

Abstract
Since its introduction in the early 1990s, layer-by-layer (LbL) self-assembly of films has been widely used in the fields of nanoelectronics, optics, sensors, surface coatings, and controlled drug delivery. The growth of this industry is propelled by the ease of film manufacture, low cost, mild assembly conditions, precise control of coating thickness, and versatility of coating materials. Despite the wealth of research on LbL for biomolecule delivery, clinical translation has been limited and slow. This review provides an overview of methods and mechanisms of loading biomolecules within LbL films and achieving controlled release. In particular, this review highlights recent advances in the development of LbL coatings for the delivery of different types of biomolecules including proteins, polypeptides, DNA, particles and viruses. To address the need for co-delivery of multiple types of biomolecules at different timing, we also review recent advances in incorporating compartmentalization into LbL assembly. Existing obstacles to clinical translation of LbL technologies and enabling technologies for future directions are also discussed.
1. Introduction
Biomaterials that interact with bodily tissue or fluids are primarily selected due to their properties that protect them against the patient’s immune response. The tissue–biomaterial interface is the key determinant of this biological response. Implants such as catheters, pacemakers, cochlear implants, and diagnostic sensors are designed to survive in vivo without integration into the surrounding tissue. Such implants are often coated with a polyethylene glycol film to prevent protein absorption and subsequent cell attachment.
In contrast, other implants depend on integration to survive, such as dental implants, bone screws, and hip stems. Chemical or physical surface treatments have been applied to such implants to enhance tissue integration by encouraging cell attachment and subsequent tissue ingrowth. Given the close proximity of the implant surface to the biological environment, this surface is an appropriate location for the presentation of biomolecules to further enhance or inhibit tissue interaction. For example, antimicrobial coatings on the surface of urinary catheters help prevent biofilm formation1 (a major source of infection), drug coatings on the surface of coronary stents aid in the fight against restenosis,2 and immobilization of growth factors on titanium surfaces has been applied to enhance osteointegration.3
Despite the widespread use of surface coatings, the ability to control biomolecule deposition, concentration, bioactivity, coating thickness, and the rate of release remain as significant challenges.4 Layer-by-layer (LbL) films were introduced in an effort to address many of these issues. LbL is a simple and versatile deposition process with broad application in materials science, for example in biomotors, superhydrophobic surfaces, biosensors, implant coatings, semiconductors, fiber optics, and drug-delivery devices. Previous detailed reviews of LbL assembly and applications in materials science discussed broad aspects of the technology;4–8 this review will focus specifically on LbL for controlled drug delivery.
LbL was introduced in 1992 to overcome some of the difficulties associated with other multilayer techniques, such as Langmuir–Blodgett and self-assembled monolayers.9 Langmuir–Blodgett films require expensive instrumentation and may only be used for the encapsulation of amphiphilic components,4 while self-assembled monolayers suffer from low loading efficiency and are only applicable to a limited range of surfaces.4 In contrast, LbL is a simple aqueous-based layering process that is better suited for the deposition of sensitive biomolecules on a range of material surfaces.
LbL films are created through the sequential deposition of biomolecules in solution containing functional groups that drive self-assembly.10,11 (Fig. 1) Most techniques rely on electrostatic interactions between oppositely charged polyelectrolytes during sequential deposition; however, a variety of other chemical interactions are also harnessed by LbL techniques, including hydrogen bonding,12 biomolecule recognition,13 click chemistry,14 and sol–gel reactions.15 Techniques are often combined for maximum versatility, empowering the user to customize films with maximum control over film thickness, biomolecule concentration, film stability, and release mechanisms and duration, while simultaneously protecting the functionality of the biomolecule of interest.
Fig. 1.
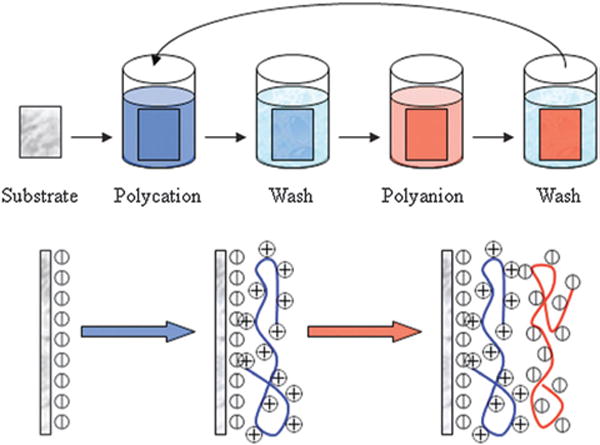
Schematic for the process of LbL assembly. The sequential deposition of positively (polycation) and negatively (polyanion) charged layers is applied to the substrate surface until the desired number of layers is achieved. Figure adapted from ref. 16.
In this review, we discussed various methods used for forming LbL assembly as thin film coatings, and the structures of the resulting films. In particular, this review focuses to review applications of LbL platforms for the delivery of various biomolecules including proteins, polypeptides, DNA, small molecules, particles and supramolecules such as viruses. The mechanisms that modulate biomolecule deposition and release were further reviewed. Most biomolecule delivery requires control over time and duration of controlled release, and recent progress in compartmentalization of LBL assembly to achieve controlled release of multiple biomolecules was highlighted.
2. Coating methods
Three methods currently exist for applying LbL coatings to a surface: dipping, spraying, and spin coating. Each method has distinct advantages and disadvantages, which are discussed below.
2.1 Dip coating
Dipping is the most commonly used method for LbL. The process is simple and does not require any specialized equipment. In a typical set-up, polyelectrolytes are stored in reservoirs, and the substrate to be coated is circulated through the reservoirs in the appropriate order. The process is repeated until the desired number of layers is achieved. As polyelectrolytes are stored in reservoirs, there is little loss of reagents, and concentration can be accurately controlled. Furthermore, the material to be coated is completely immersed in the reservoir solution, enabling the uniform coating of complex 3D structures.17 Fig. 2 depicts LBL coating on the surface of titanium rods; the LBL coating can been seen to fill the cavities of the rod surface (Fig. 2A and B) while fluorescence imaging is used to view the coating following deposition (Fig. 2C and D). Although dip coating is simple, the process is time consuming due to the time required to reach equilibrium adsorption for each coating step, especially in the case of weakly charged polyelectrolytes.18,19 The method may be automated with a simple slide strainer, allowing the accurate control of dipping time and order.20 The automated method also eliminates the likelihood of human error and enables the deposition of many layers over an extended time period (e.g. 400 layers over two days).17 Despite the use of automated equipment, more efficient techniques are required to make LbL a viable translatable technology. Spin coating is one such technique that may address the problem of lengthy coating time periods.
Fig. 2.

Scanning electron microscopy and fluorescence microscopy demonstrate surface changes of titanium rod surface before (A and C) or after LbL coating (B and D). Scanning electron microscopy showed LbL coating filled up the groves of the etched titanium surface. Fluorescent microscopy confirms the effective coating on the titanium surface with FITC-labeled protein after LbL coating. Figure adapted from ref. 21.
2.2 Spin coating
In spin coating, a liquid is deposited and spread across a planar surface through rapid spinning of the substrate. The film thickness is largely controlled by solution viscosity, angular speed, and spin time.22 The process is rapid (~30 s per layer), thereby significantly reducing the time for film construction. The major disadvantages of spin coating are the technical challenges of homogenously coating irregularly shaped 2D substrates and the inability to deposit films onto 3D substrates.23
However, spin coating is very useful for the preparation of 2D stand-alone films for drug delivery. Spin coating involves the rapid evaporation of solvent from the coating material, leading to the formation of films that are thicker than those resulting from the traditional dipping technique.6 Shear flow across the surface also leads to the formation of smoother films with less interlayer diffusion. Spin-coating LbL has been used in the development of doxorubicin-releasing thin films, which exhibited release characteristics dependent on the number of layers incorporated into the film.24,25 Despite the successful development of drug-releasing thin films manufactured through spin coating, their use in LbL assembly is limited to coatings on 2D substrates.
2.3 Spray coating
Spray coating has many advantages over dipping and spin-coating. Unlike spin coating, spraying enables homogenous coating of 3D substrates. Deposition is faster and smoother than by dipping, accelerating the process by more than 250-fold while retaining a high-quality finish.18 Dipping requires a deposition time of 15–20 min per layer to reach equilibrium, while spray-coated films only require 6 s per layer.18 Spray coating can even be performed without a rinsing step, which is always performed during dip coating.26
Dipping and spray coating, however, may lead to vastly different release profiles.27 Antibiotic-releasing films showed a linear release over 40 h after dip coating, while spray-coated samples released >90% of their cargo within 4 h. Films prepared via spray coating were consistently thinner, smoother, and contained higher drug concentrations than dip-coated films.28 Thus, spray coating is efficient and can significantly influence the controlled release of its cargo.
3. Structure
As reservoirs for controlled drug delivery, LbL coatings are used to encapsulate the payload and release it in response to an external stimulus. The coatings may be applied as a surface coating (Section 3.1) or built on a sacrificial template to create stand-alone structures (Section 3.2).
3.1 LbL surface coatings
In LbL surface coatings, a drug reservoir is applied to the surface of a material; the reservoir is designed to release the molecule of interest, such as a drug, in a controlled manner. The underlying material is often permanent and will continue to exist after the coating degrades (e.g. a cardiovascular stent).29 Surface coatings may be applied to control a biological response to the device (e.g. peri-implant tissue formation21), but may also represent the primary function of the device (e.g. drug coating on transdermal needles30). Since the coating is designed to control biomolecule release at the implant surface, it is essential that the coating remains integrated with the underlying material. Integration with the underlying material can be enhanced by pre-treatment of the surface; polycaprolactone may be plasma-etched to modify hydrophobicity,31 silicon may be exposed to warm silanol for the presentation of phosphonate groups,32 and titanium can be prepared in the presence of sodium hydroxide for the presentation of hydroxyl groups.33 To ensure adequate integration with the underlying material, foundation layers may be deposited prior to the deposition of biomolecule layers. Non-degradable materials such as polyethylenimine are useful for foundation layers, which remain intact during biomolecule release and persist after the biomolecule-containing layers are completely depleted.34 Care must be taken to ensure that surface coatings remain stable following LbL deposition, especially if the coating will experience harsh physical forces upon implantation.
3.2 LbL stand-alone structures
LbL is also useful for fabricating stand-alone structures. Such structures are created by performing LbL on a template surface; the template is then removed, leaving the layered structure intact.7 A variety of stand-alone structures have been created using LbL, including drug coated particles, microcantilivers,35 nanotubules,36 free-standing films,37,38 hollow spheres,39 and complex 3D structures.8
Drug coated particles are among the most commonly used stand-alone structures.40 The surface coating of drug particles can offer many advantages to the underlying drug including: targeted delivery, protection against degradation, a method to control release, and the possibility to arrest drug crystallization.41,42 Early studies demonstrated that LbL coating on microcrystals of ibuprofen could delay drug release by tailoring coating thickness, crystal size and material solubility.43 More recently, doxorubicin containing liposomes have been modified by the addition of PLA/siRNA multilayers on the outer surface of the nanoparticles. The dual delivery vehicles decreased tumor volume 8-fold when compared to non-treated controls.44
As an alternative to drug coating, hollow spheres may be constructed for the post-encapsulation of drug molecules. The hollow nature of the sphere creates an internal reservoir for drug loading. The layered structure in the outer coating can be used to incorporate additional biomolecules, to tailor drug release, or even to target delivery.45
Hollow-sphere fabrication begins with choosing a suitable template from the spectrum of available materials. The choice of materials depends on the final application and on restrictions due to the components of the layered structure. A major difficulty associated with stand-alone structures is the removal of the sphere’s core while preserving the layered structure and retaining the functionality of the entrapped biomolecules. Polystyrene,46 biocrystals,47 and silica beads48 are commonly used as templates for hollow-sphere construction; their removal requires solvents such as tetrahydrofurane or degradation under acidic conditions. Buffered hydrofluoric acid/ammonium fluoride (pH 5.5) was previously used to dissolve silica beads during the construction of enzyme-loaded hollow spheres.49 The buffered conditions retained the functionality of the enzyme, demonstrating that careful design of the process may yield a functional reservoir for controlled release. Crosslinking of the layered structure is often required to prevent the collapse of the coating after template removal.
4. LbL for delivery of different biomolecules
To apply LbL technologies for biomedical applications, various types of biomolecules have been explored as potential cargos for loading and release from LbL films. This section reviews the previous work on loading/release different types of biomolecules, validating their bioactivity after release using relevant assays.
4.1 Protein multi-layer films
One of the main trends in the biomedical applications of LBL technology is embedding bioactive proteins into thin films to enhance bioactivity of tissue engineering scaffolds or implantable materials. Bone morphogenetic protein 2 (BMP) is one of the most extensively studied proteins delivered using LBL films.50–54 BMP-2 is a dimeric disulfide-linked polypeptide growth factor under transforming growth factor-β superfamily, and has been approved by Food and Drug Administration (FDA) to induce bone repair. The efficacy of BMP-2 to induce bone formation in vivo is highly dependent on the release kinetics. The conventional methods used in clinic for BMP2 release often leads to rapid burst release, whereas more sustained long term delivery of BMP2 would be desirable for effective bone regeneration. This difficulty cannot be overcome satisfactorily merely by increasing the loading dose of BMP-2. Apart from the disadvantage of high cost, transient high local concentration of BMP-2 could induce various undesirable side effects such as excessive bone resorption or induction of bone formation at unintended sites.54 Using a LbL platform, the Hammond group51 reported that BMP-2 can be imbedded in LbL films and released protein retains its ability to induce osteogenic differentiation of preosteoblasts. When implanted intramuscularly in vivo, BMP-2 released from LbL coated implant surface induced bone differentiation of endogenous progenitor cells, which matured over nine weeks as measured by MicroCT imaging and histology. More recently, they also reported53 the co-delivery of osteoconductive hydroxyapatite (HAP) and BMP-2 using LbL coating acted synergistically to induce osteoblastic differentiation of endogenous progenitor cells without indications of foreign body response. In another study, Zheng et al.54 reported LbL assembled BMP2-coprecipitated BioCaP (BMP2-cop.BioCaP) particles, and monitored the in vivo responses in rats. Their results showed that LbL assembled particles led to 10-fold higher osteoinductive efficiency than the absorbed BMP-2 protein. Furthermore, their results showed that LbL formed particles reduced host foreign-body reaction to a clinically used bone-defect-filling material.
In addition to promote tissue regeneration, LbL technology has also been used for releasing proteins to modulate inflammation. 7ND is a mutant version of monocyte chemotactic protein 1 (MCP-1), and has been shown to reduce undesirable migration of macrophages by functioning as a dominant negative inhibitor of MCP-1.55 Our group has recently reported successful loading of the 7ND protein to the orthopaedic implant surface using LbL strategies with great stability. Furthermore, released 7ND from the coated implant retained its bioactivity and effectively reduced macrophage migration towards MCP-1. Such an LbL platform can be applied for controlled release of the 7ND protein from orthopedic implants in situ to reduce wear particle-induced inflammatory responses, thereby prolonging the lifetime of implants and reducing the need for revision surgeries.56
4.2 Polypeptide multilayer films
In addition to full size proteins, polypeptides represent another major category of biomolecules that holds great interest for delivery using LbL platforms. There are two major forms of secondary structure that are found in proteins, the α-helix and the β-sheet. The secondary structures of polypeptides embedded in LBL films have been explored by several groups.57–63 Haynie et al.60 immobilized poly-L-lysine (PLL) using the LbL method and found that the secondary structures of polypeptides did not change compared with those in solution. On the other hand, Müller63 and Boulmedais et al.62 showed that PLL underwent a transition from random coils to α-helixes when adsorbed from solution onto the partner PDDA, PAH, or poly(vinyl sulfate) layer because of the lower local pH in the LbL film. The difference in the observed results suggests that the interactions between polypeptides and polyelectrolytes in the LBL films, including hydrogen bonding, hydrophobic forces, and electrostatic attraction, are multifold and complex.61
4.3 LbL for DNA and oligonucleotide delivery
DNA vaccines have many potential benefits but have failed to generate robust immune responses in humans. Recently, methods such as electroporation have shown improved efficacy for DNA delivery in vivo, but a safe method for reproducible and pain-free DNA vaccination remains elusive. Sukhorukov et al.64 and Montrel et al.65 fabricated DNA-based LbL films by alternative assembly of anionic DNA strands and cationic polyelectrolytes such as PEI, PLL, and polyallylamine. Using various assays, the authors proved that the DNA conserved its double-helical structure in the LbL films. Water molecules were found to easily penetrate into all types of films and bind with DNA hydration centers (phosphate groups). In contrast to DNA films, the hydration in the LBL films did not initiate the B-to-A conformational transition of the double helix. More importantly, the DNA-containing films retained bioactivity and exhibited remarkable binding abilities with different DNA-intercalated molecules, including antitumor drugs. Demuth et al.66 also proposed an LBL-approach for rapid implantation of vaccine-loaded polymer films carrying DNA, immune-stimulatory RNA, and biodegradable polycations into the immune-cell-rich epidermis, using LbL coated microneedles with releasable polyelectrolyte multilayers. The authors demonstrated films transferred into the skin following the brief microneedle application promoted by local transfection and controlled the persistence of DNA and adjuvants in the skin from days to weeks, with kinetics being determined by film composition. Importantly, the released DNA vaccines induced immune responses against a model HIV antigen comparable to electroporation in mice, enhanced memory T-cell generation, and elicited 140-fold higher gene expression in non-human primate skin than intradermal DNA injection. These results suggest that the LbL method could provide a powerful tool for DNA delivery in situ from device coatings in a minimally invasive manner.
4.4 LbL for small molecule drug delivery
Most of all new chemical entities approved by the US Food and Drug Administration (FDA) were small molecules, many of which are not highly water-soluble. Smith et al.67 have reported nanoscale LBL-coatings for small molecule delivery using charged cyclodextrin polymers to trap a small molecular drug. The authors showed that surface-eroded films led to the release of embedded small molecule drugs within the cyclodextrin carrier with retained bioactivity. Furthermore, the release kinetics was found to be independent of the therapeutic agent and could be regulated through the choice of degradable polycations, which makes it broadly applicable for releasing different small molecules.
4.5 Particle multilayer films
Since LbL assembly forms under aqueous conditions, particles like micelles have been used to help encapsulate hydrophobic drugs. Kim et al.68 reported LbL assembly with drug-incorporated micelles in which multilayers were assembled via hydrogen-bonding rather than electrostatic interactions. In this case, the micelles were composed of poly(ethylene oxide)-block-poly(ɛ-caprolactone) (PEO-b-PCL) and were loaded with an antibacterial drug, triclosan. Due to the sensitive nature of hydrogen bonding, the film can be rapidly deconstructed to release micelles upon exposure to physiological conditions. The authors demonstrate that micelle LbL films loaded with antibacterial drug triclosan are effective in inhibiting the bacteria growth. Qi et al.69 recently reported particle multilayer films using two different polymeric micelles that have either a polycationic or polyanionic corona. Each micelle type was impregnated with dye molecules serving as model compounds. Release of the dye molecules was explored in the presence and absence of micelles in solution. The authors found that under both conditions the dye molecules were released from the film after 30 min of exposure. The LbL samples that were immersed into micelle-rich solutions released the dye molecules more rapidly than in the case of micelle deficient solutions. This suggests that the release rates for hydrophobic molecules not only depend on the degradability of the LbL films but also on the solubility of the drug in the selected solution.
4.6 Spontaneous assembly of viruses on LbL films
To examine the interactions between randomly arranged supermolecular species with LbL assembled films, Hammond and coworkers70 chose to use the LbL assembly process to incorporate genetically engineered M13 viral particles to create cohesive thin films. Their results show that M13, a highly complex biomacromolecule with a MW of about 14 000 000, could spontaneously form a two-dimensional monolayer structure of viruses atop a cohesive polyelectrolyte multilayer. They further demonstrate that such a viral-assembled monolayer can serve as a biologically tunable scaffold to nucleate, grow and align nanoparticles or nanowires over multiple length scales. This would allow coupling of virus functionality and advantage of LbL films and is highly tunable by choosing different polyions.
5. Mechanisms for deposition
5.1 Electrostatic bonding
Electrostatic bonding is by far the most commonly used LbL technique for controlled drug delivery. Interlayer bonding occurs through electrostatic interactions between positively and negatively charged polyelectrolytes (Fig. 3A). The process is performed under aqueous conditions and takes advantage of the natural charge density of biomolecules such as DNA, protein, peptides, and nanoparticles; it can even be used to incorporate multiple biomolecules into a single layered structure. The polyelectrolytes used for electrostatic bonding must be water-soluble (often a dilute acidic or basic solution is used to aid dissolution) and possess an excess positive or negative charge;72,73 commonly used polyelectrolyte couples include poly(acrylic acid) (PAA)/poly(allylamine hydrochloride) (PAH), poly(beta-amino ester) (PBAE)/chitosan, and poly(ethyleneimine)(PEI)/poly(styrene sulfonate) (PSS).
Fig. 3.
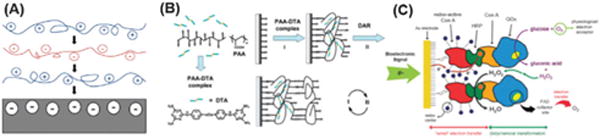
Schematic illustrates the formation of LbL coatings using different methods. (A) Electrostatic interactions; (B) hydrogen bonding; or (C) biological interactions. Figures adapted from ref. 13 and 71.
Polyelectrolytes with excess amine groups are often chosen for the formation of cationic layers; the choice of anionic polyelectrolytes can vary, but these molecules always possess a positive electron-to-proton ratio due to their negative charge. As layering occurs via electrostatic bonding, the template material must also possess a surface charge. If no surface charge exists, a variety of surface treatment options exist to achieve a surface charge as discussed earlier (Section 3.1). In the absence of surface charge, there are other options for LbL assembly, such as hydrogen bonding (Section 5.2). Fig. 4 highlights the formation and release of proteins from a LBL coating build via electrostatic interaction. Tailored release profiles are achievable by choosing appropriate polyelectrolytes and layering order.
Fig. 4.
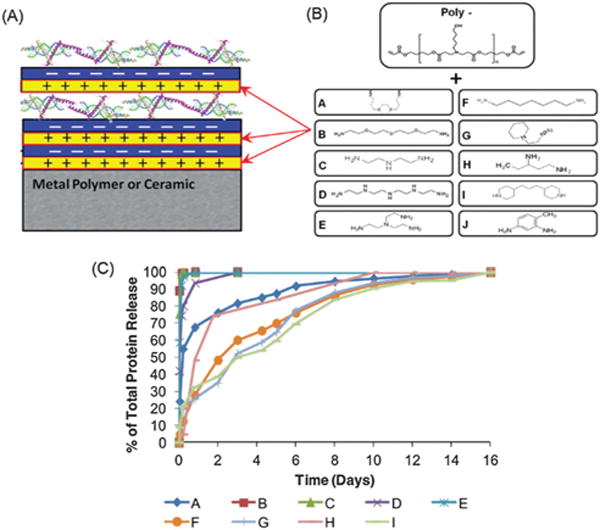
(A) Schematic of LbL assembly; (B) chemical structures of the end group of the cationic polymer used in constructing the LbL coating; (C) a broad range of the release profile from LBL coating can be achieved by tuning the chemical structure of the cationic polymer used for LBL assembly. Figure adapted from ref. 72.
5.2 Hydrogen bonding
Hydrogen bonding occurs when sequential layers consist of hydrogen bond donors and acceptors (Fig. 3B)12. The resulting bond is a dipole–dipole attraction and should not be confused with a covalent bond. Hydrogen-bonded LbL films expand the spectrum of LbL applications. Hydrogen bonds are sensitive to changes in the local environment, including temperature and pH.74 Therefore, LbL films constructed in such a manner may be used for a range of environment-sensitive drug-delivery applications. Hydrogen bonding also facilitates the incorporation of polymers with low glass transition temperatures (e.g. poly(ethylene oxide) (PEO)), which are particularly useful for flexible stand-alone structures. Finally, since hydrogen bonding involves polymers that are electrically neutral, materials that are perhaps unsuitable for electrostatic bonding can be incorporated into the multilayered structure. Common hydrogen-bonded couples include PEO/PAA and poly(vinyl methyl ether)/poly(methacrylic acid). PEO is a hydrogen bond donor and PAA is an acceptor; thus, hydrogen bonds form at the layer junction. This bonding mechanism has been exploited for the construction of PEO-containing block co-polymer micelles, enabling the incorporation of hydrophobic drugs into LbL systems.75 In perhaps the most useful application of hydrogen-bonded LbL films, temperature-sensitive polymers such as poly(N-isopropylacrylamide) (PNIPAM) and poly(N-vinylcaprolactam) serve as hydrogen donors. The temperature-induced swelling of these polymers enables drug loading and release in a controlled manner; additionally, the bonds formed by these polymers are reversible at high pH. Taken together, hydrogen-bonded LbL films greatly expand the set of available methods for drug loading and release.
5.3 Bonding through biomolecule recognition
In nature, spontaneous reactions bind molecules through a mechanism known as biorecognition (Fig. 3C). An example of such recognition is the highly specific interaction between antibodies and antigens. Sandwich enzyme-linked immunosorbent assay is a highly efficient yet simple example of bonding through biorecognition. In this technique, a surface-absorbed primary antibody acts as highly specific recognition sites for an antigen; the antigen then correspondingly acts as a biorecognition site for a secondary antibody. LbL based on biorecognition generally proceeds through a biotin/streptavidin interaction, which labels an antibody for detection purposes. Avidin is a tetramer protein consisting of four identical polypeptide chains, all of which have a high affinity for biotin. Its multiple recognition sites make avidin an ideal linker for biotin-labelled biomolecules such as biopolymers, cells, proteins, DNA, and lipids.76 Although this bonding mechanism has found little use in controlled drug release, a broad range of applications currently rely upon highly specific and efficient multilayer bonding e.g. enzyme-linked immunosorbent assays, immunohistology and biosensors13
Complementary DNA recognition is another biomolecule recognition phenomenon that can be used for LbL construction. 3-D tetrahedral DNA nanocages were constructed using spacer motifs with DNA tails.77 Adjacent DNA tails were designed with complementary sequences to foster hybridization. Depending on the length of the spacer and the location of DNA sequences, precise 3-D structures could be created. Furthermore, by introducing an ATP aptamer into the DNA tail region, the linkages could be dissociated through competitive binding when ATP was introduced into the system.77 This novel system highlights the possibility of using biological recognition for both the construction and dissociation of LbL systems.
6. Mechanisms of biomolecule release
The release of biomolecules from LbL-assembled films is dependent on the underlying mechanisms that catalyze degradation of the coating. Here we discuss degradation based on hydrolysis (Section 6.1), temperature (Section 6.2), pH (Section 6.3), enzymatic degradation (Section 6.4), and light (Section 6.5).
6.1 Hydrolysis
Many LbL platforms rely on hydrolysis to degrade entire polyelectrolyte layers and crosslinkers to release biomolecules of interest. Often, polyanions or molecules of interest are distributed between hydrolyzable polycation layers. As a result, a nondegradable biomolecule is released as the polycation is hydrolyzed.
During in vitro experimentation, LbL-coated materials are often dried to prevent premature, solvent-based degradation. Upon exposure to an aqueous solution, the coating undergoes hydrolysis and degradation. Initially, degradation via hydrolysis competes with swelling of the coating layers. Swelling, which occurs almost immediately after the addition of aqueous solvent, is dependent on both temperature and pH.78
A commonly used hydrolytic polycation is PBAE 1, also referred to as Polymer 1. Polymer 1 is able to undergo hydrolysis as a function of its ester linkages.79 Furthermore, due to its amine functionality and slow degradation rate in acidic environments,78 Polymer 1 is preferentially used as the primary cation for LbL. The kinetics of this degradation reaction explains the pH-dependent degradation rates for Polymer 1.79 It also has been hypothesized that the hydrolysis of Polymer 1 and other similar polycations occurs as a result of nucleophilic attack by its own amine groups.78,80,81
In addition to its extensive characterization,79,82,83 Polymer 1 has been successfully coupled to a broad spectrum of polyanions, such as the model polyanions PSS and PAA84 and DNA plasmids.34 Release profiles for other polyanions of interest, such as chondroitin sulfate and heparin, have been characterized using Polymer 1 as the hydrolytically degradable loading layer;78 various polyanions demonstrated pH-dependent swelling and linear degradation.78 Furthermore, by controlling the hydrophobicity of the cationic polymer, the rate of hydrolysis, and hence biomolecule release, can be easily modified.83
Alternatively, hydrolysis can drive biomolecule release via crosslinkers. For a crosslinker comprised of a dextran backbone and azide and alkyne moieties, the azide and alkyne groups connect to the backbone via hydrolyzable carbonate esters. Hydrolysis of the crosslinkers triggers the degradation of the outer LbL coating and the release of biomolecules stored inside the microsphere.85 In this specific case, the shell of the microsphere containing the biomolecule of interest was created by LbL coating, rather than embedding the biomolecule directly in the layers of the LbL coating.
6.2 Temperature
Temperature changes can be used to control biomolecule loading and release (Fig. 5A). This allows the user to achieve “on-demand” drug delivery with external heating/cooling sources. Two approaches that use temperature to stimulate or inhibit release are: (1) the incorporation of a thermoresponsive polymer and (2) heat-induced shrinking and expansion of LbL films. The thermoresponsive polymer PNIPAM is a particularly attractive biomaterial for this purpose because it has a lower critical solution temperature (LCST) of ~32 °C, which is close to physiological temperature. PNIPAM is hydrophilic below its LCST and hydrophobic above it.86 Block copolymer micelles assembled with tannic acid and poly(N-vinylpyrrolidone)-b-PNIPAM, which encapsulated the drug doxorubicin, showed retention of doxorubicin in the block copolymer micelle core at 37 °C (above PNIPAM’s LCST) and rapid release at 20 °C (below PNIPAM’s LCST).87 Heat treatment has also been shown88 to control release through inhibition, rather than stimulation. An increase in external temperature above 35 °C, the glass transition temperature in this case, resulted in decreased permeability, wall thickening, and densification of polydiallyldimethylammonium chloride/PSS and poly(arginine)/DS capsules, leading to the entrapment of biomolecules.88 Biomolecules of varying hydrodynamic radius (fluorescein isothiocyanate (FITC)-dextran and PAA) were both successfully entrapped through heat treatment.88
Fig. 5.
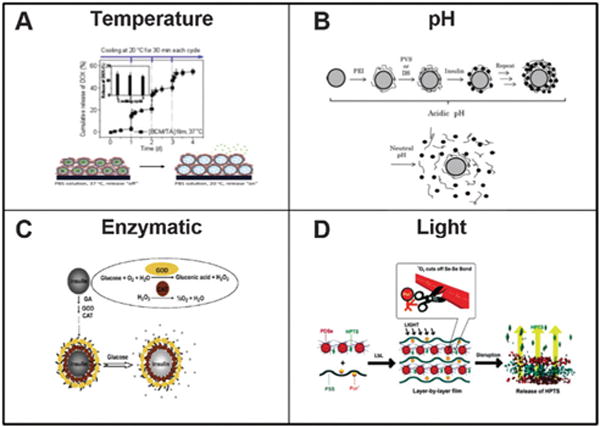
Various methods to trigger LbL degradation. (A) Temperature; (B) pH; (C) enzymatic or (D) light. Figures adapted from ref. 87, 90, 95 and 103.
6.3 pH
pH has been shown to modulate the release of biomolecules from LbL films through two distinct mechanisms: (1) loss of electrostatic forces and (2) induction of porosity in multilayer films (Fig. 5B). The pH under which film assembly occurs affects the charge of polyelectrolytes upon deposition and highly influences layer interaction.86 However, once assembled, external pH changes in the microenvironment can cause weak polyelectrolytes to undergo charge reversal, especially in the range of a species’ pKa. Charge transition leads to a loss of electrostatic interaction between the layers, disassembling the film. Mesoporous silica nanoparticles were coated with FITC-chondroitin sulphate/sodium alginate followed by PEGylation.89 Doxorubicin was then absorbed into the nanoparticles as a model chemotherapeutic drug. The resulting nanoparticles demonstrated pH sensitive release of doxorubicin due to decreased electrostatic forces between adjacent polyelectrolytes. Such drug delivery systems are particularly lucrative for targeting the low pH of a tumor microenvironment.
Insulin-containing LbL films were shown to undergo a charge transition, either from positive to negative or negative to positive, depending on the associated polyelectrolyte, assembly method, and external pH.90 When insulin was paired with a polyanion (poly(vinylsulfate) or dextran sulfate (DS)) prepared at pH 4, it was released in solutions with pH 5.0–7.4 due to a positive-to-negative charge shift.90 Conversely, when insulin was paired with the polycation PAH with assembly at pH 7.4, it was released upon exposure to solutions of pH ≤ 5.91 This disassembly was attributed to a negative-to-positive insulin charge shift.91
The second mechanism through which changes in pH trigger biomolecule release is the induction of porosity in multilayer films. Exposure of films to acidic conditions for as few as 30 s has been shown to create micropores and nanopores in the films,92 which may affect the permeability of biomolecules contained within these films. In addition to controlled release, microporous and nanoporous polymers can be useful for anti-reflection coatings. PAH/PAA films formed reversible, pH-responsive pores upon immersion in a solution of pH 1.8 for 30 s.93 The proposed mechanism for pore formation is the protonation of PAA’s carboxylic acid groups at low pH, which cleaves ionic bonds between PAH and PAA and reorganizes the film.94
6.4 Enzymatic degradation
Another method for releasing biomolecules from LbL platforms relies on enzymes to catalyze film degradation and subsequent biomolecule release (Fig. 5C). Enzymes can be an internal component of the LbL film or an external mechanism that triggers degradation upon exposure.
As enzymatic components of the LbL film, catalase and glucose oxidase were used to coat capsules loaded with insulin.95 The permeability of the enzymatic multilayer changed in response to glucose concentration and the degradation of glutaraldehyde (GA) crosslinks. The interaction between glucose and the GA crosslinker lowered the pH of the solution, catalyzing a reaction in the enzymatic LbL shell, which consisted of glucose oxidase and catalase. As a result, shell permeability increased and facilitated the release of insulin.95
Enzymes can be introduced into the LbL film to induce degradation. For example, FITC-dextran was encapsulated in poly-L-arginine (pARG)/DS LbL-coated capsules.96 Exposure to enzymes (in vitro, pronase, a mixture of proteases; in vivo, proteases from VERO-1 cells) catalyzed the degradation of the outer LbL coating and facilitated FITC-dextran release.96
Enzymatic triggered release can be a useful method to achieve targeted drug delivery. Doxorubicin (DOX) and indocyanine green coated nanoparticles were coated with a layer of casein.97 In vivo studies demonstrated protection of the drug load through the low pH gastric environment, however enzymatic destruction of the casein layer in the small intestines resulted in the release of the payload.
Current work95 with LbL platforms catalyzed via enzymatic release often employs microspheres or other capsules to release the biomolecule of interest. This technique is well suited for translational applications in which the catalytic enzyme is location or target specific.
6.5 Light
Light can also induce the disassembly of multilayer films; visible, near-infrared, and ultraviolet (UV) light each trigger biomolecule release (Fig. 5D). Light is an attractive trigger for release due to its spatial and temporal precision and its ability to be applied remotely, rendering it noninvasive.98 Visible light was shown to produce reactive oxygen species in vivo that can cleave diselenide bonds in a diselenide-containing polycation layered with a PSS polyanion, resulting in controlled release of 8-hydroxy-1,3,6-pyrenetrisulfonic acid, a fluorophore.98 The application of near-infrared radiation successfully resulted in the controlled release of doxycycline from an Ag-nanocage surrounded by mesoporous SiO2 and coated with PNIPAM,99 and UV light triggered the aggregation of poly(diallyldimethylammonium chloride) (PDADMAC)/poly(1-[4-(3-carboxy-4-hydroxyphenylazo) benzenesulfonamido]-1,2-ethanediyl, sodium salt) (PAZO) polyelectrolytes for microcapsule breakage and the release of bovine serum albumin.100
Aside from responding to different types of light, the aforementioned examples utilized light in mechanistically different ways for biomolecule release, justifying the choice of biomaterials used in LbL assembly for each application. The visible light example relied on photochemical cleavage of polymer bonds to liberate the fluorophore trapped between layers.98 In the near-infrared example,99 light was used to elicit a thermal response that subsequently triggered biomolecule release. Heat was released upon light absorption by metal and metal-oxide nanoparticles and dyes. In this near-infrared example, Ag-nanocages were used as “heaters” to control the release of doxycycline from the Ag-nanocage and PNIPAM-coated silica shells.99,101 Gold and silver nanoparticles absorb visible light,102 while titanium-oxide nanoparticles absorb UV light.102 Fluorescent and porphyrinoid dyes, which absorb light in the visible spectrum, have also been used instead of high-energy metal nanoparticles because of their ability to produce a more controlled optical response.102 This strategy may be beneficial for controlled release, rather than a burst release, of the encapsulated material.102 In the PDADMAC/PAZO system, UV light-induced azo aggregation followed by capsule breakage was attributed to a photoisomerization reaction.100 The azobenzene derivative PAZO, which consists of two phenyl rings connected by an azo (N=N) bond, responds to UV light through cis–trans isomerization.100 In this case, the steric hindrance of azo aggregates inhibited full cis–trans isomerization, and capsule breakage was irreversible;100 however, other studies100–102 have shown that isomerization leads to membrane disruption and content release in a reversible fashion. In addition to photochemical cleavage, photothermal effects, and photoisomerization, other mechanisms of light-triggered release include photocrosslinking and decrosslinking, photo-induced oxidation, and photochemical hydrophobicity changes.101 Overall, the photo-responsiveness of an LbL apparatus is highly dependent on the type of light and the photo-sensitivity and reactivity of the materials used.
7. Controlling compartmentalization
Efficient compartmentalization is a useful mechanism to control biomolecule release, or even to incorporate triggers for the timed release of multiple biomolecules. For example, crosslinking various components of the LbL construct during coating, as done in methods based on pH and covalent chemical crosslinking, alters the release profile of biomolecules. Modifying polyelectrolyte layers and/or introducing additional coating layers allow for adjustments to the compartmentalization20 and release profiles104 for biomolecules of interest. In this section, we further explore the utility of structural and functional coating modifications in blocked (Section 7.1) and sequential (Section 7.2) release.
7.1 Blocking layers
Blocking layers within LbL films can be used to create compartmentalized films as a means to regulate interlayer diffusion and further tailor release profiles, especially in terms of the order of biomolecule release. The build-up of LbL-assembled films can either follow a linear or an exponential growth curve depending on properties such as the diffusivity of and electrostatic forces between polyelectrolytes. Polyelectrolytes exhibiting linear growth are often highly charged and non-diffusive, such as PAH, PAA, and PSS. On the other hand, polyelectrolytes that exhibit exponential growth are weakly charged and highly diffusive, for example poly(L-lysine), sodium alginate, and poly(lactide-co-glycolides).92
Alternation of polyelectrolytes with linear and exponential growth profiles can yield stratified, multicompartmentalized films,105 allowing the release of multiple drug types in an ordered and temporally controlled manner. Less-diffusive polyelectrolytes form a “barrier” layer between highly diffusive polyelectrolytes, which form a “reservoir.”105 Blocking layers, which often consist of linearly grown and less-diffusible polyelectrolytes, are commonly comprised of materials such as PAH/PAA,20 clay,106,107 and graphene oxide.108 A single covalently crosslinked PAH/PAA barrier layer was shown to delay release of linearly growing DS, resulting in sequential release of heparin and DS. However, the highly diffusible heparin was not inhibited by the PAH/PAA blocker, highlighting the limitations of sequential release order in such a system.20
One of the first blocking-layer studies employed the clay mineral montmorillonite as a barrier for Ca2+ ion diffusion.106 Clay, a charged, inorganic replacement for a polyelectrolyte, enhances the mechanical durability of the resulting films.106,107 Clay barriers remain a material of active interest; a recent study used a LAPONITE® clay barrier to achieve temporally controlled release of a recombinant human bone morphogenic protein (rhBMP-2) and gentamicin (GS) (GS release is depicted in Fig. 6).107 The clay barrier successfully delayed the release of the diffusive molecule gentamicin, while the release of non-diffusive rhBMP-2 was delayed through superior stacking of gentamicin alone.107
Fig. 6.
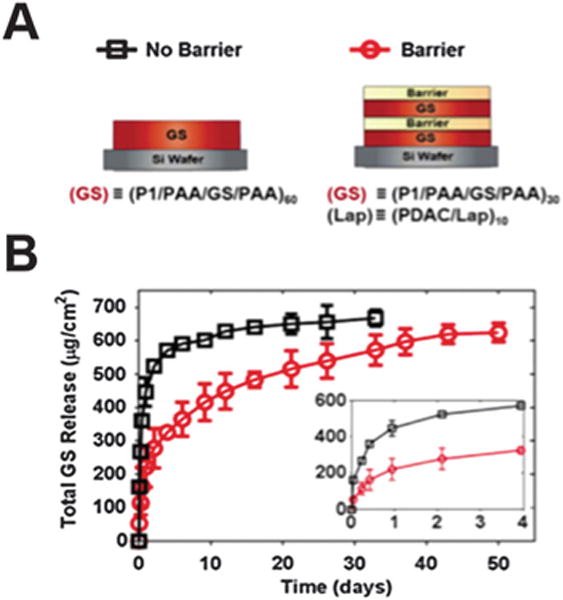
Delaying biomolecule release from LbL using clay barrier layers. (A) The layering structure is depicted highlighting the location of the clay barrier layer. (B) Release profile of gentamicin (GS) with and without the inclusion of the barrier layer. Figure adapted from ref. 107.
Graphene oxide has also been used as a barrier layer because of its low permeability and its ability to be charged via the introduction of carboxylic acid groups with strong acid and amine groups in a reaction with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide.108 The number of graphene oxide layers was found to be proportional to the time delay of ovalbumin release.108
7.2 Sequential release
The degradation rates of many LbL platforms depend upon interlayer diffusion within the coating complex. Current work investigates the compartmentalization of the various layers of LbL coating to allow for multiagent, sequential release of biomolecules from a single platform. Loading multiple biomolecules onto a single platform in a controlled manner using LbL empowers many translational applications to release biomolecules along various time scales.
Sequential release of plasmid DNA for transfection relied on the modification of side-chain functionality of the PBAE, Polymer 2.109 This additional amine functionality facilitated stratification of the plasmid DNA as a function of loading order; it prevented interlayer rearrangement and the diffusion that is present with common PBAEs.109 The resulting release curve indicated that release was loading-dependent, since pDsRed-N1 plasmid DNA in the top layers was released before pEGFP-N1 plasmid DNA in the bottom layers and vice versa.109 The different release rates for the different plasmid DNA molecules demonstrated that, sequential diffusion was achieved without chemical or pH-dependent crosslinking, which could affect biomolecule functionality (Fig. 7).109
Fig. 7.
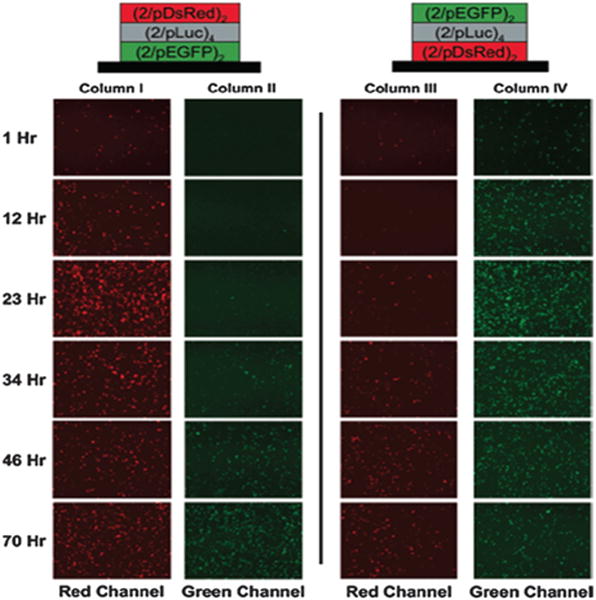
Sequential release of plasmid DNA can be achieved by staggering the deposition of each plasmid DNA. A blocker layer (2/pLuc) can be used further delay the release of second DNA. Successful sequential DNA release was demonstrated by sequential transfection of cells with EGFP or DsRed encoding plasmid DNA. Figure adapted from ref. 109.
Similarly, altering the charge density of polyelectrolytes already used in LbL coating alters the release profile and facilitates the compartmentalization of biomolecules of interest.104 PAA has a higher charge density than chondroitin sulfate, thus yielding more ionic crosslinking. As a result, PAA and chondroitin sulfate can be coupled within the same LbL coating such that PAA is used to load biomolecules for more long-term release and chondroitin sulfate is used for biomolecules with shorter desired release profiles.104 BMP-2 and vascular endothelial growth factor (VEGF) are two proteins that are important for bone regeneration and have been shown to exhibit synergy in bone healing response both in vitro and in vivo.110 These proteins were released from the same polyelectrolyte multilayer film, with BMP-2 released over 2 weeks and VEGF released over 8 days.104 In order to achieve the different release patterns, the polyanion with higher charge density, PAA, was used to load BMP-2, while VEGF was released via chondroitin sulfate.104
Multiagent release of heparin followed by dextran sulfate employed a single, covalently crosslinked barrier layer of PAA and PAH (Section 7.1). The release of two different polysaccharides from the same surface with Polymer 1 as the polycation demonstrated the top-down degradation pattern of LbL platforms.20 In this study, different rates of release for different barrier layer conditions also emphasized that barrier layers are subject to extensive manipulation.20 Ionic crosslinking was not as effective at compartmentalizing different biomolecules as covalent bond-based layers.20
In sum, investigations into sequential release are heavily coupled with current research on blocker layers. Physical barriers between compartments of different biomolecules empower sequential release, while other methods rely on modification of the polycation and polyanion components of the LbL coating itself. The mechanism underlying sequential release is heavily influenced by the biomolecule of interest and the conditions needed to preserve its functionality and structure. Release from a single platform over different time scales20,109 promises greater translational application and better mimicry of physiological healing conditions, as shown for BMP-2 and VEGF release for bone regeneration.104
8. Current challenges and future directions
LbL is a highly versatile platform with applications ranging from microelectronics to implant coatings.4,5,111 Many applications require the deposition of at least 5–10 layers to alter surface properties (e.g. modification of light path112). As biomolecule release is often required to occur over days or weeks, a large reservoir of biomolecules must be deposited on the material surface. In order to prevent the rapid diffusion of biomolecules across the layered structure, many layers must encase the reservoir, resulting in long production time periods and high variability. For example, in order to deposit BMP-2 on the surface of polycaprolactone/tricalcium phosphate scaffolds, 400 layers of BMP-2 were deposited over two days.17
New polymers enable greater control over biomolecule binding and subsequent release. Protein binding and release were both affected by small-molecule end groups located on the termini of PBAE polymers.72 Polymer end groups could be selected to modify protein release over hours or weeks.72 Novel polymers allowed the deposition and controlled release of growth factors using as few as 10 layers,72 and a similar strategy drove the deposition of anti-inflammatory molecules on the surface of titanium rods.21 It was shown that layering order and chemical make-up of the polycation lead to drastically different release characteristics, with release over one week after the deposition of as few as 15 layers.72 Reductions in the layer number, however, must be accompanied by increased binding affinity for the biomolecule of interest in order to maintain a high reservoir concentration. It should also be noted that as the layer number decreases, the barrier for diffusion also decreases; therefore, controlled release and (especially) compartmentalization are difficult to achieve.
The main obstacle to achieving compartmentalization is the inability to control interlayer diffusion. A solid understanding of the fundamental mechanisms underlying layer formation will help guide material selection, yielding films with controlled and predictable release properties. As an example, during linear layer growth, absorbing species are deposited on the upper surface and remain kinetically locked, with little interlayer diffusion. Polymeric chains typically interpenetrate with adjacent layers and are found in the 3–4 layers above or below the point of absorption.6 However, during exponential growth, polymers readily diffuse throughout the bulk of the layered structure and only return to the surface during deposition of a complementary charged molecule.6 Given the highly diffusive nature of polyelectrolytes, compartmentalization is difficult to achieve. Interdiffusion occurs when polyanions of low charge density and high mobility are incorporated into the layered structure. As charge density increases, polyanions undergo less diffusion through the bulk structure.6 Similarly, charged molecules with low molecular weight diffuse more readily than similar molecules with higher molecular weight.6 Understanding how materials are deposited and diffuse in the layered structure will help model and predict release characteristics, thus facilitating the development of thin films with ordered release of multiple biomolecules.
Another obstacle to the clinical translation of LbL coatings is the ability to form coatings in a time- and cost-effective manner. Spin coating (Section 2.2) and spray coating (Section 2.3) lead to shorter production time periods, which also decrease production costs. Shorter production and layering time periods not only speed up the rate of layer formation, but also minimize the diffusion of biomolecules from the coating surface during deposition (5–10 min per layer). It has been demonstrated that spray coating decreases the production time 250-fold over traditional dip coating (Section 2.1);18 however, the mechanism of layer formation can differ greatly, especially when depositing weakly charged molecules. For example, vancomycin was previous deposited via dipping or spray coating. Since vancomycin is a weakly charged molecule, deposition through dip coating resulted in significant interdiffusion.28 However, one spray cycle occurs over a time scale that is shorter than that of interdiffusion, and thus the drug remains at the surface of the layered structure. This phenomenon yields thick, low-concentration films via dip coating and thin, high-concentration films via spray coating; these films exhibit different release profiles.28 This example highlights the advantage of spray coating and the necessity to fully understand the kinetics of deposition in the production of controlled release devices. Another approach to improve upon time and cost effectiveness is the use of microfluidics to form microcapsules. A microfluidic chip was used to deposit six hydrogen-bonded layers of PEM on an oil core in less than 3 minutes.113 Such chips are scalable, reduce material usage, open to automated production and can incorporate in-process screening for quality control (e.g. DLS for size analysis and UV absorbance for content verification).
Of particular future interest is the development of crystalline arrays of colloidal particles, which may be applied as templates for the construction of porous, 3D, layered structures. The high surface area of crystalline arrays may be useful to increase drug-loading concentration or even to spatially control drug presentation.8 Titanium-dioxide nanoarrays fabricated through a simple electrochemical process have been used to control drug release based on the nanotube diameter and length.114 A similar concept may be achieved using LbL on crystalline array templates, with the added advantage of accurately controlling mechanical stability, wall thickness, biomolecule affinity, and drug loading both within the walls and inside the porous array after template removal. LbL-coated transdermal microneedles are also seen as having strong translation potential.115 Coating can be performed through line-of-sight deposition, making spray coating a realistic strategy. Transdermal needles often require the rapid release of cargo, thus negating the need for barrier layers, crosslinking, or complex binding strategies to delay biomolecule diffusion.116 Finally, stent coatings are normally applied using a spray-coating technique similar to that used for LbL deposition,117 and may therefore be easily transitioned to LbL deposition. A proof of concept study demonstrated that LbL coated siRNA on cardiovascular stents withstood ethylene oxide sterilization and could deliver siRNA nanoparticles to porcine arteries ex vivo.118 Simple adjustments to the current equipment would enable LbL coating with greater control over release kinetics, allow for changes in dosage, and include biomolecules that were previously difficult to deposit.
Despite extensive research on LbL technology for drug delivery over the past 20 years, clinical translation of the technology remains lacking. There are some promising indications, however, that clinical translation is on the horizon. Artificial Cell Technologies Inc. (ACT) is currently developing artificial LbL assembly vaccines through the incorporation of immunogenic epitopes into nanofilms assembled through electrostatic interaction. ACT is currently preparing an IND filing to conduct Phase I human trials of its RSV vaccine. LayerBio is also developing controlled release solutions for ophthalmology and wound care utilizing LbL technology developed at Massachusetts Institute of Technology.
In conclusion, as our understanding of LbL coating evolves in tandem with the development of faster, more economical, and reproducible coating techniques, we increase our ability to build films suitable for clinical translation. A bright future awaits LbL biomolecule delivery, with goals now set on reaching the end user.
Acknowledgments
The authors would like to acknowledge National Institute of Health (Grants 2R01AR055650 and 1R01AR063717) and Stanford Department of Orthopaedic Surgery for funding. F. Y. would like to thank NIH (R01DE024772-01), California Institute of Regenerative Medicine Tools and Technologies Awards, the National Science Foundation (NSF) CAREER Award, and Stanford Child Health Research Institute Faculty Scholar Award for support. X. Y. Jiang would like to thank Stanford Child Health Research Institute postdoctoral fellowship for support.
Biographies

M. Keeney
Michael Keeney received his PhD in Biomedical Engineering from the National University of Ireland in 2010. He joined Prof. Fan Yang’s lab at Stanford University in 2010 where he worked as a postdoctoral fellow for 4 years with research focus on novel materials and systems for controlled release. During his postdoctoral experience he authored over 15 peer reviewed publications. He entered the industrial sector in 2014 where he currently works as a scientist.

X. Y. Jiang
Xinyi Jiang received his PhD in Pharmaceutics from Fudan University (China) in 2012. He joined Prof. Fan Yang’s lab at Stanford University in 2013. He has authored over 20 peer reviewed publications from 2010 up to now. He received the Stanford Child Health Research Institute (CHRI) Grant & Postdoctoral Award in 2014 and Stanford Dean’s postdoctoral fellowships in 2013 in the School of Medicine at Stanford University, where he is now working as a postdoctoral fellow with a research focus on stem cell-based cancer gene therapy and layer-by-layer coating implants for drug delivery applications.

M. Yamane
Maya Yamane joined Professor Fan Yang’s research group in 2012, where she worked on developing a temporally tunable growth factor delivery platform using layer-by-layer nanocoating. She received her BA in Human Biology from Stanford University in 2014 with a concentration in Biotechnology and Infectious Diseases. She is currently an MD candidate at Columbia University, College of Physicians and Surgeons.

M. Lee
Meelim J. Lee is an undergraduate at Stanford University studying bioengineering. She joined the Stanford Fan Yang Group in 2013 and has worked on sustained protein release for bone regeneration using layer-by-layer nanocoatings.

S. Goodman
Stuart B. Goodman is the Robert L. and Mary Ellenburg Professor of Surgery, and Professor with Tenure in the Department of Orthopaedic Surgery at Stanford University. Dr Goodman received his BSc, MD and MSc (Institute of Medical Science) from the University of Toronto, and his PhD in Orthopedic Medical Science from Lund University in Sweden. His basic science interests center on biocompatibility of orthopaedic implants, inflammation, and musculoskeletal tissue regeneration and repair. Dr Goodman has published over 380 peer-reviewed manuscripts in medical and bioengineering journals. Dr Goodman and co-workers have received awards for their research from the Society for Biomaterials, Orthopaedic Research Society, the American Orthopaedic Association, Western Orthopaedic Association, and the Association of Bone and Joint Surgeons.

F. Yang
Fan Yang is currently an Assistant Professor at Stanford University in the Departments of Orthopaedic Surgery and Bioengineering, and Director of Stem Cells and Biomaterials Engineering Laboratory. Prior to joining Stanford, Prof. Yang received her PhD in Biomedical Engineering from the Johns Hopkins University School of Medicine, and conducted her postdoctoral Fellowship in the laboratory of Prof. Robert Langer at MIT, sponsored by the Ruth L. Kirschstein National Research Service Award. Her research seeks to understand how microenvironmental cues regulate stem cell fate, and to develop novel biomaterials and stem cell-based therapeutics for treating musculoskeletal diseases, cardiovascular diseases and cancer.
Notes and references
- 1.Darouiche RO, Raad, Heard SO, Thornby JI, Wenker OC, Gabrielli A, Berg J, Khardori N, Hanna H, Hachem R, Harris RL, Mayhall G. N Engl J Med. 1999;340:1–8. doi: 10.1056/NEJM199901073400101. [DOI] [PubMed] [Google Scholar]
- 2.Fattori R, Piva T. Lancet. 2003;361:247–249. doi: 10.1016/S0140-6736(03)12275-1. [DOI] [PubMed] [Google Scholar]
- 3.Puleo DA, Kissling RA, Sheu MS. Biomaterials. 2002;23:2079–2087. doi: 10.1016/s0142-9612(01)00339-8. [DOI] [PubMed] [Google Scholar]
- 4.Tang ZY, Wang Y, Podsiadlo P, Kotov NA. Adv Mater. 2007;19:906. [Google Scholar]
- 5.Ariga K, Hill JP, Ji QM. Phys Chem Chem Phys. 2007;9:2319–2340. doi: 10.1039/b700410a. [DOI] [PubMed] [Google Scholar]
- 6.Hammond PT. AIChE J. 2011;57:2928–2940. [Google Scholar]
- 7.Jiang CY, Tsukruk VV. Adv Mater. 2006;18:829–840. [Google Scholar]
- 8.Wang Y, Angelatos AA, Caruso F. Chem Mater. 2008;20:848–858. [Google Scholar]
- 9.Decher G, Hong JD, Schmitt J. Thin Solid Films. 1992;210:831–835. [Google Scholar]
- 10.Lvov Y, Decher G, Sukhorukov G. Macromolecules. 1993;26:5396–5399. [Google Scholar]
- 11.Lvov Y, Ariga K, Ichinose I, Kunitake T. J Am Chem Soc. 1995;117:6117–6123. [Google Scholar]
- 12.Kharlampieva E, Kozlovskaya V, Sukhishvili SA. Adv Mater. 2009;21:3053–3065. [Google Scholar]
- 13.Pallarola D, von Bildering C, Pietrasanta LI, Queralto N, Knoll W, Battaglini F, Azzaroni O. Phys Chem Chem Phys. 2012;14:11027–11039. doi: 10.1039/c2cp41225j. [DOI] [PubMed] [Google Scholar]
- 14.Such GK, Quinn JF, Quinn A, Tjipto E, Caruso F. J Am Chem Soc. 2006;128:9318–9319. doi: 10.1021/ja063043+. [DOI] [PubMed] [Google Scholar]
- 15.Ichinose I, Senzu H, Kunitake T. Chem Mater. 1997;9:1296–1298. [Google Scholar]
- 16.Xiang Y, Lu S, Jiang SP. Chem Soc Rev. 2012;41:7291–7321. doi: 10.1039/c2cs35048c. [DOI] [PubMed] [Google Scholar]
- 17.Macdonald ML, Samuel RE, Shah NJ, Padera RF, Beben YM, Hammond PT. Biomaterials. 2011;32:1446–1453. doi: 10.1016/j.biomaterials.2010.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Izquierdo A, Ono SS, Voegel JC, Schaaf P, Decher G. Langmuir. 2005;21:7558–7567. doi: 10.1021/la047407s. [DOI] [PubMed] [Google Scholar]
- 19.Kim SY, Hong J, Kavian R, Lee SW, Hyder MN, Shao-Horn Y, Hammond PT. Energy Environ Sci. 2013;6:888–897. [Google Scholar]
- 20.Wood KC, Chuang HF, Batten RD, Lynn DM, Hammond PT. Proc Natl Acad Sci U S A. 2006;103:10207–10212. doi: 10.1073/pnas.0602884103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keeney M, Waters H, Barcay K, Jiang XY, Yao ZY, Pajarinen J, Egashira K, Goodman SB, Yang F. Biomaterials. 2013;34:10287–10295. doi: 10.1016/j.biomaterials.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Norrman K, Ghanbari-Siahkalia A, Larsena NB. Annu Rep Prog Chem, Sect C: Phys Chem. 2006;101:174–201. [Google Scholar]
- 23.Yu L, Lee YY, Tay FEH, Iliescu C. J Phys: Conf Ser. 2006;34:937. [Google Scholar]
- 24.Serpe MJ, Yarmey KA, Nolan CM, Lyon LA. Biomacromolecules. 2005;6:408–413. doi: 10.1021/bm049455x. [DOI] [PubMed] [Google Scholar]
- 25.Nolan CM, Serpe MJ, Lyon LA. Biomacromolecules. 2004;5:1940–1946. doi: 10.1021/bm049750h. [DOI] [PubMed] [Google Scholar]
- 26.Schaaf P, Voegel JC, Jierry L, Boulmedais F. Adv Mater. 2012;24:1001–1016. doi: 10.1002/adma.201104227. [DOI] [PubMed] [Google Scholar]
- 27.Zhuk I, Jariwala F, Attygalle AB, Wu Y, Libera MR, Sukhishvili SA. ACS Nano. 2014;8:7733–7745. doi: 10.1021/nn500674g. [DOI] [PubMed] [Google Scholar]
- 28.Shukla A, Avadhany SN, Fang JC, Hammond PT. Small. 2010;6:2392–2404. doi: 10.1002/smll.201001150. [DOI] [PubMed] [Google Scholar]
- 29.Meng S, Liu Z, Shen L, Guo Z, Chou LL, Zhong W, Du Q, Ge J. Biomaterials. 2009;30:2276–2283. doi: 10.1016/j.biomaterials.2008.12.075. [DOI] [PubMed] [Google Scholar]
- 30.Saurer EM, Flessner RM, Sullivan SP, Prausnitz MR, Lynn DM. Biomacromolecules. 2010;11:3136–3143. doi: 10.1021/bm1009443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee M, Chen TT, Iruela-Arispe ML, Wu BM, Dunn JC. Biomaterials. 2007;28:1862–1870. doi: 10.1016/j.biomaterials.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 32.Lee H, Kepley LJ, Hong HG, Mallouk TE. J Am Chem Soc. 1988;110:618–620. [Google Scholar]
- 33.Chen JL, Li QL, Chen JY, Chen C, Huang N. Appl Surf Sci. 2009;255:6894–6900. [Google Scholar]
- 34.Zhang J, Chua LS, Lynn DM. Langmuir. 2004;20:8015–8021. doi: 10.1021/la048888i. [DOI] [PubMed] [Google Scholar]
- 35.Hua F, Tianhong C, Lvov YM. Nano Lett. 2004;4:823–825. [Google Scholar]
- 36.Liang Z, Susha AS, Yu A, Caruso F. Adv Mater. 2003;15:1849–1853. [Google Scholar]
- 37.Mamedov AA, Kotov NA. Langmuir. 2000;16:5530–5533. [Google Scholar]
- 38.Estephan ZG, Qian ZX, Lee D, Crocker JC, Park SJ. Nano Lett. 2013;13:4449–4455. doi: 10.1021/nl4023308. [DOI] [PubMed] [Google Scholar]
- 39.Réthoré G, Pandit A. Small. 2010;6:488–498. doi: 10.1002/smll.200901253. [DOI] [PubMed] [Google Scholar]
- 40.Amancha KP, Balkundi S, Lvov Y, Hussain A. Int J Pharm. 2014;466:96–108. doi: 10.1016/j.ijpharm.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 41.Strydom SJ, Otto DP, Stieger N, Aucamp ME, Liebenberg W, de Villiers MM. Powder Technol. 2014;256:470–476. [Google Scholar]
- 42.Wu T, Sun Y, Li N, de Villiers MM, Yu L. Langmuir. 2007;23:5148–5153. doi: 10.1021/la070050i. [DOI] [PubMed] [Google Scholar]
- 43.Qiu XP, Leporatti S, Donath E, Mohwald H. Langmuir. 2001;17:5375–5380. [Google Scholar]
- 44.Deng ZJ, Morton SW, Ben-Akiva E, Dreaden EC, Shopsowitz KE, Hammond PT. ACS Nano. 2013;7:9571–9584. doi: 10.1021/nn4047925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cortez C, Tomaskovic-Crook E, Johnston APR, Radt B, Cody SH, Scott AM, Nice EC, Heath JK, Caruso F. Adv Mater. 2006;18:1998–2003. [Google Scholar]
- 46.Wang L, Sasaki T, Ebina Y, Kurashima K, Watanabe M. Chem Mater. 2002;14:4827–4832. [Google Scholar]
- 47.Caruso F, Trau D, Möhwald H, Renneberg R. Langmuir. 2000;16:1485–1488. [Google Scholar]
- 48.Zelikin AN, Li Q, Caruso F. Angew Chem, Int Ed Engl. 2006;45:7743–7745. doi: 10.1002/anie.200602779. [DOI] [PubMed] [Google Scholar]
- 49.Yu AM, Wang YJ, Barlow E, Caruso F. Adv Mater. 2005;17:1737–1741. [Google Scholar]
- 50.Crouzier T, Ren K, Nicolas C, Roy C, Picart C. Small. 2009;5:598–608. doi: 10.1002/smll.200800804. [DOI] [PubMed] [Google Scholar]
- 51.Macdonald ML, Samuel RE, Shah NJ, Padera RF, Beben YM, Hammond PT. Biomaterials. 2011;32:1446–1453. doi: 10.1016/j.biomaterials.2010.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Min J, Braatz RD, Hammond PT. Biomaterials. 2014;35:2507–2517. doi: 10.1016/j.biomaterials.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shah NJ, Hyder MN, Moskowitz JS, Quadir MA, Morton SW, Seeherman HJ, Padera RF, Spector M, Hammond PT. Sci Transl Med. 2013;5:191ra183. doi: 10.1126/scitranslmed.3005576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zheng Y, Wu G, Liu T, Liu Y, Wismeijer D, Liu Y. Clin Implant Dent Relat Res. 2014;16:643–654. doi: 10.1111/cid.12050. [DOI] [PubMed] [Google Scholar]
- 55.Jiang X, Sato T, Yao Z, Keeney M, Pajarinen J, Lin Th, Loi F, Egashira K, Goodman S, Yang F. J Orthop Res. 2015 doi: 10.1002/jor.22977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keeney M, Waters H, Barcay K, Jiang X, Yao Z, Pajarinen J, Egashira K, Goodman SB, Yang F. Biomaterials. 2013;34:10287–10295. doi: 10.1016/j.biomaterials.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Antunes JC, Pereira CL, Molinos M, Ferreira-da-Silva F, Dessì M, Gloria A, Ambrosio L, Gonçalves RM, Barbosa MrA. Biomacromolecules. 2011;12:4183–4195. doi: 10.1021/bm2008235. [DOI] [PubMed] [Google Scholar]
- 58.Barrantes A, Santos O, Sotres J, Arnebrant T. J Colloid Interface Sci. 2012;388:191–200. doi: 10.1016/j.jcis.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 59.Westwood M, Noel TR, Parker R. Carbohydr Polym. 2013;94:137–146. doi: 10.1016/j.carbpol.2012.12.065. [DOI] [PubMed] [Google Scholar]
- 60.Haynie DT, Balkundi S, Palath N, Chakravarthula K, Dave K. Langmuir. 2004;20:4540–4547. doi: 10.1021/la036330p. [DOI] [PubMed] [Google Scholar]
- 61.Tang Z, Wang Y, Podsiadlo P, Kotov NA. Adv Mater. 2006;18:3203. [Google Scholar]
- 62.Boulmedais F, Ball V, Schwinte P, Frisch B, Schaaf P, Voegel JC. Langmuir. 2003;19:440–445. [Google Scholar]
- 63.Müller M. Biomacromolecules. 2001;2:262–269. doi: 10.1021/bm005628g. [DOI] [PubMed] [Google Scholar]
- 64.Sukhorukov GB, Montrel MM, Petrov AI, Shabarchina LI, Sukhorukov BI. Biosens Bioelectron. 1996;11:913–922. doi: 10.1016/0956-5663(96)89440-1. [DOI] [PubMed] [Google Scholar]
- 65.Montrel M, Sukhorukov G, Petrov A, Shabarchina L, Sukhorukov B. Sens Actuators, B. 1997;42:225–231. [Google Scholar]
- 66.DeMuth PC, Min Y, Huang B, Kramer JA, Miller AD, Barouch DH, Hammond PT, Irvine DJ. Nat Mater. 2013;12:367–376. doi: 10.1038/nmat3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smith RC, Riollano M, Leung A, Hammond PT. Angew Chem. 2009;121:9136–9139. doi: 10.1002/anie.200902782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim BS, Park SW, Hammond PT. ACS Nano. 2008;2:386–392. doi: 10.1021/nn700408z. [DOI] [PubMed] [Google Scholar]
- 69.Qi B, Tong X, Zhao Y. Macromolecules. 2006;39:5714–5719. [Google Scholar]
- 70.Yoo PJ, Nam KT, Qi J, Lee SK, Park J, Belcher AM, Hammond PT. Nat Mater. 2006;5:234–240. doi: 10.1038/nmat1596. [DOI] [PubMed] [Google Scholar]
- 71.Zeng G, Gao J, Chen S, Chen H, Wang Z, Zhang X. Langmuir. 2007;23:11631–11636. doi: 10.1021/la702054d. [DOI] [PubMed] [Google Scholar]
- 72.Keeney M, Mathur M, Cheng E, Tong XM, Yang F. Biomacromolecules. 2013;14:794–800. doi: 10.1021/bm3018559. [DOI] [PubMed] [Google Scholar]
- 73.Bhalerao UM, Valiveti AK, Acharya J, Halve AK, Kaushik MP. Colloids Surf, B. 2015;125:151–159. doi: 10.1016/j.colsurfb.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 74.Zhou L, Chen M, Tian LL, Guan Y, Zhang YJ. ACS Appl Mater Interfaces. 2013;5:3541–3548. doi: 10.1021/am4008787. [DOI] [PubMed] [Google Scholar]
- 75.Kim BS, Park SW, Hammond PT. ACS Nano. 2008;2:386–392. doi: 10.1021/nn700408z. [DOI] [PubMed] [Google Scholar]
- 76.Sato K, Takahashi S, Anzai J. Anal Sci. 2012;28:929–938. doi: 10.2116/analsci.28.929. [DOI] [PubMed] [Google Scholar]
- 77.Liu ZY, Tian C, Yu JW, Li YL, Jiang W, Mao CD. J Am Chem Soc. 2015;137:1730–1733. doi: 10.1021/ja5101307. [DOI] [PubMed] [Google Scholar]
- 78.Wood KC, Boedicker JQ, Lynn DM, Hammond PT. Langmuir. 2005;21:1603–1609. doi: 10.1021/la0476480. [DOI] [PubMed] [Google Scholar]
- 79.Lynn DM, Langer R. J Am Chem Soc. 2000;122:10761–10768. [Google Scholar]
- 80.Lim Y-b, Choi YH, Park J-s. J Am Chem Soc. 1999;21:5633–5639. [Google Scholar]
- 81.Lim YB, Kim CH, Kim K, Kim SW, Park JS. J Am Chem Soc. 2000;122:6524–6525. [Google Scholar]
- 82.Anderson DG, Akinc A, Hossain N, Langer R. Mol Ther. 2005;11:426–434. doi: 10.1016/j.ymthe.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 83.Zhang J, Fredin NJ, Janz JF, Sun B, Lynn DM. Langmuir. 2006;22:239–245. doi: 10.1021/la052360b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vázquez E, Dewitt DM, Hammond PT, Lynn DM. J Am Chem Soc. 2002;124:13992–13993. doi: 10.1021/ja026405w. [DOI] [PubMed] [Google Scholar]
- 85.De Geest BG, Van Camp W, Du Prez FE, De Smedt SC, Demeester J, Hennink WE. Macromol Rapid Commun. 2008;29:1111–1118. [Google Scholar]
- 86.Wohl BM, Engbersen JFJ. J Controlled Release. 2012;158:2–14. doi: 10.1016/j.jconrel.2011.08.035. [DOI] [PubMed] [Google Scholar]
- 87.Zhu ZC, Gao N, Wang HJ, Sukhishvili SA. J Controlled Release. 2013;171:73–80. doi: 10.1016/j.jconrel.2013.06.031. [DOI] [PubMed] [Google Scholar]
- 88.Kohler K, Sukhorukov GB. Adv Funct Mater. 2007;17:2053–2061. [Google Scholar]
- 89.Du P, Zhao X, Zeng J, Guo J, Liu P. Appl Surf Sci. 2015;345:90–98. [Google Scholar]
- 90.Hashide R, Yoshida K, Hasebe Y, Takahashi S, Sato K, Anzai J. Colloids Surf, B. 2012;89:242–247. doi: 10.1016/j.colsurfb.2011.09.023. [DOI] [PubMed] [Google Scholar]
- 91.Yoshida K, Hashide R, Ishii T, Takahashi S, Sato K, Anzai J. Colloids Surf, B. 2012;91:274–279. doi: 10.1016/j.colsurfb.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 92.Detzel CJ, Larkin AL, Rajagopalan P. Tissue Eng, Part B. 2011;17:101–113. doi: 10.1089/ten.teb.2010.0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhai L, Nolte AJ, Cohen RE, Rubner MF. Macromolecules. 2004;37:6113–6123. [Google Scholar]
- 94.Mendelsohn JD, Barrett CJ, Chan VV, Pal AJ, Mayes AM, Rubner MF. Langmuir. 2000;16:5017–5023. [Google Scholar]
- 95.Qi W, Yan X, Fei J, Wang A, Cui Y, Li J. Biomaterials. 2009;30:2799–2806. doi: 10.1016/j.biomaterials.2009.01.027. [DOI] [PubMed] [Google Scholar]
- 96.De Geest BG, Vandenbroucke RE, Guenther AM, Sukhorukov GB, Hennink WE, Sanders NN, Demeester J, De Smedt SC. Adv Mater. 2006;18:1005–1009. [Google Scholar]
- 97.Huang J, Shu Q, Wang LY, Wu H, Wang AY, Mao H. Biomaterials. 2015;39:105–113. doi: 10.1016/j.biomaterials.2014.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ren HF, Wu YT, Li Y, Cao W, Sun ZW, Xu HP, Zhang X. Small. 2013;9:3981–3986. doi: 10.1002/smll.201300628. [DOI] [PubMed] [Google Scholar]
- 99.Yang JP, Shen DK, Zhou L, Li W, Li XM, Yao C, Wang R, El-Toni AM, Zhang F, Zhao DY. Chem Mater. 2013;25:3030–3037. [Google Scholar]
- 100.Yi QY, Sukhorukov GB. Soft Matter. 2014;10:1384–1391. doi: 10.1039/c3sm51648b. [DOI] [PubMed] [Google Scholar]
- 101.Fomina N, Sankaranarayanan J, Almutairi A. Adv Drug Delivery Rev. 2012;64:1005–1020. doi: 10.1016/j.addr.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bedard MF, De Geest BG, Skirtach AG, Mohwald H, Sukhorukov GB. Adv Colloid Interface Sci. 2010;158:2–14. doi: 10.1016/j.cis.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 103.Ren H, Wu Y, Li Y, Cao W, Sun Z, Xu H, Zhang X. Small. 2013;9:3981–3986. doi: 10.1002/smll.201300628. [DOI] [PubMed] [Google Scholar]
- 104.Shah NJ, Macdonald ML, Beben YM, Padera RF, Samuel RE, Hammond PT. Biomaterials. 2011;32:6183–6193. doi: 10.1016/j.biomaterials.2011.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Garza JM, Schaaf P, Muller S, Ball V, Stoltz JF, Voegel JC, Lavalle P. Langmuir. 2004;20:7298–7302. doi: 10.1021/la049106o. [DOI] [PubMed] [Google Scholar]
- 106.Struth B, Eckle M, Decher G, Oeser R, Simon P, Schubert DW, Schmitt J. Eur Phys J E: Soft Matter Biol Phys. 2001;6:351–358. [Google Scholar]
- 107.Min J, Braatz RD, Hammond PT. Biomaterials. 2014;35:2507–2517. doi: 10.1016/j.biomaterials.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hong J, Shah NJ, Drake AC, DeMuth PC, Lee JB, Chen JZ, Hammond PT. ACS Nano. 2012;6:81–88. doi: 10.1021/nn202607r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang J, Montañez SI, Jewell CM, Lynn DM. Langmuir. 2007;23:11139–11146. doi: 10.1021/la702021s. [DOI] [PubMed] [Google Scholar]
- 110.Samee M, Kasugai S, Kondo H, Ohya K, Shimokawa H, Kuroda S. J Pharmacol Sci. 2008;108:18–31. doi: 10.1254/jphs.08036fp. [DOI] [PubMed] [Google Scholar]
- 111.Zhang X, Chen H, Zhang HY. Chem Commun. 2007:1395–1405. doi: 10.1039/B615590a. [DOI] [PubMed] [Google Scholar]
- 112.Ahn JS, Hammond PT, Rubner MF, Lee I. Colloids Surf, A. 2005;259:45–53. [Google Scholar]
- 113.Kantak C, Beyer S, Yobas L, Bansal T, Trau D. Lab Chip. 2011;11:1030–1035. doi: 10.1039/c0lc00381f. [DOI] [PubMed] [Google Scholar]
- 114.Peng LL, Mendelsohn AD, LaTempa TJ, Yoriya S, Grimes CA, Desai TA. Nano Lett. 2009;9:1932–1936. doi: 10.1021/nl9001052. [DOI] [PubMed] [Google Scholar]
- 115.Su X, Kim BS, Kim SR, Hammond PT, Irvine DJ. ACS Nano. 2009;3:3719–3729. doi: 10.1021/nn900928u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Daddona PE, Matriano JA, Mandema J, Maa YF. Pharm Res. 2011;28:159–165. doi: 10.1007/s11095-010-0192-9. [DOI] [PubMed] [Google Scholar]
- 117.Acharya G, Park K. Adv Drug Delivery Rev. 2006;58:387–401. doi: 10.1016/j.addr.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 118.Hossfeld S, Nolte A, Hartmann H, Recke M, Schaller M, Walker T, Kjems J, Schlosshauer B, Stoll D, Wendel HP, Krastev R. Acta Biomater. 2013;9:6741–6752. doi: 10.1016/j.actbio.2013.01.013. [DOI] [PubMed] [Google Scholar]