

Abstract
Autophagy, a self-catabolic process, has been found to be involved in abrogating the proliferation and metastasis of breast cancer. SLC9A3R1 (solute carrier family 9, subfamily A [NHE3, cation proton antiporter 3], member 3 regulator 1), a multifunctional scaffold protein, is involved in suppressing breast cancer cells proliferation and the SLC9A3R1-related signaling pathway regulates the activation of autophagy processes. However, the precise regulatory mechanism and signaling pathway of SLC9A3R1 in the regulation of autophagy processes in breast cancer cells remains unknown. Here, we report that the stability of BECN1, the major component of the autophagic core lipid kinase complex, is augmented in SLC9A3R1-overexpressing breast cancer MDA-MB-231 cells, subsequently stimulating autophagy by attenuating the interaction between BECN1 and BCL2. Initially, we found that SLC9A3R1 partially stimulated autophagy through the PTEN-PI3K-AKT1 signaling cascade in MDA-MB-231 cells. SLC9A3R1 then attenuated the interaction between BECN1 and BCL2 to stimulate the autophagic core lipid kinase complex. Further findings revealed that SLC9A3R1 bound to BECN1 and subsequently blocked ubiquitin-dependent BECN1 degradation. And the deletion of the C-terminal domain of SLC9A3R1 resulted in significantly reduced binding to BECN1. Moreover, the lack of C-terminal of SLC9A3R1 neither reduced the ubiquitination of BECN1 nor induced autophagy in breast cancer cells. The decrease in BECN1 degradation induced by SLC9A3R1 resulted in the activity of autophagy stimulation in breast cancer cells. These findings indicate that the SLC9A3R1-BECN1 signaling pathway participates in the activation of autophagy processes in breast cancer cells.
Keywords: breast cancer cells; protein interaction; protein stability; SLC9A3R1 signaling pathway, stimulating autophagy; ubiquitination degradation
Abbreviations
- BCL2
B-cell CLL/lymphoma 2
- CHX
cycloheximide
- CQ
chloroquine
- EGFR
epidermal growth factor receptor
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- PI3K
phosphatidylinositol-4,5-bisphosphate 3-kinase
- PIK3C3
phosphatidylinositol 3-kinase catalytic subunit type 3
- PIK3R4
phosphoinositide-3-kinase regulatory subunit 4
- PTEN
phosphatase and tensin homolog
- SLC9A3R1
solute carrier family 9, subfamily A (NHE3, cation proton antiporter 3), member 3 regulator 1
- UB
ubiquitin.
Introduction
Autophagy, a self-catabolic process that maintains intracellular homeostasis and determines cell fate under stress, is involved in regulating the proliferation and metastasis of breast cancer. The autophagic core lipid kinase complex, which contains BECN1, PIK3C3 (phosphatidylinositol 3-kinase catalytic subunit type 3), PIK3R4 (phosphoinositide-3-kinase regulatory subunit 4), and possibly AMBRA1, UVRAG, or ATG14, plays a crucial role in the activation of autophagy.1 The upregulation of autophagy reduces the proliferation of breast carcinoma cells by stimulating BECN1.2 Autophagy maintains cellular homeostasis by limiting endoplasmic reticulum (ER) and oxidative stress, which is related to the autophagy-mediated suppression of mammary tumorigenesis.3 Loss of autophagy also induces the DNA damage response in mammary tumors, and cooperates with defective apoptosis to increase mammary tumorigenesis.4 Moreover, autophagy promotes the degradation of SNAI/SNAIL and TWIST to inhibit the process of epithelial-to-mesenchymal transition in breast cancer cells.5 These studies show that autophagy abrogates the proliferation and metastasis of breast cancer via multiple pathways.
SLC9A3R1 (solute carrier family 9, subfamily A [NHE3, cation proton antiporter 3], member 3 regulator 1) composed of 2 tandem PDZ domains and a C-terminal ERM-binding domain, functions as a multifunctional scaffold protein that participates in cellular activities. SLC9A3R1 influences a variety of protein functions, including those of ion channels, receptors, signaling, and nuclear proteins.6 Moreover, SLC9A3R1 has been found to be involved in the regulation of the proliferation of breast cancer. Dai et al. report that a SLC9A3R1 allele is disrupted in more than 58% of breast cancer cell lines.7 In addition, SLC9A3R1 overexpression suppresses the growth of MDA-MB-231 breast cancer cells,8 whereas SLC9A3R1 knockdown results in enhanced growth in both MCF-7 and T47D breast cancer cell lines.9 Therefore, SLC9A3R1 has potential antitumor effects in breast cancer. However, the precise regulatory mechanism and signaling pathway of SLC9A3R1 in the regulation of autophagy processes remains unknown.
The SLC9A3R1-related signaling pathway participates in the activation of autophagy. SLC9A3R1 increases the expression of PTEN (phosphatase and tensin homolog) by stabilizing the PTEN protein,10 whereas a lack of PTEN suppresses autophagy.11 Moreover, SLC9A3R1 functions as a brake of the phosphatidylinositol-4, 5-bisphosphate 3-kinase (PI3K)-AKT1 pathway,12 which controls autophagy by stimulating the MTOR (mechanistic target of rapamycin [serine/threonine kinase]) complex. Hence, it is possible that SLC9A3R1 participates in the activation of autophagy in breast cancer cells.
In the present study, we show that the SLC9A3R1 activates autophagy in breast cancer cells, which is regulated by the interaction of SLC9A3R1-BECN1 and subsequent-mediated protein ubiquitination degradation. SLC9A3R1 stimulates autophagy partially through PTEN-PI3K-AKT1 pathways. Moreover, SLC9A3R1 binds to BECN1 and hinders the ubiquitination and degradation of BECN1 to trigger autophagy in these cells. Our studies provide insight into the underlying molecular mechanism and reveal a novel signaling pathway for SLC9A3R1-activated autophagy in breast cancer cells.
Results
SLC9A3R1 stimulates autophagy in breast cancer cells
To investigate the effects of SLC9A3R1 on the regulation of autophagy in breast cancer cells, the relationship between SLC9A3R1 expression and autophagy was examined in various breast cancer cells. It has been reported that SLC9A3R1 is expressed at a low level in MDA-MB-231 cells and at a high level in MCF-7 cells.9 We found that the SLC9A3R1 overexpression increased the expression of PIK3C3, BECN1, p-BCL2/BCL2 and the ratio of MAP1LC3B (microtubule-associated protein 1 light chain 3 β)-II/MAP1LC3B-I (LC3B-II/LC3B-I) and decreased the levels of p-MTOR/MTOR and SQSTM1 in MDA-MB-231 cells (Fig. 1A). The knockdown of SLC9A3R1 reduced the expression of PIK3C3, BECN1, p-BCL2/BCL2, and LC3B-II/I and increased the levels of p-MTOR/MTOR and SQSTM1 in MCF-7 cells (Fig. S1A).
Figure 1.

Figure 1 (See previous page). SLC9A3R1 stimulates autophagy activity in MDA-MB 231 cells. (A) Overexpression of SLC9A3R1 upregulated the expression of autophagy-related proteins. MDA-MB-231 cells stably expressing vector or MYC-SLC9A3R1 were harvested and autophagy-related proteins were detected by western blot analysis. ACTB served as the loading control. The value for the vector was set to 1.0 and the other values were normalized. (B) Overexpression of SLC9A3R1 aggregated LC3 foci. MDA-MB-231 cells stably expressing vector or MYC-SLC9A3R1 were fixed in 4% paraformaldehyde, stained with fluorochromes, and imaged by confocal microscopy, Scale bar: 20 μm. (C) Statistical analysis of the numbers of LC3 dots per cell in (B). Quantification of LC3 dots per cell was done as described in the Materials and Methods. (D) Electron microscopy of MDA-MB-231 cells stably expressing vector or MYC-SLC9A3R1. Typical autophagosome observed in SLC9A3R1-overexpressing breast cancer cells. The lysosome is indicated by the arrow, and the autophagosome is indicated by the arrowhead. Magnification × 12, 000–30, 000. (E) Statistical analysis of the numbers of autophagosomes per 100 μm2 in (D). (F) Treatment with CQ augmented the expression of LC3B-II. MDA-MB-231 cells expressing either vector or MYC-SLC9A3R1 were treated with or without CQ (50 μM) for 12 h, and the expression of LC3B-II was measured by western blotting. Data are presented as the mean ± SE of 4 independent assays. *, P < 0.05; **, P < 0.01; ***, P < 0.001; N, nucleus; APs, autophagosomes; CQ, chloroquine.
Conversion of the soluble form of LC3 (LC3-I) to a lipidated form (LC3-II) is a marker of autophagy.13 Our results showed that SLC9A3R1 augmented LC3 foci in MDA-MB-231 cells (Fig. 1B and C), and the knockdown of SLC9A3R1 inhibited the starvation-induced increase in LC3 foci in MCF-7 cells (Fig. S1B and C). In addition, electron microscopy revealed double-membrane vacuolar structures with the morphological features of autophagosomes in SLC9A3R1-overexpressing MDA-MB-231 cells (Fig. 1D and E). Chloroquine (CQ), a lysosomal protease inhibitor, also enhanced the SLC9A3R1-induced accumulation of LC3B-II in MDA-MB-231 cells (Fig. 1F). Taken together, our data indicate that SLC9A3R1 upregulates the autophagy activity in breast cancer cells.
SLC9A3R1 stimulates autophagy partially via the PTEN-PI3K-AKT1 pathway
To investigate the mechanism by which SLC9A3R1 regulates on autophagy, we assessed the distribution of SLC9A3R1 in breast cancer cells. We found that SLC9A3R1 was mostly distributed in the cytoplasm of the studied breast cancer cells (Fig. 2A-2D), indicating that SLC9A3R1 regulates autophagy probably by affecting cytoplasmic components. PTEN is the major suppressor of the PI3K-AKT1-MTOR signaling pathway in a large number of cancers.14 In addition, SLC9A3R1 promotes the expression of PTEN by controlling the degradation of this protein,10 probably that SLC9A3R1 stimulates autophagy by augmenting the level of PTEN and then abrogating the PI3K-AKT1-MTOR pathway. Indeed, the expression of PTEN was increased and the phosphorylation of AKT1 was decreased in the SLC9A3R1-overexpressing breast cancer cells (Fig. 2E); furthermore, the changes in the above-mentioned proteins were reversed in SLC9A3R1-silenced breast cancer cells (Fig. 2F). Nonetheless, SLC9A3R1 still enhanced the level of BECN1 and the ratio of LC3B-II/I following the silencing of PTEN (Fig. 2G). These data indicate that SLC9A3R1 activates autophagy partially via the PTEN-PI3K-AKT1 pathway in breast cancer cells.
Figure 2.

(See previous page). SLC9A3R1 partially stimulates autophagy through the PTEN-PI3K-AKT1 pathway. (A–D) SLC9A3R1 is mostly distributed in the cytoplasm of breast cancer cells. For panels (A and C), breast cancer cells were harvested, and the subcellular fraction was extracted for immunoblotting. ACTB and LMNA were used as loading controls. For panels (B and D), breast cancer cells in dishes were fixed in 4% paraformaldehyde, stained with fluorochromes, and imaged by confocal microscopy, Scale bar: 20 μm. For panels (A and B), the experiments were carried out in MDA-MB-231 cells stably expressing vector or MYC-SLC9A3R1. For panels (C and D), the experiments were carried out in MCF-7 cells stably expressing control-shRNA or sh-SLC9A3R1. (E) SLC9A3R1 stimulates the PTEN-PI3K-AKT1 pathway in MDA-MB-231 cells. MDA-MB-231 cells stably expressing vector or MYC-SLC9A3R1 were harvested, and the expression of the indicated proteins was determined by immunoblotting. The value for the vector was set to 1.0 and the other values were normalized. (F) Silencing of SLC9A3R1 suppresses the PTEN-PI3K-AKT1 pathway in MCF-7 cells. MCF-7 cells stably expressing control-shRNA or sh-SLC9A3R1 were harvested, and the expression of the indicated proteins was detected by immunoblotting. The value for the control-shRNA was set to 1.0 and the other values were normalized. (G) PTEN is unnecessary for the activation of autophagy when SLC9A3R1 stimulates. The MDA-MB-231 cells were transfected with control-siRNA or si-PTEN. Cell lysates were collected, and the expression of autophagy-related proteins was analyzed by western blotting. The value for the vector was set to 1.0 and the other values were normalized. Data are presented as the mean ± SE (n = 4). *, P < 0.05; ***, P < 0.001.
SLC9A3R1 enhances the expression of BECN1 and blocks the interaction of BECN1 and BCL2
SLC9A3R1 inhibits the proliferation of breast cancer cells by inhibiting EGF (epidermal growth factor)-induced EGFR (epidermal growth factor receptor) and the phosphorylation of downstream molecules,15 suggesting that SLC9A3R1 is a negative regulator of EGFR signaling. As an EGFR inhibitor induces autophagy by promoting the expression of BECN1 in the cytoplasm,16 we suspected that SLC9A3R1 might induce autophagy by augmenting the expression of BECN1. MYC-SLC9A3R1 plasmids were transfected into MDA-MB-231 cells, and the increase in SLC9A3R1 induced BECN1 expression in a dose-dependent manner (Fig. 3A). Similarly, silencing of SLC9A3R1 decreased the expression of BECN1 in a dose-dependent manner in MCF-7 cells (Fig. 3B). In addition to the expression of BECN1, the dissociation of BECN1 and BCL2 is important for the activation of autophagy.17 Our data showed that SLC9A3R1 reduced the binding of BECN1 and BCL2 (Fig. 3C and D). Taken together, these data suggest that SLC9A3R1 induces the expression of BECN1 and blocks the interaction between BECN1 and BCL2.
Figure 3.
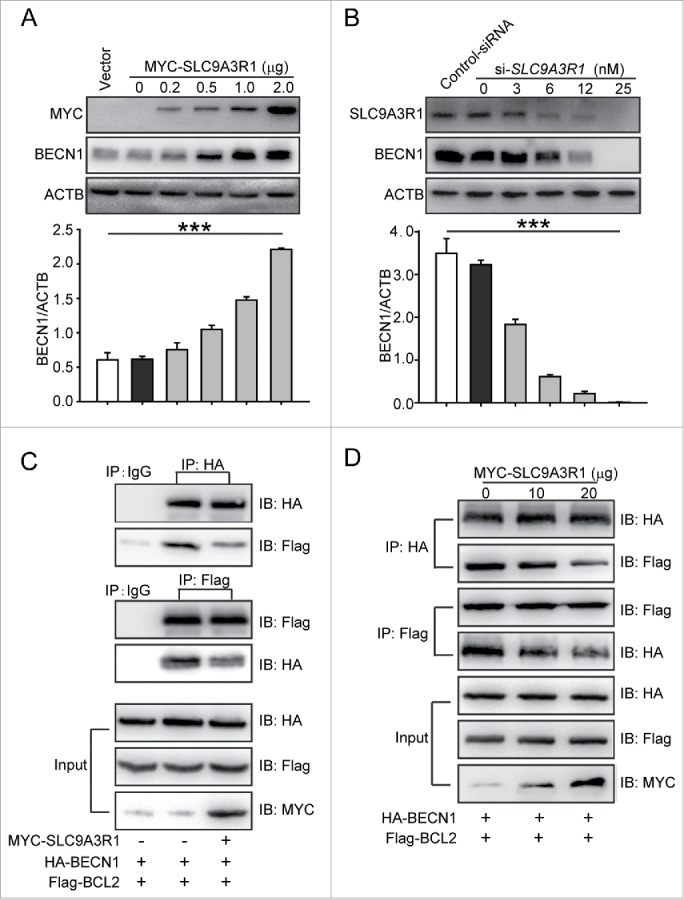
SLC9A3R1 activates the autophagic core lipid kinase complex by inducing the expression of BECN1. (A) SLC9A3R1 increases the expression of BECN1 in MDA-MB-231 cells. MDA-MB-231 cells were transfected with the indicated concentrations of MYC-SLC9A3R1 or vector and protein lysates were analyzed by immunoblotting with the indicated antibodies. (B) Knockdown of SLC9A3R1 decreases the expression of BECN1 in MCF-7 cells. MCF-7 cells were transfected with the indicated concentrations of si-SLC9A3R1 or control-siRNA, and protein lysates were analyzed by immunoblotting with the indicated antibodies. (C, D) SLC9A3R1 reduces the binding between BECN1 and BCL2. MDA-MB-231 cells were transfected with HA-BECN1, Flag-BCL2, and MYC-SLC9A3R1 plasmids for 24 h; cell lysates were then prepared and immunoprecipitated with anti-HA antibody or equal amount of mouse IgG, and the precipitates were detected using an anti-Flag antibody. Data are presented as the mean ± SE (n = 4). ***, P < 0.001.
SLC9A3R1 inhibits the degradation of BECN1
Because SLC9A3R1 was found to augment the expression of BECN1 to stimulate autophagy in breast cancer cells (Fig. 3), we further explored whether SLC9A3R1 induces BECN1 by regulating the transcription or degradation pathway. The results showed that the levels of BECN1 mRNA were not changed in either SLC9A3R1-overexpressing or SLC9A3R1-silenced cells compared to their control cells (Fig. 4A and B). However, SLC9A3R1 overexpression suppressed the degradation of BECN1 in cycloheximide (CHX)-treated MDA-MB-231 cells (Fig. 4C). We found that the knockdown of SLC9A3R1 promoted the degradation of BECN1 in MCF-7 cells (Fig. 4D). Taken together, these results indicate that SLC9A3R1 suppresses the degradation of BECN1.
Figure 4.

SLC9A3R1 inhibits the degradation of BECN1. (A, B) SLC9A3R1 did not affect BECN1 mRNA in breast cancer cells. Cells were transfected with the indicated concentrations of MYC-SLC9A3R1 (A) or si-SLC9A3R1 (B), and total RNA was isolated. The BECN1 mRNA was analyzed by fluorescent quantitative RT-PCR, as indicated in Materials and Methods. (C) SLC9A3R1 inhibits the degradation of BECN1 in MDA-MB-231 cells. Cells were transfected with HA-BECN1 for 24 h, subsequently the cells were then treated with CHX (20 μmol/L) for the indicated times. The cell lysates were then examined by western blotting using an anti-HA antibody. Cells were also transfected with an equal amount of pEGFP-N1 plasmid to monitor the transfection efficiency. (D) Knockdown of SLC9A3R1 promotes the degradation of BECN1 in MCF-7 cells. MCF-7 cells overexpressing HA-tagged BECN1 by lentivirus vector were treated with CHX (20 μmol/L) for the indicated times. The cell lysates were then analyzed by western blotting using anti-HA antibodies. Data are presented as the mean ± SE (n = 4). NS, nonsignificant; CHX, cycloheximide; GFP, green fluorescent protein.
SLC9A3R1 inhibits the ubiquitination of BECN1
Most cytosolic proteins are degraded by either the ubiquitin-proteasome pathway or the autophagy-lysosomal pathway. We next investigated which pathway participates in the regulation of BECN1 degradation that is suppressed by SLC9A3R1. We detected the expression of PTEN as the positive control for the inhibitory effects of MG132 on proteasomal-dependent degradation (Fig. S2A). After treatment with the specific proteasome inhibitor MG132, the degradation of BECN1 was significantly attenuated in both control-shRNA and SLC9A3R1-shRNA MCF-7 cells (Fig. 5A). And we detected the expression of SQSTM1 as the positive control for the inhibitory effects of CQ on lysosomal degradation (Fig. S2B). However, blockage of the lysosomal pathway by CQ did not alter the degradation rate of BECN1 (Fig. 5B). We further examined whether SLC9A3R1 affects the ubiquitination of BECN1. Using an anti-HA (to detect BECN1-HA) antibody, fewer ubiquitinated proteins were immunoprecipitated from SLC9A3R1-overexpressing cells compared with control cells (Fig. 5C). Taken together, these data suggest that SLC9A3R1 suppresses the degradation of BECN1 via the ubiquitin-proteasome pathway.
Figure 5.
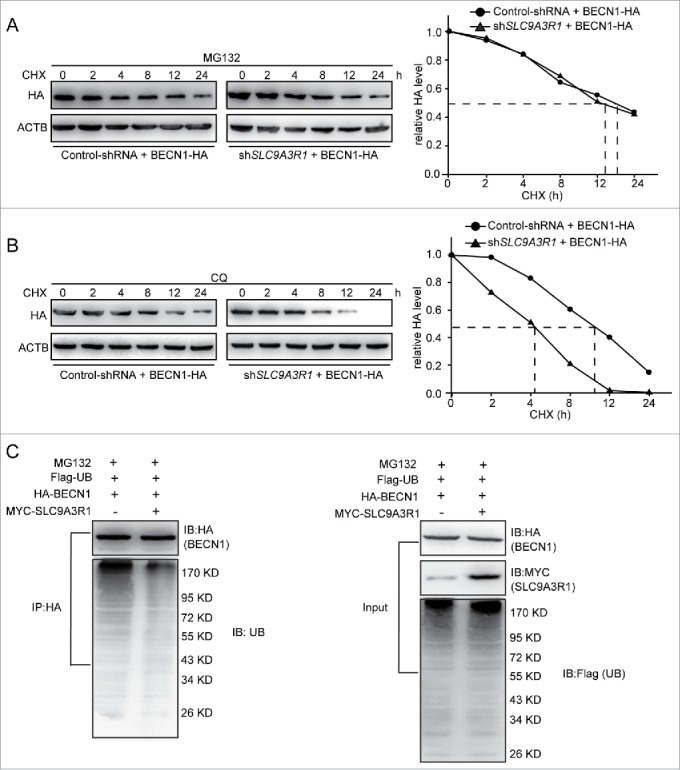
SLC9A3R1 inhibits the proteasomal degradation of BECN1. (A) MG132 inhibition of the proteasome blocks the degradation of BECN1 induced by SLC9A3R1 depletion. MCF-7 cells were overexpressed HA-tagged BECN1 by lentivirus vector and treated with MG132 (10 μmol/L) for 2 h and then treated with CHX (20 μmol/L) for the indicated times. (B) CQ inhibition of the lysosome does not affect the degradation of BECN1 induced by SLC9A3R1 depletion. MCF-7 cells were overexpressed HA-tagged BECN1 by lentivirus vector and treated with CQ (100 μmol/L) for 12 h and then treated with CHX (20 μmol/L) for the indicated times. (C) Overexpression of SLC9A3R1 eliminates the ubiquitination of BECN1. MDA-MB-231 cells were transfected with Flag-ubiquitin, HA-BECN1 and MYC-SLC9A3R1 expression plasmids for 24 h and then treated with MG132 (10 μmol/L) for 2 h; the cell lysates were immunoprecipitated using an anti-HA antibody, and the precipitates were probed with anti-ubiquitin and anti-HA antibodies. CHX, cycloheximide; CQ, chloroquine; UB, ubiquitin.
SLC9A3R1 inhibits the ubiquitin-dependent degradation of BECN1 by binding to BECN1
SLC9A3R1 functions as a scaffold protein by interacting with several proteins, such as CTNNB118,19 and PTEN,10 and regulating their degradation. As shown in Fig. 4, SLC9A3R1 downregulated the degradation of BECN1, indicating a potential interaction between SLC9A3R1 and BECN1. Co-immunoprecipitation and GST affinity isolation assays showed that BECN1 could bind to SLC9A3R1 both in vivo and in vitro (Fig. 6A, B and Fig. S3). Thus, to characterize the interaction between SLC9A3R1 and BECN1, the domains of BECN1 that interact with SLC9A3R1 were determined. Different truncated mutants of BECN1 were coexpressed with SLC9A3R1, and the integration of SLC9A3R1 with different truncated mutants of BECN1 was assessed by coimmunoprecipitation. Deletion of the BCL2-binding domain of BECN1 (BECN1BBDΔ) resulted in significantly reduced binding to SLC9A3R1, suggesting that the BCL2-binding domain of BECN1 is important for the interaction with SLC9A3R1 (Fig. 6C). Moreover, overexpression of SLC9A3R1 reduced the ubiquitination of BECN1 (Fig. 5C), whereas a lack of the BCL2-binding domain of BECN1 did not alter the ubiquitination of BECN1 in SLC9A3R1-overexpressing cells (Fig. 6D). These data indicate that SLC9A3R1 inhibits the ubiquitin-dependent degradation of BECN1 by binding to the protein.
Figure 6.
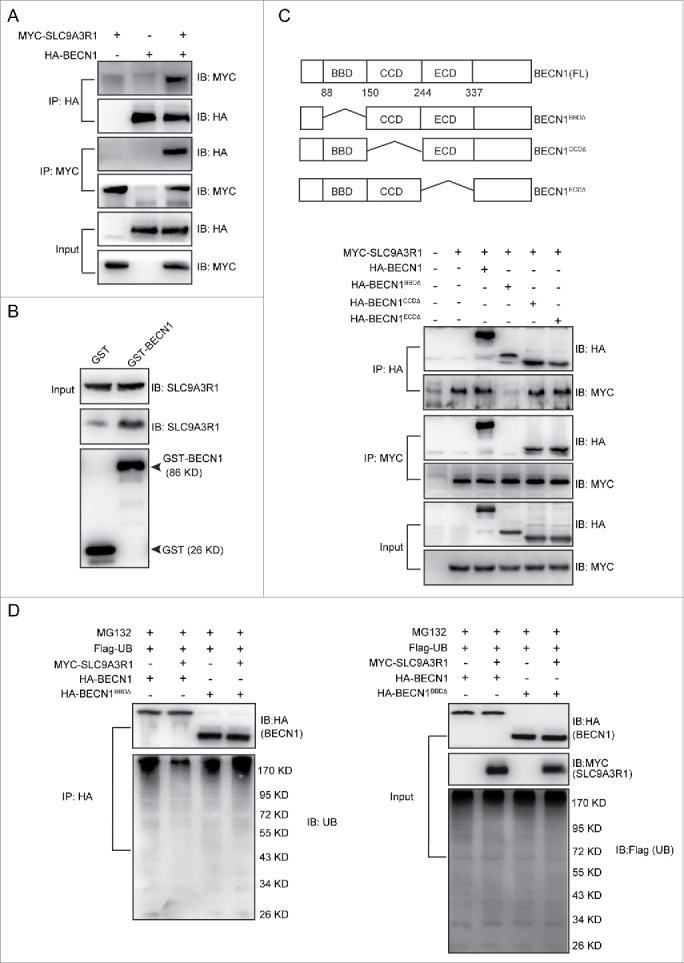
SLC9A3R1 inhibits the ubiquitin-dependent degradation of BECN1 by binding to BECN1. (A) SLC9A3R1 interacts with BECN1. MYC-SLC9A3R1 was cotransfected with HA-BECN1 into MDA-MB-231 cells. Whole-cell lysates were immunoprecipitated with an anti-HA antibody and blotted with an anti-MYC antibody. (B) In vitro binding between SLC9A3R1 and BECN1. The recombinant SLC9A3R1 full-length protein was incubated with an equal amount of GST or GST-BECN1 and analyzed by western blotting using an anti-SLC9A3R1 antibody. The presence of the GST fusion proteins was confirmed by western blotting using an anti-GST antibody. (C) Deficiency of the BCL2-binding domain of BECN1 inhibits the interaction between SLC9A3R1 and BECN1. Top: deletion mutants of BECN1. Below: MDA-MB-231 cells were cotransfected with MYC-SLC9A3R1 and the indicated constructs of HA-BECN1. Whole-cell extracts were immunoprecipitated with an anti-HA antibody. (D) SLC9A3R1 does not affect the ubiquitination of BECN1 lacking a BCL2-binding domain. MDA-MB-231 cells were cotransfected with Flag-ubiquitin, MYC-SLC9A3R1 and the indicated constructs of HA-BECN1 and then treated with MG132 (10 μmol/L) for 2 h. The cell lysates were extracted and immunoprecipitated using an anti-HA antibody. The precipitates were then examined with anti-ubiquitin and anti-HA antibodies. UB, ubiquitin.
SLC9A3R1 stimulates autophagy by binding and stabilizing BECN1
Based on SLC9A3R1-stimulated autophagy in breast cancer cells (Fig.1), we further investigated whether SLC9A3R1 increases the autophagy via interacting with BECN1 and then stabilizing it. SLC9A3R1 protein is consisted of 3 unique domains that include PDZ1, PDZ2, and C-terminal domain (Fig. 7A). To investigate which domain of SLC9A3R1 was required for interaction with BECN1, the different truncated mutants of SLC9A3R1 were coexpressed with BECN1, and assessed by coimmunoprecipitation. The deletion of the C-terminal domain of SLC9A3R1 (SLC9A3R1CDΔ) resulted in significantly reduced binding to BECN1 (Fig. 7A), suggesting that the C-terminal domain of SLC9A3R1 was important to the interaction with BECN1. Further, we found that the lack of the C-terminal domain of SLC9A3R1 neither reduced the ubiquitination of BECN1 (Fig. 7B) nor induced the expression of BECN1 or the ratio of LC3B-II/LC3B-I (Fig. 7C) in breast cancer cells. These data suggested that the lack of C-terminal of SLC9A3R1 did not induce autophagy because it did not delay the ubiquitination of BECN1. Taken together, these data indicated that SLC9A3R1 stimulates autophagy by binding and stabilizing BECN1 in breast cancer cells (Fig. 7D).
Figure 7.
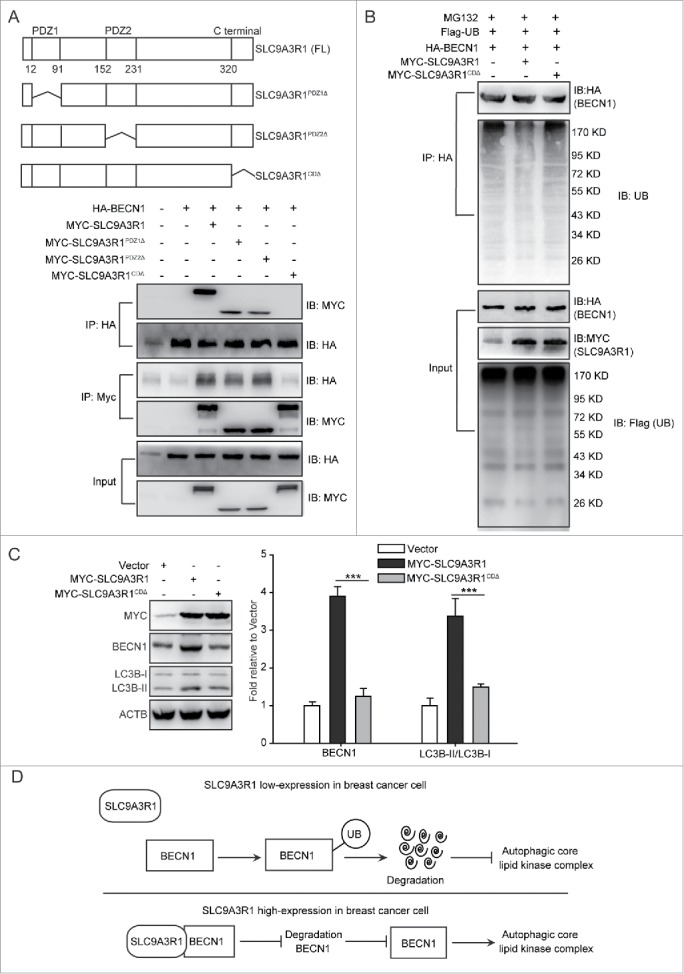
SLC9A3R1 stimulates autophagy by binding and stabilizing BECN1 in breast cancer cells. (A) Deficiency of the C-terminal domain of SLC9A3R1 inhibits the interaction between SLC9A3R1 and BECN1. Top: deletion mutants of SLC9A3R1. Below: MDA-MB-231 cells were cotransfected with HA-BECN1 and the indicated constructs of MYC-SLC9A3R1. Whole-cell extracts were immunoprecipitated with an anti-HA antibody. (B) Deficiency of the C-terminal domain of SLC9A3R1 does not affect the ubiquitination of BECN1. MDA-MB-231 cells were cotransfected with Flag-ubiquitin, HA-BECN1 and the indicated constructs of MYC-SLC9A3R1. The cells were then treated with MG132 (10 μmol/L) for 2 h. The cell lysates were extracted and immunoprecipitated using an anti-HA antibody. The precipitates were then analyzed with anti-ubiquitin and anti-HA antibodies. (C) Deficiency of the C-terminal domain of SLC9A3R1 does not induce autophagy. MDA-MB-231 cells stably expressing MYC-SLC9A3R1 or the indicated constructs of MYC-SLC9A3R1 were harvested and autophagy-related proteins were detected by western blot analysis. (D) Schematic diagram of the mechanism of SLC9A3R1-mediated autophagy stimulation in breast cancer cells. SLC9A3R1 interacts with BECN1 and inhibits the ubiquitin degradation of BECN1. Thus, enhanced BECN1 expression stimulates the autophagic core lipid kinase complex in breast cancer cells. Data are presented as the mean ± SE (n = 4). **, P < 0.01; ***, P < 0.001. UB, ubiquitin.
Discussion
Numerous factors regulate the activity of autophagy, particularly intracellular receptor and scaffold proteins,20-22 which are directly involved in the process of cargo selection and autophagosome formation.20,22 The receptor protein SQSTM1 recognizes and binds to ubiquitinated, undegraded cellular materials22 and the yeast scaffold protein Atg11 triggers the degradation of mitochondria by autophagy.20 In addition to autophagy-related proteins, other scaffold proteins can also stimulate autophagy; for example, the scaffold protein MAPK8IP1 (mitogen-activated protein kinase 8 interacting protein 1) regulates autophagosome maturation by binding to LC3.21 In the current study, scaffold protein SLC9A3R1 was found to stimulate autophagy by binding to BECN1, supplementing the effects by which scaffold proteins regulate autophagy activation. Moreover, the binding of BECN1 and PIK3C3 is necessary for BECN1 to organize the phagophore assembly site in the process of stimulating autophagy.23 In our studies, SLC9A3R1 induces the expression of PIK3C3 in breast cancer cells (Fig. 1A and Fig. S1A), and it is possible that SLC9A3R1 regulates the expression of PIK3C3 and then affects the stability of the BECN1-PIK3C3 complex. It suggests that SLC9A3R1 may regulate autophagy by affecting more than one autophagy regulator.
Indeed, SLC9A3R1 participates in several signaling pathways that are related to the regulation of autophagy, such as PTEN-PI3K-AKT1.12 SLC9A3R1 increases the expression of PTEN via interaction with PTEN,10 PTEN increases autophagy,24 whereas the loss of PTEN results in the inhibition of autophagy.11 Rajaa et al. have found that PTEN inhibits the ubiquitin-proteasome pathway,24 which is the main degradation pathway of BECN1 (Fig. 5A and B). Hence, it is possible that PTEN increases the expression of BECN1 to activate autophagy through delaying the degradation of BECN1 and SLC9A3R1 increases the expression of BECN1 through regulating the PTEN-mediated BECN1 degradation. Moreover, PTEN upregulates the expression of LC324 and suppresses the activity of the PI3K-AKT1 pathway by decreasing AKT1 phosphorylation.25 Thus, it is possible that SLC9A3R1 stimulates autophagy by increasing the expression of PTEN to inhibit the PI3K-AKT1 pathway. Our current results reveal that SLC9A3R1 regulates autophagy partially through the PTEN-PI3K-AKT1 pathway, which is consistent with the results of the abovementioned studies. However, Ueno et al. report that the loss of PTEN causes the strong inhibition of autophagy without affecting LC3 lipidation in mouse liver,11 which is contradictory to our current finding. Since our experimental system is human breast cancer cells, the diverging observations could be caused by the differences of tissues and cell lines.
The autophagic core lipid kinase complex plays a crucial role in the activation of autophagy and is involved in the process of autophagy vesicle nucleation.1 BCL2, an inhibitory factor in autophagy, binds to the BCL2-binding domain of BECN1 to suppress this complex.26 In our study, SLC9A3R1 was found to reduce the integration between BECN1 and BCL2 via its interaction with BECN1 (Fig. 4). In addition, the site of BCL2 binding to BECN1 was essential for the interaction between SLC9A3R1 and BECN1 (Fig. 6), suggesting that SLC9A3R1 suppresses the interaction between BCL2 and BECN1 by competitively occupying the site of BCL2 binding to BECN1. Taken together, our results show that SLC9A3R1 functions as an autophagic core lipid kinase complex-activating factor by competing with BCL2 to interact with BECN1.
SLC9A3R1 regulates protein stability by binding to various proteins: SLC9A3R1 inhibits the degradation of PTEN by interacting with this protein10 and prevents the degradation of CTNNB1 by interacting with it.18,19 Our findings show that SLC9A3R1 delays the degradation of BECN1 by binding to the protein (Fig. 4), which is consistent with the results of the abovementioned reports. In our study, we found that SLC9A3R1 also delayed the degradation of PTEN in MDA-MB-231 cells (Fig. S2C), which suggested that the effects of SLC9A3R1 on delaying protein degradation are not specific for BECN1. Moreover, our observations revealed that the BCL2-binding domain of BECN1 is essential for the interaction between SLC9A3R1 and BECN1 (Fig. 6). It has been reported that there are multiple ubiquitin recognition sites present in the BCL2-binding domain of BECN1,27 and it is possible that SLC9A3R1 hinders ubiquitin recognition when it binds to BECN1. These data suggest that SLC9A3R1 plays a role in the suppression of ubiquitin recognition by interaction with BECN1 to trigger autophagy in breast cancer cells.
In summary, SLC9A3R1 upregulates the expression of BECN1 by stabilizing BECN1 to stimulate autophagy in breast cancer cells. The interaction between SLC9A3R1 and BECN1 to increase the stability of the latter is necessary for autophagy in breast cancer cells. Our findings define a novel signaling pathway and the molecular mechanism underlying the regulation of autophagy induced by SLC9A3R1 in breast cancer cells.
Materials and Methods
Reagents
CHX (01810) and MG132 (C2211) were obtained from Sigma. Anti-human SLC9A3R1 (3394), BCL2 (2870), BECN1 (3495 and 4122), PIK3C3 (4263), MTOR (2983), AKT1 (4691), PTEN (9188), p-BCL2 (S70) (2827), p-MTOR (S2448) (2971), p-AKT1 (S437) (4060), HA (3724), MYC (2278), Flag (2908) and ACTB (4970) antibodies and mouse IgG (5414) were obtained from Cell Signaling Technology. LC3B (L8918) and SQSTM1 (P0076) antibodies were purchased from Sigma. SLC9A3R1 siRNA (sc-63330), PTEN siRNA (sc-29459) and control siRNA (sc-37007) were purchased from Santa Cruz Biotechnology. Anti-human SLC9A3R1 (ab9526) and recombinant SLC9A3R1 full-length protein (ab112385) were purchased from Abcam. Other materials were purchased from commercial sources.
Plasmid construction
MYC-tagged SLC9A3R1 and its deletion mutants (range of deletions indicated by amino acid residue numbers) 12 to 91Δ, 152 to 231Δ and 320 to 358Δ were created in the pCMV6-AN-MYC (Origene, PS100012) vector by standard subcloning. The SLC9A3R1-shRNA-targeting sequence (5′ -CACCGACCAGAAACGCAGCAGCAAACTT-CAAGAGAGTTTGCTGCTGCG-TTTCTGGTTTTTTTG-3′) was subcloned into the pRS shRNA cloning plasmid (Origene, TR20003). A control shRNA oligonucleotide that did not match any known human coding cDNA was used as a control. HA-tagged BECN1 and its deletion mutants (88 to 150Δ, 151 to 244Δ and 245 to 337Δ) were constructed in the pCMV6-AC-HA vector (Origene, PS100004) by standard subcloning. The ubiquitin gene was synthesized as described previously28 and cloned into the pFLAG-CMV-2 (Sigma, E7033) expression plasmid. Full-length BECN1 was subcloned in-frame to pGEX4T-1 to generate GST fusion proteins. The overexpression lentiviral vector (Origene, TR30022) carrying HA-tagged BECN1 gene was constructed following the manufacturer's instructions.
Cell culture and transfection
Breast cancer cell lines were purchased from American Type Culture Collection (HTB-26 and HTB-22) and maintained as previously.5,29 Transient transfection was performed using Lipofectamine2000 (Invitrogen, 11668019) according to the manufacturer's instructions. To generate MDA-MB-231 cells stably expressing the pCMV6-AN-MYC and the MYC-tagged SLC9A3R1 plasmids were both transfected into MDA-MB-231 cells. After 24 h of transfection, stable transfectants were selected using neomycin (Invitrogen, 10131035),5 and a single clone was amplified using a dilution cloning technique. To generate MCF-7 cells stably expressing the control-shRNA and SLC9A3R1-shRNA plasmids were both transfected into MCF-7 cells. After 24 h of transfection, stable transfectants were selected using puromycin (Invitrogen, A1113802), and a single clone was amplified by a dilution cloning technique.
Immunoprecipitation and western blot analysis
Cells were washed 3 times with phosphate-buffered saline (Invitrogen, 20012–027) and harvested. The cell lysates were extracted in co-immunoprecipitation buffer.28 As described previously,28 total cell lysates (5 mg protein) were subjected to immunoprecipitation with the appropriate antibodies overnight at 4°C and then incubated with protein A/G Plus-agarose (Santa Cruz Biotechnology, sc-2003) for 24 h at 4°C. After washing 3 times, the immunocomplex was mixed with 2×SDS loading buffer and boiled for 5 min. For western blot analysis, coprecipitates or whole-cell extracts were electrophoresed by SDS-PAGE and blotted on PVDF membranes (Millipore, Immobilon-P). The membranes were immunoblotted with the indicated antibodies and developed using a ChemiImager 5500 imaging system (Alpha Innotech Co., San Leandro, CA).
Electron microscopy
As described previously,13 breast cancer cells were immediately fixed with 2.5% glutaraldehyde containing 0.1 mol/L sodium cacodylate and stored at 4°C. The samples were then embedded, and ultrathin (50 to 60 nm) sections were cut using an ultramicrotome (LEICA EM UC7, Wetzlar, Germany). The samples were examined with a JEM-1400 electron microscope (JEOL USA, Inc., Peabody, MA) at 80 kV, and the images were captured with a Veleta TEM camera (Olympus, Shinjuku, Japan).
Confocal assay
Standard protocols for immunofluorescence microscopy were used as described previously.30,31 The cells were planted on coverglass-bottom dishes, washed twice, fixed in 4% paraformaldehyde for 20 min, and then washed 3 times. The cells on the dishes (0.5-μm thick) were prepared and stained with the indicated primary antibodies overnight at 4°C. The sections were washed twice, incubated with fluorochrome-labeled secondary antibodies (1:200) for 30 min, and then washed 3 times. Images were obtained using a Leica SP2 confocal microscope (Leica Microsystems, Exton, PA) and analyzed with Leica confocal software.
For quantification of the number of LC3 dots, a total of 100 cells were recorded by the Leica SP2 confocal microscope and analyzed using the ImageJ analysis software. LC3 puncta with a diameter between 0.3 and 1 μm were scored as positive.32
Real-time PCR
Total RNA was extracted from MDA-MB-231 and MCF-7 cells using TRIzol Reagent (Invitrogen, 15596026). As described in the manual, total RNA (100 ng) was detected using the SYBR Green One-Step qRT-PCR Kit (Invitrogen, 11736059). The following primers were used: ACTB, 5′-CAA CTG GGA CGA CAT GGA GAA AAT-3′ and 5′-CCA GAG GCG TAC AGG GAT AGC AC-3′; BECN1, 5′-TCT GCC TTC CTC TGT AG-3′ and 5′-TTC CAC GGG AAC ACT G-3′.
GST affinity isolation assay
As described previously,28 equal amounts of GST or GST fusion proteins bound to glutathione-Sepharose beads (Amersham Bioscience, 20182003–2) were incubated with recombinant SLC9A3R1 full-length protein. The beads were washed 3 times, and interacting proteins were detected by immunoblotting.
Statistics
Data are represented as the mean ± SE. The statistical analyses were performed using the Student t test to compare 2 groups. The P value was set at 0.05. All statistics were performed using SPSS 17.0 software.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was supported by the National Natural Scientific Foundation (81473249, 31401186, and 81321004), National Mega-project for Innovative Drugs (2014ZX09201042) and Institute of Medicinal Biotechnology foundation (IMBF201407).
References
- 1.He C, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol 2010; 22:140-9; PMID:20097051; http://dx.doi.org/ 10.1016/j.ceb.2010.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672-6; PMID:10604474; http://dx.doi.org/ 10.1038/45257 [DOI] [PubMed] [Google Scholar]
- 3.Kongara S, Kravchuk O, Teplova I, Lozy F, Schulte J, Moore D, Barnard N, Neumann CA, White E, Karantza V. Autophagy regulates keratin 8 homeostasis in mammary epithelial cells and in breast tumors. Mol Cancer Res 2010; 8:873-84; PMID:20530580; http://dx.doi.org/ 10.1158/1541-7786.MCR-09-0494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karantza-Wadsworth V, White E. Role of autophagy in breast cancer. Autophagy 2007; 3:610-3; PMID:17786023; http://dx.doi.org/ 10.4161/auto.4867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lv Q, Wang W, Xue J, Hua F, Mu R, Lin H, Yan J, Lv X, Chen X, Hu ZW. DEDD interacts with PI3KC3 to activate autophagy and attenuate epithelial-mesenchymal transition in human breast cancer. Cancer Res 2012; 72:3238-50; PMID:22719072; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3832 [DOI] [PubMed] [Google Scholar]
- 6.Chen JY, Lin YY, Jou TS. Phosphorylation of EBP50 negatively regulates β-PIX-dependent Rac1 activity in anoikis. Cell Death Differ 2012; 19:1027-37; PMID:22301917; http://dx.doi.org/ 10.1038/cdd.2012.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai JL, Wang L, Sahin AA, Broemeling LD, Schutte M, Pan Y. NHERF (Na+/H+ exchanger regulatory factor) gene mutations in human breast cancer. Oncogene 2004; 23:8681-7; PMID:15467753`; http://dx.doi.org/ 10.1038/sj.onc.1207962 [DOI] [PubMed] [Google Scholar]
- 8.Zheng JF, Sun LC, Liu H, Huang Y, Li Y, He J. EBP50 exerts tumor suppressor activity by promoting cell apoptosis and retarding extracellular signal-regulated kinase activity. Amino Acids 2010; 38:1261-8; PMID:20012548; http://dx.doi.org/ 10.1007/s00726-009-0437-2 [DOI] [PubMed] [Google Scholar]
- 9.Pan Y, Wang L, Dai JL. Suppression of breast cancer cell growth by Na+/H+ exchanger regulatory factor 1 (NHERF1). Breast Cancer Res 2006; 8:R63; PMID:17078868; http://dx.doi.org/ 10.1186/bcr1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang L, Wang Y, Chen P, Hu J, Xiong Y, Feng D, Liu H, Zhang H, Yang H, He J. Na(+)/H(+) exchanger regulatory factor 1 (NHERF1) is required for the estradiol-dependent increase of phosphatase and tensin homolog (PTEN) protein expression. Endocrinology 2011; 152:4537-49; PMID:21990315; http://dx.doi.org/ 10.1210/en.2011-1207 [DOI] [PubMed] [Google Scholar]
- 11.Ueno T, Sato W, Horie Y, Komatsu M, Tanida I, Yoshida M, Ohshima S, Mak TW, Watanabe S, Kominami E. Loss of Pten, a tumor suppressor, causes the strong inhibition of autophagy without affecting LC3 lipidation. Autophagy 2008; 4:692-700; PMID:18424911; http://dx.doi.org/ 10.4161/auto.6085 [DOI] [PubMed] [Google Scholar]
- 12.Georgescu MM. NHERF1: molecular brake on the PI3K pathway in breast cancer. Breast Cancer Res 2008; 10:106; PMID:18430260; http://dx.doi.org/ 10.1186/bcr1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke AW, Wang XY, Dai Z, Peng YF, Gu CY, Qiu SJ, Fan J. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 2011; 7:1159-72; PMID:21691147; http://dx.doi.org/ 10.4161/auto.7.10.16818 [DOI] [PubMed] [Google Scholar]
- 14.Georgescu MM. PTEN Tumor Suppressor Network in PI3K-Akt Pathway Control. Genes Cancer 2010; 1:1170-7; PMID:21779440; http://dx.doi.org/ 10.1177/1947601911407325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao W, Feng D, Bian W, Yang L, Li Y, Yang Z, Xiong Y, Zheng J, Zhai R, He J. EBP50 inhibits EGF-induced breast cancer cell proliferation by blocking EGFR phosphorylation. Amino Acids 2012; 43:2027-35; PMID:22476347; http://dx.doi.org/ 10.1007/s00726-012-1277-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han J, Hou W, Lu C, Goldstein LA, Stolz DB, Watkins SC, Rabinowich H. Interaction between Her2 and Beclin-1 proteins underlies a new mechanism of reciprocal regulation. J Biol Chem 2013; 288:20315-25; PMID:23703612; http://dx.doi.org/ 10.1074/jbc.M113.461350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marquez RT, Xu L. Bcl-2: Beclin 1 complex: multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am J Cancer Res 2012; 2:214-21; PMID:22485198 [PMC free article] [PubMed] [Google Scholar]
- 18.Kreimann EL, Morales FC, de Orbeta-Cruz J, Takahashi Y, Adams H, Liu TJ, McCrea PD, Georgescu MM. Cortical stabilization of β-catenin contributes to NHERF1/EBP50 tumor suppressor function. Oncogene 2007; 26:5290-9; PMID:17325659; http://dx.doi.org/ 10.1038/sj.onc.1210336 [DOI] [PubMed] [Google Scholar]
- 19.Shibata T, Chuma M, Kokubu A, Sakamoto M, Hirohashi S. EBP50, a β-catenin-associating protein, enhances Wnt signaling and is over-expressed in hepatocellular carcinoma. Hepatology 2003; 38:178-86; PMID:12830000; http://dx.doi.org/ 10.1053/jhep.2003.50270 [DOI] [PubMed] [Google Scholar]
- 20.Mao K, Wang K, Liu X, Klionsky DJ. The scaffold protein Atg11 recruits fission machinery to drive selective mitochondria degradation by autophagy. Dev Cell 2013; 26:9-18; PMID:23810512; http://dx.doi.org/ 10.1016/j.devcel.2013.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu MM, Nirschl JJ, Holzbaur EL. LC3 binding to the scaffolding protein JIP1 regulates processive dynein-driven transport of autophagosomes. Dev Cell 2014; 29:577-90; PMID:24914561; http://dx.doi.org/ 10.1016/j.devcel.2014.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geetha T, Zheng C, Vishwaprakash N, Broderick TL, Babu JR. Sequestosome 1/p62, a scaffolding protein, is a newly identified partner of IRS-1 protein. J Biol Chem 2012; 287:29672-8; PMID:22761437; http://dx.doi.org/ 10.1074/jbc.M111.322404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011; 18:571-80; PMID:21311563; http://dx.doi.org/ 10.1038/cdd.2010.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Errafiy R, Aguado C, Ghislat G, Esteve JM, Gil A, Loutfi M, Knecht E. PTEN increases autophagy and inhibits the ubiquitin-proteasome pathway in glioma cells independently of its lipid phosphatase activity. PLoS One 2013; 8:e83318; PMID:24349488; http://dx.doi.org/ 10.1371/journal.pone.0083318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wan X, Helman LJ. Levels of PTEN protein modulate Akt phosphorylation on serine 473, but not on threonine 308, in IGF-II-overexpressing rhabdomyosarcomas cells. Oncogene 2003; 22:8205-11; PMID:14603261; http://dx.doi.org/ 10.1038/sj.onc.1206878 [DOI] [PubMed] [Google Scholar]
- 26.Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex-at the crossroads of autophagy and beyond. Trends Cell Biol 2010; 20:355-62; PMID:20356743; http://dx.doi.org/ 10.1016/j.tcb.2010.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal 2010; 3:ra42; PMID:20501938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hua F, Mu R, Liu J, Xue J, Wang Z, Lin H, Yang H, Chen X, Hu Z. TRB3 interacts with SMAD3 promoting tumor cell migration and invasion. J Cell Sci 2011; 124:3235-46; PMID:21896644; http://dx.doi.org/ 10.1242/jcs.082875 [DOI] [PubMed] [Google Scholar]
- 29.Gong Y, He H, Liu H, Zhang C, Zhao W, Shao RG. Phosphorylation of myofibrillogenesis regulator-1 activates the MAPK signaling pathway and induces proliferation and migration in human breast cancer MCF7 cells. FEBS Lett 2014; 588:2903-10; PMID:25066297; http://dx.doi.org/ 10.1016/j.febslet.2014.07.018 [DOI] [PubMed] [Google Scholar]
- 30.Hua F, Zhou J, Liu J, Zhu C, Cui B, Lin H, Liu Y, Jin W, Yang H, Hu Z. Glycogen synthase kinase-3beta negatively regulates TGF-beta1 and Angiotensin II-mediated cellular activity through interaction with Smad3. Eur J Pharmacol 2010; 644:17-23; PMID:20599907; http://dx.doi.org/ 10.1016/j.ejphar.2010.06.042 [DOI] [PubMed] [Google Scholar]
- 31.Zhang J, Liang Q, Lei Y, Yao M, Li L, Gao X, Feng J, Zhang Y, Gao H, Liu DX, Lu J, Huang B. SOX4 induces epithelial-mesenchymal transition and contributes to breast cancer progression. Cancer Res 2012; 72:4597-608; PMID:22787120; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-1045 [DOI] [PubMed] [Google Scholar]
- 32.Guo Y, Chang C, Huang R, Liu B, Bao L, Liu W. AP1 is essential for generation of autophagosomes from the trans-Golgi network. J Cell Sci 2012; 125:1706-15; PMID:22328508; http://dx.doi.org/ 10.1242/jcs.093203 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.