell proliferation, colony survival, and migration, and the expression of Stat3-regulated genes.

(A) Single-cell cultures of U251MG, SF295, MDA-MB-231, MCF7 and NIH3T3 cells treated once with 0–5 μM 6, 10 or 22 and allowed to grow until large colonies were visible, which were stained with crystal violet, counted and plotted. U251MG colony formation was completely inhibited by 1 μM 6 or 22; (B) 5-bromo-2′-deoxyuridine (BrdU) incorporation analysis for proliferating U251MG cells treated with 0 or 5 μM 6 or 22 for 3–24 h. Images were captured under a fluorescence microscope and analyzed with ImageJ software; (C) cultured U251MG cells were wounded, treated once with 0–5 μM 6, 10 or 22 and allowed to migrate to the denuded area over 19 h and imaged; (D) cell cycle distribution analysis of U251MG cells treated or untreated (DMSO) with 15 μM Cmpd1 for 24 or 72 h, processed by propidium iodide (PI) staining, and analyzed by flow cytometry for DNA content, which is plotted; and (D) immunoblots of pStat3, Stat3, c-Myc, Bcl-2, Bcl-xL, Mcl-1 or GAPDH from whole-cell lysates from U251MG cells treated with 5 μM 6 for 0–24 h. Positions of proteins in gel are shown; control lane (0) represents 0.025% DMSO-treated cells or whole-cell lysate preparation from 0.025% DMSO-treated cells. Values are the mean ± S.D., n=3–6. Data are representative of 2–3 independent determinations.
Immunostaining with confocal microscopy imaging showed, compared to control, untreated U251MG cells (Fig. S5H, DMSO, yellow arrowheads), treatment with 6 or 22 for 15 h caused defects in both the chromatid structures and the sister-chromatids segregation, characterized by more than two poles (Fig. S5I, 6, 22, yellow arrowheads), indicating a dysfunctional mitosis or a mitotic catastrophe 34. For controls, the poorly-bioactive 11 showed minimal effect (Fig. S5I, 11), while paclitaxel (microtubule stabilizer), caused strong defects (Fig. S5I, paclitaxel, Pax). Cell cycle distribution analysis revealed that 22 induced G2/M accumulation of U251MG cells following 15h treatment (Fig. 5D). Further, treatment with 5 μM 6 down regulated c-Myc, Bcl-2, Mcl-1 and Bcl-xL expression in U251MG cells subsequent to the inhibition of pStat3 (Fig. 5E).
However, unlike paclitaxel or staurosporine that induced apoptosis as measured by poly ADP ribose polymerase (PARP) or caspase cleavage, treatment of U251MG cells with 22 for 24 or 48 h did not (Fig. S5J). Glioma patient-derived xenograft cells, G22, growing as 3D spheroids formed over 48 h, were treated once with 5 μM 6, 10 or 22 for 24 h, before the addition of matrigel. Images show decreased 3D spheroid growth by 24-h treatment with 6 or 22 in the absence of matrigel (Fig. S5K, 24 h vs. 0, 6 and 22). Moreover, strong growth impairment and extensive cell disaggregation (green arrowheads) of spheroids in matrigel occurs at 48–96 h post-treatment with 6 or 22 (Fig. S5I, 48–96 h, 6 and 22), while 10 was modest (Fig. S5K, 24–96 h, 10 vs. DMSO). 6 and 22 also attenuated the outward cell migration from the spheroid periphery into the matrigel (Fig. S5K, 48–96 h). Results together show the hirsutinolides induce antitumor cell response in vitro of tumor cells harboring aberrantly-active Stat3 and to some extent of cells that do not, suggesting that they might modulate additional intracellular events together with inhibiting Stat3 signaling.
Modulation of Hsp105, vimentin, TNAP2, translational activator GCN1, G6PD isoform a, TrxR1 cytoplasmic isoform 3, MAP1B and importin subunit α-2 expression
We hypothesized that the inhibition of Stat3 alone does not completely explain the differential effects of hirsutinolides on tumor cells. Therefore, we pursued a global proteomics with Mass Spectrometry analysis 35 of lysates from U251MG cells treated for 1 h with 5 μM 6 or 22 to determine additional possible molecular events that may contribute to the antitumor responses to the hirsutinolides. Results showed 2–3-fold lower Hsp105, vimentin and TNAP2 expression in cells treated with 6, 2–3-fold decrease in translational activator GCN1, G6PD isoform a, and TrxR1 cytoplasmic isoform 3 levels upon treatment with 22, and 2-fold lower MAP1B and over 2-fold higher importin subunit α-2 expressions in cells treated with 6 or 22 (Table 3). Immunoblotting analysis to validate these changes shows the proteins are variably expressed across diverse human tumor and patient-derived xenograft cells (Fig. S6A). Notably, the expression of these proteins in the glioma cell lines, U251MG and U373MG or the patient tumor-derived xenograft cells, G22 are differentially inhibited by treatment with 6 or 22 (Fig. S6B–D). The strongest changes were observed in MAP1B expression across the three cells. In addition, vimentin, G6PD, GCN1 and TrxR1 expression were strikingly altered in U251MG following treatment with 6 or 22 (Fig. S6B), G6PD was strongly repressed in U373MG cells treated with 22 (Fig. S6C), while vimentin expression was measurably suppressed by 6 in G22 cells (Fig. S6D). The combined modulation of these putative targets would contribute to the antitumor responses induced by the active hirsutinolides.
Table 3.
Altered proteins in U251MG cells treated with 6 or 22 (5 μM, 1h).
| U251MG + 6 Out of 31 | ||
|---|---|---|
| Gene identifier | Levels relative to DMSO-treated cells | |
| gi|42544159 | heat shock protein 105 kDa [Homo sapiens] | 0.5012 |
| gi|62414289 | vimentin [Homo sapiens] | 0.3076 |
| gi|26051240 | tumor necrosis factor alpha-induced protein 2 [Homo sapiens] | 0.4487 |
| gi|153945728 | microtubule-associated protein 1B [Homo sapiens] | 0.6486 |
| gi|4504897 | importin subunit alpha-2 [Homo sapiens] | 2.0137 |
|
U251MG + 22 Out of 21 | ||
| Gene identifier | Levels relative to DMSO-treated cells | |
| gi|54607053 | translational activator GCN1 [Homo sapiens] | 0.5546 |
| gi|148277071 | thioredoxin reductase 1, cytoplasmic isoform 3 [Homo sapiens] | 0.3048 |
| gi|109389365 | glucose-6-phosphate 1-dehydrogenase isoform a [Homo sapiens] | 0.2754 |
| gi|153945728 | microtubule-associated protein 1B [Homo sapiens] | 0.413 |
| gi|4504897 | importin subunit alpha-2 [Homo sapiens] | 2.421 |
Active hirsutinolides inhibit growth of subcutaneous glioma xenografts in mice
Oral gavage dosing of 6 or 22 inhibited growth of subcutaneous mouse xenografts of human glioma U251MG tumors that harbor aberrantly-active Stat3 (Fig. 6A). The tumor growth inhibition is associated with lower pStat3 and Stat3 levels in residual tumor tissues in four-to-five cases out of six (Fig. 6B), consistent with the auto-regulation by Stat3 of its own promoter 36. No significant changes in body weights (Fig. S7) or obvious signs of toxicity, such as loss of appetite, decreased activity, or lethargy were observed.
Discussion
Presented herein are hirsutinolide natural products and their semi-synthetic analogs that inhibit Stat3 activation and functions and induce anti-proliferative effects in vitro and anti-tumor effects in vivo. Data suggest the decrease in cell viability and growth is in part due to cell cycle arrest at G2/M phase and mitotic catastrophe. The semi-synthetic analogs and the ability of the hirsutinolides to inhibit Stat3 activity and suppress glioma cell proliferation and tumor growth are all novel. The Michael acceptor reactions promoted by the olefinic and the exomethylene carbons of the hirsutinolides alone appear insufficient for a robust inhibition of Stat3 activity or induction of antitumor effects. This is supported by the low-to-moderate effects of 8, 9, 14, 18, 20, 19 and 21 on both Stat3 activity and tumor cell growth, and the lack of impact of soluble cysteine on the Stat3-inhibitory activity of 22. Comparatively, we note that the more reactive NEM induced stronger chemical shift changes in the Stat3 NMR spectra and exerted Stat3-inhibitory effect 32. The antitumor efficacy of 6 and 22 as orally bioavailable agents against glioma xenografts provides proof-of-concept for their therapeutic potential as anticancer drug leads.
With respect to the inhibition of Stat3 activity, each of the hirsutinolides, 6, 7, 11, 10, 20 and 22 and the semi-synthetic analogs, 30, 31, 32, 33, 38, 39, 40, 35, 37 and 36 present a critical side chain at the position 13, with or without a supporting appendage at position 8, which promotes Stat3 inhibition, as measured by cell-free EMSA analysis. The intracellular activities of these agents are further influenced by additional factors that likely impact cell membrane permeability. The inhibition of Stat3 signaling by the hirsutinolides partly occurs through direct binding. NMR structural evidence confirms the active hirsutinolides interact with the Stat3 DNA-binding domain, presumably via hydrophobic interactions between the position 13 ester side chain, complemented by the position 8 ester group, and the Stat3 Val375 and/or Leu378 or Lys383 residues, in addition to H-bonding with Arg379, Gly380, and/or Arg423. Accordingly, the hirsutinolides directly block Stat3:Stat3 DNA-binding complexation. The inhibition of the Stat3 functions in tumor cells is consistent with the decreased Bcl-2, c-Myc, Mcl-1, and Bcl-xL expression 5, 37, which altogether represent part of the underlying mechanisms for the antitumor responses in vitro and in vivo.
Modulation of the NFκB pathway 38, and the induction of anti-carcinogenic 39 and anti-inflammatory 40, 41 effects have previously been reported in response to treatment with compound 6 and other hirsutinolides or sesquiterpene lactones, suggesting the potential of the hirsutinolides to modulate other targets 38, 39, 41. It is noted that the inhibition of Stat3 activity alone is insufficient to explain all the biological responses to the hirsutinolides. Our study and others 38, 39, 41 together support the ability of the hirsutinolides to modulate multiple targets, including Hsp105, vimentin, GCN1, G6PDH isoform a, TrxR1 cytoplasmic isoform 3, MAP1B, and importin subunit α-2 expression. The rapid alterations in the expression and function of these proteins contribute to the inhibition of glioma cell proliferation, survival, and migration in vitro, and of tumor growth in vivo. However, our studies show that the bioactive hirsutinolides do not modulate Stat1, Stat5, Erk1/2, Akt, SOCS or PTP1B induction.
The effects of 6, 7, 10, 20 and 22 against the Stat3 pathway and glioma phenotype suggest the potential that these agents could be developed as part of treatment strategies tailored to malignant glioma and potentially other tumors harboring aberrantly-active Stat3. Additional knowledge of the structural basis for the activities of the hirsutinolide series could facilitate a therapeutic development campaign, supported by semi-synthesis using the relatively abundant series member, 14 as a starting material.
Materials and Methods
Source of hirsutinolide library
The hirsutinolides were derived from Vernonia cinerea plant. Their isolation from V. cinerea, structural elucidation, and the characterization of the hirsutinolides have been previously reported 40.
Semi-synthesis of hirsutinolide analogs
The semi-synthesis of analogs of 22 using 14, 8 or 17 as starting materials, and the rationale are described in detail in Supplementary Materials “Methods” section. All the semi-synthetic derivatives were obtained with >96.0% purity by HPLC (UV detection at 254 and 284 nm).
Cell lines and reagents
Normal mouse fibroblasts (NIH3T3), their counterparts transformed by v-Src (NIH3T3/v-Src) or overexpressing the human EGF receptor (NIH3T3/hEGFR), and the human breast cancer (MDA-MB-231 and MFC7) cells have all been previously reported 23, 42, 43. These cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% heat-inactivated fetal bovine serum (FBS). The human glioma lines, U251MG, SF-295 and U373MG were obtained from the Division of Cancer Treatment and Diagnosis, Tumor Repository of the National Cancer Institute (Frederick, MD) and cultured in RPMI or DMEM containing 10% FBS and supplemented with 1% nonessential amino acids (Corning Inc., Corning, NY). The human glioma patient tumor-derived mouse xenograft cells (G6 - G102) were obtained from Jann N. Sarkaria (Mayo Clinic) and cultured in DMEM containing 5–10% heat-inactivated FBS. Antibodies against GCN1, MAP1B, Hsp105, G6PD, TrxR1, Karyopherin (importin) α2, Stat1, Stat3 (C20X), β-actin, GAPDH and β-tubulin (Santa Cruz Biotechnology, Inc., Dallas, Texas), and against FLAG (Sigma-Aldrich, St. Louis, MO) were purchased from the indicated sources. All other antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA).
Cell proliferation assay
CyQuant cell proliferation assay (Invitrogen/Life Technologies Corp, Carlsbad, CA) of 72-h-treated cultured cells was performed as previously reported 26 and following the manufacturer’s instructions. Relative cell viability of the treated cells was normalized to the DMSO-treated control cells.
Trypan Blue Cell Counting
Cells in culture in 6-well plates were untreated or treated once with increasing concentrations of compounds for 0–96 h. Cells were harvested every 24 h and viable cells were counted by trypan blue exclusion/phase-contrast microscopy.
Transient transfection and SDS-PAGE/Western blotting analysis
U251MG cells in 10 cm dishes were transiently transfected with 7.5–21 μg of the appropriate FLAG-Stat3 plasmids (Stat3 NTD, CCD, DBD and CTD) or pcDNA3 (mock). Twenty-four hours after transfection, cells were trypsinized and re-seeded in two 6-cm dishes. Eighteen hours thereafter, cells were treated or untreated with 5 μM 6 for 1.5 h. Cultured cells were harvested and whole-cell lysates prepared for SDS-PAGE and immunoblotting analysis, as previously reported 26. Primary antibodies used were anti- Stat3, pY705Stat3, pErk1/2, Erk1/2, pJAK2, JAK2, SOCS3, PTP1B, pS-Akt, Akt, Hsp105, importin α-2, vimentin, G6PD, TrxR1, MAP1B, c-Myc, Bcl-xL, Bcl-2, Mcl-1, FLAG, GCN1, pY701Stat1, Stat1, pY694Stat5, Stat5, Caspase 3, PARP, β-tubulin, GAPDH and β-actin.
Colony survival assay
This assay was performed as previously reported 26. Briefly, cells were seeded as single-cell in 6-well plates (250 cells per well), treated once the next day with compounds for 72 h, and allowed to grow for 1 week (NIH3T3), 2 weeks (MDA-MB-231, U251MG) or 4 weeks (MCF-7) when large colonies were visible. Colonies were stained with crystal violet for 4 h and counted with FluorChem imaging system (Protein Simple, Santa Clara, CA).
Nuclear extract preparation and gel shift assays
Nuclear extract preparations and electrophoretic mobility shift assay (EMSA) were carried out as previously described 26. The 32P-labeled oligonucleotide probes used were hSIE (high-affinity sis-inducible element from the c-fos gene, m67 variant (5′-AGCTTCATTTCCCGTAAATCCCTA-3′) that binds Stat3 and Stat1 and MGFe (mammary gland factor element from the bovine β-casein gene promoter, 5′-AGATTTCTAGGAATTCAA) for Stat1 and Stat5 binding. U251MG cells in sub-confluent cultures were pretreated with different concentrations of the compounds for the indicated period and then harvested for nuclear extract preparation for EMSA analysis. Except where indicated, nuclear extracts prepared from NIH3T3/v-Src cells containing activated Stat3 or EGF-stimulated NIH3T3/hEGFR fibroblasts containing activated Stat1, Stat3 and Stat5 were pre-incubated with compounds for 30 min at room temperature prior to incubation with the radiolabeled probe for 30 min before subjecting to EMSA analysis.
Site-directed mutagenesis
The Stat3 gene encompassing amino acid residues 127–711 was cloned into the pET28 plasmid to create wild-type pET28-Stat3 (127–711). Three substitution mutations, V375A, L378A and V375A/L378A, were generated using the pET28-Stat3 (127–711) construct as a template, with the QuikChange mutagenesis protocol (Stratagene, La Jolla, CA). Primers used (V375A, 5′-CAAAGACTCTGGGGACGCTGCAGCTCTCAGAGGATC-3′, L378A, 5′-GACTCTGGGGACGTTGCAGCTGCAAGAGGATCCCGGAAATTTAAC-3′, and V375A/L378A, 5′-CAAAGACTCTGGGGACGCTGCAGCTGCAAGAGGATC-3′) were synthesized by Integrated DNA Technologies (Coralville, IA). Mutations were verified by DNA sequencing.
NMR studies of Stat3 interactions with compounds
Soluble His-tagged wild-type pET28-Stat3 (127–711-His6) and its mutants were expressed and purified by nickel affinity chromatography (Ni-NTA agarose from Qiagen, Valencia, CA) using previously described protocols 44. The expression constructs were transformed into E. coli BL21(DE3) codon+ competent cells (Agilent Technologies, Waltham, MA). For the expression of ILV (Ile, Leu and Val)-selectively labeled sample, 5 ml Luria Bertani (LB) broth overnight pre-culture was inoculated into 1L LB, followed by 37 °C incubation until Optical Density (OD)600 reached 0.6–0.7. Cells were gently harvested through centrifugation at 3,000 rpm for 20 min, and then resuspended in 1L M9 minimal wash media that contained 6.6 g Na2HPO4, 3.0 g KH2PO4 and 1.5 g NaCl in H2O. Cells were harvested again and resuspended in 250 ml M9 media with 100% D2O. Cells were grown at 37 °C for 30 min and then the temperature was lowered to 22 °C and incubated for another 30 min. Amino acid precursors for ILV-labeling (100 mg alpha-ketoisovaleric acid, 60 mg alpha-ketobutyric acid) were added, and 1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added for induction. After 16–18 h of culture, cells were harvested and stored at −80 °C. The frozen cell pellets were thawed and resuspended in 15 ml lysis buffer (20 mM Tris-HCl, 0.5 M NaCl, 10 mM imidazole, pH 7.7). BugBuster protein extraction reagent and Benzonase were added to lyse cells according to manufacturer’s (EMD Millipore, Billerica, MA) instruction. The cell lysates were centrifuged at 15,000 rpm for 30 min, then the supernatant was applied to a Ni-NTA affinity column (Qiagen Inc, Valencia, CA). After extensive washing of the column with 20 column volume of washing buffer (20 mM Tris-HCl, 0.5 M NaCl, 20 mM imidazole, pH 7.7), Stat3 was eluted with 3.5 column volume of elution buffer (20 mM Tris-HCl, 0.5 M NaCl, 250 mM imidazole, pH 8.0). A PD-10 desalting column (GE Healthcare Bio-Sciences, Piscataway, NJ) was then used to exchange the buffer to 10 mM MES, 0.5 mM TCEP, 0.03% NaN3 at pH 5.5 in 100% D2O, which was used in all subsequent NMR experiments. Protein sample concentrations were determined by Bradford Assay (BioRad Laboratories, Hercules, CA) and by UV absorbance at 280 nm.
NMR studies were performed using the pure, labeled wild-type human Stat3 protein (127–711) and its three mutant counterparts. NMR samples contained deuterated versions of all buffer reagents in 100% D2O (20 μM protein, 10 mM MES, pH 5.5, 0.5 mM TCEP in 100% D2O). The compounds were dissolved in DMSO-d6 and added into Stat3 solution. 1H-13C heteronuclear multiple quantum correlation (HMQC) spectra were acquired at 35 °C on the Burker Ascend 700 spectrometer equipped with a cryogenic probe (Bruker BioSpin, Germany). 1H-13C correlation spectra for methyl-labeled Stat3 were obtained using the 2D 1H-13C HMQC experiment to monitor the interaction between Stat3 and compounds. The data were processed with NMRPipe, and the spectra were analyzed with the program SPARKY.
BrdU Assay
U251MG cells were grown on glass cover slips in 24-well plates, treated with DMSO, 5 μM 6 or 22 for 3–24 h and analyzed for 5-bromo-2′-deoxyuridine (BrdU) incorporation. Ten micromolar of BrdU was added to the culture medium in the last 2 h of treatment. Cells were immediately fixed in 4% paraformaldehyde for 10 min and stained with an anti-BrdU antibody (Cell Signaling Technology, Beverly, MA) following the manufacturer’s instructions. The cover slips were counterstained with DAPI (4′,6-diamidino-2-phenylindole) and imaged under a fluorescence microscope (Olympus IX71, Japan). Cells were counted with ImageJ software.
Cell cycle analysis
U251MG cells were synchronized by double thymidine block (2 mM), released for 9 h and untreated (DMSO) or treated with 8 μM 22 for 15 h. Cells were trypsinized, centrifuged at 1,000 rpm at 4 °C for 10 min, and washed with 1x PBS. Cells were fixed with fixation buffer (70% ethanol and 3% FBS in PBS) overnight at −20 °C. Subsequently, cells were washed two-times with 1x PBS, each followed by centrifugation at 1,000 rpm at 4 °C for 10 min, and cell pellet was re-suspended in the incubation PI/RNase buffer (BD Biosciences, San Jose, CA) at room temperature for 15 min for staining. Samples were placed on ice and analyzed using the BD Accuri C6 flow cytometer (BD Biosciences). Data were analyzed with FlowJo (Ashland, OR).
3D spheroid assay
Glioma patient-derived xenograft cells, G22 were seeded on ultra-low binding 96-well round bottom plate (1,000 cells/well), incubated for 48 h, and treated with DMSO, or 5 μM 6, 10 or 22. After 24 h, matrigel was added and the spheroids were visualized at a 5× magnification using an Axiovert 200 inverted fluorescence microscope (Zeiss, Germany). Pictures were taken using a mounted Canon Powershot A640 digital camera (Melville, NY) at every 24 h for up to 120 h.
Immunofluorescence staining
U251MG cells growing on glass cover slips were synchronized by double thymidine block (2 mM), released for 9 h and treated with DMSO, 100 nM paclitaxel, or 8 μM 6, 11 or 22 for 15 h. Cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.1% Digitonin/PBS for 10 min and washed three times with PBS-T (PBS Tween-20). Slides were then blocked in 0.2% bovine serum albumin for 30 min and incubated with anti-β-tubulin and MAP1B antibodies at 1:350 and 1:300 dilution, respectively, in PBS-T for 1 h at room temperature. Subsequently, cells were rinsed three times with PBS-T and incubated for 1 h with Alexa Fluor 546 (donkey anti-mouse) and Alexa Fluor 633 (donkey anti-goat) (Molecular Probes/Invitrogen, Carlsbad, CA) secondary antibodies for β-tubulin and MAP1B detection, respectively, at room temperature in the dark. Specimens were then washed three times with PBS-T, mounted on slides with DAPI Fluoromount-G mounting medium (Southern Biotech, Birmingham, AL), and examined under a Leica TCS SP5 confocal microscope (Leica, Germany). Images were captured and processed using Leica LAS AF Lite software.
Scratch assay for migration
This assay was performed as previously reported 26. Briefly, sub-confluent cultures of U251MG cells in 6-well plates were wounded using pipette tips, treated with compound or vehicle (DMSO), and allowed to migrate into the denuded area over a 19-h period. The migration of cells was visualized at a 10× magnification using an Axiovert 200 inverted fluorescence microscope (Zeiss, Germany), with pictures taken using a mounted Canon Powershot A640 digital camera.
Mice and in vivo tumor studies
Four-week old female athymic nude mice were purchased from Harlan Laboratories (Indianapolis, IN) and maintained in the institutional animal facilities approved by the American Association for Accreditation of Laboratory Animal Care. Mice were injected sub-cutaneously in the left flank area with 7.3 ×106 U251MG cells in 200 μL of equal mixture of PBS and matrigel. After 20 days, tumors of a 170–250 mm3 were established. Tumor-bearing mice were grouped so that the mean tumor sizes in all groups were nearly identical and given 6 or 22 (2 mg/kg, oral gavage, 100 μL) three times a week for 33 days. Animals were monitored daily, tumor sizes were measured with calipers, and the body weights were taken every 3 days. Tumor volumes were calculated according to the formula V = 0.52 × a2 × b, where a is the smallest superficial diameter and b is the largest superficial diameter. For each treatment group, the tumor volumes for each set of measurements were statistically analyzed in comparison with the control (vehicle-treated) group using a paired t test.
Statistical analysis
Statistical analysis was performed on mean values using Prism GraphPad Software, Inc. (La Jolla, CA). The significance of differences between groups was determined by the paired t-test at p <0.05*, <0.01**, and < 0.001***.
Supplementary Material
Acknowledgments
We thank all colleagues and members of our laboratory for the stimulating discussions. This work was supported by the National Cancer Institute Grant CA161931 (JT), University of Hawaii start-up funds (JT), NCRR INBRE Program P20 RR016467 (LCC), NIMHD Program U54MD008149 (LCC), and the University of Hawaii Daniel K. Inouye College of Pharmacy start-up funds (LCC). We also thank the Mass Spectrometry and Proteomics Facility at Penn State College of Medicine for performing 2D-LC/MALDI-MS/MS.
Abbreviations Used
The abbreviations used are
- STAT
signal transducer and activator of transcription
- EMSA
electrophoretic mobility shift assay
- MAPK
mitogen-activated protein kinase
- ERK
extracellular signal-regulated kinases
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- MAP1B
microtubule-associated protein1B
- TrxR1
thioredoxin reductase 1 cytoplasmic isoform 3
- G6PD
glucose-6-phosphate 1-dehydrogenase isoform a
- Hsp105
heat shock protein105
- TNAP2
tumor necrosis factor α-induced protein 2
- iTRAQ
isobaric tags for relative and absolute quantification
Footnotes
Supporting Information Availability. Supplementary information on “Materials and Methods”. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Priester M, Copanaki E, Vafaizadeh V, Hensel S, Bernreuther C, Glatzel M, Seifert V, Groner B, Kögel D, Weissenberger J. STAT3 silencing inhibits glioma single cell infiltration and tumor growth. Neuro Oncol. 2013;15:840–852. doi: 10.1093/neuonc/not025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 3.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 4.Yu H, Jove R. The STATS of Cancer-New molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 5.Miklossy G, Hilliard TS, Turkson J. Therapeutic modulators of STAT signaling for human diseases. Nat Rev Drug Discov. 2013;12:611–629. doi: 10.1038/nrd4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, Dalton W, Jove R, Pardoll D, Yu H. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2004;10:48–54. doi: 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- 7.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang X, Yue P, Fletcher S, Zhao W, Gunning PT, Turkson J. A novel small-molecule disrupts Stat3 SH2 domain-phosphotyrosine interactions and Stat3-dependent tumor processes. Biochem Pharmacol. 2010;79:1398–1409. doi: 10.1016/j.bcp.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu Y, Li X, Zhang S, Shen D, Li H, Wu Y, Qiu Y, Ji Y, Chen F. Targeting Stat3 suppresses growth of U251 cell-derived tumours in nude mice. J Clin Neurosci. 2012;19:443–446. doi: 10.1016/j.jocn.2011.04.017. [DOI] [PubMed] [Google Scholar]
- 11.Chen J, Bai L, Bernard D, Nikolovska-Coleska Z, Gomez C, Zhang J, Yi H, Wang S. Structure-based design of conformationally constrained, cell-permeable STAT3 inhibitors. ACS Med Chem Lett. 2010;1:85–89. doi: 10.1021/ml100010j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin L, Hutzen B, Li PK, Ball S, Zuo M, DeAngelis S, Foust E, Sobo M, Friedman L, Bhasin D, Cen L, Li C, Lin J. A novel small molecule, LLL12, inhibits STAT3 phosphorylation and activities and exhibits potent growth-suppressive activity in human cancer cells. Neoplasia. 2010;12:39–50. doi: 10.1593/neo.91196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siddiquee K, Glenn M, Gunning P, Katt WP, Zhang S, Schroeck C, Jove R, Sebti S, Hamilton AD, Turkson J. An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem Biol. 2007;2:787–798. doi: 10.1021/cb7001973. [DOI] [PubMed] [Google Scholar]
- 14.Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence H, Yip MLR, Jove R, McLaughlin M, Lawrence N, Sebti S, Turkson J. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A. 2007;104:7391–7396. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J, Nikolovska-Coleska Z, Yang C-Y, Gomez C, Gao W, Krajewski K, Jiang S, Roller P, Wang S. Design and synthesis of a new, conformationally constrained, macrocyclic small-molecule inhibitor of STAT3 via ‘click chemistry’. Bioorg Med Chem Lett. 2007;17:3939–3942. doi: 10.1016/j.bmcl.2007.04.096. [DOI] [PubMed] [Google Scholar]
- 16.Shuai K, Horvath CM, Huang LH, Qureshi SA, Cowburn D, Darnell JE., Jr Interferon activation of the transcription factor Stat91 involves dimerization through SH2-phosphotyrosyl peptide interactions. Cell. 1994;76:821–828. doi: 10.1016/0092-8674(94)90357-3. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Sun Y, Pireddu R, Yang H, Urlam MK, Lawrence HR, Guida WC, Lawrence NJ, Sebti S. A novel inhibitor of Stat3 homodimerization slectively suppresses STAT3 activity and malignant transformation. Cancer Res. 2013;73:1922–1933. doi: 10.1158/0008-5472.CAN-12-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bill MA, Nicholas C, Mace TA, Etter JP, Li C, Schwartz EB, Fuchs JR, Young GS, Lin L, Lin J, He L, Phelps M, Li PK, Lesinski GB. Structurally modified curcumin analogs inhibit STAT3 phosphorylation and promote apoptosis of human renal cell carcinoma and melanoma cell lines. PLoS One. 2012;7:e40724. doi: 10.1371/journal.pone.0040724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim JE, Kim HS, Shin YJ, Lee CS, Won C, Lee SA, Lee JW, Kim Y, Kang JS, Ye SK, Chung MH. LYR71, a derivative of trimeric resveratrol, inhibits tumorigenesis by blocking STAT3-mediated matrix metalloproteinase 9 expression. Exp Mol Med. 2008;40:514–522. doi: 10.3858/emm.2008.40.5.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee K-H. Discovery and development of natural product-derived chemotherapeutic agents based on a medicinal chemistry approach. J Nat Prod. 2010;73:500–516. doi: 10.1021/np900821e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003;63:1270–1279. [PubMed] [Google Scholar]
- 22.Turkson J, Kim JS, Zhang S, Yuan J, Huang M, Glenn M, Haura E, Sebti S, Hamilton AD, Jove R. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol Cancer Ther. 2004;3:261–269. [PubMed] [Google Scholar]
- 23.Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E, Laudano A, Sebti S, Hamilton AD, Jove R. Phosphotyrosyl peptides block Stat3-mediated DNA-binding activity, gene regulation and cell transformation. J Biol Chem. 2001;276:45443–45455. doi: 10.1074/jbc.M107527200. [DOI] [PubMed] [Google Scholar]
- 24.Turkson J, Bowman T, Adnane J, Zhang Y, Djeu JY, Sekharam M, Frank DA, Holzman LB, Wu J, Sebti S, Jove R. Requirement for Ras/Rac1-mediated p38 and c-Jun N-terminal kinase signaling in Stat3 transcriptional activity induced by the Src oncoprotein. Mol Cell Biol. 1999;19:7519–7528. doi: 10.1128/mcb.19.11.7519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turkson J, Bowman T, Garcia R, Caldenhoven E, De Groot RP, Jove R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol Cell Biol. 1998;18:2545–2552. doi: 10.1128/mcb.18.5.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X, Yue P, Page BDG, Li T, Zhao W, Namanja A, Paladino D, Zhao J, Chen Y, Gunning PT, Turkson J. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc Natl Acad Sci U S A. 2012;109:9623–9628. doi: 10.1073/pnas.1121606109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim JE, Patel M, Ruzevick J, Jackson CM, Lim M. STAT3 activation in glioblastoma: Biochemical and therapeutic implications. Cancers (Basel) 2014;6:376–395. doi: 10.3390/cancers6010376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahaman SO, Harbor PC, Chernova O, Barnett GH, Vogelbaum MA, Haque SJ. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene. 2002;21:8404–8413. doi: 10.1038/sj.onc.1206047. [DOI] [PubMed] [Google Scholar]
- 29.Ayala I, Sounier R, Use N, Gans P, Boisbouvier J. An efficient protocol for the complete incorporation of methyl-protonated alanine in perdeuterated protein. J Biomol NMR. 2009;43:111–119. doi: 10.1007/s10858-008-9294-7. [DOI] [PubMed] [Google Scholar]
- 30.Goto NK, Gardner KH, Mueller GA, Willis RC, Kay LE. A robust and cost-effective method for the production of Val, Leu, Ile (delta 1) methyl-protonated 15N-, 13C-, 2H-labeled proteins. J Biomol NMR. 1999;13:369–374. doi: 10.1023/a:1008393201236. [DOI] [PubMed] [Google Scholar]
- 31.Hu W, Namanja AT, Wong S, Chen Y. Selective editing of Val and Leu methyl groups in high molecular weight protein NMR. J Biomol NMR. 2012;53:113–124. doi: 10.1007/s10858-012-9629-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li L, Cheung S-h, Evans LE, Shaw PE. Modulation of gene expression and tumor cell growth by redox modification of Stat3. Cancer Res. 2010;70:8222–8232. doi: 10.1158/0008-5472.CAN-10-0894. [DOI] [PubMed] [Google Scholar]
- 33.Avonto C, Taglialatela-Scafati O, Pollastro F, Minassi A, Di Marzo V, De Petrocellis L, Appendino G. An NMR spectroscopic method to identify and classify thiol-trapping agents: revival of Michael acceptors for drug discovery? Angew Chem Int Ed Engl. 2011;50:467–471. doi: 10.1002/anie.201005959. [DOI] [PubMed] [Google Scholar]
- 34.Kimura M, Yoshioka T, Saio M, Banno Y, Nagaoka H, Okano Y. Mitotic catastrophe and cell death induced by depletion of centrosomal proteins. Cell Death Dis. 2013;4:e603. doi: 10.1038/cddis.2013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 36.Ichiba M, Nakajima K, Yamanaka Y, Kiuchi N, Hirano T. Autoregulation of the Stat3 gene through cooperation with a cAMP-responsive element-binding protein. J Biol Chem. 1998;273:6132–6138. doi: 10.1074/jbc.273.11.6132. [DOI] [PubMed] [Google Scholar]
- 37.Turkson J. STAT proteins as novel targets for cancer drug discovery. Expert Opin Ther Targets. 2004;8:409–422. doi: 10.1517/14728222.8.5.409. [DOI] [PubMed] [Google Scholar]
- 38.Youn UJ, Park E-J, Kondratyuk TP, Simmons CJ, Borris RP, Tanamatayarat P, Wongwiwatthananukit S, Toyama O, Songsak T, Pezzuto JM, Chang LC. Anti-inflammatory sesquiterpene lactones from the flower of Vernonia cinerea. Bioorg Med Chem Lett. 2012;22:5559–5562. doi: 10.1016/j.bmcl.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 39.Chen X, Zhan Z-J, Zhang X-W, Ding J, Yue J-M. Sesquiterepene lactones with potent cytotoxic activities from Vernonia chinensis. Planta Med. 2005;71:949–954. doi: 10.1055/s-2005-871212. [DOI] [PubMed] [Google Scholar]
- 40.Youn UJ, Miklossy G, Chai X, Wongwiwatthananukit S, Toyama O, Songsak T, Turkson J, Chang LC. Bioactive sesquiterpene lactones and other compounds isolated from Vernonia cinerea. Fitoterapia. 2014;93:194–200. doi: 10.1016/j.fitote.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohnishi M, Yoshimi N, Kawamori T, Ino N, Hirose Y, Tanaka T, Yamahara J, Miyata H, Mori H. Inhibitory effects of dietary protocatechuic acid and costunolide on 7,12-dimethylbenz[a]anthracene-induced hamster cheek pouch carcinogenesis. Jpn J Cancer Res. 1997;88:111–119. doi: 10.1111/j.1349-7006.1997.tb00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garcia R, Bowman TL, Niu G, Yu H, Minton S, Muro-Cacho CA, Cox CE, Falcone R, Fairclough R, Parson S, Laudano A, Gazit A, Levitzki A, Kraker A, Jove R. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene. 2001;20:2499–2513. doi: 10.1038/sj.onc.1204349. [DOI] [PubMed] [Google Scholar]
- 43.Johnson PJ, Coussens PM, Danko AV, Shalloway D. Overexpressed pp60c-src can induce focus formation without complete transformation of NIH 3T3 cells. Mol Cell Biol. 1985;5:1073–1083. doi: 10.1128/mcb.5.5.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Namanja AT, Li YJ, Su Y, Wong S, Lu JJ, Colson LT, Wu C, Li SS, Chen Y. Insights into high affinity small ubiquitin-like modifier (SUMO) recognition by SUMO-interacting motifs (SIMs) revealed by a combination of NMR and peptide array analysis. J Biol Chem. 2012;287:3231–3240. doi: 10.1074/jbc.M111.293118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.