

Abstract
The essential enzyme CYP121 is a target for drug development against antibiotic resistant strains of Mycobacterium tuberculosis. A triazol-1-yl phenol fragment 1 was identified to bind to CYP121 using a cascade of biophysical assays. Synthetic merging and optimization of 1 produced a 100-fold improvement in binding affinity, yielding lead compound 2 (KD = 15 μM). Deconstruction of 2 into its component retrofragments allowed the group efficiency of structural motifs to be assessed, the identification of more LE scaffolds for optimization and highlighted binding affinity hotspots. Structure-guided addition of a metal-binding pharmacophore onto LE retrofragment scaffolds produced low nanomolar (KD = 15 nM) CYP121 ligands. Elaboration of these compounds to target binding hotspots in the distal active site afforded compounds with excellent selectivity against human drug-metabolizing P450s. Analysis of the factors governing ligand potency and selectivity using X-ray crystallography, UV–vis spectroscopy, and native mass spectrometry provides insight for subsequent drug development.
Introduction
Tuberculosis (TB) is responsible for the death of 1.5 million people annually.1 The development of antibiotic resistant strains of the causal pathogen Mycobacterium tuberculosis (Mtb) and comorbidity with immunosuppressive conditions such as HIV and diabetes are major contributors to the current crisis and there is an urgent need for the development of new drugs with novel mechanisms of action.
Cytochrome P450 enzymes (P450s or CYPs) are a currently unexploited class of protein targets for TB therapeutics. The Mtb genome encodes genes for 20 P450 isoforms, which is more than 200 times as many as humans for the relative size of the genome.2 The large proportion of genetic material dedicated to P450s suggests that these enzymes might have important functions for the survival and virulence of the pathogen.3 Biochemical and genetic studies over the past 15 years have provided evidence of essential roles for Mtb P450s in bacterial viability, the catabolism of host cholesterol, establishment of infection, persistence in the granuloma, cellular stress response mechanisms, and drug susceptibility.4−14 A number of azole-containing antifungal compounds that have potent antimycotic activity against Mtb H37Rv both in vitro and in murine models of TB, have tight binding affinity (KD) for a number of the Mtb P450s, which provides further support for these enzymes as potential drug targets.12,15,16 There is correlation between the minimum inhibitory concentration (MIC) values of the most potent azole compounds clotrimazole, econazole, and miconazole, which have MICs of 11, 8, and 8 μg/mL, respectively, against Mtb H37Rv,12 and their KD’s for the most strongly inhibited P450 CYP121 (KD = 73–136 nM).12,17 In addition, evidence that the gene encoding CYP121 (rv2276) is essential for Mtb H37Rv viability12 implies that this P450 is the major target responsible for the efficacy of these compounds. While azole antifungal drugs have promise as treatments for TB, the specific compounds that have been shown to have efficacy against Mtb suffer from either low oral bioavailability or cause significant toxic side effects because of their broad spectrum activity against human P450s or steroidogenic inhibition.18,19 These properties make the azole drugs incompatible with long-term dosing regimens required for TB treatment. Consequently, there is interest in developing more potent and selective inhibitors of Mtb P450s.
CYP121 is a soluble, monomeric enzyme, which has limited similarity (≤34% protein sequence identity) to other Mtb P450s.11 The enzyme catalyzes the cyclization of the dipeptide cyclo-(l-Tyr-l-Tyr) (cYY) to form the diketopiperazine mycocyclosin.11 While the function of mycocyclosin has not been determined, diketopiperazine secondary metabolites often have antimicrobial or cytotoxic activity, properties which might be of importance for Mtb virulence.10 This combination of gene essentiality, low sequence similarity to other P450s, and tight azole binding affinity has made the development of CYP121 inhibitors an area of interest in the search for new TB drugs and the focus of the research presented here.
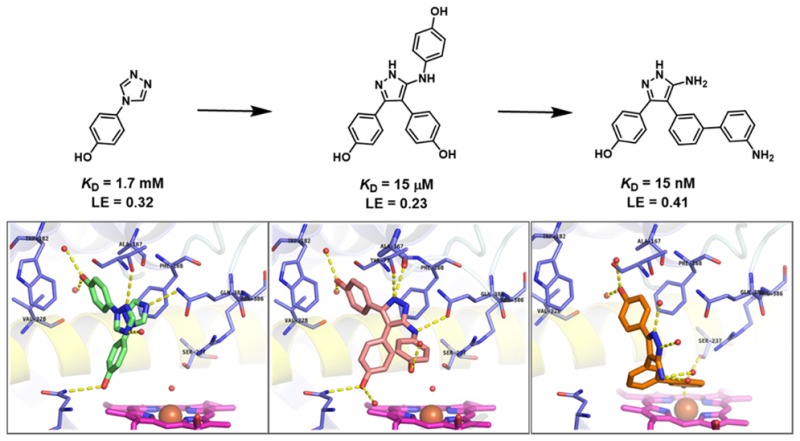
Fragment-based drug discovery (FBDD) is an established technique in both academia and industry that has been applied to a wide range of molecular targets.20−25 The availability of high resolution crystal structures of CYP121, in both the substrate-free and substrate/ligand-bound forms, make it amenable to a fragment-based approach.26 We have previously reported the development of low micromolar affinity CYP121 inhibitors, developed from fragments that were identified in a biophysical screen of our fragment library.27,28 Six fragment hits were crystallized with CYP121, one of which was 4-(1H-1,2,4-triazol-1-yl)phenol 1 (KD = 1.7 mM) (Figure 1). Fragment 1 bound in two different overlapping orientations in the CYP121 active site, suggesting an approach based on fragment merging. Synthesis of a range of analogues, guided by in silico screening to optimize the binding interactions of the heterocyclic fragment core, resulted in the identification of the triphenol pyrazole-amine 2 (KD = 15 μM). An X-ray crystal structure of 2 bound to CYP121 revealed that 2 maintained the binding mode of the original fragment 1. The optimized hydrogen bonding interactions between the 5-aminopyrazole core of 2 and distal pocket residues Gln385, Ala167, and Thr77 represented a privileged binding mode for an azole-containing compound, which generally bind directly to the heme cofactor of P450s using a heterocyclic nitrogen atom. The combination of the low micromolar affinity of 2 and its unique binding mode, which was conserved with fragment 1, led us to pursue further optimization of this compound series.
Figure 1.

Fragment merging and in silico directed optimization of fragment 1 (green, PDB 4G47) led to the discovery of low micromolar CYP121 inhibitor 2 (salmon, PDB 4KTL). Fragment 1 and lead 2 retained a conserved binding mode distal to the heme cofactor (magenta). Hydrogen bonding interactions between the 5-aminopyrazole ring and CYP121 residues Gln385 and Ala167, and the phenol group with Asn85 and active site water molecules are shown as yellow dashed lines. Figures prepared using PyMOL v1.7.4 (Schrödinger, LLC).
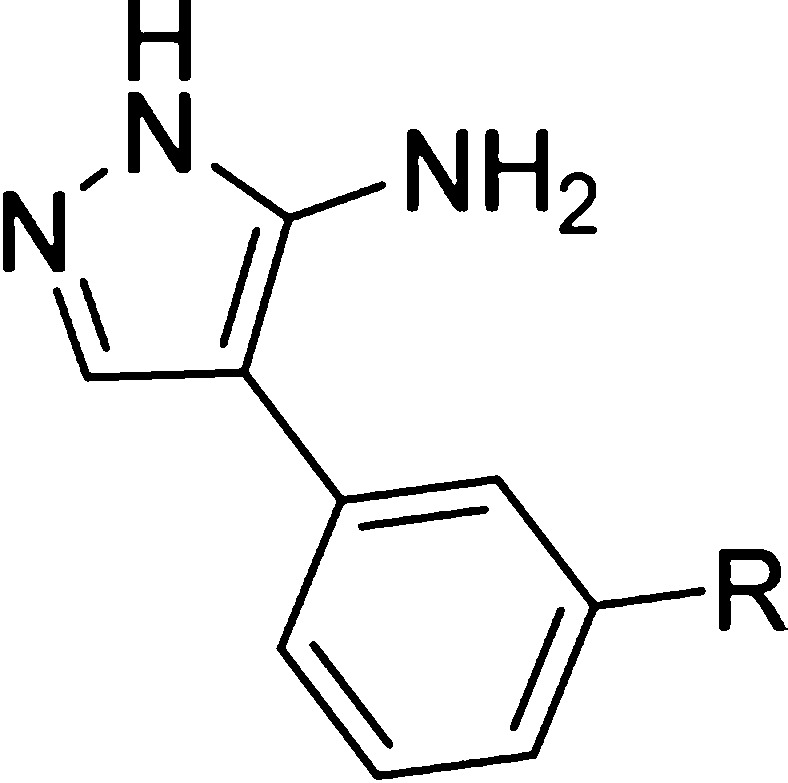
Despite a 100-fold improvement in binding affinity between fragment 1 and lead 2, the low ligand efficiency (LE)29,30 of 2 (LE = 0.23) led us to undertake a deconstruction–reconstruction approach to identify more efficient scaffolds for subsequent optimization. Retrofragmentation of 2 to its component “fragments” was used to assess the binding contribution, or group efficiency (GE),31 of the three aromatic rings that decorate the amino-pyrazole core, hereafter referred to as Ar1, Ar2, and Ar3, as defined in Figure 3.
Figure 3.

Deconstruction of lead compound 2 into retrosynthetic fragments 3–6 was used to assess the binding affinity, ligand efficiency (LE), and group efficiency (GE) contribution of the individual components. The three aromatic rings decorating the aminopyrazole core of lead compound 2 have been labeled Ar1, Ar2, and Ar3 as illustrated, which refers to the orientation the rings occupy in the X-ray crystal structure of 2 in complex with CYP121 (Figure 1). Retrofragments 3 and 4 represent Ar1 and Ar2, respectively. The lower panel tabulates the binding affinity (KD) and LE and GE properties of compounds from the sequential reconstruction of lead 2 from component monoaryl (3 and 4) and biaryl (5 and 6) retrofragments. KD values were determined using ITC. LE was calculated from the Gibbs free energy of binding divided by the number of heavy atoms (ΔG/HA).29,30 GE contributions of the three aromatic rings Ar1, Ar2, and Ar3 were calculated from the change in Gibbs free energy per added heavy atom (−ΔΔG/ΔHA) as the retrofragments were progressively recombined to form lead compound 2.31KD previously reported.28
GE analysis provided an assessment of the change in binding affinity per heavy atom that could be attributed to each of the Ar1, Ar2, and Ar3 rings of lead 2, as the compound was sequentially rebuilt from a series of retrofragments 3–6.31 X-ray crystal structures of CYP121 in complex with retrofragments 3–6 were used to validate our GE analysis, enabled the identification of conserved binding hotspots within the enzyme active site and was used to guide our subsequent optimization of 2 using fragment growing and merging strategies. The significant improvements in binding affinity and LE that can be achieved by the structure-guided incorporation of a metal binding pharmacophore into the newly identified minimal scaffolds is illustrated. In contrast, fragment growing toward pockets in the distal active site produced only modest improvements in binding affinity and resulted in the development of a second series of compounds with a binding mode that was distinct from the metal-coordinating ligands. Investigation of the SAR governing these interactions and knowledge of binding hot-spots were subsequently exploited to improve the isoform selectivity of inhibitors against human P450 enzymes, and also identified regions of the CYP121 active site that were tolerant to structural modification and which could be exploited during the future optimization of these compounds.
Results
Retrofragmentation
Retrosynthetic fragmentation of lead compound 2 indicated that the relative binding contributions of Ar1, Ar2, and Ar3 could be determined from a combination of monophenol and biphenol fragments 3, 4, 5, and 6 (Figure 3).32 The sequential reconstruction of 2 from these fragments enabled the GE of each aromatic ring to be calculated from the difference in the free energy of binding per heavy atom added at each stage of reconstruction (GE = −ΔΔG/ΔHA).31 Retrofragments 3 and 5 were synthesized according to the previously reported literature procedure,28,33 which was also used to access fragment 4. Biphenol fragment 6 was synthesized according to the general route for accessing 3,5-disubstituted aminopyrazoles described by Johnson et al.34 X-ray crystal structures of fragments 3–628 in complex with CYP121 (Figure 2a–c) were obtained in order to aid the interpretation of binding affinity and GE trends. Retrofragments 3–6 each reproduced the privileged non-heme binding mode of lead compound 2, which is unusual for P450 ligands containing azole motifs. Analysis of the conserved and variable binding interactions made by the retrofragments highlighted structural features likely to be important for driving the binding affinity of 2.32,35 Monophenol 4 bound in an orientation that overlapped with that of Ar2 of the lead compound 2 (Figure 2a). The water-bridged hydrogen bonding network between Asn85, Thr229, and the heme propionate group with the 4-hydroxy substituent of 4 was conserved, while the 5-aminopyrazole ring was shifted toward the I-helix (highlighted yellow in Figure 2) to enable hydrogen bonding interactions with Val228 and aromatic interactions with Phe168 and Trp182, mimicking the function originally fulfilled by Ar1 of lead compound 2. The small shift in position of the aminopyrazole ring of 4 indicated that interactions made at the Ar1 site might contribute more significantly to the affinity of lead compound 2 than do the hydrogen-bonding interactions made by 2 with Gln385.
Figure 2.

X-ray crystal structures of lead compound 2 and component retrofragments 4–6 in complex with CYP121. The I- (yellow), and F-, and G- (blue) helices are shown in cartoon representation. (a) Overlaid structures of retrofragments 4 (purple) (PBD 4KTJ), 5 (yellow) (PDB 4KTF), and 6 (cyan) (PDB 5IBJ) with lead compound 2 (salmon) (PDB 4KTL), illustrating the conserved binding mode distal to the heme cofactor (magenta).28 (b) Structures, binding affinity, and ligand efficiency (LE) of compound 2 and retrofragments 4, 5, and 6. (c–e) X-ray crystal structures of CYP121 in complex with retrofragments 4, 5, and 6, respectively, indicating polar interactions (yellow dashes) with active site residues (blue) and solvent (red spheres). Co-crystallized sulfate (yellow lines) is also indicated in (d). The associated omit FoFc electron density map of fragment 6 contoured to 3σ has been provided in the Supporting Information, Figure S5. Figures prepared using PyMOL v1.7.4 (Schrödinger, LLC).
A 1.65 Å X-ray crystal structure of retrofragment 3 in complex with CYP121 was obtained, however incomplete ligand occupancy prevented adequate refinement of the structure. The poorly resolved density of 3 suggested the fragment likely acquired multiple different binding orientations, as had been previously observed for the original hit fragment 1 (Figure 1). Analysis of the ligand density suggested that 3 bound CYP121 in an orientation that overlapped with Ar1 of lead 2. However, the 5-aminopyrazole ring was rotated 180°, further indicating that hydrogen bonding to Gln385 probably contributed less to binding affinity than other conserved interactions (Figure S1, Supporting Information). The fused Ar1–Ar2 fragment 5 accurately recapitulated the binding mode of 2, satisfying the offset and edge-face π-stacking interactions with Phe168 and Trp182 that were identified as important in the region occupied by Ar1, as well as the hydrogen bonding networks formed by the Ar2 phenol and between the aminopyrazole ring and Gln385 (Figure 2b). The conserved binding mode of 5 in the absence of Ar3 suggested a minimal contribution to binding from this motif and was consistent with observations that Ar3 resided in a large, water-filled pocket in the X-ray structure of 2 (Figure 1). This lack of binding contribution was reflected in the low GE calculated for Ar3 on reforming 2 from 5 (Figure 3).
The X-ray crystal structure of the Ar1–Ar3 fused fragment 6 showed that the fragment bound CYP121 in an orientation that was flipped 180° from that expected in lead 2. The 5-amino-phenol ring that distinguished 6 from retrofragment 3 was oriented toward the Ar1 site, instead of the Ar3 pocket, and the original phenol of 3 was shifted into the Ar2 site (Figure 2c). In this orientation, the pyrazole core formed new hydrogen bonding interactions with I-helix residue Thr229, similar to those observed for fragment 4. This change in binding mode accounts for the good GE values calculated when elaborating fragment 3 into 6 (vide infra). The 100-fold improvement in binding affinity obtained for 6 was thus attributed to optimizing interactions at the Ar1 and Ar2 sites, demonstrating the importance of obtaining structural data for the accurate interpretation of GE analysis. From these data, it was concluded that Ar1 and Ar2 formed highly conserved interactions that could be exploited in the design of subsequent analogues. In contrast, Ar3 contributed minimally to binding and would likely be tolerant to structural modifications. As such, investigating the SAR of Ar3 was a priority in order to identify more GE replacements for the original phenol ring of lead compound 2.
The binding affinity and LE of the retrofragments for CYP121 was determined using ITC (Figure 3). Fragments 3 (KD = 1.3 mM, LE = 0.30) and 4 (KD = 1.9 mM, LE = 0.29) were found to have comparable binding affinities and LE to that of the original fragment hit 4-(1H-1,2,4-triazol-1-yl)phenol 1, indicating that substitution of the original triazole core of 1 for a 5-aminopyrazole ring resulted in no significant difference in binding affinity. The combination of any two phenol rings to satisfy binding interactions at the Ar1 and Ar2 sites, as exemplified by biphenol retrofragment 5 (KD = 40 μM) or 6 (KD = 10.6 μM), resulted in an almost additive improvement in binding affinity. The small difference in the binding affinity of 5 or 6 (ΔG ≈ −0.9–1.7 kJ mol–1) compared to that calculated for the theoretical sum of 3 and 4 (ΔG3+4 (theor) ≈ −7.7 kJ mol–1) can be attributed in part to binding contributions from the 5-aminopyrazole motif that is common to the structures of the monophenol fragments. The additive binding affinity was reflected in the good GE values (GE = 0.29–0.41) calculated for the addition of an aromatic ring to a monophenol scaffold 3 or 4. Reduced torsional strain between the phenol rings was suggested to account for the slightly lower KD value of retrofragment 6 versus 5.28 The addition of a third aromatic ring to fragment 5 or 6 to give lead 2 had a negligible effect on binding affinity, as reflected in the small GE values of −0.03–0.08 calculated for occupation of the Ar3 pocket.
The LE of retrofragments (LE ≈ 0.3) was comparable to that of the original fragment hit 1 (LE = 0.32), indicating that both mono- and biphenol scaffolds represented better starting points for the optimization of CYP121 inhibitors than did lead compound 2 (LE = 0.24). Retrofragments 4 and 5 were selected as the most favorable scaffolds for further optimization because of the consistency of their binding modes relative to lead compound 2 and because of the opportunities these scaffolds provided for elaboration toward the heme iron from Ar2 and from the exocyclic pyrazole-5-amino group to explore the Ar3 pocket. The change in the binding mode of retrofragment 6 relative to lead 2 led us to deprioritize further optimization based on this scaffold despite its slightly better binding affinity compared to 5. This combination of GE analysis, assessment of LE, and interrogation of X-ray crystal structures facilitated a rational strategy for the independent optimization of each aromatic ring Ar1, Ar2, and Ar3 of lead 2, guided by insights into the key binding interactions and activity hotspots.
Analogue Design Rationale
Our approach to developing potent CYP121 inhibitors focused on three independent series of analogues, each optimizing the interactions of the Ar1, Ar2, and Ar3 rings of lead compound 2, respectively (Figure 4a–c). Analogues were designed as derivatives of either the biaryl scaffold 5, which had been identified as the most LE retrofragment that also retained the original binding mode of 2, or the monoaryl 4 scaffold, which had been identified to participate in a conserved hydrogen-bonding network and bound to CYP121 in close proximity to the heme cofactor.
Figure 4.

Proposed optimization strategies for Ar1, Ar2, and Ar3 of lead CYP121 inhibitor 2. (a) Overlaid X-ray crystal structures of biaryl fragment 5 (yellow) (PDB 4KTF) and benzamide fragment 7 (green) (PDB 5EDT) bound to CYP121. The associated omit Fo–Fc electron density map of fragment 7 contoured to 3σ can be found in Supporting Information Figure S5. (b) Overlaid X-ray crystal structures of CYP121 in complex with retrofragment 5 (yellow) and with heme binding fragments 8 (copper) (PDB 4G44) and 9 (cyan) (PDB 4G45),27 indicating the proposed meta-attachment point for substituents and distance (6.5 Å) to the heme coordinating position (yellow dashed line). (c) Docking of Ar3 analogues of lead 2, elaborating from the 5-aminopyrazole group of retrofragment 5 (gray) with carbonyl (orange) or methylene (green) linkages to enhance binding interactions with the distal active site pocket (Asn74, Thr65, gray sticks) of CYP121. Figures prepared using PyMOL v1.7.4 (Schrödinger, LLC).
Ar1 Analogues
Analogues in the Ar1 series were designed to probe the effect of electronics and hydrogen bonding on the conserved π-stacking interactions formed between this motif and Phe168 and Trp182 and the potential to form polar interactions with Thr77 and Asp185, residues residing on the substrate selective F/G-helices.36 The crystal structure of a substituted benzamide fragment 7 (KD = 3.4 mM), which had been identified during our initial fragment screen on CYP121 (PDB 5EDT),27 indicated that meta-substitution or elaboration from the para-position would be most favorable (Figure 4a). Ligand docking simulations37 were used to support our design and indicated that changing the position of the aromatic substituent was unlikely to cause conformational strain or perturb the global geometry of the ligands (Figure S2, Supporting Information).
Ar2 Analogues
The proximity and orientation of Ar2 in relation to the heme cofactor, and the conserved hydrogen bonding interactions made by the 4-hydroxy substituent with Asn85 and the water-bridged heme propionate group, indicated that Ar2 could provide an ideal anchor for the incorporation of a metal-binding functional group. Compounds which interact with both active site residues and the heme cofactor are generally considered to be more effective than inhibitors that only utilize one form of interaction and it was proposed that exploiting the high enthalpic contribution of metal coordination could significantly improve the potency of CYP121 ligands.36 Comparison of the X-ray crystal structures of 2, 4, and 5 with those of heme binding fragments 8 and 9, which had been identified during our original fragment screen on CYP121,27 indicated that extending from the meta-position of Ar2 with substituents 5.5–6.5 Å in length would provide the appropriate proximity for metal binding (Figure 4b). A range of substituents containing aromatic or basic nitrogen atoms were selected as functional groups commonly occurring in heme binding P450 inhibitors. Primary anilines were prioritized because of the prevalence of this chemotype in fragment hits identified during our original fragment screen on CYP121 and also from our subsequent optimization attempts which had yielded several co-crystal structures of CYP121 with aniline ligands.27
Analogues derived from both monoaryl 4 and biaryl 5 fragment scaffolds were proposed to maximize LE and to allow the importance of Ar1 on the binding mode, affinity, and selectivity of the compounds to be investigated (Figure 4b). A range of analogues in which the 4-hydroxy group of Ar2 was removed were also designed in order to determine whether torsional strain resulting from interactions between the 4-hydroxy group and metal-binding substituents introduced ortho to this might perturb the geometry required for metal coordination. It was hypothesized that the loss of enthalpic contributions from hydrogen bonds made by the 4-hydroxy group of Ar2 was likely to be small in comparison to that gained by metal-coordination.38
Ar3 Analogues
A fragment growing approach was devised to explore the SAR of Ar3 and to improve the GE of this motif by potentially introducing binding interactions with residues in the back of the active site. Interrogation of the X-ray crystal structure of lead 2 and ligand docking studies37 were used to select an appropriate linker-length for growing, with methylene or carbonyl linkers being prioritized. A range of five- and six-membered (hetero)aromatic groups were selected to incorporate onto these linkers, including phenols as direct mimics of lead 2 and the natural cYY substrate, phenol bioisosteres, and substituents that were predicted by docking simulations to form favorable cation−π interactions with a nearby Arg386 residue, or polar interactions with amide groups of the protein backbone (Figure 4c).37
Synthetic Chemistry
Analogues varying at Ar1 were synthesized according the published procedure for biaryl retrofragment 5 (Scheme 1).28,33 In brief, the Claisen condensation of 4-methoxyphenylacetonitrile 11 with the appropriately substituted benzoate ester 10a–d afforded the β-ketonitrile intermediates 12a–d. Reaction of 12a–d with hydrazine hydrate and concentrated hydrochloric acid gave the 5-aminopyrazoles 13a–d in excellent yields (91–97%). Subsequent deprotection of the aryl-methyl ether(s) with BBr3 (1.0 M in DCM) gave the desired phenol analogues 5 and 14b–d in near quantitative yields.
Scheme 1. Synthesis of Biaryl-aminopyrazole Analogues (Ar1).

Reagents and conditions: (a) NaH, THF, 60 °C, 20–48 h; (b) HCl (37%), NH2NH2·xH2O, EtOH, 90 °C, 14 h; (c) BBr3 (1.0 M in DCM), DCM, 0 °C, 6 h. Synthesis and characterization of 14d previously described.28
The syntheses of analogues incorporating a metal binding functional group at the meta position of Ar2 are summarized in Scheme 2 (i) and (ii) for analogues based on the monoaryl 4 or biaryl 5 retrofragment scaffolds, respectively. Analogues retaining the 4-hydroxy substituent of Ar2 were synthesized from the appropriately protected 4-benzyloxy 15b or 4-methoxy 15c substituted 3-bromophenylacetonitrile starting materials, which were either obtained directly from commercial sources or synthesized in two steps by deprotection of the aryl methyl ether using BBr3 followed by benzyl protection under Finkelstein conditions. Synthesis of the common 5-aminopyrazole ring followed a similar route to that employed to access Ar1 analogues (Scheme 1). Claisen condensation of phenylacetonitriles 15a–c with either ethyl formate or methyl 4-methoxybenzoate afforded the 3-oxopropanenitrile 16a–c or β-ketonitrile 21a–b intermediates, respectively. The crude products were treated with hydrazine hydrate at acidic pH and heated under reflux to give the 5-aminopyrazole compounds 17a–c and 22a–b in yields of 48–67% and 77–95%, respectively, over two reaction steps. A microwave-assisted Suzuki–Miyaura cross-coupling reaction was used to introduce a range of nitrogen-containing heterocycles 18e–i or nitro-substituted aromatics 18a–d, 23a–b from their boronic acid precursors. Nitroarenes were subsequently reduced using tin(II) chloride to reveal the heme binding anilines 19a–d, 24a–b in yields up to 77%. Removal of the methyl or benzyl protecting groups using BBr3 afforded the 4-hydroxyphenyl Ar2 analogues 20a and 25a–b, which were purified by flash chromatography to give 46–50% yields. However, the presence of inorganic contaminants in 20a prevented the testing of this compound.
Scheme 2. Synthesis of Ar2 Analogues Based on the Core Scaffold of (i) Monoaryl Fragment 4 or (ii) Biaryl Fragment 5.

Reagents and conditions: (a) ethyl formate, NaOEt, EtOH, 0–90 °C, 22 h; (b) AcOH, NH2NH2·xH2O, EtOH, 75 °C, 3 h; (c) aryl-boronic acid, Pd(PPh3)4, Na2CO3, 1,4-dioxane:H2O (1:1 v/v), μw, 180 min; (d) SnCl2·2H2O, EtOH, 75 °C, 15 h; (e) BBr3 (1.0 M in DCM), DCM, 0 °C, 4 h; (g) methyl 4-methoxybenzoate, NaH, THF, 60 °C, 48 h; (h) HCl (37%), NH2NH2·xH2O, EtOH, 90 °C, 16 h.
Ar3 analogues containing a methylamine-linked substituent 26a–f were synthesized from the reductive alkylation of biaryl retrofragment 5 with the respective aldehyde (Scheme 3). Reaction conditions, including solvent polarity, acid catalyst, and use of either sodium triacetoxyborohydride or sodium cyanoborohydride reducing reagents were optimized for individual substrates.39,40 Analogues 26g and 26h were synthesized in good yields (78–95%) under similar conditions by reductive alkylation of the dimethoxy protected precursor 13a. Subsequent deprotection of intermediates 27a–b with BBr3 revealed the diphenols 26g–h in 61–68% yields.
Scheme 3. Synthesis of Benzyl-amine Analogues.

Reagents and conditions: (a) aldehyde (0.9–1.1 equiv), AcOH*, Na(OAc)3BH, DMF, DCE, rt, 24–48 h (b) aldehyde (1 equiv), AcOH, NaCNBH3, MeOH, rt, 15–24 h; (c) BBr3 (1.0 M in DCM), DCM, 0 °C, 2–24 h. *No AcOH with 26d.
A series of 5-amidopyrazole analogues 29a–c containing a carbonyl linker were synthesized from 5-aminopyrazole 13a and the respective benzoyl chloride to give 28a–c (Scheme 4). Benzoyl chloride electrophiles were either purchased or synthesized in situ from the corresponding benzoic acid precursor using oxalyl chloride and catalytic DMF. Yields of the desired 5-amido regioisomers were low (27–34%), but significant polarity differences enabled easy separation from endocyclic 1N-substituted byproducts using column chromatography. Subsequent deprotection of the aryl-ethers with BBr3 afforded the analogues 29a–c.
Scheme 4. Synthesis of Benzamide Analogues.

Reagents and conditions: (a) (4-OMe)-benzoyl chloride, Et3N, DCM, 0 °C–rt, 3.5 h; (b) nicotinic acid, oxalyl chloride, DMF, DCM, 0 °C–rt, 2 h, then Et3N, DCM, 0 °C–rt, 14 h; (c) BBr3 (1.0 M in DCM), DCM, −78 °C–rt, 14 h.
Inhibitor Binding Mode and Selection of Heme Binding Functional Groups for Ar2 Analogues
The binding mode of analogues was initially characterized using UV–vis spectroscopy to allow the identification of ligands that interacted with the ferric iron, or with the axial water ligand of the heme cofactor, from changes in the optical spectrum of CYP121. P450 inhibitors which displace the axial water ligand and directly coordinate to the ferric iron of the heme cofactor, typically by the lone pair electrons of a nitrogen atom, stabilize an inactive state of the enzyme by sterically blocking the active site and usually lowering the reduction potential of the enzyme.36 This binding mode (type II) produces a red-shift in the Soret band of the P450 optical spectrum, the magnitude (Δλmax, nm) of which is dependent on both the affinity (KD) and concentrations of the ligand and P450 under specific assay conditions.41 In contrast, ligands which interact with the heme cofactor via water-bridged hydrogen bonding to the axial water ligand, or which displace the axial water but do not directly coordinate to the ferric iron, can produce reverse type I and type I spectra, respectively.42
A single shot UV–vis absorbance assay was used to rapidly identify Ar2 substituents with the appropriate properties and coordination geometry for heme binding interactions (Table 1). A preliminary series of Ar2 analogues, substituted at the meta-position with aniline or heteroaromatic groups, were synthesized based on the monoaryl scaffold 4 (Scheme 2 (i)). Only primary 3- or 4-aminophenyl analogues 19a and 19d were found to cause a significant red-shift (Δλmax > 1 nm) in the Soret band of CYP121, which occurs at 416.5 nm in the water-ligated resting state.17 Indole 18g,h, pyrazole 18i, and 3-pyridyl 18e substituents showed no heme binding interactions (Δλmax = 0 nm), and only minor perturbation of the Soret band was observed for the 4-pyridyl isomer 18f (Δλmax = 1 nm).
Table 1. Identification of Heme Binding Functional Groups Using UV–Vis Spectroscopya.

Analogues were prepared as 0.1–1 mM stocks in DMSO-d6. The change in the wavelength of the Soret band (Δλmax, nm) of CYP121 (5 μM) was calculated as the difference of that found in the presence of analogues compared to 1% v/v DMSO-d6 alone (λmax = 416.5 nm). Binding affinities (KD) for selected compounds were calculated from the titration of compounds (0.2 μL aliquots of 10–100 mM stocks) into CYP121 (5 μM). Full details are provided in the Experimental Section.
The binding affinities of the 3-aminophenyl 19a (KD = 14 μM) and 4-aminophenyl 19d (KD = 230 μM) substituted analogues were determined by optical titration according to the previously described procedure.17,41 The 16-fold difference in affinity between the two regioisomers clearly indicated that the 3-aminophenyl substituent provided a better geometry for heme ligation, and consequently this functional group was incorporated into all subsequent analogues in the Ar2 series.
All subsequent Ar2 analogues containing a 3-aminophenyl substituent (19b,c, 24a, 25a,b) caused type II shifts in the CYP121 optical spectrum, allowing their binding affinity to be determined by optical titration in addition to ITC (Table 2 and Figure 5). The magnitude of the type II shift decreased with increasing steric bulk ortho to the 3-aminophenyl substituent. These results supported the hypothesis that torsional strain in the biphenyl system, or the steric bulk of the benzyl or methyl hydroxy protecting groups, in analogues 19b and 19c, respectively, prevented the ligands approaching the cofactor for heme coordination.
Table 2. Binding Affinity and Ligand Efficiency Values of CYP121 Inhibitorsc.

Binding affinity of 14d previously reported.28
Poor solubility prevented KD being determined for 24a using ITC.
Binding affinity (KD, μM) values for all analogues were measured by ITC. The KD values for the Ar2 series of heme binding compounds were also determined by UV–vis optical titration as described previously.17 Ligand efficiency (LE) was calculated from the Gibbs free energy of binding divided by number of heavy atoms (ΔG/HA).29,30 Full details are provided in the Experimental Section.
Figure 5.

Optical titration difference spectra (top panel) and concentration-dependent changes in CYP121 absorbance (bottom panel) for representative heme binding ligands (a) 24a, (b) 25b, and (c) 19a. Absorption spectra of CYP121 (5 μM) were collected between 250 and 800 nm after sequential additions of 0.2 μL aliquots of DMSO-d6 stock solutions (0.15–83 mM) of ligands. Difference spectra were obtained from the subtraction of the ligand-free CYP121 spectra from those obtained over the course of the titration. The maximum change in absorbance from each difference spectrum was plotted against ligand concentration and data were fitted using a modified version of the Morrison equation for tight binding ligands 24a and 25b(17) or a hyperbolic function for 19a.
The only analogue in the Ar1 or Ar3 series of compounds to perturb the Soret band was the 3(4)-imidazole-methylamine substituted compound 26h. The type II shift observed was small (Δλmax26h (100 μM) = +1.5 nm) compared to that observed for the 3-aminophenyl substituted Ar2 analogues (Δλmax25a [100 μM] = +5.0 nm), which was consistent with results observed for heteroaromatic nitrogen substituents during our initial selection of a heme binding functional group for Ar2 analogues (Table 1). A 2.0 Å X-ray crystal structure of 26h in complex with CYP121 indicated that the compound must adopt multiple different binding modes, as the 3(4)-imidazole substituent of the crystallized compound resided in the Ar3 pocket, distal to the heme cofactor (Figure 8b). These competing binding modes might account for the weak affinity of 26h (KD = 274 μM) compared to the structurally similar 3-thiophenyl 26c or 2-furanyl 26d methylamine analogues, which did not perturb the optical spectrum of CYP121 (Table 2). Owing to a lack of spectral perturbation, the binding affinity of all other Ar1 and Ar3 analogues was assumed to result from binding interactions made with residues in the distal active site pocket and provided support that these analogues retained a similar binding mode to lead 2.
Figure 8.

X-ray crystal structures of Ar3 analogues (a) 26a (PDB 5IBI) and (b) 26h (PDB 5IBH) in complex with CYP121. Both compounds closely recapitulated the binding mode of lead 2. Ar3 substituents remained flexible, interacting only with active site water molecules for 26a or forming novel interactions with active site residues Ala75 and Asn74 for 26h. The omit Fo – Fc electron density maps of ligands 26a and 26h contoured to 3σ have been provided in Supporting Information Figure S5. Figures prepared using PyMOL v1.7.4 (Schrödinger, LLC).
Binding Affinity and X-ray Crystallography
The binding affinity (KD) of analogues for CYP121 was determined by ITC (Table 2, Figure 6a–c). The KDs of heme binding Ar2 analogues were also determined by UV–vis optical titration (Table 2, Figure 5).43−45 Comparison of the KDs obtained for the Ar2 series by ITC and optical titration enabled robust comparisons to be made with the affinity of the non-heme binding Ar1 and Ar3 series of analogues, as well as with previously reported type II inhibitors in the literature.12,27,28 In general, KD values obtained by optical titration were 10–100-fold lower than those measured in ITC, consistent with previous reports in the literature.45 Variations in affinity data generated using different biophysical techniques have been previously attributed to the kinetically significant but spectrally silent, multistep binding interactions of P450s with ligands, the potential presence of heterogeneous enzyme populations in solution, requirement for conformational change, and/or enzyme cooperativity.45,46 High resolution (1.6–2.2 Å) X-ray crystal structures of CYP121 in complex with a number of analogues were obtained, allowing inhibitor binding modes to be confirmed and enabling robust comparison of the SAR contributing to binding affinity (Figure 7,8).
Figure 6.

Representative ITC binding isotherms and integrated enthalpy plots for (a) fused Ar1–Ar3 fragment 6 (500 μM); (b) non-heme binding Ar3 analogue 26a (1 mM); (c) heme binding analogue 25a (500 μM). The isotherm of each compound titrated into buffer alone was subtracted from that containing CYP121 (50 μM). The integrated enthalpy change for each injection plotted as a function of molar ratio of ligand-to-CYP121 was fitted using a one-site equilibrium binding model to calculate binding affinity (KD). The binding stoichiometry (N) was allowed to vary for analogues 6, 26a, and 25a.
Figure 7.

X-ray crystal structures of heme binding Ar2 analogues 25b (PDB 5IBJ), 25a (PDB 5IBE), and 19a (PDB 5IBF) in complex with CYP121. (a) 25b (cyan) closely recapitulated the binding of lead 2, forming polar contacts (yellow dashes) with amino acids (blue sticks) in binding hotspots as well as additional binding contacts to Ser237, Val228, and the heme iron. (b) 25a (orange) was rotated away from the I-helix (yellow cartoon), placing the 5-aminopyrazole ring in the Ar3 pocket. Polar contacts to the heme iron and Ser237 are indicated (yellow dashes). (c) Two molecules of 19a bound per active site, each ligand fulfilling a subset of the interactions identified as binding hotspots. Inset: Heme binding 19a recapitulated the binding mode of 25a while the non-heme binding 19a ligand satisfied aromatic interactions in the Ar1 pocket, as well as the hydrogen bonding interactions of fragment 7. (d) Extracted structures of 25b, 25a, and 19a annotated to show metal coordination (yellow dashes) distance, angle of approach to the porphyrin ring (yellow angle), and dihedral torsion in the biphenyl-aniline motif. Removal of the Ar2 4-hydroxy group reduced the dihedral angle of the biphenyl aniline system in 19a and 25a by 30° compared to 25b and allowed the analogues to approach the plane of the porphyrin at a sharper angle (31–33°). The omit Fo – Fc electron density maps of ligands 19a, 25a, and 25b contoured to 3σ have been provided in Supporting Information Figure S5. Figures prepared using PyMOL v1.7.4 (Schrödinger, LLC).
Ar1 Analogues
Changing the position of the original 4-hydroxy group to the 3-position 14b resulted in a moderate reduction in binding affinity, while substitution with a 3-fluoro substituent increased the KD of 14c 5-fold compared to 5. These results suggest that aromatic stacking interactions made by Ar1 contribute to binding affinity in this region. The change in π-electron density from the introduction of m-hydroxy or m-fluoro substituents would likely weaken both offset face-to-face aromatic interactions with Phe168 and edge-to-face interactions with Trp182, in addition to disrupting hydrogen bonding interactions with two conserved water molecules shown to solvate the p-hydroxy substituent of fragments 1, 5, and lead compound 2 (Figure 1 and 2d).47 Disubstitution of Ar1 with hydroxy groups in the 2- and 4-positions, 14d, also lowered the binding affinity (KD = 180 μM) despite introducing polar interactions with residues Thr77 and Ala167 as shown in the X-ray crystal structure of 14d.28 Intramolecular hydrogen bonding between the 2-hydroxy group and the 1-NH-pyrazole heterocycle might account for this result by favoring the 3-aminopyrazole tautomer and thus weaken bonding interactions to Gln385. No overall change in the conformation of the ligands was observed due to changes made to Ar1 substituents, consistent with our initial docking results.28 The sensitivity of Ar1 to even minor structural changes confirmed our previous predictions from retrofragmentation and GE analysis that binding interactions in this region represented a key affinity hotspot for CYP121 that might be of use during later stages of compound development. We thus directed our attention toward investigating the Ar2 and Ar3 aromatic rings.
Ar2 Analogues
Incorporation of a 3-aminophenyl substituent onto the Ar2 ring of the monoaryl scaffold 4 or the biaryl scaffold 5 resulted in a more than 100-fold improvement in binding affinity. Analogue 19a had an equivalent KD of 14 μM (LE = 0.35) by optical titration to that of lead compound 2 and was made significantly more LE by removal of the Ar3 motif. The reincorporation of the Ar1 phenol ring to give compound 25b (KD = 0.29 μM) resulted in a further 50-fold improvement in binding affinity, as had been previously observed during our lead-deconstruction analysis when the Ar1 and Ar2 fragments 3 and 4 were fused to give biaryl retrofragment 5 (Figure 3). Removal of the Ar2 4-hydroxy group of 25b to relieve torsional strain in the heme binding biphenyl-amine system improved CYP121 affinity a further 20-fold to produce the low nanomolar, high LE compounds 24a and 25a. While the 4-hydroxy analogue of 19a was not analyzed, the effect of ortho-substitution on weakening heme binding interactions was made apparent through assessment of the 4-benzyloxy and 4-methoxy substituted analogues 19b and 19c. While both 19b and 19c caused type II shifts in the CYP121 Soret band at high concentrations (100–1000 μM), the bulky protecting groups clearly prevented ligands obtaining the optimal geometry for heme ligation and a KD could not be obtained for these compounds by optical titration within solubility limits. The relatively good binding affinity of 19b (KD = 28 μM) determined by ITC suggested that this compound might instead adopt an alternative “heme-silent” binding mode in the distal active site.45
The improved LE of 19a, 24a, and 25a–b compared to lead 2 (LE = 0.24) was reflected in the good GE value of 0.42 calculated for the 3-aminophenyl substituent based on the introduction of seven heavy atoms to retrofragment 5.31,48 Comparison with the low GE of 0.08 calculated previously for the Ar3 phenol (Figure 3), which had the same number of heavy atoms, demonstrated the utility of our lead deconstruction-elaboration approach in allowing the identification of maximally efficient scaffolds and in targeting binding hotspots.
The 2.1 Å crystal structure of 25b (Figure 7a), the structurally most similar compound to retrofragment 5, in complex with CYP121 revealed that the analogue accurately replicated the conserved binding mode of the lead 2 (Figure 1). All intermolecular interactions that had been identified as binding hotspots, such as aromatic stacking of Ar1 with Phe168 and Trp182, interaction of the Ar2 4-hydroxy group with a water-bridged heme propionate network, and hydrogen bonding between the aminopyrazole ring and Gln385 were satisfied. The 3-aminophenyl substituent was located 2.5 Å from the heme-iron and at an angle of 36°, consistent with that required for tetrahedral NH2–Fe(III) coordination.49 Hydrogen bonding interactions were also identified between the 3-amino group of 25b and Ser237, which is thought to be the conserved alcohol residue of CYP121 required for the catalysis of proton transfer to the heme iron and for organization of the axial water ligand.12,36
The 1.6 Å crystal structure of 25a confirmed that removal of the Ar2 4-hydroxy group resulted in a 30° reduction in the dihedral angle of the biphenylamine motif (Figure 7b,d). As for analogue 25b, the 3-aminophenyl substituent of 25a bound in close proximity (2.3 Å) and at an appropriate orientation (34°) for heme coordination. However, reduced rotation in the biphenyl system allowed a slightly sharper angle of approach to the heme ring. Close overlap between the aniline ring of 25a and the sterically unhindered aniline-containing fragments 8 and 9 suggested that aromatic stacking interactions were optimized by the removal of the 4-hydroxy substituent from 25b. These improved interactions might account for the 9-fold improvement in potency between the two analogues and illustrate the utility of fragments as tools for elucidating preferred binding motifs (Figure 4b, and Figure S3, Supporting Information). The combination of improved heme interactions and removal of functional groups identified to interact with binding hotspots, such as the Ar2 4-hydroxy group, resulted in a change in orientation of 25a compared to the conserved position of 25b and lead 2. Analogue 25a was tilted sideways to place the exocyclic 5-amino group of the pyrazole ring into the pocket normally occupied by Ar3, resulting in a loss of interactions with Gln385. The fact that 24a retained good affinity despite this loss of interactions with Gln385 was consistent with observations made about the binding of retrofragments during lead deconstruction (vide supra). The equipotent (KD = 33 nM) 4-methoxy protected precursor 24a was found to bind in an identical orientation to 25a, demonstrating some tolerance of the Ar1 group to substituent variation (Figure S4, Supporting Information).
Two molecules of analogue 19a, which can be considered a fragment of 25a (lacking only the Ar1 ring), were found to bind within each active site of the CYP121 crystallographic dimer (Figure 7c). The orientation of the two overlapping molecules of 19a illustrated both the importance of the Ar1 region as a binding hotspot and also demonstrated optimization strategies which might further improve selectivity. One molecule of 19a ligated the heme iron as observed for 25a, with a coordination distance of 2.4 Å and a 32° angle of approach to the heme ring. The 5-aminopyrazole group similarly occupied the water-ligated Ar3 site. The second molecule of 19a satisfied the requirements of the remaining binding hotspots, including hydrogen-bonding interactions with Gln385 and aromatic stacking interactions in the Ar1 site. Extension of the 3-aminobiphenyl system into the Ar1 pocket demonstrated a route to achieving the additional hydrogen bonding interactions with the F/G-helix residues Thr77, Ala178, and Asp185 that had earlier been suggested by the identification of hit fragment 7 (Figure 7c, inset and Figure 2a).
Ar3 Analogues
The binding affinity of Ar3 analogues was largely unaffected by the structural changes made to the 5-aminopyrazole substituent. Analogues substituted with a range of aromatic or heteroaromatic groups maintained low micromolar binding affinities (KD < 100 μM), and a number of compounds (26b–d) had comparable potency to lead 2 (KD = 15 μM) (Table 2). Efforts to grow further out into the active site pocket occupied by Ar3 by the addition of a methylene linker resulted in a small improvement in binding affinity with a KD of 6.3 μM determined for the 4-hydroxybenzyl analogue 26a, making it the most potent non-heme binding CYP121 ligand described to date. The poorly defined electron density of the 4-hydroxybenzyl ring in the X-ray crystal structure of 26a suggested that it was conformationally flexible and only interactions with active site solvent molecules were observed (Figure 8a). All Ar3 analogues containing methylamine-linked aromatic substituents were more potent than their carbonyl-linked amido-pyrazole counterparts 29a–c. For example, comparison of 4-hydroxy-benzyl 26a and benzamide 29b analogues or 4-pyridinyl-methylamine 26g and isonicotinamide 29c analogues revealed a 2.5–16-fold loss in affinity, respectively, for the carbonyl-linked analogues. Increased conformational rigidity enforced by the 5-amido group or changes to the hydrogen bond donor–acceptor interactions of the 5-amino-NH with Gln385 was used to rationalize these SAR. A preference for 4-hydroxy substituted aromatics was observed in both the methylamine (26) and amide-linked (29) analogues, which was unsurprising considering the structure of the natural cYY CYP121 substrate.10,11 The addition of bromine ortho to the 4-hydroxy group in analogue 26b had little effect on binding affinity and indicated that larger substituents could be tolerated in this region of the active site. Indole 26e and indazole 26f bioisosteres of the 4-hydroxyphenyl group resulted in a 3–4-fold increase in KD, possibly indicating that the greater acidity or hydrogen bonding ability of the hydroxyl group was favorable for binding. Further supporting this assumption, analogues containing basic aromatic nitrogens, such as the pyridinyl 26g or 3(4)-imidazole 26h methylamine-linked analogues, significantly weakened CYP121 binding affinity.
As previously highlighted, a 2.0 Å X-ray crystal structure of 26h in complex with CYP121 was obtained that showed the imidazole ring binding distal to the heme cofactor in the Ar3 pocket (Figure 8b). Hydrogen bonding interactions with Asn74 and Ala75 had not been previously observed in other CYP121–ligand structures and could provide a new route for compound optimization. Despite these novel interactions the binding affinity of 26h was weak (KD = 274 μM), which could have been the result of the mixed binding modes suggested by UV–vis spectral analysis (vide supra). The consistency of the binding mode of 26a and 26h with lead 2 demonstrated in X-ray crystallography provided confidence when interpreting the SAR of the remainder of the Ar3 analogues. The binding affinity of thiophenyl–26c or furanyl–26d methylamine-linked analogues was comparable to that of lead 2 and tolerance to these smaller, hydrophobic heterocycles might enable the pharmacokinetic properties of subsequent analogues to be modulated during later phases of drug development. The insensitivity of the Ar3 pocket to structural modifications could indicate a functional adaption of the enzyme to enable the accommodation of different substrates. Recent evidence that CYP121 binds a number of other cyclic dipeptides, including cYF and cYW, supports this proposal.50
Inhibitor Affinity and Stoichiometry
The relative affinity of lead compounds, their binding stoichiometry to CYP121 and their effect on the oligomerization state of the enzyme were investigated using native mass spectrometry. Most bacterial P450s are monomeric and soluble cytosolic proteins. However, some form higher homomeric oligomerization states, heterodimers with their cognate reductase proteins, or are expressed as fusion products covalently linked to a reductase domain.36,51 We have previously reported that CYP121 exists predominantly as a dimer in solution, with a small percentage of monomeric protein present.52 Dimeric CYP121 forms complexes with two ligand molecules simultaneously and the dimer interface does not appear to occlude the substrate entry channel. The stability of the dimer is reduced by the binding of azole antifungal compounds. In contrast, other small molecule CYP121 inhibitors and the CYP121 substrate (cYY) form stable complexes with the protein and do not affect the oligomerization state.52
Native mass spectra were collected for CYP121 with the most potent Ar3 analogue 26a, Ar2 analogues 19a, 24a, 25a, and 25b and the biaryl retrofragment 6 (Figure 9a–c). All analogues formed stable complexes with the CYP121 monomer and dimer. The proportion of ligand-bound CYP121 increased in a concentration-dependent manner as the ratio of ligand-to-protein was increased from sub- (0.5:1) to superstoichiometric (25:1) values. The relative occupancy of monomeric and dimeric CYP121 and binding stoichiometry observed at three different ligand concentrations allowed analogues to be ranked in terms of their relative binding affinities (Table S1, Supporting Information). The rank order of affinities correlated well with KD values calculated previously by ITC and UV–vis optical titration (Table 2). Both the CYP121 monomer and dimer were entirely in the bound state at stoichiometric ligand-to-protein concentrations of Ar2 analogues 25a and 25b, consistent with the low nanomolar affinities of these compounds. In contrast, complete occupancy of the CYP121 dimer by the more weakly binding analogues 19a and 26a was not achieved until ligands were present in 25-fold excess ligand-to-protein concentrations. Despite the comparable KD of retrofragment 6 to Ar2 analogue 19a, the CYP121 dimer remained singly bound even at 25-fold excess concentration of 6. This apparent difference in binding strength between 6 and 19a observed by native mass spectrometry might reflect the different binding mode of the two analogues. Heme binding interactions made by 19a would likely be strengthened in the gas phase of the mass spectrometer, while the closer proximity of 6 to the entrance of the active site could allow the ligand to be displaced more easily during ionization. The double-occupancy of the CYP121 monomer observed for analogue 19a in X-ray crystallography was not detected within the concentration ranges tested in native mass spectrometry experiments (2.5–218 μM ligand).
Figure 9.

Representative native mass spectra of CYP121 and selected compounds 26a, 25a, and 19a demonstrated that compounds formed stable complexes with both monomeric (m/z 5250–6750) and dimeric (m/z 5250–6750) CYP121. The number of bound ligands (orange spheres) increased in a concentration-dependent manner and was proportionate to the binding affinity (KD) of the compounds. Spectra were collected for CYP121 (8.7 or 5 μM) at three different ligand-to-protein ratios (0.5:1, 1:1, and 25:1) and in the presence of DMSO-d6 (2.2% or 5% v/v) alone. The concentration of protein or DMSO-d6 did not affect the quality of the spectra or ligand binding interactions, but higher concentrations of DMSO-d6 reduced the charge state of the protein. Ligand binding stoichiometry was calculated from the difference in mass of ligand-bound and unbound (orange dashed lines) protein peaks divided by the molecular weight of ligands (Table S1, Supporting Information).
No significant change in the oligomerization state of CYP121 was observed for any of the analogues compared to that previously noted with the azole antifungals (Table S2, Supporting Information).52 The different effects of Ar2 analogues and azole antifungal compounds on the oligomerization state might reflect the nature of the metal binding functional group in each series. Greater conformational reorganization of the CYP121 active site might be expected in order to accommodate axial coordination of an sp2-hybridized imidazole ring (∼75°),49 while the primary aniline present in the current analogues approaches the porphyrin at a sharper angle (∼31–36°, Figure 7d).46,49
Selectivity against Mtb P450s
Selectivity screening against off-target microbial P450s is far less common during pharmaceutical development than screening against mammalian P450s and consequently the functions and promiscuity of these enzymes remain largely uncharacterized. All of the Mtb P450s that have been studied to date bind with varying affinity to compounds from the azole series of antifungal drugs, but there have been few reports detailing their interactions with non-imidazole/triazole ligands.4,7,9,12,49,53−55 Compounds from our development of CYP121 inhibitors were screened against a panel of Mtb P450s using UV–vis spectroscopy (Table 3) to both assess their selectivity and also to aid the development of chemical tools which could be used to help characterize these P450 systems in Mtb.
Table 3. Binding of Select Retrofragments and CYP121 Ligands to Mtb P450 Isoformsa.
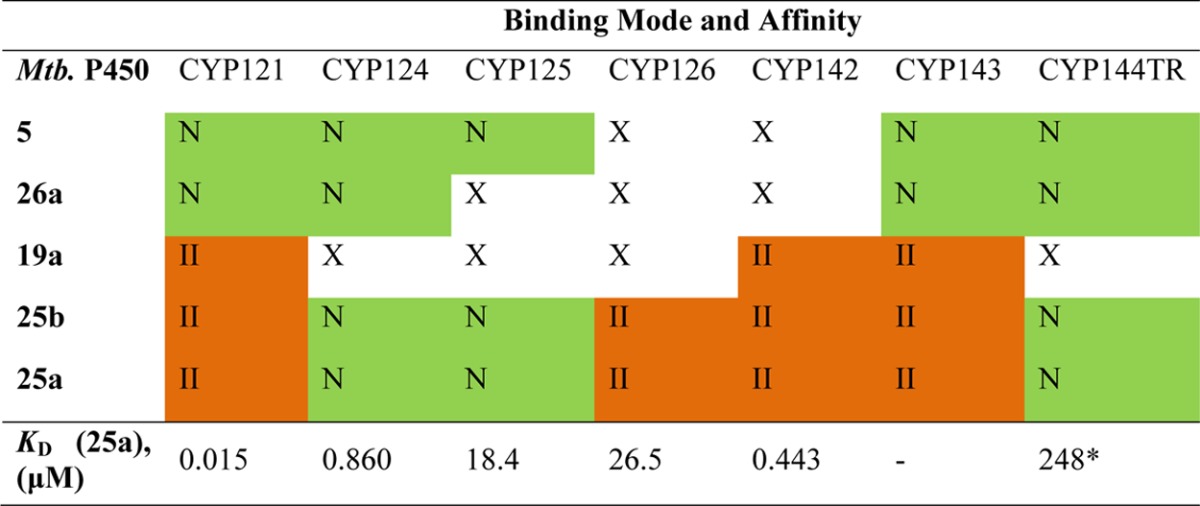
The binding mode of compounds is indicated as type II heme binding (II, orange), binding to the distal pocket (N, green), or nonbinding (X, unshaded). Binding was determined using a single shot UV–vis assay in which analogues (100 μM or 1 mM) were added to a solution of each P450 enzyme (2.5–5 μM) and the absorbance spectrum was collected between 800 and 250 nm. Compounds were prepared as DMSO-d6 stocks and added to give a maximum concentration of 1% v/v DMSO-d6. The change in the maximum wavelength of the Soret band (Δλmax, nm) compared to that measured for each P450 in buffer containing 1% v/v DMSO-d6 was used to assess the binding mode and the relative strength of binding. Compounds that did not change the Soret λmax were tested in competition with known type II inhibitors (50 μM), with compounds that decreased the Δλmax compared to that observed in the presence of the type II inhibitor alone being designated as “non-heme binding” (N) competitive ligands. The binding affinity of analogue 25a was calculated by direct or competitive optical titration, as described previously, depending on the mode of binding to each P450 respectively.17,58,59 *KD calculated by ITC for analogue 25b.
A qualitative assessment of the binding promiscuity of the CYP121 ligands developed in this study was obtained by measuring the change in the absorbance maximum (Δλmax, nm) of the characteristic Soret band of each P450 isoform when incubated with the ligands (100 μM to 1 mM) compared to a DMSO (1% v/v) control. Binding interactions made by the compounds with the distal part of the P450 active site, which do not directly perturb the heme environment or λmax of the Soret band, were characterized by measuring the decrease in the Δλmax caused by known type II ligands when Mtb P450s were coincubated with mixtures of the type II ligand and the novel CYP121 inhibitors. The binding affinity of the most potent CYP121 compound 25a has been provided to give a quantitative assessment of ligand selectivity across the different P450 isoforms. Experiments were performed as direct titrations for Mtb P450s CYP121, CYP126, and CYP142, or in competition with a known type II inhibitor where 25a exhibited a binding mode that did not influence the λmax of the heme absorbance spectrum, as in the case of Mtb P450 isoforms CYP124 and CYP125. Owing to the weak binding affinity of 25a for the Mtb P450 CYP144, a KD was obtained instead for the related compound 25b by ITC.
Subsequent to commencing work on these compounds, the original 4-(1H-1,2,4-triazol-1-yl)phenol fragment hit 1 for CYP121 had been identified to bind to all of the P450s reported here in ligand-observed NMR experiments (unpublished data). The biaryl retrofragment 5, which consisted of the fused Ar1 and Ar2 motifs of lead 2 and could be seen to represent a “merged” analogue of hit fragment 1, was less promiscuous, only binding to five of the seven P450s screened. Elaboration of 5 to include an Ar3 substituent, as seen in the 4-hydroxybenzyl substituted analogue 26a, further enhanced CYP121 selectivity. In contrast, the incorporation of a heme binding 3-aminophenyl substituent onto the scaffold of 5, as in 25a and 25b, reduced the selectivity for inhibitors for CYP121, as might be expected from the introduction of a binding group targeting a conserved active site feature. While type II spectral changes were identified for 4 of the 7 P450s studied, 25a and 25b bound in positions that did not perturb the P450 heme spectrum in 3 of the 7 isoforms tested, illustrating the preference of different P450s for certain metal-binding pharmacophores and the impact of protein microenvironment on metal chelation.49,56 The KD of 25a varied between 0.44 and 27 μM across the different P450 isoforms, which gave a 30–1800-fold selectivity ratio (SR = off-target binding affinity/on-target binding affinity) for CYP121.57 The low solubility of Ar2 analogue 19a limited testing to the lower concentration of 100 μM which, in combination with the small size of the compound, possibly accounted for the apparently greater selectivity observed in the single-shot UV–vis assay. These results demonstrated how optimizing ligand affinity by targeting binding interactions made in the distal pocket, as through the introduction of Ar1 or Ar3 motifs, can enhance drug selectivity between P450s.
Selectivity against Human P450s
Drug–drug interactions resulting from the inhibition of human P450s, or toxicities resulting from the metabolic activation of drugs by human P450s, are leading causes of attrition during pharmaceutical development. Thus, understanding the SAR influencing P450 promiscuity and isoform selectivity is a topic of significant interest. The five human P450s CYP1A (CYP1A1 and CYP1A2 isoforms), CYP2C9, CYP2C19, CYP2D6, and CYP3A4 are responsible for the phase I metabolism of more than 80% of marketed drugs.36 Compounds 19a, 25a, and 26a were screened for inhibitory activity against these five human P450s and were found to have good selectivity profiles, showing weak or no inhibition (IC50 > 10 μM) against all five human P450 isoforms (Table 4).60 The exception was compound 19a, which was a moderately potent (1 μM < IC50 < 10 μM) inhibitor of CYP1A (IC50 = 1.22 μM), an isoform which has a documented preference for aromatic and heterocyclic amine substrates.36 The reincorporation of Ar1 into analogue 19a to give potent analogue 25a resulted in a 20-fold increase in IC50 against CYP1A (IC5025a = 21.4 μM). When the corresponding improvement in CYP121 binding affinity for the two compounds was also taken into account, this amounted to an approximate 15000-fold better apparent selectivity ratio (app SR = off-target potency/on-target potency).57 The 4-hydroxybenzyl substituted Ar3 analogue 26a did not inhibit any of the human P450s by 50% within the tested concentration range. Taken together, the improved selectivity from reintroducing Ar1 to the heme binding analogue 19a to give 25a, and the high selectivity of the non-heme binding analogue 26a, supported our hypothesis that targeting variable residues and hotspots in the distal pocket of the CYP121 active site could be used to achieve P450 isoform selectivity. Aromatic amines, such as those present in the potent CYP121 ligands 25a, 24a, 25b, and 19a, are commonly identified as potential toxicophores. However, considering 13% of marketed drugs contain aniline functional groups, it is premature to exclude all compounds with this functional group from drug-like chemical space.61 The toxicity of aniline structures requires metabolic activation, which is primarily mediated by human P450s.62 The good selectivity of CYP121 inhibitors, particularly compound 25a, demonstrated here against human P450s, is promising for the development of this series of CYP121 inhibitors, however further assessment against other P450, monooxygenase, and peroxidase enzymes is warranted. The selectivity of compounds derived from lead 2 against human P450s was good compared to that of other classes of P450 inhibitors that have been reported recently63,64 and suggests that the careful optimization of metal binding functional groups and targeting active site residues are methods which could reduce P450 liability during drug development.
Table 4. Selectivity of CYP121 Ligands against Drug-Metabolizing Human P450 Isoformsa.
| IC50 (μM) |
KD (μM) | app SR | |||||
|---|---|---|---|---|---|---|---|
| CYP1A | CYP2C9 | CYP2C19 | CYP2D6 | CYP3A4 | CYP121 | (IC50/KD) | |
| 19a | 1.22 | >25 | >25 | >25 | >25 | 14.0 | 0.09 |
| 25a | 21.4 | >25 | >25 | >25 | 11.3 | 0.015 | 1427 |
| 26a | >25 | >25 | >25 | >25 | >25 | 6.3 | >4 |
The concentration of analogues 19a, 25a, and 26a required to inhibit the activity of human P450s by 50% (IC50, μM) was calculated from a seven-point dose–response curve. Compounds that did not achieve 50% inhibition over the tested concentration range 0–25 μM were considered non-inhibitors.60 Studies were performed in human liver microsomes. Turnover of human P450 substrates (CYP1A, ethoxyresorufin; CYP2C9, tolbutamide; CYP2C19, mephenytoin; CYP2D6, dextromethorphan; CYP3A4, midazolam) was detected by LC-MS/MS or fluorescence for CYP1A. Reported CYP1A activity represents the combined activities of both the CYP1A1 and CYP1A2 isoforms. The apparent selectivity ratio (app SR = off-target potency (IC50)/on-target potency (KD)) was calculated as a ratio of the IC50 (μM) of each compound for CYP1A versus their respective CYP121 KD (μM).57 The IC50 values of known inhibitors were determined as positive controls and are listed in Table S3 of the Supporting Information. Experiments were conducted by Cyprotex (Cheshire, UK).
Cellular Potency
The antimycobacterial activity of the most potent analogues 19a, 24a, 25a, 25b, 26a, and biaryl retrofragment 5 was assessed in vitro against Mtb H37Rv. No significant inhibition of bacterial growth was detected for any of the lead compounds (MIC90 ≥ 50 μM) over the tested concentration range (0–100 μM) (Table S4, Supporting Information). Achieving adequate intracellular drug exposure is a particular challenge for TB therapeutics because of the unique cell-wall structure of the bacterium and the expression of numerous multisubstrate efflux transporters.65,66 Efforts to determine whether low membrane permeability or efflux might account for the poor cellular activity of the potent CYP121 ligands described here are currently in progress.
Conclusions
Using a fragment-based approach, a novel series of potent and isoform selective inhibitors of the essential Mtb enzyme CYP121 have been developed. An initial fragment screen and synthetic optimization strategy yielded the previously reported non-heme binding lead 2, which had micromolar affinity for CYP121 but low LE. The deconstruction of lead compound 2 and synthesis of component retrofragments 3–6 allowed the GE of individual structural motifs Ar1, Ar2, and Ar3 to be assessed. Analysis of the binding mode of retrofragments enabled the identification of activity hotspots and the selection of more LE scaffolds for synthetic optimization. The rational incorporation of a metal-binding functional group onto Ar2, guided by X-ray crystallography and UV–vis spectroscopy, was used to achieve a 1000-fold improvement in binding affinity over lead compound 2, while the incorporation of structural groups that projected into the Ar1 or Ar3 regions of the distal active site was shown to modulate the isoform selectivity of inhibitors against human P450s. Extensive characterization of analogues by X-ray crystallography, UV–vis spectroscopy, and native mass spectrometry validated our design strategy, demonstrating that analogues maintained binding interactions in identified activity hotspots and formed stable complexes with the native CYP121 dimer. Analogues with low nanomolar potency (KD = 15 nM) for CYP121 are reported, and these molecules have good selectivity against human drug metabolizing P450s and a 30-fold selectivity ratio for CYP121 over other Mtb P450 isoforms. Analysis of selectivity data from this series provides insight into factors governing the selectivity of P450s, including the effect of the protein microenvironment and preference for specific metal-binding pharmacophores,56,67 which has relevance more broadly for drug development. Further structural optimization to improve the cellular activity of these analogues is ongoing with the aim of developing novel classes of anti-TB agents.
Experimental Section
General Chemistry
All reagents were purchased from Sigma-Aldrich Company Ltd. (Poole, UK) or Alfa Aesar (Heysham, UK) unless otherwise specified. Unless otherwise stated, reactions were conducted under positive pressure of a dry nitrogen atmosphere. Temperatures of 0 °C, −10 °C, and −78 °C were obtained by cooling the reaction vessel in a bath of ice, salt, and ice or acetone and dry ice (CO2(s)), respectively. Anhydrous DCM was prepared by distillation over calcium hydride. THF was freshly distilled over calcium hydride and lithium aluminum hydride, using triphenylmethane as an indicator. Other anhydrous solvents were purchased directly from commercial sources and used without further purification.
Analytical thin layer chromatography (TLC) was performed using Merck glass-backed silica (Kieselgel 60 F254 0.25 mm) plates. Compounds were visualized using short wave (254 nm) or long wave (365 nm) ultraviolet light or potassium permanganate stain. Retention factors (Rf) are quoted with respect to the solvent system used to develop the plate. Flash column chromatography was performed using an Isolera Spektra One/Four purification system and the appropriately sized Biotage SNAP column containing KP-silica gel (50 μm). Solvents are reported as volume/volume eluent mixture where applicable. Reactions were monitored by TLC and LCMS to determine consumption of starting materials.
Infrared absorption spectra were recorded on a Bruker ALPHA FT-IR or Spectrum One FT-IR (PerkinElmer) spectrometer by attenuated total reflectance (ATR) using a diamond crystal. Data are reported as vibrational frequency (cm–1) and intensity (strong, medium, weak, or broad) for the assigned functional group.
Nuclear magnetic resonance (NMR) spectra were recorded at 300 K unless otherwise stated, using either a Bruker 400 MHz AVANCE III HD Smart Probe, 400 MHz QNP cryoprobe, or 500 MHz DCH cryoprobe spectrometer. Spectra acquired on the 500 MHz DCH cryoprobe have been converted to analogue and back to reduce baseline roll. All spectra were recorded in the deuterated solvent indicated. Data are reported as chemical shift in parts per million (δ ppm) relative to the residual protonated solvent resonance peak. The relative integral, multiplicity, coupling constants (J Hz), and assignment of peaks has been provided where possible. Assignment of 1H NMR and 13C NMR spectra was assisted by DEPT, homonuclear (COSY), and heteronuclear (edited 1H–13C-HSQC and 1H–13C HMBC) 2D-NMR experiments. 13C NMR spectra were processed with line broadening (LB) of 1–12 Hz for individual compounds to enable resolution of signals from slowly relaxing quaternary pyrazole carbons C3, C4, and C5 and resolution of closely overlapping aromatic signals. LB has been noted for relevant spectra and 13C signals requiring LB for resolution have been noted as broad (br). 13C signals that were obscured or were only identified through proton relaxation in 2D-NMR experiments are also noted.
Liquid chromatography mass spectrometry (LCMS) was carried out using an AQUITY UPLC H-class system (Waters, Manchester UK). Samples were run using a gradient of water (1–5%) (+0.1% formic acid) in acetonitrile over a period of 4 min. High resolution mass spectra (HRMS) were recorded using a Micromass Quadrapole-Time-of-Flight (Q-Tof) spectrophotometer or an Orbitrap LCMS spectrometer attached to a Dionex Ultimate 3000 HPLC. The mass to charge ratio (m/z) of the molecular ion and difference from calculated mass (δ ppm) have been quoted.
All final compounds had a purity greater than 98% by LCMS analysis, except for compounds 14b, 18g, 18f, 19c, and 26b, which were 92–98% pure by LCMS analysis.
Synthesis of Retrofragment (3)
4-(5-Amino-1H-pyrazol-3-yl)phenol
Step 1: Synthesis of 3-(4-methoxyphenyl)-3-oxopropanenitrile. Methyl 4-methoxybenzoate (831 mg, 5 mmol) and acetonitrile (1 mL) were added to a stirred suspension of NaH (60% dispersion in mineral oil, 490 mg, 12.25 mmol) in anhydrous THF (25 mL). The reaction was stirred at 60 °C for 40 h, and then the volatiles were removed under reduced pressure. The crude residue was diluted with H2O (15 mL) and acidified with 1 M HCl, and then the product was extracted into DCM (3 × 20 mL). The combined organic fractions were washed with brine (5 mL), dried over anhydrous Na2SO4, and the solvent removed under reduced pressure. The crude product was purified by flash chromatography (40–100% v/v DCM in petroleum ether) to yield 3-(4-methoxyphenyl)-3-oxopropanenitrile as a white solid (578 mg, 3.30 mmol, 66%), Rf 0.30 (1:2 v/v EtOAc in petroleum ether). LCMS (ESI−) m/z 174.1 [M – H]−, retention time 1.68 min (100%). HRMS (ESI+): m/z (calcd C10H10NO2 [M + H]+ = 176.0712), obsd 176.0703 (δ ppm = 5.1). 1H NMR (500 MHz, CDCl3) δ 7.90 (d, J = 9.0 Hz, 2H), 6.98 (d, J = 9.0 Hz, 2H), 4.02 (s, 2H), 3.90 (s, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ 185.5, 164.9, 131.1, 127.4, 114.5, 114.2, 55.8, 29.2 ppm. IR (thin film) vmax 3083, 3020, 2942, 2844 (w, C–H), 2259 (w, nitrile C–N), 1685 (s, C=O), 1623 (w, C=C), 1596 (s), 1514 (s), 1465, 1450 (s, CH3), 1439 (m, C=C), 1326 (s, CH3), 1268 (s, C–O, CH2), 1224, 1172 (s, C–O), 1011 (s, C–O, CH2), 930, 828, 814 (s, C–H) cm–1.
Step 2: Synthesis of 3-(4-methoxyphenyl)-1H-pyrazol-5-amine. Concentrated HCl (37% v/v, 2.4 mL) was added slowly to a stirred solution of 3-(4-methoxyphenyl)-3-oxopropanenitrile (552 mg, 3.15 mmol) in absolute EtOH (20 mL). The reaction was heated to reflux, and then hydrazine hydrate (750 μL, 15.5 mmol) was added dropwise over 10 min. The reaction was heated under reflux for 5 h and then concentrated under reduced pressure (5 mL), diluted with H2O (10 mL), and brought to pH 8.0 using saturated NaHCO3. The product was extracted into EtOAc (3 × 20 mL), and the combined organic fractions were washed with brine (5 mL), dried over anhydrous Na2SO4, and the solvent removed under reduced pressure. The crude product was purified by flash chromatography (0–5% v/v MeOH in DCM) to yield 3-(4-methoxyphenyl)-1H-pyrazol-5-amine as a white solid (512 mg, 2.71 mmol, 86%), Rf 0.18 (5% v/v MeOH in DCM). LCMS (ESI+) m/z 190.2 [M + H]+, retention time 1.27 min (100%). HRMS (ESI+): m/z (calcd C10H12N3O [M + H]+ = 190.0975), obsd 190.0970 (δ ppm = 2.4). 1H NMR (500 MHz, MeOD) δ 7.50 (br, s, 1H), 7.38 (d, J = 8.8 Hz, 2H), 6.93 (d, J = 8.8 Hz, 2H), 3.79 (s, 3H) ppm. 13C NMR (125 MHz, MeOD) δ 159.4, 152.7, 128.9 (2 × C), 127.0, 115.2, 109.6, 55.7 ppm. IR (thin film) vmax 3397, 3148 (m, br, N–H), 2962, 2839 (m, C–H), 1615 (m, N–H), 1505, 1479, 1462 (s, N–H, C–H, C=C), 1439 (m, C=C), 1284 (m, C–N, C–O), 1244 (s, C–N, C–O), 1177, 1111 (m, C–H), 1028 (s, C–O), 831, 817, 769 (s, C–H), 710 (m, C–H) cm–1.
Step 3: Synthesis of 4-(5-Amino-1H-pyrazol-3-yl)phenol (3). A solution of 1 M BBr3 in DCM (12.5 mL, 12.5 mmol) was added dropwise over a period of 10 min to a stirred solution of 3-(4-methoxyphenyl)-1H-pyrazol-5-amine (473 mg, 2.5 mmol) in anhydrous DCM (22 mL) at 0 °C. The reaction was stirred at 0 °C and allowed to come to room temperature over 14 h. When complete, the reaction was quenched with saturated NaHCO3 and extracted into EtOAc (3 × 30 mL). The organic phase was combined, washed with brine (10 mL), dried over anhydrous Na2SO4, and the solvent removed under reduced pressure to yield 4-(5-amino-1H-pyrazol-3-yl)phenol 3 as an off-white solid (342 mg, 1.95 mmol, 75%), Rf 0.35 (10% v/v MeOH in EtOAc). LCMS (ESI+) m/z 176.2 [M + H]+, retention time 0.81 min (100%). HRMS (+ ESI): m/z (calcd C9H10N3O [M + H]+ = 176.0824), obsd 176.0823 (δ ppm = 0.6). 1H NMR (500 MHz, DMSO-d6) δ 9.49 (s, 1H), 7.44 (d, J = 9.0 Hz, 2H), 6.76 (d, J = 8.5 Hz, 2H), 5.63 (s, 1H) ppm. 13C NMR (125 MHz, DMSO-d6) δ 156.9, 153.6, 145.2, 126.1, 122.7, 115.4, 86.9 ppm. IR (thin film) vmax 3305, 3141 (w, br, N–H, O–H), 1615 (m, N–H, C=C), 1519 (s, N–H), 1489 (s, N–H), 1377 (w, O–H), 1279, 1244 (m, C–N, C–O), 1179 (m, C–O), 1111, 1014 (m, C–H), 834 (s, C–H), 765 (s, C–H) cm–1
Synthesis of Retrofragment (6)
4-((3-(4-Hydroxyphenyl)-1H-pyrazol-5-yl)amino)phenol
Step 1: Synthesis of methyl 3-(4-methoxyphenyl)-3-oxopropanoate. Dimethyl carbonate (1.26 mL, 15 mmol) was added to a stirred suspension of NaH (60% dispersion in mineral oil, 600 mg, 15 mmol) in anhydrous toluene (2 mL). The reaction was brought to reflux, and a solution of 4-methoxyacetophenone (0.90 g, 6 mmol) in anhydrous toluene (3 mL) was added dropwise. The reaction was heated at reflux for a further 15 min and then allowed to cool to room temperature. The reaction was quenched with a solution of AcOH (0.5 mL) in water (2 mL). The organic layer was separated, and the aqueous fraction was extracted with Et2O (2 × 5 mL). The organics were combined, washed with a solution of saturated K2CO3, dried over anhydrous Na2SO4, and the solvent removed under reduced pressure. The crude product was purified by flash chromatography (10–50% v/v EtOAc in petroleum ether) to yield methyl 3-(4-methoxyphenyl)-3-oxopropanoate as a yellow oil (1.05 g). The product was 90% pure by 1H NMR integration and was taken forward without further purification; Rf 0.18 (20% v/v EtOAc in petroleum ether). LCMS (ESI+) m/z 207.2 [M – H]−, retention time 1.75 min (96%). HRMS (ESI+): m/z (calcd C11H13O4 [M + H]+ = 209.0808), obsd 209.0803 (δ ppm = 2.4). 1H NMR (500 MHz, CDCl3) δ 7.92 (d, J = 9.0 Hz, 2H), 6.94 (d, J = 9.0 Hz, 2H), 3.96 (s, 2H), 3.87 (s, 3H), 3.74 (s, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ 190.9, 168.3, 164.2, 130.0, 129.2, 114.1, 55.7, 52.6, 45.7 ppm. IR (thin film) vmax 2954, 2842 (w, C–H), 1739 (s, C=O), 1675 (s, C=O), 1599 (s, C=C), 1575, 1511 (m, C=C), 1437, 1421 (m, CH2, CH3), 1323 (m, C–O, C–H), 1258 (s, C–O), 1215 (m), 1147, 1170 (s, C–O, C–H), 1025 (m, C–O), 986 (m), 840 (m, C–H) cm–1.
Step 2: Synthesis of N,3-Bis(4-methoxyphenyl)-3-oxopropanamide. A solution of 4-methoxyaniline (406 mg, 3.3 mmol) in m-xylene (6 mL) was added to a stirred solution of methyl 3-(4-methoxyphenyl)-3-oxopropanoate (623 mg, 3 mmol) in m-xylene (6 mL), and the reaction was heated to 150 °C for 20 h. The product precipitated on cooling to room temperature and was purified by trituration at 0 °C with ice-cold EtOAc:hexane (1:5 v/v). The precipitate was collected by vacuum filtration, washed with 1:5 EtOAc:hexane followed by 1:3 EtOAc:hexane, and dried under reduced pressure to give N,3-bis(4-methoxyphenyl)-3-oxopropanamide as a brown powder (567 mg, 1.9 mmol, 63%), Rf 0.06 (1:2 EtOAc:petroleum ether). LCMS (ESI+) m/z 322.0 [M + Na]+, retention time 1.87 min (100%). HRMS (ESI+): m/z (calcd C17H18NO4 [M + H]+ = 300.1236), obsd 300.1251 (δ ppm = 5.0). 1H NMR (500 MHz, CDCl3) δ 9.26 (s, 1H), 8.02 (d, J = 8.9 Hz, 2H), 7.48 (d, J = 9.0 Hz, 2H), 6.97 (d, J = 8.9 Hz, 2H), 6.86 (d, J = 9.0 Hz, 2H), 4.04 (s, 2H), 3.89 (s, 3H), 3.79 (s, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ 195.1, 164.7, 164.0, 156.6, 131.2, 130.9, 129.2, 122.0, 114.3, 114.2, 55.8, 55.6, 45.1 ppm. IR (thin film) vmax 3279 (w, N–H), 3144, 2963, 2840 (w, C–H), 1685 (m, C=O), 1598 (s, C=C), 1556 (m, C=C), 1509 (s, N–H), 1465, 1419 (w, CH2, CH3), 1323, 1263, 1246, 1217, 1170 (m, C=C, C–O, C–N), 1028, 1013 (C–O), 826 (s, C–H) cm–1.
Step 3: Synthesis of N,3-Bis(4-methoxyphenyl)-1H-pyrazol-5-amine. Lawesson’s reagent (419 mg, 1.04 mmol) was added to a stirred solution of N,3-bis(4-methoxyphenyl)-3-oxopropanamide (120 mg, 0.4 mmol) in dry THF (20 mL). The reaction was heated to reflux for 4 h and then concentrated under reduced pressure to give a red oil. The sulphonamide intermediate was redissolved in absolute EtOH (5 mL), and glacial AcOH (69 μL, 1.3 mmol) was added. The flask was evacuated, flushed with N2 gas, and then hydrazine monohydrate (66 μL, 1.3 mmol) added. The reaction was heated to reflux for 3 h and then allowed to stir at room temperature for 24 h. When complete, the reaction was concentrated under reduced pressure. The crude orange oil was diluted with DCM (20 mL) and made basic with a satd NaHCO3 solution (5 mL). The phases were separated, and the aqueous layer was extracted with DCM (2 × 2 mL). The organic fractions were combined, dried over anhydrous Na2SO4, and the crude product dried directly onto silica gel. Flash chromatography (25–80% v/v EtOAc in petroleum ether) yielded an off-white solid that was further purified by trituration with Et2O to give N,3-bis(4-methoxyphenyl)-1H-pyrazol-5-amine as a white powder (41 mg, 0.14 mmol, 35%), Rf 0.11 (2:1 v/v EtOAc in petroleum ether). LCMS (ESI+) m/z 296.3 [M + H]+, retention time 1.95 min (100%). HRMS (ESI+): m/z (calcd C17H18N3O2 [M + H]+ = 296.1399), obsd 296.1393 (δ ppm = 2.0). 1H NMR (500 MHz, acetone-d6) δ 11.42 (s, 1H), 7.67 (d, J = 8.7 Hz, 2H), 7.35 (m, 2H), 6.99 (d, J = 8.9 Hz, 2H), 6.81 (d, J = 9.0 Hz, 2H), 6.13 (d, J = 1.2 Hz, 1H), 3.83 (s, 3H), 3.73 (s, 3H) ppm. 13C NMR (125 MHz, acetone-d6) δ 160.9, 154.2, 144.7, 139.2, 127.7, 124.6, 118.0, 117.97, 115.42, 115.40, 91.0, 56.1, 56.0 ppm. 13C peak at 144.7 identified from 1H–13C-HMBC spectrum. IR (thin film) vmax 3402 (m, N–H), 3270 (br, w, N–H), 2968, 2840 (w, C–H), 1612, 1598, 1587 (m, C=C), 1554, 1505, 1464, 1427 (s, C–H, C=N), 1294 (m, C–N), 1239 (s, C–O), 1182 (s, C–N), 1111 (m), 1026 (s, C–O), 959 (m), 823, 762 (s, C–H) cm–1.
Step 4: 4-((3-(4-Hydroxyphenyl)-1H-pyrazol-5-yl)amino)phenol (6). A 1 M solution of BBr3 in DCM (1.4 mL, 1.4 mmol) was added dropwise over a period of 15 min to a stirred suspension of N-3-bis(4-methoxyphenyl)-1H-pyrazol-5-amine (104 mg, 0.35 mmol) in dry DCM (7.5 mL) at 0 °C. The reaction was allowed to come slowly to room temperature and stirred for 8 h, then quenched with ice water (40 mL). EtOAc (20 mL) was added, and the aqueous phase was made slightly basic with satd NaHCO3. The phases were separated, and the aqueous fraction was extracted with EtOAc (3 × 15 mL). The organic fractions were combined, dried over anhydrous Na2SO4, and the solvent removed under reduced pressure. The crude product was purified by flash chromatography (5–10% v/v MeOH in DCM, followed by 5% MeOH in EtOAc) to yield 4-((3-(4-hydroxyphenyl)-1H-pyrazol-5-yl)amino)phenol (6) as an off-white solid (43 mg, 0.16 mmol, 46%), Rf 0.52 (10% v/v MeOH in EtOAc). LCMS (ESI+) m/z 268.2 [M + H]+, retention time 1.37 min (100%). HRMS (ESI+): m/z (calcd C15H14N3O2 [M + H]+ = 268.1081), obsd 296.1070 (δ ppm = 4.0). 1H NMR (500 MHz, MeOD) δ 7.49 (d, J = 8.5 Hz, 2H), 7.05 (d, J = 8.0 Hz, 2H), 6.82 (d, J = J = 8.7 Hz, 2H), 6.70 (d, J = 8.9 Hz, 2H), 6.03 (s, 1H) ppm. 13C NMR (125 MHz, MeOD) δ 158.9, 155.5 (br), 152.0, 145.5 (br), 137.9, 127.8, 122.8, 119.2, 116.6, 116.6, 90.7 (br) ppm. IR (thin film) vmax 3417 (s, br, O–H), 3292 (s, br, N–H), 1607, 1615 (w, C=C), 1574 (s, C=C, N–H), 1522 (s, C=C, N–H), 1383 (w, C–O), 1254 (m, br, C–O, C–N), 1107 (w, C–H), 830 (s, C–H), 759 (m, O–H) cm–1.
General Procedure A
Synthesis of β-Ketonitriles 12a–c and 21a–b
2,3-Bis(4-methoxyphenyl)-3-oxopropanenitrile (12a)
2-(4-Methoxyphenyl)acetonitrile 11 (2.32 mL, 17.1 mmol) and methyl 4-methoxybenzoate 10a (2.58 g, 15.5 mmol) were added to a stirred suspension of NaH (60% in mineral oil, 1.34 g, 33.6 mmol) in anhydrous THF (30 mL). The reaction was heated to 60 °C for 20 h then cooled to 0 °C and quenched with H2O (10 mL). The suspension was concentrated under reduced pressure then diluted with dichloromethane (50 mL) and acidified with 3 M HCl to pH 2–3. The organic layer was separated off, and the aqueous phase was extracted with dichloromethane (3 × 50 mL). The organic fractions were combined, washed with brine (10 mL), dried over anhydrous MgSO4, and the solvent removed under reduced pressure to yield 2,3-bis(4-methoxyphenyl)-3-oxopropanenitrile 12a as a yellow gum (3.84 g, 13.7 mmol, 88%). The crude oxopropanenitriles 12a was used in subsequent reactions without further purification, although other analogues were purified by flash chromatography eluting with the solvent system specified. LCMS (+ ESI): m/z 280.2 [M + Na]+; retention time 2.11 min (93%). HRMS (+ ESI): m/z (calcd C17H16NO3 [M + H]+ = 282.1130), obsd 282.1135 (δ ppm =1.8). 1H NMR (500 MHz, CDCl3) δ 7.92 (d, J = 9.0 Hz, 2H), 7.34 (d, J = 8.8 Hz, 2H), 6.92 (d, J = 7.8 Hz, 2H), 6.90 (d, J = 7.8 Hz, 2H), 5.50 (s, 1H), 3.85 (s, 3H,), 3.78 (s, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ 187.6, 164.5, 160.1, 131.8, 129.5, 126.5, 122.7, 117.2, 115.1, 114.3, 55.7, 55.5, 45.8 ppm. IR (thin film) vmax 3008, 2936, 2839 (w, C–H), 2248 (w, nitrile CN), 1677 (m, C=O), 1597, 1510 (s, C=C), 1462, 1422 (w), 1305 (m, C–O), 1254, 1237, 1173 (s, C–O), 1027 (m), 939, 822, 780 (m) cm–1.
3-(3-Methoxyphenyl)-2-(4-methoxyphenyl)-3-oxopropanenitrile (12b)
Prepared as an orange oil (0.703 g, 2.50 mmol, 83%) according to general procedure A. The crude product was purified by flash chromatography (0–50% v/v EtOAc in petroleum ether), Rf 0.55 (50% v/v EtOAc in petroleum ether). LCMS (ESI−) m/z 294.2 [M + H]+, retention time 2.13 min. 1H NMR (400 MHz, CDCl3) δ 7.49 (d, J = 7.6 Hz, 1H), 7.45 (app t, J = 2.4, 1.6 Hz, 1H), 7.34 (m, 3H), 7.11 (d, J = 8.4, 2.0 Hz, 1H), 6.90 (d, J = 8.8 Hz, 2H), 5.53 (s, 1H), 3.82 (s, 3H), 3.78 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 189.1, 160.2, 160.1, 135.1, 130.0, 129.6, 122.3, 121.8, 121.0, 116.9, 115.2, 113.7, 55.6, 55.5, 46.2 ppm.
3-(3-Fluorophenyl)-2-(4-methoxyphenyl)-3-oxopropanenitrile (12c)
Prepared as an orange solid (229 mg, 0.852 mmol, 85%) according to general procedure A. The crude product was purified by flash chromatography (10–50% v/v EtOAc in petroleum ether). LCMS (ESI+) m/z 270.2 [M + H]+, retention time 2.08 min (89%). HRMS (ESI+) m/z (calcd C16H13FNO2 [M + H]+ = 270.0930), obsd 270.0933 (δ ppm = 1.1). 1H NMR (500 MHz, CDCl3) δ 7.71 (d, J = 8.0 Hz, 1H), 7.61 (d, J = 9.0 Hz, 1H), 7.43 (m, 1H), 7.33 (d, J = 8.8 Hz, 2H), 7.29 (m, 1H), 6.91 (d, J = 8.9 Hz, 2H), 5.49 (s, 1H), 3.79 (s, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ 188.1, 162.8 (d, JC–F = 248 Hz), 160.4, 135.6 (JC–F = 6.4 Hz), 130.8 (d, JC–F = 8 Hz), 129.5, 125.1 (d, JC–F = 2 Hz), 121.6 (d, JC–F = 10 Hz), 121.5, 116.5, 116.0 (d, JC–F = 23 Hz), 115.3, 55.5, 46.4 ppm. 13C NMR processed with LB = 3 Hz. IR (thin film) vmax 3076, 2959, 2936, 2840 (w, C–H), 2248, 2207 (w, nitrile CN), 1694 (m, C=O), 1608, 1588 (m, C=C), 1510 (s), 1439, 1304 (m), 1252 (s), 1149, 1030 (m, C–O), 885 (w), 834, 803, 788 (m) cm–1.
2-(3-Bromophenyl)-3-(4-methoxyphenyl)-3-oxopropanenitrile (21a)
Prepared as a yellow semisolid (2.18 g) according to general procedure A and used in subsequent reactions without further purification. The crude product contained 15% unreacted starting material by 1H NMR integration; Rf 0.18 (50% v/v EtOAc in petroleum ether). LCMS (ESI−) m/z 328.1 [M – H]−, retention time 2.23 min (59%). 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 8.9 Hz, 2H), 7.62 (s, 1H), 7.49 (d, J = 8.0 Hz, 1H), 7.8 (d, J = 7.8 Hz, 1H), 7.27 (m, 1H), 6.95 (d, J = 8.9 Hz, 2H), 5.53 (s, 1H), 3.89 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 186.7, 164.9, 132.9, 132.4, 131.9, 131.3, 127.0, 126.3, 123.6, 121.7, 116.4, 114.6, 55.8, 45.7 ppm. IR (thin film) vmax 2937, 2842 (w, C–H), 2252 (w, CN), 1679 (m, C=O), 1597 (s), 1572, 1512, 1424, 1323 (m, C=C, C–H), 1264, 1238, 1172 (s, C–N, C–O), 1026 (m), 941, 833, 775, 692 (m, C–H) cm–1.
2-(4-(Benzyloxy)-3-bromophenyl)-3-(4-methoxyphenyl)-3-oxopropanenitrile (21b)
Prepared as an orange semisolid (0.845 g, 1.56 mmol, quant.) according to general procedure A and either used directly in subsequent reactions or further purified by flash chromatography (0–10% v/v MeOH in DCM). LCMS (ESI+): m/z 436.1 [M + H]+, retention time 2.44 min (94%). HRMS (ESI+): m/z (calcd C23H19BrNO3 [M + H]+ = 436.0548), obsd 436.0561 (δ ppm = 3.0). 1H NMR (500 MHz, CDCl3) δ 7.92 (d, J = 8.9 Hz, 2H), 7.63 (d, J = 2.4 Hz, 1H), 7.44 (d, J = 7.4 Hz, 2H), 7.38 (appt, J = 7.6, 7.2 Hz, 2H), 7.34–7.30 (m, 2H), 6.93 (m, 3H), 5.45 (s, 1H), 5.13 (s, 2H), 3.87 (s, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ 186.8, 164.6, 155.6, 136.0, 133.1, 131.8, 128.7, 128.3, 128.1, 126.9, 126.2, 124.2, 116.7, 114.5, 114.3, 113.5, 70.9, 55.7, 44.9 ppm. IR (thin film) vmax 3064, 3033, 2935, 2840 (w, C–H), 2249 (w, nitrile C–N), 1678 (m, C=O), 1597 (s), 1572, 1510 (m, C=C), 1492 (s), 1454 (m), 1382 (w, C–N), 1261, 1220, 1173 (s, C–N, C–O), 1052, 1024 (m, C–O), 808, 735 (m) cm–1.
General Procedure B
Synthesis of Bisphenyl-1H-pyrazol-5-amines 13a–c and 22a–b
3,4-Bis(4-methoxyphenyl)-1H-pyrazol-5-amine (13a)28,33
Concentrated HCl (37% v/v, 6.25 mL) was added slowly to a stirred solution of crude 2,3-bis(4-methoxyphenyl)-3-oxopropanenitrile 12a (4.16g, ∼11.84 mmol, 1 equiv) in absolute EtOH (63 mL). The reaction was then heated to reflux, and hydrazine hydrate (3.6 mL, 74 mmol, 5 equiv) was added dropwise over 10 min. After heating at reflux until all starting material had been consumed (typically 5–15 h), the reaction was cooled to room temperature and concentrated under reduced pressure (20 mL). The reaction was diluted in water (15 mL) and brought to pH 8.0 using saturated NaHCO3. The aqueous emulsion was extracted with EtOAc (3 × 20 mL). The organic fractions were combined and washed with brine (10 mL), dried over anhydrous MgSO4, and the solvent removed under reduced pressure. The crude product was purified by flash chromatography (40% v/v EtOAc in petroleum ether, then 0–10% v/v MeOH in EtOAc) to yield 3,4-bis(4-methoxyphenyl)-1H-pyrazol-5-amine 13a as a yellow solid (3.171 g, 10.7 mmol, 91%), Rf 0.29 (10% v/v MeOH in DCM). LCMS (ESI+): m/z 296.1 [M + H]+; retention time 2.01 min (95%). HRMS (ESI+): m/z (calcd C17H18N3O2 [M + H]+ = 296.1399), obsd 296.1392 (δ ppm = 2.4). 1H NMR (500 MHz, DMSO-d6) δ 11.77 (s, br, 1H), 7.22 (d, J = 8.8 Hz, 2H), 7.10 (d, J = 8.5 Hz, 2H), 6.89 (d, J = 8.8 Hz, 2H), 6.86 (d, J = 8.8 Hz, 2H), 4.39 (s, br, 2H), 3.75 (s, 3H), 3.74 (s, 3H) ppm. 13C NMR (125 MHz, DMSO-d6) δ 158.6, 157.4, 152.6, 139.4, 130.5, 128.3, 125.9, 123.8, 114.0, 113.8, 103.0, 55.6, 55.0 ppm. 13C NMR processed with LB = 12 Hz. IR (solid) vmax 3432, 3349 (w, N–H), 3210 (w, br, N–H), 3932, 2833 (w, C–H), 1622 (w, C=C), 1513, 1489 (m, N–H, C–H), 1303, 1284 (w), 1241 (s, C–O, C–N), 1179 (s, C–O), 1111 (w), 1020 (s, C–O), 964 (w, C–H), 834 (s, C–H), 798, 735 (m, C–H) cm–1.
3-(3-Methoxyphenyl)-4-(4-methoxyphenyl)-1H-pyrazol-5-amine (13b)
Prepared as an orange amorphous solid (680 mg, 2.30 mmol, 92%) according to general procedure B. The crude material was carried forward without further purification. LCMS (ESI+) m/z 296.2 [M + H]+, retention time 1.80 min (93%). HRMS (ESI+) m/z (calcd C17H18N3O2 [M + H]+ = 296.1394), obsd 296.1381 (δ ppm = 4.4). 1H NMR (500 MHz, MeOD) δ 7.19–7.12 (m, 3H), 6.90 (m, 3H), 6.85 (dd, J = 2.6, 1.5 Hz, 1H), 6.80 (ddd, J = 8.3, 2.6, 1.0 Hz, 1H), 3.77 (s, 3H), 3.62 (s, 3H) ppm. 13C NMR (125 MHz, MeOD) δ 161.0, 160.0, 153.1 (br), 143.3 (br), 133.4 (br), 132.2, 130.6, 126.4, 120.8, 115.3, 115.0, 113.8, 106.3 (br), 55.7, 55.5 ppm. 13C NMR spectrum processed with LB = 12 Hz. IR (thin film) vmax 3422, 3336, 3182 (m, br, N–H), 2999, 2938, 2834 (m, C–H), 1607, 1578, 1525, 1488, 1464, 1285 (m, C=C), 1243 (s, C–N), 1177, 1040, 1022 (m, C–O), 834, 788, 749, 694 (m) cm–1.
3-(3-Fluorophenyl)-4-(4-methoxyphenyl)-1H-pyrazol-5-amine (13c)
Prepared as an orange solid (142 mg, 0.50 mmol, 97%) according to general procedure B. The crude product was purified by flash chromatography (0–5% v/v MeOH in DCM). LCMS (ESI+) m/z 284.2 [M + H]+, retention time 1.82 min (100%). HRMS (ESI+) m/z (calcd C16H15FN3O [M + H]+ = 284.1199), obsd 284.1188 (δ ppm = 3.9). 1H NMR (500 MHz, CDCl3) δ 7.24 (m, 1H), 7.19 (d, J = 8.7 Hz, 2H), 7.12 (dt, J = 7.7, 1.3 Hz, 1H), 7.03 (ddd, J = 9.8, 2.6, 1.6 Hz, 1H), 6.97 (app tdd, J = 8.4, 2.6, 1.0 Hz, 1H), 6.91 (d, J = 8.7 Hz, 2H), 3.83 (s, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ 162.9 (d, JC–F = 249 Hz), 158.7, 152.8, 140.2, 132.7 (d, JC–F = 8 Hz), 130.9, 130.4 (d, JC–F= 8 Hz), 124.4, 123.0 (d, JC–F = 3 Hz), 115.2 (d, JC–F = 21 Hz), 114.6, 114.3 (d, JC–F = 23 Hz), 106.3, 55.4 ppm. 19F-NMR (376 MHz, CDCl3) δ −112.4 ppm. IR (thin film) vmax 3310, 3190 (m, br, N–H), 3000, 2934, 2836 (m, C–H), 1613 (m, C=C), 1579, 1524 (m, N–H), 1488 (m, C=C), 1286 (m), 1245 (s, C–N, C–O), 1176, 1021 (m), 864, 835 (m, C–H), 788, 751 (m, C–H), 690, 605 (w) cm–1.
4-(3-Bromophenyl)-3-(4-methoxyphenyl)-1H-pyrazol-5-amine (22a)
Prepared as a brown amorphous solid (1.86 g, 5.42 mmol, 77% over two reaction steps from 3-bromophenylacetonitrile). The crude product was purified by flash chromatography (50–100% v/v EtOAc in petroleum ether), Rf 0.08 (50% v/v EtOAc in petroleum ether). LCMS (ESI+) m/z 346.1 [M (81Br) + H]+, retention time 1.96 min (100%). HRMS (ESI+) m/z (calcd C16H1579BrN3O [M + H]+ = 344.0393), obsd 344.0385 (δ ppm = 2.5). 1H NMR (500 MHz, MeOD) δ 7.37 (m, 2H), 7.24–7.19 (m, 4H), 6.88 (d, J = 7.2 Hz, 2H), 3.78 (s, 3H) ppm. 13C NMR (125 MHz, MeOD) δ 161.3, 152.6 (br), 144.2 (br), 137.1, 133.4, 131.4, 130.2 (2C), 129.5, 124.2 (br), 123.6, 115.1, 104.6 (br), 55.7 ppm. 13C NMR processed with LB = 12 Hz. IR (thin film) vmax 3063, 2936, 2841 (w, C–H), 2250 (w, nitrile C–N), 1677 (m, C=O), 1596 (s), 1574, 1510, 1474, 1423 (m, C=C), 1322 (m), 1264, 1239 (s, C–O), 1173 (s), 1027 (m, C–O), 941 (m), 832, 772, 693 (m) cm–1.
4-(4-(Benzyloxy)-3-bromophenyl)-3-(4-methoxyphenyl)-1H-pyrazol-5-amine (22b)
Prepared as a yellow foam (881 mg, 1.96 mmol, 95%) according to general procedure B. No further purification by flash chromatography was required. LCMS (ESI+) m/z 450.2, 452.2 [M + H]+, retention time 2.17 min (98%). HRMS (ESI+): m/z (calcd C23H21BrN3O2 [M + H]+ = 450.0817), obsd 450.0811 (δ ppm = 1.3). 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 2.1 Hz, 1H), 7.49 (d, J = 7.3 Hz, 2H), 7.40 (app t, J = 7.5 Hz, 2H), 7.33 (t, J = 7.3 Hz, 1H), 7.24 (d, J = 8.7 Hz, 2H), 7.11 (dd, J = 8.4, 2.1 Hz, 1H), 6.91 (d, J = 8.4 Hz, 1H), 6.84 (d, J = 8.8 Hz, 2H), 5.16 (s, 2H), 3.80 (s, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ 159.9, 153.9, 152.8, 141.4, 136.6, 134.3, 129.9, 128.8, 128.7, 128.1, 127.2, 126.9, 122.3, 114.4, 114.2, 112.9, 104.1, 71.0, 55.4 ppm. 13C NMR processed with LB = 3 Hz. IR (thin film) vmax 3419, 3197 (w, br, N–H), 2933, 2836 (w, C–H), 1613 (m, C=C), 1523 (s, N–H), 1454 (m), 1381 (w), 1280, 1249 (s, C–N), 1178, 1031 (m, C–O), 834, 737, 697 (m) cm–1.
General Procedure C
Synthesis of Oxopropanenitriles 16a–c
2-(3-Bromophenyl)-3-oxopropanenitrile (16a)
2-(3-Bromophenyl)acetonitrile 15a (1.09 g, 5.5 mmol) was added to a freshly prepared solution of sodium metal (139 mg, 6.05 mmol) in EtOH (7.5 mL) at 0 °C. After the reaction was stirred on ice for 5 min, ethyl formate (1.11 mL, 13.8 mmol) was added and the reaction was heated to reflux for 15–24 h. When no starting material remained, the reaction was cooled and concentrated under reduced pressure. The residue was diluted with H2O (5 mL) and acidified to pH 4 using 1 M HCl. The aqueous phase was extracted with Et2O or EtOAc (3 × 10 mL), and the combined organic fractions were washed with brine (5 mL), dried over anhydrous Na2SO4, and the solvent evaporated to yield an off-white solid (1.18 g, 5.26 mmol, 96%). The crude product was carried forward into subsequent reaction steps as a mixture of enantiomers and some acetonitrile starting material; Rf 0.08 (50% v/v EtOAc in petroleum ether). LCMS (ESI−) m/z 222.0 [M – H]−, retention time 2.01 min (97%). HRMS (ESI−): m/z (calcd C9H6BrNO [M – H]− = 221.9560), obsd 221.9556 (δ ppm = 1.7). 1H NMR (400 MHz, MeOD) δ 7.94 (s, 1H), 7.90 (s, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.59 (m, 1H), 7.57 (s, 1H), 7.54 (m, 1H), 7.51 (d, J = 7.9 Hz, 1H), 7.44–7.33 (m, 4H), 7.33–7.20 (m, 3H), 4.76 (d, J = 7.4 Hz, 1H) ppm. IR (thin film) vmax 3103, 2977 (m, C–H), 2725 (w, aldehyde C–H), 2223 (m, nitrile C–N), 1644 (m, C=O), 1594, 1558 (m, C=C), 1473 (m), 1378 (s, aldehyde C–H), 1253 (s), 1194, 1073 (m), 874 (w), 771 (s), 680 (m) cm–1.
Synthesis of 2-(4-(Benzyloxy)-3-bromophenyl)acetonitrile (15b)
Step 1: Synthesis of 2-(3-bromo-4-hydroxyphenyl)acetonitrile.10 A solution of BBr3 in DCM (1 M, 6.64 mL) was added dropwise to a cooled (−78 °C) solution of 2-(3-bromo-4-methoxyphenyl)acetonitrile (500 mg, 2.2 mmol) in DCM (2.2 mL). The reaction was allowed to warm to room temperature and stirred for 14 h or until all starting material had been consumed. The reaction was quenched by slowly pouring into ice–water (15 mL). The aqueous phase was extracted with DCM (3 × 10 mL). The organic fractions were combined, washed with water (10 mL) and brine (10 mL), and then dried over anhydrous MgSO4. The solvent was evaporated under reduced pressure to yield 2-(3-bromo-4-hydroxyphenyl)acetonitrile as white crystals (0.460 g, 2.17 mmol, 98%), Rf 0.23 (1:2 EtOAc:hexane). LCMS (ESI−) m/z 210.0 [M – H]−; retention time 1.76 min (95%). 1H NMR (500 MHz, CDCl3) δ 7.45 (d, J = 1.8 Hz, 1H), 7.18 (dd, J = 8.2 Hz, 1.9 Hz, 1H), 7.02 (d, J = 8.4 Hz, 1H), 5.57 (s, 1H), 3.67 (s, 2H) ppm. 13C NMR (125 MHz, CDCl3) δ 152.4, 131.5, 129.0, 123.4, 117.6, 116.8, 110.7, 22.6 ppm. IR (thin film) vmax 3305 (s, br, O–H), 2924, 2904 (w, C–H), 2272 (w, nitrile CN), 1613 (w), 1511, 1423, 1410 (s, C=C), 1356 (m, C–N), 1279 (s), 1219 (m, C–O), 802 (s), 658 (m) cm–1.
2-(4-(Benzyloxy)-3-bromophenyl)acetonitrile (15b)
Potassium carbonate (1.94 g, 14.0 mmol), potassium iodide (1.28 g, 7.70 mmol), and benzyl bromide (1.67 mL, 14.0 mmol) were added to a stirred solution of 2-(3-bromo-4-hydroxyphenyl)acetonitrile (1.48 g, 7.00 mmol) in acetone (65 mL). The suspension was heated to reflux for 15 h, at which point all of the starting material had been consumed. The reaction was concentrated in vacuo, and the resulting residue was diluted with EtOAc (120 mL). The organic fraction was washed with water (3 × 50 mL) and brine (50 mL), then dried over anhydrous MgSO4. The solvent was evaporated under reduced pressure to yield a brown oil. The crude product was purified by flash chromatography (0–30% v/v EtOAc in petroleum ether) to yield 2-(4-(benzyloxy)-3-bromophenyl)acetonitrile 15b as a pale-yellow oil (1.960 g, 6.49 mmol, 93%), Rf 0.44 (1:2 EtOAc:hexane). LCMS (ESI+) m/z 306, 308 [M + H, reduced nitrile]+; retention time 2.60 min (98%). HRMS (ESI+): m/z (calcd C15H13BrNO [M + H]+ = 302.0181), obsd 302.0166 (δ ppm = 5.0). 1H NMR (400 MHz, CDCl3) δ 7.53 (d, J = 2.2 Hz, 1H), 7.45 (m, 2H), 7.39 (app t, J = 7.4 Hz, 6 Hz, 2H), 7.34 (t, J = 7.3 Hz, 1H), 7.20 (dd, J = 8.5 Hz, 2.2 Hz, 1H), 6.92 (d, J = 8.4 Hz, 1H), 5.18 (s, 2H), 3.60 (s, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ 154.9, 136.2, 133.0, 128.8, 128.2, 128.1, 127.1, 123.6, 117.7, 114.2, 113.2, 71.0, 22.6 ppm. IR (thin film) vmax 3089, 3064, 2915, 2869 (w, C–H), 2253 (w, nitrile CN), 1604 (m), 1493 (s), 1454, 1413, 1382 (m, C–N), 1282, 1254 (s), 1157 (w), 1051 (s, C–O), 1006 (m), 806 (m), 737, 696 (s, C–Br) cm–1.
2-(4-(Benzyloxy)-3-bromophenyl)-3-oxopropanenitrile (16b)
Prepared as an orange residue (334 mg, 1.01 mmol, 51%) according to general procedure C. The crude product was purified by flash chromatography (0–5% v/v MeOH in DCM) to give a mixture of isomers; Rf 0.07 (1:2 v/v EtOAc in petroleum ether). LCMS (ESI−) m/z 330.0 [M – H]−, retention time 2.28 min (100%). HRMS (ESI+): m/z (calcd C16H13BrNO2 [M + H]+ = 330.0130), obsd 330.0140 (δ ppm = 3.0). 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 2.2 Hz, 1H), 7.54 (m, 2H), 7.46 (m, 5H), 7.43–7.36 (m, 5H), 7.33 (m, 2H), 7.21 (dd, J = 8.6, 2.4 Hz, 1H), 6.94 (d, J = 9.0 Hz, 1H), 6.91 (d, J = 8.4 Hz, 1H), 5.17 (m, 4H), 3.52 (s, 1H), 3.45 (d, J = 3.2 Hz, 1H) ppm. IR (thin film) vmax 3142 (br), 3034 (w, C–H), 2722 (w, C–H aldehyde), 2216 (w, nitrile, C–N), 1644 (m, C=O), 1599 (w, C=C), 1494 (s), 1379 (w, C–H aldehyde), 1285 (s), 1079, 1003, 808 (m), 731 (s), 694 (s, C–Br) cm–1.
2-(3-Bromo-4-methoxyphenyl)-3-oxopropanenitrile (16c)
Prepared as a cream-colored solid (508 mg) according to general procedure C. The crude product was taken forward without further purification; Rf 0.11 (1/3 v/v EtOAc in petroleum ether). LCMS (ESI−) m/z 253.1 [M – H]−, retention time 1.96 min (100%). HRMS (ESI+): m/z (calcd C10H9BrNO2 [M + H]+ = 253.9817), obsd 253.9829 (δ ppm = 4.7). 1H NMR (400 MHz, MeOD) δ 7.98 (d, J = 2.2 Hz, 1H), 7.77 (s, 1H), 7.66 (dd, J = 8.7, 2.2 Hz, 1H), 7.60 (dd, J = 6.3, 2.2 Hz, 1H), 7.56 (s, 1H), 7.49 (s, 1H), 7.38 (dt, J = 8.8, 2.4 Hz, 1H), 7.34 (dt, J = 8.8, 2.4 Hz, 1H), 7.06 (d, J = 8.8 Hz, 3H), 4.73 (d, J = 6.6 Hz, 1H), 3.95–3.80 (m, 10H) ppm. IR (solid) vmax 3243 (m, br), 3063, 2968, 2838 (w, C–H), 2216 (m, CN), 1845 (w), 1654 (m, C=O), 1602 (m), 1499 (s, C–N, C–H), 1438, 1375, 1349 (w, C=C, N–H), 1289 (m), 1256 (s, C–O), 1208 (m), 1056, 1013 (s, C–O), 870 (m), 803 (s, C–H), 686 (m) cm–1.
General Procedure D
Synthesis of Phenyl-1H-pyrazol-5-amines 17a–c
4-(3-Bromophenyl)-1H-pyrazol-5-amine (17a)
Glacial AcOH (679 μL, 12.9 mmol) followed by hydrazine hydrate (500 μL, 10.3 mmol) was added to a stirred solution of the crude 2-(3-bromophenyl)-3-oxopropanenitrile 16a (1.15 g, 5.14 mmol) in EtOH (7 mL) at room temperature. The reaction was heated to 85 °C for 3–5 h, and then the solvent was removed under reduced pressure. The residue was diluted with water (10 mL) and Et2O or EtOAc (10 mL), and brought to pH 8.0 with a satd NaHCO3 solution. The phases were separated, and the aqueous phase was extracted with Et2O or EtOAc (3 × 10 mL). The combined organic fractions were washed with brine (5 mL), dried over anhydrous Na2SO4, and the solvent removed under reduced pressure. The product was purified by trituration with petroleum ether and DCM to yield 4-(3-bromophenyl)-1H-pyrazol-5-amine 17a (865 mg, 3.70 mmol, 67% over two reactions steps from 3-bromophenylacetonitrile) as a pink solid; Rf 0.09 (2:1 v/v EtOAc in petroleum ether). LCMS (ESI+) m/z 240.1 [M (81Br) + H]+, retention time 1.61 min (100%). HRMS (ESI+) m/z (calcd C9H9BrN3 [M + H]+ = 237.9980), obsd 237.9984 (δ ppm = 1.7). 1H NMR (500 MHz, MeOD) δ 7.67 (s, 1H), 7.62 (s, br, 1H), 7.46 (d, J = 7.4 Hz, 1H), 7.32 (d, J = 7.8 Hz, 1H), 7.26 (app t., J = 7.8 Hz, 1H) ppm. 13C NMR (125 MHz, MeOD) δ 153.2 (br), 139.6 (br), 137.2, 131.5, 130.1, 129.4, 126.0 (br), 123.8, 108.4 (br) ppm. 13C NMR processed with LB = 12 Hz. IR (thin film) vmax 3392, 3227 (m, br, N–H), 2900 (w, C–H), 1600, 1567, 1496 (m, C=C, C–N), 1409 (w), 1079 (w), 872 (m), 779 (s), 687 (m) cm–1.
4-(4-(Benzyloxy)-3-bromophenyl)-1H-pyrazol-5-amine (17b)
Prepared as a cream solid (648 mg, 1.88 mmol, 94%) according to general procedure D. The crude product was used without further purification; Rf 0.23 (10% v/v MeOH in DCM). LCMS (ESI+): m/z 346.1 [M + H]+, retention time 1.94 min (100%). HRMS (ESI+): m/z (calcd C16H15N3OBr [M + H]+ = 344.0398), obsd 344.0387 (δ ppm = −3.2). 1H NMR (500 MHz, MeOD) δ 7.69 (d, J = 2.2 Hz, 1H), 7.54 (s, 1H), 7.49 (d, J = 7.5 Hz, 2H), 7.36 (m, 3H), 7.30 (t, J = 7.4 Hz, 1H), 7.09 (d, J = 8.5 Hz, 1H), 5.17 (s, 2H) ppm. 13C NMR (125 MHz, MeOD) δ 154.4, 138.3, 132.3, 129.5, 129.1, 128.9, 128.3, 127.7, 115.6, 113.6, 107.3, 71.9 ppm. 13C NMR processed with LB = 3 Hz. IR (thin film) vmax 3400, 3318 (m, N–H), 3165 (m, br, C–H), 3061, 3036 (m, C–H), 2889 (m, C–H), 1600, 1565, 1500 (m, C=C), 1486 (s, N–H), 1396, 1381, 1367 (w), 1292, 1257 (s, C–N), 1047 (s, C–O), 1013, 872 (m), 802 (s), 726, 704, 641 (m) cm–1.
4-(3-Bromo-4-methoxyphenyl)-1H-pyrazol-5-amine (17c)
Prepared as a white solid (339 mg, 1.26 mmol, 66% over two reaction steps) according to general procedure D. The crude material was purified by flash chromatography (2–10% v/v MeOH in EtOAc), Rf 0.05 (50% v/v EtOAc in petroleum ether). LCMS (ESI+) m/z 268.1 [M + H]+, retention time 1.59 min (100%). HRMS (ESI+): m/z (calcd C10H11BrN3O [M + H]+ = 268.0080), obsd 268.0071 (δ ppm = 3.4) 1H NMR (500 MHz, acetone-d6) δ 11.05 (br, s), 7.72 (d, J = 2.1 Hz, 1H), 7.63 (s, 1H), 7.49 (dd, J = 8.5, 2.2 Hz, 1H), 7.07 (d, J = 8.5 Hz, 1H), 4.38 (br, s, 2H), 3.88 (s, 3H) ppm. 13C NMR (126 MHz, Acetone-d6) δ 153.6, 150.0 (br), 130.6, 128.4, 126.3, 124.3, 112.6, 112.5, 111.4, 105.5 (br), 55.7 ppm. One 13C signal unresolved. 13C NMR processed with LB = 3.0 Hz. IR (solid) vmax 3407, 3333, 3225 (m, br, N–H), 2937, 2841 (w, C–H), 1619 (w, C=C), 1574 (m, N–H), 1516 (m), 1482 (s, C=C, C–H), 1383 (w, CH3), 1286 (m, C–N/C-O), 1250 (s, C–N, C–O), 1184 (w), 1045, 1015 (m, C–O, C–H), 744 (m, C–H), 680 (s, C–Br) cm–1.
General Procedure E
Suzuki–Miyaura Coupling to Synthesize Biphenyl-1H-pyrazol-5-amines 18a,b, 18d–i and 23a–b
4-(3′-Nitro-[1,1′-biphenyl]-3-yl)-1H-pyrazol-5-amine (18a)
4-(3-Bromophenyl)-1H-pyrazol-5-amine 17a (238 mg, 1 mmol), 3-nitrophenyl boronic acid (150 mg, 1.5 mmol) (1–2 equiv), Na2CO3 (318 mg, 3 mmol), and Pd(PPh3)4 (116 mg, 0.1 mmol) were combined in a microwave reaction vessel in a 1:1 v/v mixture of 1,4-dioxane:water (12 mL). The reaction was heated at 120 °C in a microwave reactor for 1.5–3 h. The reaction was then allowed to cool to room temperature and diluted with DCM (20 mL) and water (5 mL). The phases were separated, and the aqueous phase was extracted with DCM (3 × 3 mL). The organic fractions were combined, washed with brine (3 mL), dried over anhydrous MgSO4, and the solvent removed under reduced pressure. The crude product was purified by flash chromatography (0–5% v/v MeOH in DCM) to yield 4-(3′-nitro-[1,1′-biphenyl]-3-yl)-1H-pyrazol-5-amine 18a as a yellow amorphous solid (152 mg, 0.50 mmol, 54%), Rf 0.28 (10% v/v MeOH in DCM). LCMS (ESI+) m/z 281.2 [M + H]+, retention time 1.82 min (100%). HRMS (ESI+) m/z (calcd C15H13N4O2 [M + H]+ = 281.1033), obsd 281.1023 (δ ppm = 3.6). 1H NMR (500 MHz, CDCl3) δ 8.46 (app t, J = 2.0, 2.0 Hz, 1H), 8.21 (ddd, J = 8.3, 2.0, 2.0 Hz, 1H), 7.93 (ddd, J = 7.3, 1.5, 1.0 Hz, 1H), 7.70 (s, 1H), 7.62 (app t, J = 8.0, 7.5 Hz, 1H), 7.59 (s, 1H), 7.53–7.48 (m, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ 151.2 (br), 148.8, 142.8, 139.6, 134.1, 133.2, 129.9, 129.9, 128.8 (br), 126.9, 125.7, 125.1, 122.3, 122.1, 108.5 (br) ppm. 13C NMR processed with LB = 3 Hz. IR (thin film) vmax 3182 (m, br, N–H), 2952 (m, C–H), 1606 (m, N–H), 1524 (s, NO2), 1470, 1438 (w, C=C), 1345 (s, NO2), 1290, 1265 (w, C–N), 1087, 1055, 1026 (w, C–H), 793, 738, 685 (m, C–H) cm–1.
4-(6-(Benzyloxy)-3′-nitro-[1,1′-biphenyl]-3-yl)-1H-pyrazol-5-amine (18b)
Prepared as a yellow–brown foam (76 mg, 0.20 mmol, 70%) according to general procedure E. The crude product was purified by flash chromatography (0–5% v/v MeOH in DCM). LCMS (ESI+): m/z 387.0 [M + H]+, retention time 2.07 min (94%). HRMS (ESI+): m/z (calcd C22H19N4O3 [M + H]+ = 387.1457), obsd 387.1456 (δ ppm = 0.3). 1H NMR (500 MHz, MeOD) δ 8.51 (app t, J = 2.0, 2.0 Hz, 1H), 8.18 (ddd, J = 8.3, 2.0, 1.0 Hz, 1H), 7.97 (ddd, J = 8.0, 1.5, 1.0 Hz, 1H), 7.63 (app t, J = 8.0, 7.0 Hz, 1H, 16-CH), 7.52–7.48 (m, 2H), 7.35 (m, 2H), 7.30–7.26 (m, 2H), 7.24 (d, J = 8.4 Hz, 1H), 5.15 (s, 2H) ppm. One aromatic 1H unresolved. 13C NMR (125 MHz, MeOD) δ 155.0, 152.8, 149.3, 141.6, 138.4, 136.9, 130.3, 130.2, 129.9, 129.5 (2C), 129.1, 128.48, 128.4, 128.3, 125.6, 122.6, 115.1, 71.8 ppm. IR (thin film vmax 3415, 3325, 3193 (br, m, N–H), 3033, 2941, 2880 (w, C–H), 1613, 1578 (w, C=C), 1525 (s, NO2), 1525 (m), 1380 (s, NO2), 1287, 1260 (m, C–N, C–O), 1055, 1021 (w, C–O), 890 (w), 811, 735, 688 (m) cm–1.
4-(6-Methoxy-3′-nitro-[1,1′-biphenyl]-3-yl)-1H-pyrazol-5-amine (18c)
Prepared as a yellow amorphous solid (165 mg, 0.53 mmol, 53%) according to general procedure E, except that the reactions was carried out at 120 °C using conventional heating over 14 h. The crude product was purified by flash chromatography (0–5% v/v MeOH in DCM), Rf 0.21 (10% v/v MeOH in DCM). LCMS (ESI+) m/z 311.2 [M + H]+, retention time 1.83 min (100%). HRMS (ESI+): m/z (calcd C16H15N4O3 [M + H]+ = 311.1144), obsd 311.1139 (δ ppm = 1.6). 1H NMR (500 MHz, MeOD) δ 8.41 (d, J = 2.2 Hz, 1H), 8.17 (d, J = 8.2 Hz, 1H), 7.93 (d, J = 7.9 Hz, 1H), 7.68–7.55 (m, 2H), 7.51 (d, J = 8.6 Hz, 1H), 7.47 (s, 1H), 7.14 (d, J = 8.5 Hz, 1H), 3.84 (s, 3H) ppm. 13C NMR (125 MHz, MeOD) δ 156.0, 152.8, 149.4, 141.7, 136.9, 130.2, 129.9, 129.7, 129.2, 128.8, 127.7, 125.3, 122.6, 113.2, 109.2, 56.2 ppm. 13C NMR processed with LB = 3.0 Hz. IR (thin film) vmax 3421, 3182 (m, br, N–H), 2952 (m, C–H), 1613, 1577 (w, N–H, C=C), 1525 (s, NO2), 1348 (s, NO2), 1260 (m, C–O, C–N), 1181 (w, C–H), 1022 (m, C–O), 814, 747, 688 (m, C–H) cm–1.
4-(4′-Nitro-[1,1′-biphenyl]-3-yl)-1H-pyrazol-5-amine (18d)
Prepared according to general procedure E and crudely purified by elution through a short silica plug (0–5% v/v MeOH in DCM) to remove inorganic byproducts. The product mixture was then carried forward with further purification and characterization performed after reduction of the aromatic nitro group. LCMS (ESI+): m/z 281.4 [M + H]+, retention time 3.89 min (52%).
4-(3-(Pyridin-3-yl)phenyl)-1H-pyrazol-5-amine (18e)
Prepared according to general procedure E. The crude product was purified by flash chromatography (0–5% v/v MeOH in DCM) to yield 18e (0.030 g, 0.13 mmol, 61%). LCMS (ESI+): m/z 237.3 [M + H]+, retention time 1.12 min (100%). 1H NMR (400 MHz, CDCl3): δ 8.84 (dd, J = 2.4, 0.8 Hz, 1H), 8.58 (dd, J = 4.9, 1.4 Hz, 1H), 7.86 (ddd, J = 7.9, 2.3, 1.7 Hz, 1H), 7.64 (m, 1H), 7.54 (s, 1H), 7.46 (m, 2H), 7.42 (m, 1H), 7.35 (ddd, J = 8.0, 4.8, 0.8 Hz, 1H), 5.65 (br s, 3H) ppm. 13C NMR (100 MHz, CDCl3): δ 150.8, 148.5, 148.2, 138.5, 136.6, 134.6, 134.0, 129.7, 129.1, 126.4, 125.5, 124.8, 123.7, 108.2 ppm. IR (neat) vmax 3365 (w, N–H), 3149 (w, br, N–H), 2932 (w, br, C–H), 1608 (m), 1506 (w), 1463 (m), 1400, 1380 (w), 1188, 1123, 1059, 1044, 1027 (w), 843 (m), 774, 708, 695 (s, C–H) cm–1.
4-(3-(Pyridin-4-yl)phenyl)-1H-pyrazol-5-amine (18f)
Prepared according to general procedure E. The crude product was purified by flash chromatography (0–5% v/v MeOH in DCM) to yield 18f (0.014 g, 0.060 mmol, 29%). LCMS (ESI+): m/z 237.2 [M + H]+ (calcd for C14H12N4, 237.1), retention time 1.04 min (96%). 1H NMR (500 MHz, MeOD): δ 8.58 (d, J = 1.6 Hz, 1H), 8.57 (d, J = 1.6 Hz, 1H), 7.87 (t, J = 1.6 Hz, 1H), 7.77 (d, J = 1.6 Hz, 1H), 7.76 (d, J = 1.6 Hz, 1H), 7.70 (s, 1H), 7.56–7.61 (m, 2H), 7.51 (t, J = 7.6 Hz, 1H) ppm. 13C NMR (125 MHz, MeOD) δ 151.3, 151.2, 150.3, 139.3, 135.9, 132.0, 128.5, 126.0, 125.3, 123.4, 108.2 ppm. one unresolved quaternary 13C. IR (thin film) vmax 3458, 3159 (m, br, N–H), 291 (m, C–H), 1618 (m, C=C), 1594 (s, N–H, C–H)), 1561 (m), 1508, 1402 (s, C–N), 1334, 1223, 1054, 1023 (m), 789, 701 (s, C–H) cm–1.
4-(3-(1H-Indol-5-yl)phenyl)-1H-pyrazol-5-amine (18g)
Prepared according to general procedure E. The crude product was purified by flash chromatography (0–5% v/v MeOH in DCM) to yield 18g (0.029 g, 0.11 mmol, 50%). LCMS (ESI+): m/z 275.3 [M + H]+, retention time 1.74 min (94%). HRMS (ESI+): m/z (calcd for C17H15N4 [M + H]+ = 275.1297), obsd 275.1297 (δ ppm = 0). 1H NMR (500 MHz, MeOD): δ 7.82 (app s, 1H), 7.75 (app t, 1H), 7.65 (s, 1H), 7.37–7.49 (m, 5H), 7.25 (d, J = 3.1 Hz, 1H), 6.49 (d, J = 3.2 Hz, 1H) ppm. 13C NMR (125 MHz, MeOD) δ 151.2, 144.8, 137.3, 134.8, 133.7, 131.5, 130.1, 130.0, 126.4, 126.3, 125.6, 125.3, 122.0, 119.7, 112.4, 109.2, 102.8 ppm. IR (thin film) vmax 3405, 3181 (m, br, N–H), 2936 (m, C–H), 1602 (s), 1504, 1462, 1415, 1307 (m, C=C, N–H), 1016 (s), 879 (m), 787, 763, 728, 667 (s, C–H) cm–1.
4-(3-(1H-Indol-6-yl)phenyl)-1H-pyrazol-5-amine (18h)
Prepared according to general procedure E. The crude product was purified by flash chromatography (0–5% v/v MeOH in DCM) to yield 18h (0.026 g, 0.095 mmol, 45%). LCMS (ESI+): m/z 275.3 [M + H]+, retention time 1.82 min (100%). HRMS (ESI+): m/z (calcd C17H14N4 [M + H]+ = 275.1297), obsd 275.1288 (δ ppm = 3.3). 1H NMR (500 MHz, MeOD): δ 7.76 (app t, J = 1.8 Hz, 1H), 7.65–7.66 (m, 2H), 7.60 (dd, J = 8.3, 0.6 Hz, 1H), 7.49–7.33 (dt, J = 7.0, 1.8 Hz; 1H), 7.44 (m, 2H), 7.33 (dd, J = 8.2, 1.6 Hz, 1H), 7.25 (d, J = 3.2 Hz, 1H), 6.45 (dd, J = 3.2, 0.9 Hz, 1H) ppm. 13C NMR (125 MHz, MeOD): δ 151.1, 144.5, 138.3, 135.9, 134.8, 131.6, 130.2, 128.9, 126.4, 126.3, 125.7, 125.6, 121.4, 119.8, 110.6, 109.2, 102.2 ppm. IR (thin film) vmax 3384 (s, N–H), 3175 (m, br, N–H), 1602 (s, N–H, C=C), 1567, 1455 (s, C=C), 1350, 1310 (m, C–N), 1227, 1198 (w), 1096, 1055, 1015 (m, C–N), 863 (m), 785, 722 (s, C–H) cm–1.
4-(3-(1H-Pyrazol-4-yl)phenyl)-1H-pyrazol-5-amine (18i)
Prepared according to general procedure E. The crude product was purified by flash chromatography (0–5% v/v MeOH in DCM) to yield 18i (0.012 g, 0.05 mmol, 37%). LCMS (ESI+): m/z 226.2 [M + H]+, retention time 1.30 min (100%). HRMS (ESI+): m/z (calcd for C12H12N5 [M + H]+ = 226.1093), obsd 226.1104 (δ ppm = 4.9). 1H NMR (400 MHz, MeOD): δ 7.99 (s, 2H), 7.69 (s, 1H), 7.65 (s, 1H), 7.42 (dt, J = 7.0, 1.9 Hz, 1H), 7.39–7.30 (m, 2H) ppm. 13C NMR (100 MHz, MeOD): δ 151.1, 135.1, 134.5, 131.6, 130.3, 125.5, 124.6, 123.9, 123.7, 108.9 ppm. One 13C signal unresolved. IR (neat) vmax 3131 (w, br, N–H), 2917 (w, C–H), 2547, 2358, 2247 (m, br), 1607, 1588 (m, N–H, C=C), 1503 (s), 1437, 1373, 1299 (w, C–N), 1034, 903 (m), 860, 787, 694, 667 (s, C–H) cm–1.
3-(4-Methoxyphenyl)-4-(3′-nitro-[1,1′-biphenyl]-3-yl)-1H-pyrazol-5-amine (23a)
Prepared according to general procedure E. The crude product was purified by flash chromatography (0–5% v/v MeOH in DCM) and then triturated with petroleum ether and DCM to yield 23a as a cream-colored solid (434 mg, 1.12 mmol, 37%). LCMS (ESI+) m/z 387.3 [M + H]+, retention time 2.08 min (100%). HRMS (ESI+): m/z (calcd C22H19N4O3 [M + H]+ = 387.1452), obsd 387.1443 (δ ppm = 2.2). 1H NMR (500 MHz, CDCl3) δ 8.30 (app t, J = 2.0, 2.0 Hz, 1H), 8.17 (ddd, J = 8.2, 1.3, 1.0 Hz, 1H), 7.81 (ddd, J = 7.7, 1.6, 1.1 Hz, 1H), 7.57 (app t, J = 8.0, 8.0 Hz, 1H), 7.49 (m, 3H), 7.38 (app dt, J = 6.9, 2.0, 1.7 Hz, 1H), 7.30 (d, J = 8.9 Hz, 2H), 6.89 (d, J = 8.9 Hz, 2H), 3.82 (s, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ 160.0, 153.0, 148.7, 142.6, 141.4, 139.2, 133.9, 133.0, 129.7, 129.3, 128.9, 128.2, 125.2, 122.3, 122.1, 122.0, 114.4, 105.3, 55.3 ppm. 13C NMR processed with LB = 3 Hz. IR (thin film) vmax 3500–3194 (w, br, N–H), 3056, 2970, 2835 (w, C–H), 1605 (m, N–H), 1524, 1506 (s, NO2), 1436 (m, C=C), 1347 (s, NO2, C–N), 1246 (s, C–N, C–O), 1176 (s), 1119 (m), 1029 (m, C–O), 835, 795 (m C–H), 735, 721 (s, C–H) cm–1.
4-(6-(Benzyloxy)-3′-nitro-[1,1′-biphenyl]-3-yl)-3-(4-methoxyphenyl)-1H-pyrazol-5-amine (23b)
Prepared according to general procedure E. The crude product was purified by flash chromatography (0–5% v/v MeOH in DCM) to yield 23b as a brown amorphous solid (459 mg, 0.93 mmol, 46%). LCMS (ESI+) m/z 493.3 [M + H]+, retention time 2.28 min (100%). HRMS (ESI+): m/z (calcd C29H25N4O4 [M + H]+ = 493.1876), obsd 493.1890 (δ ppm = 2.8). 1H NMR (500 MHz, CDCl3) δ 8.29 (app t, J = 2.0, 1.9 Hz, 1H), 8.05 (ddd, J = 8.3, 2.3, 1.0 Hz, 1H), 7.70 (dt, J = 7.8, 1.5, 1.2 Hz, 1H), 7.41 (app t, J = 8.0, 8.0 Hz, 1H), 7.35 (m, 4H), 7.33–7.29 (m, 4H), 7.28 (m, 1H), 7.23 (m, 1H), 7.21 (m, 3H), 7.19 (s, 1H), 7.00 (d, J = 8.5 Hz, 1H), 6.81 (d, J = 8.8 Hz, 1H), 5.13 (s, 2H), 3.83 (s, 3H) ppm. 13C NMR (125 Hz, CDCl3) δ 159.9, 154.2, 153.2, 148.0, 141.1, 139.7, 136.5, 135.6, 131.8, 130.7, 128.9, 128.8, 128.7, 128.6, 128.0, 127.1, 126.0, 124.7, 122.4, 121.8, 114.3, 113.5, 104.9, 70.6, 55.3 ppm. 13C NMR processed with LB = 3 Hz. IR (thin film) vmax 3422, 3337, 3202 (w, br, N–H), 2955, 2932, 2853 (w, C–H), 1614 (m), 1523 (s, NO2), 1348 (s, NO2), 1250 (s), 1178, 1028 (m, C–O), 835, 734, 690, 629 (m) cm–1.
General Procedure F
Reduction of Nitrophenyl Analogues 19a–d, 24a,b
4-(3′-Amino-[1,1′-biphenyl]-3-yl)-1H-pyrazol-5-amine (19a)
Tin(II) chloride dihydrate (564 mg, 2.5 mmol) was added to a solution of 4-(3′-nitro-[1,1′-biphenyl]-3-yl)-1H-pyrazol-5-amine 18a (140 mg, 0.5 mmol) in absolute EtOH (50 mL). The reaction was heated to 85 °C for 1.5–16 h until all starting material had been consumed. The reaction was then concentrated under reduced pressure (∼10 mL) and then diluted with EtOAc (50 mL), brine (50 mL), and saturated sodium potassium tartrate solution (40 mL). The product was extracted into EtOAc (2 × 20 mL), the combined organic fractions were dried over anhydrous Na2SO4, and the solvent removed under reduced pressure. The crude material was purified by flash chromatography (0–10% v/v MeOH in DCM) to give 4-(3′-amino-[1,1′-biphenyl]-3-yl)-1H-pyrazol-5-amine 19a as a yellow amorphous solid (63 mg, 0.25 mmol, 52%), Rf 021 (10% v/v MeOH in DCM). LCMS (ESI+) m/z 251.2 [M + H]+, retention time 1.21 min (100%). HRMS (ESI+) m/z (calcd C15H15N4 [M + H]+ = 251.1291), obsd 251.1284 (δ ppm = 2.9). 1H NMR (500 MHz, MeOD3) δ 7.68 (d, J = 1.5 Hz, 1H), 7.63 (s, 1H), 7.42 (m, 3H), 7.17 (app t, J = 8.0, 7.5 Hz, 1H), 7.02 (app t, J = 2.0, 2.0 Hz, 1H), 6.98 (appdt, J = 7.5, 1.5, 1.0 Hz, 1H), 6.72 (ddd, J = 8.0, 2.3, 1.0 Hz, 1H) ppm. 13C NMR (125 MHz, MeOD) δ 151.3 (br), 149.0, 143.5, 143.4, 135.0, 131.5 (br), 130.5, 130.1, 126.3, 126.1, 125.3, 118.2, 115.7, 115.2, 108.9 ppm. 13C NMR processed with LB = 3 Hz. IR (thin film) vmax 3341, 3206 (m, br, NH2), 2972, 2940 (w, C–H), 1602 (s, N–H), 1506, 1475 (m, C=C, N–H), 1314 (w, C–N), 1213 (w, C–N, C–H), 1165, 1052 (w, C–H), 782, 700 (m, C–H) cm–1.
4-(3′-Amino-6-(benzyloxy)-[1,1′-biphenyl]-3-yl)-1H-pyrazol-5-amine (19b)
Prepared according to general procedure F. The crude product was purified by flash chromatography (0–10% v/v MeOH in DCM) to yield 19b as a cream-colored solid (99 mg, 0.28 mmol, 58%), Rf 0.33 (10% v/v MeOH in DCM). LCMS (ESI+): m/z 357.2 [M + H]+, retention time 1.64 min (100%). HRMS (ESI+): m/z (calcd C22H21N4O [M + H]+ = 357.1715), obsd 357.1724 (δ ppm = 2.5). 1H NMR (500 MHz, acetone-d6) δ 10.99 (br, s, 2H), 7.62 (s, 1H), 7.47 (d, J = 2.4 Hz, 1H), 7.45–7.39 (m, 3H), 7.33 (app t, J = 7.6 Hz, 2H), 7.27 (t, J = 7.3 Hz, 1H), 7.12 (d, J = 8.4 Hz, 1H), 7.08 (app t, J = 7.8 Hz, 1H), 6.95 (app t, J = 2.0 Hz, 1H), 6.86 (d, J = 7.6 Hz, 1H), 6.63 (dd, J = 7.7, 2.2 Hz, 1H), 5.12 (s, 2H), 4.58 (br, s, 2H), 4.32 (br, s, 2H) ppm. 13C NMR (125 MHz, d6-acetone) δ 154.7, 151.3 (br), 149.2, 140.8, 139.1, 133.5, 129.8, 129.6, 129.5, 128.7, 128.6, 128.3, 127.2, 125.4 (br), 119.6, 117.1, 115.2, 114.2, 108.0 (br), 71.5 ppm. 13C NMR spectrum processed with LB = 1–10 Hz. IR (thin film) vmax 3334, 3196 (m, br, N–H), 3033, 2944 (m, C–H), 1602, 1584 (s, C=C), 1498, 1481, 1452 (m, C=C), 1379 (s, C–N), 1280 (s, C–N), 1145 (w), 1021 (m, C–O), 871 (w), 787, 734 (m), 696, 655 (s) cm–1
4-(3′-Amino-6-methoxy-[1,1′-biphenyl]-3-yl)-1H-pyrazol-5-amine (19c)
Prepared according to general procedure F. The crude product was purified by flash chromatography (0–10% v/v MeOH in DCM) to yield 19b as a white amorphous solid (103 mg, 0.38 mmol, 77%), Rf 0.34 (10% v/v MeOH in EtOAc). LCMS (ESI+) m/z 282.3 [M + H]+, retention time 1.30 min (100%). HRMS (ESI+): m/z (calcd C16H17N4O [M + H]+ = 281.1397), obsd 281.1388 (δ ppm = 3.3). 1H NMR (500 MHz, MeOD) δ 7.53 (s, 1H), 7.39 (dd, J = 8.4, 2.3 Hz, 1H), 7.36 (d, J = 2.4 Hz, 1H), 7.12 (app t, J = 7.8 Hz, 1H), 7.06 (d, J = 8.5 Hz, 1H), 6.91 (app t, J = 2.0 Hz, 1H), 6.86 (d appt, J = 7.6, 1.3 Hz, 1H), 6.69 (ddd, J = 7.9, 2.4, 1.0 Hz, 1H), 3.78 (s, 3H) ppm. 13C NMR (125 MHz, MeOD) δ 156.2, 152.7, 148.1, 140.8, 132.9, 130.1, 129.5, 128.6 (br), 127.7, 127.2, 121.0, 118.1, 115.4, 113.2, 109.4, 56.2 ppm. IR (thin film) vmax 3340, 3210 (m, br, N–H), 2953 (m, C–H), 1603, 1584 (s, N–H), 1500 (m, N–H, C=C), 1482 (s, N–H), 1454 (m, C=C), 1282 (m, C–N), 1246 (s, C–N, C–O), 1380 (w, CH3), 1055, 1021 (m, C–O), 873 (w, C–H), 814, 798, 700 (m, C–H) cm–1.
4-(4′-Amino-[1,1′-biphenyl]-3-yl)-1H-pyrazol-5-amine (19d)
Prepared according to general procedure F. The crude product was purified by flash chromatography (0–8% v/v MeOH in DCM) to yield 19d (0.005 g, 0.020 mmol, 10%, over two steps). LCMS (ESI+): m/z 251.2 [M + H]+; retention time 1.30 min (100%). HRMS (ESI+): m/z (calcd C15H15N4 [M + H]+ = 251.1297), obsd 251.1300 (δ ppm = 1.2). 1H NMR (500 MHz, MeOD): δ 7.66–7.59 (m, 2H), 7.42 (d, J 8.6 Hz, 1H), 7.39–7.32 (m, 3H), 6.79 (d, J = 8.5 Hz, 2H) ppm. 13C NMR (125 MHz, MeOD): δ 148.4, 143.2, 134.9, 132.2, 130.1, 128.7, 125.31, 125.28, 124.6, 116.8, 109.1 (br) ppm. Two 13C signals unresolved, 13C NMR processed with LB = 1–12 Hz.
4-(3′-Amino-[1,1′-biphenyl]-3-yl)-3-(4-methoxyphenyl)-1H-pyrazol-5-amine (24a)
Prepared according to general procedure F. The crude product was purified by flash chromatography (0–5% v/v MeOH in DCM) to yield 24a as a brown amorphous solid (65 mg, 0.18 mmol, 22%). Impure fractions (157 mg), which contained approximately 90% product, were also retained for use in subsequent synthetic steps, with easier chromatographic purification achieved after deprotection of the aryl methyl ether; Rf 0.32 (10% v/v MeOH in DCM). LCMS (ESI+) m/z 357.2 [M + H]+, retention time 1.60 min (100%). HRMS (ESI+): m/z (calcd C22H21N4O [M + H]+ = 357.1710), obsd 357.1698 (δ ppm = 3.5). 1H NMR (500 MHz, MeOD) δ 7.43 (m, 2H), 7.37 (dd, J = 7.8, 7.5 Hz, 1H), 7.29 (d, J = 8.8 Hz, 2H), 7.20 (d, J = 7.6 Hz, 1H), 7.10 (app t, J = 7.8 Hz, 1H), 6.87 (m, 3H), 6.81 (d, J = 7.4 Hz, 1H), 6.68 (ddd, J = 7.9, 2.3, 1.0 Hz, 1H), 3.77 (s, 3H) ppm. 13C NMR (125 MHz, MeOD) δ 161.2, 154.4 (br), 149.0, 143.3, 143.2, 142.1 (br), 134.8, 130.5, 130.2, 130.1, 129.4, 129.3, 126.0, 123.8 (br), 118.1, 115.7, 115.1 (2C), 106.7 (br), 55.8 ppm. 13C NMR processed with LB = 3 Hz. IR (thin film) vmax 3341 (br, m, N–H), 3207 (br, m, N–H), 2833 (w, C–H), 1601 (s, N–H, C=C), 1520 (s, C=C, N–H), 1506 (s, C=C, N–H), 1436 (m, C=C), 1290 (w, C–N), 1247 (s, C–O), 1178 (m, C–H), 1113 (m, C–H), 1029 (m, C–O), 972 (m), 834 (m, C–H), 783 (m, C–H), 701 (m, C–H) cm–1.
4-(3′-Amino-6-(benzyloxy)-[1,1′-biphenyl]-3-yl)-3-(4-methoxyphenyl)-1H-pyrazol-5-amine (24b)
Prepared according to general procedure F. The crude product was purified by flash chromatography (0–5% v/v MeOH in DCM) to yield 24b as a brown amorphous solid (370 mg, 0.80 mmol, 65%), Rf 0.16 (10% v/v MeOH in DCM). LCMS (ESI+) m/z 463.3 [M + H]+, retention time 1.95 min (100%). HRMS (ESI+) m/z (calcd C29H27N4O2 [M + H]+ = 463.2134), obsd 463.2141 (δ ppm = 1.5). 1H NMR (500 MHz, MeOD) δ 7.33–7.27 (m, 6H), 7.23 (app t, J = 7.1 Hz, 1H), 7.13 (m, 2H), 7.08–7.04 (m, 2H), 6.89–6.82 (m, 3H), 6.80 (dt, J = 7.7, 1.2 Hz, 1H), 6.66 (ddd, J = 7.9, 2.3, 1.0 Hz, 1H), 5.03 (s, 2H), 3.75 (s, 3H) ppm. 13C NMR (125 MHz, MeOD) δ 161.1, 155.6, 154.4, 148.0, 141.8, 140.6, 138.8, 133.4, 133.2, 130.6, 130.1, 129.5, 129.4, 128.6, 128.2, 127.2, 123.9, 121.1, 118.1, 115.4, 115.3, 115.0, 106.3, 71.7, 55.8 ppm. 13C NMR processed with LB = 3 Hz. IR (thin film) vmax 3349, 3211 (br, w, N–H), 2929, 2833 (br, w, C–H), 1603 (s), 1585, 1507 (s, C=C, C–H), 1453 (s), 1244, 1177 (s, C–N, C–O), 1024 (s, C–O), 833, 735, 696 (s, C–H) cm–1.
General Procedure G
Reductive Amination Procedure (1) for the Synthesis of Ar3 Analogues 26a–e
4,4′-(5-((4-Hydroxybenzyl)amino)-1H-pyrazole-3,4-diyl)diphenol (26a)
4-Hydroxybenzaldehyde (83 mg, 0.68 mmol) and glacial acetic acid (72 μL, 1.4 mmol) were added to a stirred solution of 4,4′-(5-amino-1H-pyrazole-3,4-diyl)diphenol 5 (200 mg, 0.75 mmol) in DMF (0.5 mL) and DCE (7 mL). The reaction was stirred at room temperature for 20–60 min then cooled to 0 °C, and Na(OAc)3BH (360 mg, 1.7 mmol) was added as a single portion. The reaction was allowed to warm to room temperature and stirred for 20 h. For some substrates, an additional equivalent of Na(OAc)3BH was then added and the reaction was stirred for a further 7 h at room temperature until all aldehyde had been consumed. The reaction was then diluted with EtOAc (50 mL) and water (10 mL) and brought to pH 7–8 using saturated NaHCO3 solution. The phases were separated, and the aqueous fraction was extracted with EtOAc (2 × 5 mL). The combined organic fractions were dried over anhydrous Na2SO4, and the solvent was removed under reduced pressure. The crude material was purified by flash chromatography (50–100% EtOAc in petroleum ether) to yield 4,4′-(5-((4-hydroxybenzyl)amino)-1H-pyrazole-3,4-diyl)diphenol 26a as a cream-colored powder (184 mg, 0.49 mmol, 72%), Rf 0.13 (10% v/v MeOH in DCM). LCMS (ESI+): m/z 374.2 [M + H]+, retention time 1.25 min (100%). HRMS (ESI+): m/z (calcd C22H20N3O3 [M + H]+ = 374.1505), obsd 374.1495 (δ ppm = 2.7). 1H NMR (500 MHz, MeOD) δ 7.19–7.11 (m, 4H), 7.01 (d, J = 8.5 Hz, 2H), 6.76 (d, J = 8.5 Hz, 2H), 6.70 (m, 4H), 4.24 (s, 2H) ppm. 13C NMR (125 MHz, MeOD) δ 158.6, 157.5, 157.3, 156.1 (br), 142.3 (br), 132.3 (2 × CH), 129.9, 125.3, 123.1 (br), 116.7, 116.2, 116.1, 105.7 (br), 49.9 ppm. One quaternary 13C signal obscured. 13C NMR processed with LB = 1–9 Hz. IR (thin film) vmax 3270 (s, br, O–H, N–H), 2940 (w, C–H), 1612, 1596 (m, C=C), 1511 (s, N–H), 1441 (m, C=C), 1370 (m), 1234 (s, C–O, C–N), 1171, 1102, 1011 (m, C–O), 834 (s, C–H) cm–1.
4,4′-(5-((3-Bromo-4-hydroxybenzyl)amino)-1H-pyrazole-3,4-diyl)diphenol (26b)
Prepared according to general procedure G. The crude product was purified by flash chromatography (50–100% v/v EtOAc in petroleum ether) to yield 26b as a yellow solid (229 mg, 0.51 mmol, 74%), Rf 0.11 (10% v/v MeOH in DCM). LCMS (ESI+) m/z 454.2 [M + H]+, retention time 1.59 min (92%). HRMS (ESI+): m/z (calcd C22H19BrN3O3 [M + H]+ = 452.0610), obsd 452.0603 (δ ppm = 1.5). 1H NMR (500 MHz, MeOD) δ 7.41 (d, J = 2.1 Hz, 1H), 7.16 (d, J = 8.7 Hz, 2H), 7.10 (dd, J = 1.9, 8.4 Hz, 1H), 7.03 (d, J = 8.7 Hz, 2H), 6.80 (d, J = 8.4 Hz, 1H), 6.78 (d, J = 8.6 Hz, 2H, 6.69 (d, J = 8.7 Hz, 2H), 4.24 (s, 2H) ppm. 13C NMR (125 MHz, MeOD) δ 158.6, 157.3, 155.5, 154.1, 142.6, 134.5, 133.2, 132.3, 129.9, 128.9, 125.2, 123.1, 117.0, 116.7, 116.2, 110.6, 105.8, 48.3 ppm. 13C NMR processed with LB = 12.0 Hz. IR (thin film) vmax) 3230 (br, s, N–H, O–H), 2928 (m, C–H), 1651, 1612, 1590 (m, C=C), 1527, 1494, 1432 (s, N–H), 1383, 1343 (m), 1255, 1233 (s, C–N, C–N), 1171 (m), 1105, 1010 (m, C–O), 835 (s, C–H), 666, 527 (m, C–Br) cm–1.
4,4′-(5-((Thiophen-3-ylmethyl)amino)-1H-pyrazole-3,4-diyl)diphenol (26c)
Prepared according to general procedure G. The crude product was purified by flash chromatography (50–90% v/v EtOAc in petroleum ether) to yield 26c as a white solid (156 mg, 0.43 mmol, 63%), Rf 0.33 (10% v/v MeOH in DCM). LCMS (ESI+) m/z 364.2 [M + H]+, retention time 1.71 min (100%). HRMS (ESI+): m/z (calcd C20H18N3O2S [M + H]+ = 364.1120), obsd 364.1107 (δ ppm = 3.6). 1H NMR (500 MHz, DMSO-d6DMSO-d6) δ 11.71 (s, br, 1H, 1-NH), 9.50 (s, br, 1H), 9.30 (s, br, 1H), 7.45 (dd, J = 4.8, 2.8 Hz, 1H), 7.26 (d, J = 2.0 Hz, 1H), 7.10 (m, 3H), 6.98 (d, J = 8.4 Hz, 2H), 6.72 (d, J = 8.4 Hz, 2H), 6.68 (d, J = 8.4 Hz, 2H), 4.49 (s, br, 1H), 4.27 (d, J = 4.27 Hz, 2H) ppm. 13C NMR (125 MHz, DMSO-d6) δ157.0, 155.6, 154.1, 142.7, 139.1, 130.7, 128.3, 128.0, 125.5, 123.9, 121.3, 115.6, 115.2, 103.5, 42.8 ppm. 13C NMR processed with LB = 3 Hz. IR (thin film) vmax 3306 (s, br, O–H, N–H), 3101, 2928, 2855 (w, C–H), 1613, 1591 (m, C=C), 1529, 1512 (s, C–N, C–O), 1436 (C=C),1375 (m), 1262, 1239 (s, C–N, C–O), 1172 (m, C–O), 836 (s, C–H) cm–1.
4,4′-(5-((Furan-2-ylmethyl)amino)-1H-pyrazole-3,4-diyl)diphenol (26d)
Prepared according to general procedure G, however no AcOH was added to the reaction and no NaHCO3 was used during reaction workup. The crude product was purified by flash chromatography (50–90% v/v EtOAc in petroleum ether) to yield 26d as a yellow amorphous solid (82 mg, 0.24 mmol, 38%), Rf 0.54 (10% v/v MeOH in EtOAc). LCMS (ESI+) m/z 348.2 [M + H]+, retention time 1.54 min (100%). HRMS (ESI+): m/z (calcd C20H18N3O3 [M + H]+ = 348.1348), obsd 348.1356 (δ ppm =2.3). 1H NMR (500 MHz, MeOD) δ 7.38 (d, J = 1.8 Hz, 1H), 7.16 (d, J = 8.8 Hz, 2H), 7.01 (d, J = 8.6 Hz, 2H), 6.78 (d, J = 8.7 Hz, 2H), 6.70 (d, J = 8.8 Hz, 2H), 6.30 (m, 1H), 6.18 (d, J = 3.2 Hz, 1H), 4.33 (s, 2H) ppm. 13C NMR (125 MHz, MeOD) δ 158.6, 157.3, 155.6 (br), 154.9, 142.8, 142.2 (br), 132.3, 129.9, 125.1, 122.9 (br), 116.7, 116.2, 111.2, 107.6, 106.0 (br), 42.5 ppm, 13C NMR processed with LB = 6 Hz. IR (thin film) vmax 3290 (s, br, N–H, O–H), 2935, 2816 (w, C–H), 1614 (m, C=C), 1529, 1500 (s, N–H), 1434 (m), 1378, 1343 (w), 1261, 1238 (s, C–N, C–O), 1172 (m, C–O), 1012 (w), 836 (s, C–H), 739 (m, furan, C–H) cm–1.
4,4′-(5-(((1H-Indol-5-yl)methyl)amino)-1H-pyrazole-3,4-diyl)diphenol (26e)
Prepared according to general procedure G. The product was purified by two sequential flash chromatography columns, eluting with 0–5% v/v MeOH in DCM, followed by 50–100% v/v EtOAc in petroleum ether, to yield 26e as a brown solid (52 mg, 0.13 mmol, 52%), Rf 0.14 (10% v/v MeOH in DCM). LCMS (ESI+) m/z 397.3 [M + H]+, retention time 1.53 min (100%). HRMS (ESI+): m/z (calcd C24H21N4O2 [M + H]+ = 397.1665), obsd 397.1653 (δ ppm = 3.0). 1H NMR (500 MHz, MeOD) δ 7.50 (d, J = 0.8 Hz, 1H), 7.31 (d, J = 8.3 Hz, 1H), 7.19 (d, J = 3.2 Hz, 1H), 7.17 (d, J = 8.7 Hz, 2H), 7.09 (dd, J = 8.4, 1.7 Hz, 1H), 7.01 (d, J = 8.5 Hz, 2H), 6.75 (d, J = 8.6 Hz, 2H), 6.70 (d, J = 8.7 Hz, 2H), 4.41 (s, 2H) ppm. 13C NMR (125 MHz, MeOD) δ 158.6, 157.3, 156.4 (br), 142.1 (br), 137.0, 132.3, 131.7, 130.0, 129.5, 125.9, 125.3, 123.0 (br), 122.6, 120.4, 116.7, 116.2, 112.1, 105.7 (br), 102.3, 50.5 ppm. 13C NMR processed with LB = 6 Hz. IR (thin film) vmax 3406 (s, br, N–H, O–H), 2927 (w, C–H), 1613, 1594 (m, C=C), 1529, 1511 (s) 1451, 1435, 1349 (m), 1262, 1240 (s, C–O, C–N), 1172 (m, C–O), 837 (s, C–H), 731 (m) cm–1.
General Procedure H
Reduction Amination Procedure (2) For the Synthesis of Ar3 Analogues 27a–b and 26f
3,4-Bis(4-methoxyphenyl)-N-(pyridin-4-ylmethyl)-1H-pyrazol-5-amine (27a)
Glacial AcOH (1 mL) was added to a suspension of 3,4-bis(4-methoxyphenyl)-1H-pyrazol-5-amine 13a (222 mg, 0.75 mmol) and pyridine-4-carbaldehyde (71 uL, 0.75 mmol) in dry MeOH (7.5 mL). The reaction was stirred at room temperature for 30–90 min, and then NaCNBH3 (47 mg, 0.75 mmol) was added as a single portion. The reaction was stirred at room temperature for 14–24 h and then concentrated under reduced pressure. The crude product was redissolved in EtOAc (40 mL) and washed with water (10 mL) that had been made basic with saturated NaHCO3 solution. The aqueous phase was then extracted with EtOAc (2 × 10 mL), and the combined organic fractions were washed with brine (5 mL). The solvent was removed under reduced pressure to dry the product directly onto silica gel. Flash chromatography (0–5% v/v MeOH in EtOAc) yielded 3,4-bis(4-methoxyphenyl)-N-(pyridin-4-ylmethyl)-1H-pyrazol-5-amine 27a as a yellow amorphous solid (226 mg, 0.59 mmol, 78%), Rf 0.13 (5% v/v MeOH in EtOAc). LCMS (ESI+) m/z 386.6 [M + H]+, retention time 1.49 min (100%). HRMS (ESI+) m/z (calcd C23H23N4O2 [M + H]+ = 387.1821), obsd 387.1819 (δppm = 0.5). 1H NMR (500 MHz, CDCl3) δ 8.53 (d, J = 5.5 Hz, 2H), 7.30 (d, J = 5.7 Hz, 2H), 7.22 (m, 4H), 6.91 (d, J = 8.9 Hz, 2H), 6.83 (d, J = 8.9 Hz, 2H), 4.49 (s, 2H), 3.82 (s, 3H), 3.78 (s, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ 159.8, 158.6, 154.8, 149.9, 140.8, 131.0, 128.7, 124.7, 122.5, 122.4, 114.6, 114.3, 104.7, 55.4 (2 ×C), 47.1 ppm. One quaternary 13C signal unresolved. 13C NMR processed with LB = 1 Hz. IR (thin film) vmax 3414 (br, w, N–H), 3001, 2934, 2835 (w, C–H), 1604 (w), 1525, 1511 (s), 1461, 1361 (m), 1285 (s, C–N), 1245, 1032 (m, C–O), 834 (s), 801 (m) cm–1.
N-((1H-Imidazol-4-yl)methyl)-3,4-bis(4-methoxyphenyl)-1H-pyrazol-5-amine (27b)
Prepared according to general procedure H. The crude product was purified by flash chromatography (0–20% v/v MeOH in DCM) to yield 27b as a white amorphous solid (484 mg, 1.29 mmol, 95%). LCMS (ESI+) m/z 376.3 [M + H]+, retention time 1.61 min (100%). HRMS (ESI+): m/z (calcd C21H22N5O2 [M + H]+ = 376.1773), obsd 376.1766 (δ ppm = 2.1). 1H NMR (500 MHz, MeOD) δ 7.75 (br, d, J = 1.5 Hz, 1H), 7.24 (d, J = 8.8 Hz, 2H), 7.12 (d, J = 8.8 Hz, 2H), 7.00 (br, d, J = 0.9 Hz, 1H), 6.89 (d, J = 8.7 Hz, 2H), 6.84 (d, J = 8.8 Hz, 2H), 4.34 (s, 2H), 3.78 (s, 3H), 3.77 (s, 3H) ppm. 13C NMR (125 MHz, MeOD) δ 161.0, 159.9, 155.2, 142.8, 137.2, 136.2, 132.2, 130.0, 126.3, 124.2, 118.2, 115.3, 114.9, 105.6, 55.7, 55.7, 42.0 ppm. 13C NMR processed with LB = 3 Hz. IR (thin film) vmax 3145 (w, br, N–H), 2902, 2835 (w, C–H), 1612, 1590 (w, C=C), 1526, 1511 (s, N–H), 1462 (m, C=C), 1246 (s, C–O, C–N), 1177, 1031 (m, C–O), 834 (m, C–H) cm–1.
4,4′-(5-(((1H-Indazol-4-yl)methyl)amino)-1H-pyrazole-3,4-diyl)diphenol (26f)
Prepared according to general procedure H except that 4,4′-(5-amino-1H-pyrazole-3,4-diyl)diphenol 5 was used as the amine component. The crude product was purified by flash chromatography (80–100% v/v EtOAc in petroleum ether, followed by 0–5% v/v MeOH in EtOAc) to yield 26f as a white solid (93 mg, 0.24 mmol, 47%), Rf 0.45 (10% v/v MeOH in EtOAc). LCMS (ESI+) m/z 398.1 [M + H]+, retention time 1.48 min (100%). HRMS (ESI+): m/z (calcd C23H20N5O2 [M + H]+ = 398.1617), obsd 398.1612 (δ ppm = 1.3). 1H NMR (500 MHz, MeOD) δ 8.11 (d, J = 1.0 Hz, 1H), 7.41 (d, J = 8.4 Hz, 1H), 7.30 (dd, J = 8.4, 6.9 Hz, 1H), 7.16 (d, J = 8.7 Hz, 2H), 7.09 (dd, J = 6.9, 1.0 Hz, 1H), 7.01 (d, J = 8.7 Hz, 2H), 6.75 (d, J = 8.6 Hz, 2H), 6.70 (d, J = 8.7 Hz, 2H), 4.72 (s, 2H) ppm. 13C NMR (125 MHz, MeOD) δ 158.7, 157.3, 155.7 (br), 142.8 (br), 141.9, 134.9, 133.7, 132.3, 129.9, 127.9, 125.2, 123.1, 123.0 (br), 116.7, 116.2, 109.9, 105.7, 47.6 (br) ppm. 13C NMR processed with LB = 1–12 Hz. IR (solid) vmax 3432, 3443 (m, N–H, O–H), 2986, 2797, m, C–H), 1612, 1589 (m, C=C), 1528, 1509 (s, C=C, N–H), 1450, 1383, 1262, 1240 (s, C–O, C–N), 1173, 1086, 1003 (m, C–O, C–N), 949 (m), 821 (s, C–H), 777, 739 (m, C–H) cm–1.
Procedures for the Synthesis of Pyrazol-5-yl-benzamide Analogues 28a–c
N-(3,4-Bis(4-methoxyphenyl)-1H-pyrazol-5-yl)benzamide (28a)
Benzoyl chloride (31 μL, 0.263 mmol) and triethylamine (42 μL, 0.3 mmol) were added sequentially to a stirred solution of 3,4-bis(4-methoxyphenyl)-1H-pyrazol-5-amine 13a in anhydrous DCM (1.25 mL) at 0 °C. The reaction was stirred at 0 °C for 5 min and then allowed to warm to room temperature and stirred for 3.5 h. The reaction was then diluted with DCM (10 mL), washed with water (3 mL) and brine (3 mL), and the solvent removed under reduced pressure. The crude product was purified by flash chromatography (0–5% v/v MeOH in DCM) to yield 28a as a white solid (31 mg, 0.08 mmol, 31%), Rf 0.16 (5% v/v MeOH in DCM). LCMS (ESI+) m/z 400.2 [M + H]+, retention time 2.03 min (100%). HRMS (ESI+): m/z (calcd C24H22N3O3 [M + H]+ = 400.1661), obsd 400.1662 (δ ppm = 0.2). 1H NMR (500 MHz, DMSO-d6) δ 13.00 (s, 1H), 9.95 (s, 1H), 7.88 (d, J = 6.3 Hz, 2H), 7.56 (app t, J = 7.5, 6.9 Hz, 1H), 7.47 (appt, J = 7.5, 6.9 Hz, 2H), 7.29 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.4 Hz, 2H), 6.96 (d, J = 8.4 Hz, 2H), 6.84 (d, J = 8.4 Hz, 2H), 3.76 (s, 3H), 3.69 (s, 3H) ppm. 13C NMR (125 MHz, DMSO-d6) δ 166.8, 159.1, 157.9, 144.2, 139.3, 134.0, 131.6, 130.3, 128.6, 128.4, 127.5, 124.6, 122.1, 114.2, 113.8, 113.7 (partially obscured), 55.2, 54.9 ppm. 13C NMR processed with LB = 3 Hz. IR (thin film) vmax 3216 (w, br, N–H), 3004, 2932, 2836 (w, C–H), 1657 (m, C=O), 1602, 1579 (m, C=C), 1509 (s, N–H), 1463 (m, C=C), 1247 (s, C–N), 1178, 1027 (m, C–O), 835 (m, C–H), 7–9 (m) cm–1.
N-(3,4-Bis(4-methoxyphenyl)-1H-pyrazol-5-yl)-4-methoxybenzamide (28b)
4-Methoxybenzoyl chloride (90 mg, 0.525 mmol) and triethylamine (84 μL, 0.6 mmol) were added sequentially to a stirred solution of 3,4-bis(4-methoxyphenyl)-1H-pyrazol-5-amine 13a in anhydrous DCM (2.5 mL) at 0 °C. The reaction was stirred at 0 °C for 5 min and then allowed to warm to room temperature and stirred for 5 h. The reaction was then diluted with DCM (10 mL), washed with water (5 mL) and brine (5 mL), dried over anhydrous MgSO4, and the solvent removed under reduced pressure. The crude product was purified by flash chromatography (80–100% v/v EtOAc in petroleum ether) to yield 28b as an orange solid (58 mg, 0.13 mmol, 27%), Rf 0.49 (5% v/v MeOH in EtOAc). LCMS (ESI+) m/z 430.3 [M + H]+, retention time 2.02 min (100%). HRMS (ESI+) m/z (calcd C25H24N3O4 [M + H]+ = 430.1767), obsd 430.1778 (δ ppm = 2.6). 1H NMR (500 MHz, DMSO-d6) δ 12.98 (s, 1H), 9.80 (s, 1H), 7.88 (d, J = 8.3 Hz, 2H), 7.29 (d, J = 8.6 Hz, 2H), 7.13 (d, J = 8.8 Hz, 2H), 7.01 (d, J = 8.4 Hz, 2H), 6.95 (d, J = 7.1 Hz, 2H), 6.83 (d, J = 8.2 Hz, 2H), 3.81 (s, 3H), 3.76 (s, 3H), 3.69 (s, 3H) ppm. 13C NMR (125 MHz, DMSO-d6) δ 166.3, 161.9, 159.1, 157.9, 144.5, 139.3, 130.4, 129.6, 128.7, 126.2, 124.7, 122.2, 114.2, 113.8, 113.7, 55.5, 55.2, 55.0 ppm. One quaternary 13C peak obscured. 13C NMR processed with LB = 3 Hz. IR (solid) vmax 3294, 3154 (w, br, N–H), 2938, 2837 (w, C–H), 1738, 1669, 1644, 1605 (w, C=C), 1501 (m, N–H, C–H), 1241, 1177 (s, C–N, C–O), 1019 (m), 961 (w), 834 (s, C–H), 759, 693 (w, C–H) cm–1
N-(3,4-Bis(4-methoxyphenyl)-1H-pyrazol-5-yl)isonicotinamide (28c)
Isonicotinic acid (38 mg, 0.31 mmol) was suspended in anhydrous DCM (2 mL). The solution was cooled to 0 °C, and oxalyl chloride (29 μL, 0.34 mmol) was added along with a catalytic amount of DMF. The reaction was allowed to come to room temperature and stirred for 2 h. The volatiles were removed under reduced pressure, and the resulting residue was resuspended in anhydrous DCM (1.25 mL). The solution was cooled to 0 °C and the aminopyrazole 5 (74 mg, 0.25 mmol) was added, followed by Et3N (122 μL, 0.875 mmol). The reaction was stirred at 0 °C for 1 h, then allowed to warm to room temperature and stirred overnight or until all of the aminopyrazole had been consumed. The reaction was then diluted with DCM (20 mL), washed with water (5 mL), then brine (3 mL), dried over anhydrous MgSO4, and the solvent evaporated under reduced pressure. The crude product was purified by silica gel chromatography (0–10% MeOH in DCM) to yield 30 as a white solid (34 mg, 0.086 mmol, 34%), Rf 0.20 (10% v/v MeOH in DCM). LCMS (ESI+): m/z 401.2 [M + H]+, retention time 1.76 min (98%). HRMS (ESI+): m/z (calcd C23H21N4O3 [M + H]+ = 401.1614), obsd 401.1617 (δ ppm = 0.7). 1H NMR (500 MHz, CDCl3) δ 8.76 (d, J = 4.8 Hz, 2H), 8.23 (s, 1H), 7.59 (d, J = 4.4 Hz, 2H), 7.36 (d, J = 8.2 Hz, 2H), 7.20 (d, J = 8.1 Hz, 2H), 6.95 (d, J = 8.2 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 3.84 (s, 3H), 3.79 (s, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ 163.7, 159.7, 159.2, 151.0, 145.9 (br), 140.2, 138.6 (br), 131.0, 129.1, 124.2 (br), 123.7, 120.9, 115.0, 114.1, 108.0 (br), 55.4, 55.4 ppm. 13C NMR processed with LB = 6 Hz. IR (thin film) vmax 3219 (w, br, N–H), 2967, 2836, 2371 (w, C–H) 1663 (m), 1613 (w), 1525, 1508, 1297 (m, C=C), 1248 (s, C–N), 1025 (w, C–O), 835 (m) cm–1.
General Procedure I
Deprotection of Aryl-methyl Ethers to Synthesize Analogues 5, 14b–c, 25a–b, 26g–h, and 29a–c
4,4′-(5-Amino-1H-pyrazole-3,4-diyl)diphenol (5)
A solution of 1 M BBr3 in DCM (21 mL, 21 mmol) was added dropwise over a period of 30 min to a stirred solution of 3,4-bis(4-methoxyphenyl)-1H-pyrazol-5-amine 13a (886 mg, 3.0 mmol) in anhydrous DCM (75 mL) at 0 °C. The reaction was stirred at 0 °C for 3.5–24 h, or until all starting material had been consumed. The reaction was quenched with saturated NaHCO3 at 0 °C, and the resulting precipitate was extracted into EtOAc (50 mL). The organic phase was collected, and the aqueous phase was re-extracted with EtOAc (3 × 25 mL). The organic fractions were combined, washed with brine (5 mL), dried over anhydrous MgSO4 or Na2SO4, and the solvent removed under reduced pressure. The crude product was purified by flash chromatography (0–10% v/v MeOH in DCM) to yield 4,4′-(5-amino-1H-pyrazole-3,4-diyl)diphenol 5 as a pale-brown solid (795 mg, 2.97 mmol, 99%), Rf 0.27 (10% v/v MeOH in EtOAc). LCMS (ESI+): m/z 269.2 [M + H]+; retention time 1.19 min (100%). HRMS (ESI+): m/z (calcd C15H14N3O2 [M + H]+ = 268.1086), obsd 268.1075 (δ ppm = 4.1). 1H NMR (500 MHz, DMSO-d6) δ 11.64 (br, s, 1H), 9.52 (br, s, 1H), 9.30 (s, 1H), 7.10 (d, J = 8.6 Hz, 2H), 6.97 (d, J = 8.2 Hz, 2H), 6.71 (d, J = 8.4 Hz, 2H), 6.67 (d, J = 8.4 Hz, 2H) ppm. 13C NMR (125 MHz, DMSO-d6) δ 156.8, 155.4, 153.1, 139.0, 130.5, 128.3, 124.2, 121.9, 115.5, 115.2, 103.8 ppm. 13C NMR processed with LB = 12 Hz. IR (thin film) vmax 3500–2700 (br, O–H, N–H), 2797, 2670, 2591 (w), 1613 (m), 1513 (s), 1488, 1435 (m), 1374 (w), 1261, 1234 (s), 1172 (m), 1101 (w), 1028, 969 (w), 834 (s), 756 (w) cm–1.
3-(5-Amino-4-(4-hydroxyphenyl)-1H-pyrazol-3-yl)phenol (14b)
Prepared according to general procedure I as a pale-brown foam in quantitative yield (157 mg, 0.5 mmol) and used without further purification. LCMS (ESI+) m/z 268.2 [M + H]+, retention time 1.25 min (95%). HRMS (ESI+) m/z (calcd C15H14N3O2 [M + H]+ = 268.1086), obsd 268.1075 (δ ppm = 4.1). 1H NMR (500 MHz, MeOD) δ 7.11 (app t, J = 7.9, 7.9 Hz, 1H), 7.07 (d, J = 8.6 Hz, 2H), 6.82 (ddd, J = 8.7, 1.2, 1.0 Hz, 1H), 6.79 (m, 3H), 6.71 (ddd, J = 8.2, 2.6, 1.0 Hz, 1H) ppm. 13C NMR (125 MHz, MeOD) δ 158.5, 157.3, 153.0, 143.3, 133.3, 132.0, 130.5, 125.0, 119.9, 116.7, 116.1, 115.5, 106.6 ppm. 13C NMR spectrum processed with LB = 3 Hz. IR (thin film) vmax 3396–3290 (w, br, O–H, N–H), 2923, 2853 (s, C–H), 1714, 1606, 1579 (w), 1455 (m), 1377 (w), 1260 (m, C–N), 1097, 1030 (m, C–O), 874 (w), 802 (m) cm–1.
4-(5-Amino-3-(3-fluorophenyl)-1H-pyrazol-4-yl)phenol (14c)
Prepared according to general procedure I, except that only 3.5 equiv of 1.0 M BBr3 in DCM were used. The crude product was purified by flash chromatography (0–10% v/v MeOH in DCM) to yield 14c as a cream-colored amorphous solid (255 mg, 0.95 mmol, 95%), Rf 0.22 (10% v/v MeOH in DCM). LCMS (ESI+) m/z 270.2 [M + H]+, retention time 1.56 min (100%). HRMS (ESI+): m/z (calcd C15H13FN3O [M + H]+ = 270.1037), obsd 270.1024 (δ ppm = 1.9). 1H NMR (500 MHz, MeOD) δ 7.28 (m, 1H), 7.15 (d, J = 7.7 Hz, 1H), 7.06 (m, 3H), 6.98 (app t, J = 10.0 Hz, 1H), 6.81 (d, J = 8.6 Hz, 2H) ppm. 13C NMR (125 MHz, MeOD) δ 164.1 (JC–F = 242.8 Hz), 157.6, 154.8 (br), 140.1 (br), 133.8, 132.5, 131.3, 124.6, 124.3, 116.8, 115.6, 115.0 (JC–F = 22.5 Hz), 107.7 ppm. 13C NMR processed with LB = 12 Hz. IR (thin film) vmax 3300 (br, m, O–H, N–H), 2962, 2936, 2856, 2794 (w, C–H), 1614, 1578, 1524 (s, C=C), 1488, 1445 (m), 1265, 1236, 1215 (s, C–N), 1171 (m, C–O)), 867, 838, 788, 686 (m) cm–1.
4-(5-Amino-4-(3′-amino-[1,1′-biphenyl]-3-yl)-1H-pyrazol-3-yl)phenol (25a)
Prepared according to general procedure I, except that only 3.5 equiv of 1.0 M BBr3 in DCM were used. The crude product was purified by flash chromatography (2–10% v/v MeOH in DCM) to yield 25a as a brown amorphous solid (30 mg, 0.089 mmol, 50%), Rf 0.05 (10% v/v MeOH in DCM). LCMS (ESI+) m/z 343.3 [M + H]+, retention time 1.37 min (100%). HRMS (ESI+): m/z (calcd C21H19N4O [M + H]+ = 343.1553), obsd 343.1541 (δ ppm = 3.6). 1H NMR (500 MHz, MeOD) 7.46–7.40 (m, 2H), 7.38 (app t, J = 7.6 Hz, 1H), 7.22 (d, J = 8.0 Hz, 1H), 7.20 (d, J = 8.6 Hz, 2H), 7.11 (app t, J = 7.7 Hz, 1H), 6.86 (s, 1H), 6.83 (d, J = 7.7 Hz, 1H), 6.75 (d, J = 8.6 Hz, 2H), 6.68 (dd, J = 8.0, 2.2 Hz, 1H) ppm. 13C NMR (125 MHz, MeOD) δ 158.9, 154.2 (br), 149.0, 143.2, 142.4 (br), 134.9, 130.4, 130.3, 130.1, 129.8, 129.43, 129.35, 125.8, 122.7 (br), 118.1, 116.4, 115.7, 115.2, 106.4 ppm. 13C NMR processed with LB = 6 Hz. IR (thin film) vmax 3341, 3206 (br, m, N–H, O–H), 2984 (m, C–H), 1604 (s, N–H, C=C), 1522, 1505 (C=C), 1470 (m, C=C), 1266, 1242 (C–O), 1173 (w, C–H), 837 (m, C–H), 786 (m, C–H), 701 (m, C–H) cm–1.
3′-Amino-5-(5-amino-3-(4-hydroxyphenyl)-1H-pyrazol-4-yl)-[1,1′-biphenyl]-2-ol (25b)
Prepared according to general procedure I. The crude product was purified by flash chromatography (0–10% v/v MeOH in DCM) to yield 25b as a white solid (25 mg, 0.07 mmol, 46%), Rf 0.07 (10% v/v MeOH in DCM). LCMS (ESI+) m/z 359.2 [M + H]+, retention time 1.21 min (100%). HRMS (ESI+) m/z (calcd C21H19N4O2 [M + H]+ = 359.1508), obsd 359.1505 (δ ppm = 0.8). 1H NMR (500 MHz, MeOD) δ 7.21 (d, J = 8.7 Hz, 2H), 7.12–7.06 (m, 2H), 7.04 (dd, J = 8.2, 2.2 Hz, 1H), 6.91–6.85 (m, 2H), 6.82 (d. app t, J = 7.6, 1.4 Hz, 1H), 6.75 (d, J = 8.7 Hz, 2H), 6.66 (ddd, J = 7.9, 2.3, 1.0 Hz, 1H) ppm. 13C NMR (125 MHz, MeOD) δ 158.7, 154.3 (br), 154.0, 148.1, 142.1 (br), 140.8, 133.2, 130.6, 130.1, 129.7, 125.7, 123.6 (br), 120.7, 117.9, 117.4, 116.3, 115.3, 106.0 (br) ppm. One quaternary 13C signal obscured by peaks 130–129 ppm. 13C NMR processed with LB = 3 Hz. IR (thin film) vmax 3374–3200 (br, s, O–H, N–H), 1612, 1510 (s, C=C), 1442, 1399 (m, C–N), 1268 (s, C–N), 1174 (m, C–O), 1111, 1032 (w, C–O), 836 (s), 700 (w) cm–1.
4,4′-(5-((Pyridin-4-ylmethyl)amino)-1H-pyrazole-3,4-diyl)diphenol (26g)
Prepared according to general procedure I, except that the reaction was allowed to warm to room temperature and stirred for 2 h before being quenched with saturated NaHCO3 solution. The crude product was purified by flash chromatography (0–10% v/v MeOH in DCM) to yield 26g as a white solid (51 mg, 0.14 mmol, 61%), Rf 0.10 (10% v/v MeOH in DCM). LCMS (ESI+) m/z 359.2 [M + H]+, retention time 1.65 min (100%). HRMS (ESI+) m/z (calcd C21H19N4O2 [M + H]+ = 359.1508), obsd 359.1508 (δppm = 0.0). 1H NMR (500 MHz, DMSO-d6) δ 11.63 (s, 1H), 9.54 (s, br, 1H), 9.32 (s, br, 1H), 8.44 (d, J = 6.0 Hz, 2H), 7.32 (d, J = 5.6 Hz, 2H), 7.08 (d, J = 8.4 Hz, 2H), 7.03 (d, J = 8.4 Hz, 2H), 6.75 (d, J = 8.4 Hz, 2H), 6.67 (d, J = 8.4 Hz, 2H), 5.21 (t, J = 5.6 Hz, 1H), 4.31 (d, J = 6.0 Hz, 2H) ppm. 13C NMR (125 MHz, DMSO-d6) δ 155.7, 155.8, 153.9, 151.3, 149.2, 139.2, 130.8, 128.4, 123.8, 122.5, 121.4, 115.6, 115.3, 103.4, 46.1 ppm. 13C NMR processed with LB = 2.0 Hz. IR (solid) vmax 3629, 2414, 3210 (w, N–H, O–H), 2905, 2585 (w, C–H), 1602, 1575 (m, C=C), 1537, 1513, 1479 (s, N–H, C–H), 1437, 1418 (m), 1385 (w, C–O), 1246 (s, C–O, C–N), 1157 (m, C–O), 1026, 1006 (m), 835 (s, C–H) 799, 661 (m) cm–1.
4,4′-(5-(((1H-Imidazol-4-yl)methyl)amino)-1H-pyrazole-3,4-diyl)diphenol (26h)
Prepared according to general procedure I. The crude product was purified by flash chromatography (1–20% v/v MeOH in DCM) to yield 26h as a white solid (118 mg, 0.34 mmol, 68%). LCMS (ESI+) m/z 348.2 [M + H]+, retention time 1.26 min (100%). HRMS (ESI+): m/z (calcd C19H18N5O2 [M + H]+ = 348.1461), obsd 348.1468 (δ ppm = 2.0). 1H NMR (500 MHz, MeOD) δ 7.60 (d, J = 1.2 Hz, 1H), 7.16 (d, J = 8.7 Hz, 2H), 7.02 (d, J = 8.6 Hz, 2H), 6.94 (d, J = 1.2 Hz, 1H), 6.76 (d, J = 8.6 Hz, 2H), 6.70 (d, J = 8.7 Hz, 2H), 4.31 (s, 2H, 14-CH2) ppm. 13C NMR (125 MHz, MeOD) δ 158.7, 157.3, 155.3 (br), 142.8 (br), 137.3, 136.2, 132.2, 129.9, 125.1, 123.1, 118.2 (br), 116.7, 116.2, 105.8, 41.9 ppm. 13C NMR processed with LB = 3 Hz. IR (thin film) vmax 3228 (s, br, O–H, N–H), 2929 (m, br, C–H), 2471 (m), 1613, 1589 (m, C=C), 1527, 1511 (s), 1445, 1379 (m, br, C–N), 1260 (s, br, C–N), 1171, 1104 (m, C–O), 974 (m), 836 (s), 739 (w) cm–1.
N-(3,4-Bis(4-hydroxyphenyl)-1H-pyrazol-5-yl)benzamide (29a)
Prepared as a tan powder (43 mg, 0.105 mmol, quant) according to general procedure I. No chromatographic purification was required. LCMS (ESI+) m/z 372.2 [M + H]+, retention time 1.58 min (100%). HRMS (ESI+): m/z (calcd C22H18N3O3 [M + H]+ = 372.1348), obsd 372.1344 (δ ppm = 1.1). 1H NMR (500 MHz, MeOD) δ 7.87 (d, J = 7.7 Hz, 2H), 7.56 (t, J = 7.4 Hz, 1H), 7.47 (app t, J = 7.6 Hz, 2H), 7.24 (d, J = 8.6 Hz, 2H), 7.10 (d, J = 8.1 Hz, 2H), 6.76 (d, J = 8.7 Hz, 2H), 6.71 (d, J = 8.6 Hz, 2H) ppm. 13C NMR (125 MHz, MeOD) δ 170.8, 159.0, 157.5, 145.1 (br), 142.4 (br), 135.2, 133.1, 131.9, 130.2, 129.6, 128.7, 124.5, 122.5 (br), 116.4, 116.3, 115.7 (br) ppm. 13C NMR processed with LB = 12 Hz. IR (thin film) vmax 3254 (s, br, O–H, N–H), 2973, 2927 (w, C–H), 1656 (m, C=O), 1614 (m, C=C), 1510 (s, br, N–H), 1439 (m, C=C), 1371 (m), 1263 (s, br, C–O, C–N), 1173 (m, C–O), 836 (m, C–H), 710 (w) cm–1.
N-(3,4-Bis(4-hydroxyphenyl)-1H-pyrazol-5-yl)-4-hydroxybenzamide (29b)
Prepared according to general procedure I. The crude product was purified by flash chromatography (1–10% v/v MeOH in DCM) to yield 29b as a cream-colored amorphous solid (29 mg, 0.074 mmol, 81%). LCMS (ESI+) m/z 372.2 [M + H]+, retention time 1.59 min (100%). HRMS (ESI+) m/z (calcd C22H18N3O4 [M + H]+ = 388.1297), obsd 388.1286 (δ ppm = 2.8). 1H NMR (500 MHz, MeOD) δ 7.76 (d, J = 8.4 Hz, 2H), 7.23 (d, J = 8.8 Hz, 2H), 7.09 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.4 Hz, 2H), 6.75 (d, J = 8.4 Hz, 2H), 6.71 (d, J = 8.8 Hz, 2H) ppm. 13C NMR (125 MHz, MeOD) δ 170.4, 162.5, 158.9, 157.48, 144.8 (br), 143.0 (br), 131.9, 130.9, 130.2, 125.9, 124.6, 122.8 (br), 116.4, 116.3, 116.1, 115.5 (br) ppm. 13C-NMR processed with LB = 12 Hz. IR (thin film) vmax 3204 (br, m, O–H, N–H), 1648, 1607, 1587 (m, C=C), 1529 (s), 1441 (m), 1261, 1236, 1172 (s, C–N, C–O), 1022, 1010 (w, C–O), 837 (m), 764 (w) cm–1
N-(3,4-Bis(4-hydroxyphenyl)-1H-pyrazol-5-yl)isonicotinamide (29c)
Prepared according to general procedure I, except that 1.0 M BBr3 in DCM was added at −78 °C and then the reaction was allowed to warm slowly to 0 °C and then stirred overnight at room temperature. The crude product was purified by flash chromatography (1–10% v/v MeOH in EtOAc) to yield 29c as an off-white solid (42.3 mg, 0.114 mmol, 87%). LCMS (ESI+) m/z 373.2 [M + H]+, retention time 1.30 min (98%). HRMS (ESI+): m/z (calcd C21H17N4O3 [M + H]+ = 373.1301), obsd 373.1306 (δ ppm = 1.3). 1H NMR (500 MHz, MeOD) δ 8.72 (s, br, 2H), 7.82 (d, J = 4.5 Hz, 2H), 7.23 (d, J = 8.5 Hz, 2H), 7.08 (d, J = 8.5 Hz, 2H), 6.76 (d, J = 8.5 Hz, 2H), 6.72 (d, J = 8.5 Hz, 2H) ppm. 13C NMR (125 MHz, MeOD) δ 168.3, 159.1, 157.7, 150.9, 143.6 (br), 143.0 (br), 131.9, 130.2, 124.3, 123.2, 122.4 (br), 116.5, 116.4, 115.6 (br) ppm. One quaternary 13C signal unresolved. 13C NMR processed with LB = 6 Hz. IR (thin film) vmax 3440–3230 (br, s, O–H, N–H), 1666, 1614 (m), 1526, 1508 (C=C), 1415 (m), 1264 (s, C–N), 1175 (m, C–O), 837 (s), 751, 685 (w) cm–1
Molecular Biology, Biophysical, and Biochemical Methods
Expression and Purification of CYP121
The CYP121 (Rv2276) gene was expressed using a pHAT2/CYP121 expression clone in Escherichia coli C41(DE3) cells as the His6-tagged construct, following previously described conditions,7,27 with minor modifications. Expression was typically performed on a 10 × 1 L scale. C41 (DE3) E. coli cells were transformed with pHAT/CYP121 and cultured overnight in LB medium containing ampicillin (100 mg/L). Expression cultures of terrific broth (TB) medium containing ampicillin (100 mg/L) were inoculated using an overnight culture of the transformant cells (0.4% v/v) and grown at 37 °C, 200 rpm until an OD600 = 0.5–0.6, at which point the temperature was reduced to 23 °C. CYP121 gene expression was induced with IPTG (0.15 mM), and heme synthesis was supported with δ-aminolevulinic acid (0.1 mM). Cultures were incubated for 24 h, and then cells were harvested by centrifugation (9000g, 4 °C, 20 min) and stored at −20 °C until purification. Pellets were defrosted and resuspended in chilled 50 mM potassium phosphate buffer (pH 8.0), containing 50 mM KCl, 10%v/v glycerol, and commercial protease inhibitors (cOmplete EDTA-free protease inhibitor cocktail tablets, Roche (1 tablet/50 mL), PMSF (1 mM), and benzamidine hydrochloride (1 mM)) (buffer A). Cells were lysed by sonication at 0 °C (30 s on/60 s off, 12 cycles) using a Sonics Vibra-Cell 505 sonicator (Sonics and Materials Inc., CT, USA). Cell debris was pelleted by centrifugation (40000g, 30 min, 4 °C), and the supernatant was immediately loaded onto a freshly recharged HisTrap FF affinity column (GE Healthcare, Little Chalfont UK). Affinity chromatography was performed using an ÄKTA purifier (GE Healthcare) at 18 °C. Protein and P450 heme absorbance (A280 and A417, respectively) were monitored, and CYP121 was eluted with a linear gradient of imidazole (0–200 mM) in buffer A. The purity of fractions collected was assessed by SDS-PAGE. Pure fractions were combined and dialyzed into 50 mM Tris-HCl (pH 7.2), containing 500 mM KCl and 1 mM EDTA (buffer B) over 24–48 h using a Slide-A-Lyser (ThermoScientific, Hemel Hempstead UK) dialysis cassette. Dialyzed protein was concentrated using a Vivaspin (10000 MWCO) ultrafiltration column at 6000g, 4 °C, and then stored at −80 °C until further use. Impure protein fractions were pooled, dialyzed into buffer A to remove imidazole, and repurified by a second HisTrap FF affinity column, as described above. Protein concentration and purity was determined by SDS-PAGE and amino acid analysis (Protein and Nucleic Acid Chemistry Facility [PNAC], Department of Biochemistry, University of Cambridge). Repeated affinity chromatography steps meant that the previously cited anion exchange and gel filtration chromatography steps were not required to obtain sufficiently pure protein.
Expression and Purification of Mtb CYP125, CYP126, CYP142, CYP143, and CYP144
Plasmid vectors encoding N-terminal His6-tagged CYP125 (pET15b/Rv3545c) and CYP126 (pET15b/Rv0778) were provided by Dr. Kirsty McLean (Manchester Institute of Biotechnology, University of Manchester). CYP1257 and CYP12627,68 proteins were expressed and purified in E. coli C41(DE3) cells according to the literature. CYP142,53 CYP143, and CYP144-TRV4 proteins were prepared according to the literature and provided by Dr. Kirsty McLean (Manchester Institute of Biotechnology, University of Manchester).
Ligand Screening by UV–Visible Spectroscopy
UV–vis screening was performed using a CARY400 UV–vis spectrophotometer (Varian, CA, USA). Ligands were prepared as DMSO-d6 stock solutions (1–100 mM) and added to protein (5 μM) solutions (198 μL) or to buffer alone to give a final DMSO-d6 concentration of 1% v/v. CYP121 was prepared in 50 mM Tris-HCl (pH 7.2), 1 mM EDTA buffer. CYP125, CYP126, CYP142, CYP143, and CYP144 were prepared in 50 mM Tris-HCl (pH 7.5), 100 mM KCl buffer. CYP124 was prepared in 100 mM potassium phosphate (pH 7.0), 100 mM KCl buffer. Spectra were recorded between 800 and 250 nm at 25 °C. Any spectral interference from the inherent absorbance of added ligands/solvent was removed by subtracting spectra collected for protein-free samples of the same ligands/solvent in buffer. The difference in Soret wavelength maximum (Δλmax) of the respective P450s Soret band (λmax) in the presence of ligands compared to that obtained for a 1% v/v DMSO-d6 control was used to identify type I or type II heme-binding interactions. Changes in the Soret less than ±1 nm were considered within experimental error.
Competition assays were performed to detect the binding interactions of ligands that did not perturb that heme cofactor. A known type II inhibitor, typically clotrimazole (5–50 μM) or econazole (5–50 μM), was added to assay solutions to achieve a final DMSO-d6 concentration of 1% v/v. The difference in the red-shift of the Soret band between competition assays and that found for the known type II ligand in the absence of competing ligands was used to determine competitive binding. Buffer control spectra were collected to account for any inherent absorbance of added ligands/solvent and all solutions were inspected and found to be free of precipitate. All spectra were generated using Origin software (OriginLab, Northampton, MA) and processed using Microsoft Excel (Microsoft Office, 2010) and GraphPad Prism 5.01 (GraphPad Software, San Diego, USA).
Optical Titrations
Optical titrations to determine KD values were carried out a Varian Cary 400 UV–vis spectrophotometer (Varian, CA, USA) according to a previously described procedure.17 Assays were performed in reduced volume (200 μL) quartz cuvettes with a path length of 1 cm (Starna, Essex, UK). Ligands were prepared as DMSO-d6 stock solutions, and proteins (5 μM final concentration) were diluted in the corresponding buffer as described in the ligand screening by UV–vis spectroscopy protocol above. Aliquots (0.2–1.0 μL) of ligand stock solutions were added directly to cuvettes containing either protein solutions, or buffer alone, to achieve final concentrations of 100 nM to 1 mM. The final DMSO-d6 concentration did not exceed 1% v/v of the assay solution. Spectra were recorded between 800 and 250 nm at 25 °C after the addition of each aliquot of ligand. Buffer control spectra were subtracted manually from protein spectra during processing or saved as a baseline for automatic subtraction from protein spectra. Difference spectra were generated by subtracting the initial ligand-free protein spectrum from each successive titration spectrum. The maximum change in absorbance for each difference spectrum was then plotted against ligand concentration and fitted using either a one-site binding model hyperbolic eq (eq 1), or a modified version of the Morrison eq (eq 2) for tight binding inhibitors.58 All spectral analysis was done using Origin software (OriginLab, Northampton, MA), and data were processed using Microsoft Excel (Microsoft Office, 2010). Plots and curve fitting were performed using GraphPad Prism 5.01 (GraphPad Software, San Diego, USA).
| 1 |
| 2 |
In eqs 1 and 2, Aobs is the observed change in absorbance, Amax is the maximum absorbance change at saturation, E is the total enzyme concentration, and L is the concentration of ligand.
Competition Optical Titrations for Non-heme Binding Ligands
The KD values of non-heme binding ligands were calculated from the change in the apparent KD of a known type II ligand, typically 4,4′-(5-(((1H-imidazol-4-yl)methyl)amino)-1H-pyrazole-3,4-diyl)diphenol 26h, when it was titrated into proteins (5 μM) in the presence of the non-heme binding ligand (50 μM). Optical titrations were performed and processed as described above, except that the non-heme binding ligand was added initially to the ligand-free protein and buffer control cuvettes. Data were fitted using either eqs 1 or 2 to determine the KD (apparent) of 26h, and then the KD of competitive ligand was calculated according to eq 3.59
| 3 |
In eq 3, KDA(app) is the apparent binding affinity calculated from the competition titration, KDA is the binding affinity of the known inhibitor, KDB is the binding affinity of the non-heme binding ligand, and [B] is the concentration of the non-heme binding ligand.
Isothermal Titration Calorimetry
ITC experiments were performed on a MicroCal iTC200 (Malvern Instruments, Malvern UK) instrument at 25 °C. Ligands were prepared as stock solutions in DMSO-d6, diluted to 0.5–2.5 mM depending on solubility, and titrated into CYP121 (50 μM). Both ligands and CYP121 were prepared in the same 50 mM Tris-HCl (pH 7.5) buffer, containing 1 mM EDTA and a final concentration of 10% v/v DMSO-d6. Titrations were comprised of 19 × 2.0 μL injections of ligand solution at 60–90 s intervals. An initial 0.2 μL injection was made and the data discarded during analysis. Control titrations were also performed (ligand into buffer) to measure any heats of dilution or buffer mismatch. Control titrations were subtracted from ligand titrations during data processing. Binding isotherms were integrated to give the enthalpy change of each injection and plotted against the molar ratio of ligand added to the sample cell. Titrations were fitted using a one-site binding model using Origin Analysis Software, setting the stoichiometry to N = 1 for weak binding compounds, or allowing N to vary for potent compounds which showed high goodness of fit.
Native Mass Spectrometry
The following experimental protocol was followed for ligands 6, 19a, and 25a: Protein stock solutions (10 μM) were prepared by dilution of purified proteins (300–500 μM) in 200 mM ammonium acetate buffer (pH 7.0). Samples were buffer exchanged by size exclusion chromatography using Micro Biospin 6 columns, molecular weight cutoff 6 kDa (BioRad, Hemel Hempstead, UK). Ligands were prepared as stock solutions in DMSO-d6 at 50, 100, and 2500 μM concentrations. Ligand–protein samples were prepared by diluting protein stocks (10 μL) and ligand stocks (1 μL) with ammonium acetate buffer (9 μL) to give final concentrations of 5 μM CYP121, 2.5–125 μM ligand, and 5% v/v DMSO-d6. An identical protocol was followed for ligands 24a, 25b, and 26a, except that concentrations of CYP121 (8.7 μM) and DMSO-d6 (2.175% v/v) were altered. The concentrations of ligands was adjusted to maintain the same ligand-to-protein ratios. Mass spectra were recorded on a Synapt HDMS instrument (Waters UK Ltd., Manchester, UK). Capillaries for nanoESI– were purchased from ThermoFisher (Hemel Hempstead, UK). Capillary tips were cut under a stereomicroscope to give inner diameters of 1–5 μm and then loaded with 2.5 μL of sample solutions. Given below are the general instrumental conditions used to acquire the reported spectra. However, conditions were optimized over the course of each experiment to preserve the noncovalent protein–ligand complex and minimize nonspecific interactions. All measurements were carried out in a positive ion mode with ion source temperature of 20 °C. A capillary voltage of 1.50–1.75 kV, cone voltage of 60–80 V, and extraction cone voltage of 2.3–4.1 V were applied to perform nanoESI. All reported spectra were collected with a trap collision energy 12–18 V, transfer collision energy 12 V, IMS pressure 5.02 × 10–1 mbar, and TOF analyzer pressure 1.17 × 10–6 mbar. External calibration of the spectra was achieved using cesium iodide at 100 mg mL–1 in water. Data acquisition and processing were performed using Micromass MassLynx v4.1. Mass differences resulting from ligand binding were calculated relative to the relevant 5% v/v DMSO-d6 spectra of CYP121 (5 μM). Mass differences were divided by the molecular weight of the ligand to calculate binding stoichiometry. The percentage of monomeric CYP121 was calculated from the sum of the intensities of peaks corresponding to monomeric CYP121 (m/z 3500–4750) divided by the sum of peak intensities corresponding to both monomeric and dimeric (m/z 5250–6750) CYP121.
X-ray Crystallography
Untagged CYP121 protein and crystals were prepared as described previously with the following adaptations.27 Crystals were prepared by vapor diffusion in 1.5–2.1 M ammonium sulfate and 0.1 M sodium MES or Cacodylate from pH 5.5–6.15. Additional crystallization was also conducted on the nanolitre scale using a Mosquito pipetting robot (Molecular Dimensions, Newmarket, UK), and crystals were produced in the Morpheus crystallization screen (Molecular Dimensions) in 800 nL drops with protein-to-mother liquor at a ratio of 1:1. Ligand soaks were carried out either by directly dissolving solid ligand at saturation or by the addition of 2–5 mM ligand solution in DMSO to the mother liquor and soaking was carried out for 24 h. Crystals were cryoprotected and frozen as described previously except from Morpheus conditions that were frozen directly. Data were collected on beamline I24 (wavelength 0.9173/1.0118 Å) and I02 (wavelength 0.9795 Å) at the Diamond Light Source Facility (Oxfordshire, UK). Data were processed and refined as previously described. Data tables and statistics are provided in Table S5, Supporting Information. CYP121–ligand cocrystal structures are deposited in the Protein Data Bank (http://www.rcsb.org/pdb/) under the accession codes: 6, 5IBJ, 7; 5EDT, 19a; 5IBF, 24a; 5IBD, 25a; 5IBE, 25b; 5IBG, 26a; 5IBI, and 26h; 5IBH. Images for presentation were rendered using an academic version of the PyMOL Molecular Graphics System, version 1.3, 2010, Schrödinger, LLC.
Human P450 Inhibition
Experimental determination of IC50 values for the inhibition of human P450s by 19a, 25a, and 26a were performed by Cyprotex (Cheshire, UK). Inhibition assays of CYP1A, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 were performed in human liver microsomes in quadruplicate, over a 7-point ligand concentration range (0–25 μM). The formation of metabolites from the turnover of isoform-specific substrates (CYP2C9, tolbutamide; CYP2C19, mephenytoin; CYP2D6, dextromethorphan; and CYP3A4, midazolam) was monitored by LC/MS. Turnover of the CYP1A substrate ethoxyresorufin was monitored by fluorescence. Activities reported for CYP1A refer to the combined inhibition of CYP1A1 and CYP1A2 isoforms. IC50 values were calculated relative to the vehicle controls. IC50 values for known inhibitors (α-naphthoflavone, sulfaphenazole, tranylcypromine, quinidine, ketoconazole) were determined as a positive control and were found to be within the expected range (Table S3, Supporting Information).
Antimycobacterial Activity Assay
Three M. tuberculosis (Mtb) H37Rv cultures were prepared in 7H9 liquid medium supplemented with 10% OADC enrichment (Sigma-Aldrich, Poole UK). Cultures were incubated at 37 °C until the OD600 reached 1.00. After this period, the bacterial suspensions were washed twice with fresh 7H9 medium and then used in the antimycobacterial activity assays. The minimum inhibitory concentration (MIC90) of the compounds was measure in triplicate in a 24 well plate-based format. Plates were prepared with 2 mL of 7H10 solid medium supplemented with 10% OADC enrichment (Sigma-Aldrich) per well. Test compounds were prepared as stock solutions in DMSO and diluted into the 7H10 medium to achieve final concentrations of 100, 50, 25, 12.5, and 6.25 μM of CYP121 inhibitors and a final 1% v/v DMSO concentration. Rifampicin was used as a positive control and tested at 3.00, 1.50, 0.75, 0.37, and 0.18 μM concentrations. Prepared Mtb suspensions (OD600 = 1.0, approximately 3.00 × 105 CFU) were aliquoted (10 μL) onto the compound-infused solid medium and incubated at 37 °C for 2 weeks. The MIC90 was determined as the lowest concentration of compound that resulted in 90% inhibition of bacterial growth compared to the negative control wells, which were not treated with compounds and contained either 0% or 1% v/v DMSO.
Molecular Modeling and Docking
Lead compound 2, retrofragments 5 and 6, and all Ar1, Ar2, and Ar3 analogues were prepared using the LigPrep v3.2 and Epik v3.0 functions of Schrödinger suite software (Schrödinger LLC, NY), selecting to include metal binding states when generating ligand ionization states. Ligands were docking into the crystal structure of CYP121 bound to lead 2 (PDB 4KTL) or the previously reported ligand 4-(1H-1,2,4-triazol-1-yl)quinolin-6-amine (PDB 4G1X). Proteins were prepared using the internal Protein Preparation function of the Schrödinger suite software. Ionization states were generated to be compatible with metal-binding interactions, and the heme-iron was manually adjusted to the ferric (+3) oxidation state. All water molecules were removed from the structure PDB 4G1X. Duplicate energy minimized (OPLS 2005) structures of PDB 4KTL were prepared either with all water molecules removed or retaining the axial heme–water ligand only. Ar1 and Ar3 analogues were docked using core constraints to replicate the position of the aminopyrazole ring of lead 2. Ar2 analogues were docked using core constraints to replicate the heme binding interactions of the 4-(1H-1,2,4-triazol-1-yl)quinolin-6-amine ligand in PDB 4G1X. Images were generated using the PyMOL Molecular Graphics System, version 1.3, 2010 (Schrödinger, LLC).
Acknowledgments
We acknowledge Dr. Dijana Matak-Vinkovic for assistance with the native mass spectrometry experiments and the Diamond Light Source for access to beamlines i02, i04, and i24 (Proposal MX8997) that contributed to the results presented here. M.E.K. was supported by a Commonwealth (University of Cambridge) Scholarship awarded in conjunction with the Cambridge Commonwealth Trust and Cambridge Overseas Trust. A.G.C. and K.J.M. were supported by grants from the BBSRC (grant no. BB/I019669/1 and BB/I019227/1). G.G.J. received funding from the Ogden Trust and the Isaac Newton Trust administered through the University of Cambridge Bursary Scheme. D.S.C.H. was supported by a Croucher Cambridge International Scholarship awarded in conjunction between the Croucher Foundation and the Cambridge Overseas Trust. S.A.H. was supported by an Oliphant Cambridge Australia Scholarship (app no. 10132070) awarded by the Cambridge Commonwealth Trust. The contributions of L.B.M. and L.P.S.C. were supported by funds from the Francis Crick Institute, which receives its core funding principally from Wellcome Trust, Cancer Research UK, and the UK Medical Research Council (to L.P.S.C., MC_UP_A253_1111) and funds from FAPESP, CNPq, and CAPES-PDSE (to L.B.M., 2011/21232-1, 140079/2013-0, 99999.003125/2014-09).
Glossary
Abbreviations Used
- cYY
cyclo-dityrosine (cyclo-l-Tyr-l-Tyr)
- KD
binding affinity
- IC50
half-maximal inhibitory concentration
- GE
group efficiency
- ITC
isothermal titration calorimetry
- LE
ligand efficiency
- Mtb
Mycobacterium tuberculosis
- P450
cytochrome P450 enzyme
- SAR
structure–activity relationships
- TB
tuberculosis
- UV–vis
UV–visible spectrophotometry
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b00007. Additional data related to this publication is available at the University of Cambridge data repository: https://www.repository.cam.ac.uk/handle/1810/254048
Accession Codes
6, 5IBJ; 7, 5EDT, 19a, 5IBF; 24a, 5IBD; 25a, 5IBE; 25b, 5IBG; 26a, 5IBI; and 26h, 5IBH.
Author Present Address
For S.A.H.: Research School of Chemistry, College of Physical and Mathematical Sciences, Building 137, Sullivans Creek Road, The Australian National University, Canberra 0200, Australia.
The authors declare no competing financial interest.
Supplementary Material
References
- Global Tuberculosis Report; World Health Organisation: Geneva, 2014.
- Cole S. T.; Brosch R.; Parkhill J.; Garnier T.; Churcher C.; Harris D.; Gordon S. V.; Eiglmeier K.; Gas S.; Barry C. E.; Tekaia F.; Badcock K.; Basham D.; Brown D.; Chillingworth T.; Connor R.; Davies R.; Devlin K.; Feltwell T.; Gentles S.; Hamlin N.; Holroyd S.; Hornsby T.; Jagels K.; Krogh A.; McLean J.; Moule S.; Murphy L.; Oliver K.; Osborne J.; Quail M. a; Rajandream M. A.; Rogers J.; Rutter S.; Seeger K.; Skelton J.; Squares R.; Squares S.; Sulston J. E.; Taylor K.; Whitehead S.; Barrell B. G. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- McLean K. J.; Clift D.; Lewis D. G.; Sabri M.; Balding P. R.; Sutcliffe M. J.; Leys D.; Munro A. W. The preponderance of P450s in the Mycobacterium tuberculosis genome. Trends Microbiol. 2006, 14, 220–228. 10.1016/j.tim.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Driscoll M. D.; McLean K. J.; Cheesman M. R.; Jowitt T. A.; Howard M.; Carroll P.; Parish T.; Munro A. W. Expression and characterization of Mycobacterium tuberculosis CYP144: Common themes and lessons learned in the M. tuberculosis P450 enzyme family. Biochim. Biophys. Acta, Proteins Proteomics 2011, 1814, 76–87. 10.1016/j.bbapap.2010.05.015. [DOI] [PubMed] [Google Scholar]
- Tailleux L.; Waddell S. J.; Pelizzola M.; Mortellaro A.; Withers M.; Tanne A.; Castagnoli P. R.; Gicquel B.; Stoker N. G.; Butcher P. D.; Foti M.; Neyrolles O. Probing host pathogen cross-talk by transcriptional profiling of both Mycobacterium tuberculosis and infected human dendritic cells and macrophages. PLoS One 2008, 3, e1403. 10.1371/journal.pone.0001403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsclaw C. M.; Sogi K. M.; Gilmore S. A.; Schelle M. W.; Leavell M. D.; Bertozzi C. R.; Leary J. A. Structural characterization of a novel sulfated menaquinone produced by stf3 from Mycobacterium tuberculosis. ACS Chem. Biol. 2008, 3, 619–624. 10.1021/cb800145r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean K. J.; Lafite P.; Levy C.; Cheesman M. R.; Mast N.; Pikuleva I. A.; Leys D.; Munro A. W. The structure of Mycobacterium tuberculosis CYP125: Molecular basis for cholesterol binding in a P450 needed for host infection. J. Biol. Chem. 2009, 284, 35524–35533. 10.1074/jbc.M109.032706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capyk J. K.; Kalscheuer R.; Stewart G. R.; Liu J.; Kwon H.; Zhao R.; Okamoto S.; Jacobs W. R.; Eltis L. D.; Mohn W. W. Mycobacterial cytochrome P450 125 (Cyp125) catalyzes the terminal hydroxylation of C27 steroids. J. Biol. Chem. 2009, 284, 35534–35542. 10.1074/jbc.M109.072132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J. B.; Kells P. M.; Podust L. M.; Ortiz de Montellano P. R. Biochemical and structural characterization of CYP124: a methyl-branched lipid omega-hydroxylase from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 20687–20692. 10.1073/pnas.0907398106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondry M.; Sauguet L.; Belin P.; Thai R.; Amouroux R.; Tellier C.; Tuphile K.; Jacquet M.; Braud S.; Courçon M.; Masson C.; Dubois S.; Lautru S.; Lecoq A.; Hashimoto S.; Genet R.; Pernodet J.-L. Cyclodipeptide synthases are a family of tRNA-dependent peptide bond-forming enzymes. Nat. Chem. Biol. 2009, 5, 414–420. 10.1038/nchembio.175. [DOI] [PubMed] [Google Scholar]
- Belin P.; Le Du M. H.; Fielding A.; Lequin O.; Jacquet M.; Charbonnier J.-B.; Lecoq A.; Thai R.; Courçon M.; Masson C.; Dugave C.; Genet R.; Pernodet J.-L.; Gondry M. Identification and structural basis of the reaction catalyzed by CYP121, an essential cytochrome P450 in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 7426–7431. 10.1073/pnas.0812191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean K. J.; Carroll P.; Lewis D. G.; Dunford A. J.; Seward H. E.; Neeli R.; Cheesman M. R.; Marsollier L.; Douglas P.; Smith W. E.; Rosenkrands I.; Cole S. T.; Leys D.; Parish T.; Munro A. W. Characterization of active site structure in CYP121: A cytochrome P450 essential for viability of Mycobacterium tuberculosis H37Rv. J. Biol. Chem. 2008, 283, 33406–33416. 10.1074/jbc.M802115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean K. J.; Dunford A. J.; Neeli R.; Driscoll M. D.; Munro A. W. Structure, function and drug targeting in Mycobacterium tuberculosis cytochrome P450 systems. Arch. Biochem. Biophys. 2007, 464, 228–240. 10.1016/j.abb.2007.03.026. [DOI] [PubMed] [Google Scholar]
- McLean K. J.; Belcher J.; Driscoll M. D.; Fernandez C. C.; Van D. L.; Bui S.; Golovanova M.; Munro A. W. The Mycobacterium tuberculosis cytochromes P450. Future Med. Chem. 2010, 2, 1339–1353. 10.4155/fmc.10.216. [DOI] [PubMed] [Google Scholar]
- Ahmad Z.; Sharma S.; Khuller G. K. Azole antifungals as novel chemotherapeutic agents against murine tuberculosis. FEMS Microbiol. Lett. 2006, 261, 181–186. 10.1111/j.1574-6968.2006.00350.x. [DOI] [PubMed] [Google Scholar]
- Ahmad Z.; Sharma S.; Khuller G. K. In vitro and ex vivo antimycobacterial potential of azole drugs against Mycobacterium tuberculosis H37Rv. FEMS Microbiol. Lett. 2005, 251, 19–22. 10.1016/j.femsle.2005.07.022. [DOI] [PubMed] [Google Scholar]
- McLean K. J.; Cheesman M. R.; Rivers S. L.; Richmond A.; Leys D.; Chapman S. K.; Reid G. A.; Price N. C.; Kelly S. M.; Clarkson J.; Smith W. E.; Munro A. W. Expression, purification and spectroscopic characterization of the cytochrome P450 CYP121 from Mycobacterium tuberculosis. J. Inorg. Biochem. 2002, 91, 527–541. 10.1016/S0162-0134(02)00479-8. [DOI] [PubMed] [Google Scholar]
- Ahmad Z.; Sharma S.; Khuller G. K. Chemotherapeutic evaluation of alginate nanoparticle-encapsulated azole antifungal and antitubercular drugs against murine tuberculosis. Nanomedicine 2007, 3, 239–243. 10.1016/j.nano.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Como J. A.; Dismukes W. E. Oral azole drugs as systemic antifungal therapy. N. Engl. J. Med. 1994, 330, 263–272. 10.1056/NEJM199401273300407. [DOI] [PubMed] [Google Scholar]
- Congreve M.; Chessari G.; Tisi D.; Woodhead A. J. Recent developments in fragment-based drug discovery. J. Med. Chem. 2008, 51, 3661–3680. 10.1021/jm8000373. [DOI] [PubMed] [Google Scholar]
- Murray C. W.; Rees D. C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. 10.1038/nchem.217. [DOI] [PubMed] [Google Scholar]
- Scott D. E.; Coyne A. G.; Hudson S. A.; Abell C. Fragment-based approaches in drug discovery and chemical biology. Biochemistry 2012, 51, 4990–5003. 10.1021/bi3005126. [DOI] [PubMed] [Google Scholar]
- Hall R. J.; Mortenson P. N.; Murray C. W. Efficient exploration of chemical space by fragment-based screening. Prog. Biophys. Mol. Biol. 2014, 116, 82–91. 10.1016/j.pbiomolbio.2014.09.007. [DOI] [PubMed] [Google Scholar]
- Joseph-McCarthy D.; Campbell A. J.; Kern G.; Moustakas D. Fragment-based lead discovery and design. J. Chem. Inf. Model. 2014, 54, 693–704. 10.1021/ci400731w. [DOI] [PubMed] [Google Scholar]
- Bollag G.; Tsai J.; Zhang J.; Zhang C.; Ibrahim P.; Nolop K.; Hirth P. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discovery 2012, 11, 873–886. 10.1038/nrd3847. [DOI] [PubMed] [Google Scholar]
- Leys D.; Mowat C. G.; McLean K. J.; Richmond A.; Chapman S. K.; Walkinshaw M. D.; Munro A. W. Atomic structure of Mycobacterium tuberculosis CYP121 to 1.06 Å reveals novel features of cytochrome P450. J. Biol. Chem. 2003, 278, 5141–5147. 10.1074/jbc.M209928200. [DOI] [PubMed] [Google Scholar]
- Hudson S. A.; McLean K. J.; Surade S.; Yang Y. Q.; Leys D.; Ciulli A.; Munro A. W.; Abell C. Application of fragment screening and merging to the discovery of inhibitors of the Mycobacterium tuberculosis cytochrome P450 CYP121. Angew. Chem., Int. Ed. 2012, 51, 9311–9316. 10.1002/anie.201202544. [DOI] [PubMed] [Google Scholar]
- Hudson S. A.; Surade S.; Coyne A. G.; Mclean K. J.; Leys D.; Munro A. W.; Abell C. Overcoming the limitations of fragment merging: Rescuing a strained merged fragment series targeting mycobacterium tuberculosis CYP121. ChemMedChem 2013, 8, 1451–1456. 10.1002/cmdc.201300219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz I. D.; Chen K.; Sharp K. A.; Kollman P. A. The maximal affinity of ligands. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 9997–10002. 10.1073/pnas.96.18.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- Verdonk M. L.; Rees D. C. Group efficiency: A guideline for hits-to-leads chemistry. ChemMedChem 2008, 3, 1179–1180. 10.1002/cmdc.200800132. [DOI] [PubMed] [Google Scholar]
- Stout T. J.; Sage C. R.; Stroud R. M. The additivity of substrate fragments in enzyme-ligand binding. Structure 1998, 6, 839–848. 10.1016/S0969-2126(98)00086-0. [DOI] [PubMed] [Google Scholar]
- Compton D. R.; Sheng S.; Carlson K. E.; Rebacz N. A.; Lee I. Y.; Katzenellenbogen B. S.; Katzenellenbogen J. A. Pyrazolo[1,5-a]pyrimidines: estrogen receptor ligands possessing estrogen receptor beta antagonist activity. J. Med. Chem. 2004, 47, 5872–5893. 10.1021/jm049631k. [DOI] [PubMed] [Google Scholar]
- Johnson M. D.; Teng M.; Zhu J.. Aminopyrazole compounds and use as CHK1 inhibitors. WO2005/00945 A1, 2005.
- Ciulli A.; Williams G.; Smith A. G.; Blundell T. L.; Abell C. Probing hot spots at protein-ligand binding sites: A fragment-based approach using biophysical methods. J. Med. Chem. 2006, 49, 4992–5000. 10.1021/jm060490r. [DOI] [PubMed] [Google Scholar]
- Cytochrome P450: Structure, Mechanism and Biochemistry, 3rd ed.; Ortiz de Montellano P. R., Ed.; Kluwer Academic/Plenum Publishers: New York, 2005. [Google Scholar]
- Glide, version 6.5; Schrödinger, LLC: New York, 2014.
- Smith A. J. T.; Zhang X.; Leach A. G.; Houk K. N. Beyond picomolar affinities: Quantitative aspects of noncovalent and covalent binding of drugs to proteins. J. Med. Chem. 2009, 52, 225–233. 10.1021/jm800498e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Magid A. F.; Carson K. G.; Harris B. D.; Maryanoff C. A.; Shah R. D. Reductive amination of aldehydes and ketones with sodium triacetoxyborohydride. Studies on direct and indirect reductive amination procedures. J. Org. Chem. 1996, 61, 3849–3862. 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- Vasudevan A.; Qian Y.; Vogt A.; Blaskovich M. A.; Ohkanda J.; Sebti S. M.; Hamilton A. D. Potent, highly selective, and non-thiol inhibitors of protein geranylgeranyltransferase-I. J. Med. Chem. 1999, 42, 1333–1340. 10.1021/jm9900873. [DOI] [PubMed] [Google Scholar]
- Schenkman J. B.; Remmer H.; Estabrook R. W. Spectral studies of drug interaction with hepatic microsomal cytochrome. Mol. Pharmacol. 1967, 3, 113–123. [PubMed] [Google Scholar]
- Ouellet H.; Kells P. M.; Ortiz de Montellano P. R.; Podust L. M. Reverse type I inhibitor of Mycobacterium tuberculosis CYP125A1. Bioorg. Med. Chem. Lett. 2011, 21, 332–337. 10.1016/j.bmcl.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean K. J.; Marshall K. R.; Richmond A.; Hunter I. S.; Fowler K.; Kieser T.; Gurcha S. S.; Besra G. S.; Munro A. W. Azole antifungals are potent inhibitors of cytochrome P450 mono-oxygenases and bacterial growth in mycobacteria and streptomycetes. Microbiology 2002, 148, 2937–2949. 10.1099/00221287-148-10-2937. [DOI] [PubMed] [Google Scholar]
- Yoshida Y.; Aoyama Y. Interaction of azole antifungal agents with cytochrome P-45014DM purified from Saccharomyces cerevisiae microsomes. Biochem. Pharmacol. 1987, 36, 229–235. 10.1016/0006-2952(87)90694-0. [DOI] [PubMed] [Google Scholar]
- Conner K. P.; Woods C.; Atkins W. M. Interactions of cytochrome P450s with their ligands. Arch. Biochem. Biophys. 2011, 507, 56–65. 10.1016/j.abb.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellet H.; Podust L. M.; Ortiz de Montellano P. R. Mycobacterium tuberculosis CYP130: Crystal structure, biophysical characterization, and interactions with antifungal azole drugs. J. Biol. Chem. 2008, 283, 5069–5080. 10.1074/jbc.M708734200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler S. E.; Houk K. N. Origin of substituent effects in edge-to-face aryl–aryl interactions. Mol. Phys. 2009, 107, 749–760. 10.1080/00268970802537614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A. L.; Keserü G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Podust L. M.; Ouellet H.; von Kries J. P.; Ortiz de Montellano P. R. Interaction of Mycobacterium tuberculosis CYP130 with heterocyclic arylamines. J. Biol. Chem. 2009, 284, 25211–25219. 10.1074/jbc.M109.017632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonvielle M.; Le Du M. H.; Lequin O.; Lecoq A.; Jacquet M.; Thai R.; Dubois S.; Grach G.; Gondry M.; Belin P. Substrate and reaction specificity of Mycobacterium tuberculosis cytochrome P450 CYP121: Insights from biochemical studies and crystal structures. J. Biol. Chem. 2013, 288, 17347–17359. 10.1074/jbc.M112.443853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro A. W.; Leys D. G.; McLean K. J.; Marshall K. R.; Ost T. W. B.; Daff S.; Miles C. S.; Chapman S. K.; Lysek D. A.; Moser C. C.; Page C. C.; Dutton P. L. P450 BM3: the very model of a modern flavocytochrome. Trends Biochem. Sci. 2002, 27, 250–257. 10.1016/S0968-0004(02)02086-8. [DOI] [PubMed] [Google Scholar]
- Duffell K. M.; Hudson S. A.; McLean K. J.; Munro A. W.; Abell C.; Matak-Vinković D. Nanoelectrospray ionization mass spectrometric study of Mycobacterium tuberculosis CYP121-ligand interactions. Anal. Chem. 2013, 85, 5707–5714. 10.1021/ac400236z. [DOI] [PubMed] [Google Scholar]
- Driscoll M. D.; McLean K. J.; Levy C.; Mast N.; Pikuleva I. A.; Lafite P.; Rigby S. E. J.; Leys D.; Munro A. W. Structural and biochemical characterization of Mycobacterium tuberculosis CYP142: evidence for multiple cholesterol 27-hydroxylase activities in a human pathogen. J. Biol. Chem. 2010, 285, 38270–38282. 10.1074/jbc.M110.164293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellamine A.; Mangla A. T.; Nes W. D.; Waterman M. R. Characterization and catalytic properties of the sterol 14alpha-demethylase from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 8937–8942. 10.1073/pnas.96.16.8937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean K. J.; Warman A. J.; Seward H. E.; Marshall K. R.; Girvan H. M.; Cheesman M. R.; Waterman M. R.; Munro A. W. Biophysical characterization of the sterol demethylase P450 from Mycobacterium tuberculosis, its cognate ferredoxin, and their interactions. Biochemistry 2006, 45, 8427–8443. 10.1021/bi0601609. [DOI] [PubMed] [Google Scholar]
- Martin D. P.; Blachly P. G.; McCammon J. A.; Cohen S. M. Exploring the influence of the protein environment on metal-binding pharmacophores. J. Med. Chem. 2014, 57, 7126–7135. 10.1021/jm500984b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A.Lead optimization and structure–activity relationships for reversible inhibitors. In Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2013. [Google Scholar]
- Morrison J. F. Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhbitors. Biochem. Biophys. Acta 1969, 185, 269–286. 10.1016/0005-2744(69)90420-3. [DOI] [PubMed] [Google Scholar]
- Hu D. D.; Eftink M. R. Thermodynamic studies of the interaction of trp aporepressor with tryptophan analogs. Biophys. Chem. 1994, 49, 233–239. 10.1016/0301-4622(93)E0073-E. [DOI] [PubMed] [Google Scholar]
- Inhibitor screening against human cytochrome P450s was outsourced to Cyprotex Discovery Ltd., Macclesfield, Cheshire, UK. [Google Scholar]
- Mirza A.; Desai R.; Reynisson J. Known drug space as a metric in exploring the boundaries of drug-like chemical space. Eur. J. Med. Chem. 2009, 44, 5006–5011. 10.1016/j.ejmech.2009.08.014. [DOI] [PubMed] [Google Scholar]
- Kim D.; Guengerich F. P. Cytochrome P450 activation of arylamines and heterocyclic amines. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 27–49. 10.1146/annurev.pharmtox.45.120403.100010. [DOI] [PubMed] [Google Scholar]
- Choi J. Y.; Calvet C. M.; Vieira D. F.; Gunatilleke S. S.; Cameron M. D.; McKerrow J. H.; Podust L. M.; Roush W. R. R-configuration of 4-aminopyridyl-based inhibitors of CYP51 confers superior efficacy against Trypanosoma cruzi. ACS Med. Chem. Lett. 2014, 5, 434–439. 10.1021/ml500010m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. Y.; Calvet C. M.; Gunatilleke S. S.; Ruiz C.; Cameron M. D.; McKerrow J. H.; Podust L. M.; Roush W. R. Rational development of 4-aminopyridyl-based inhibitors targeting Trypanosoma cruzi CYP51 as anti-chagas agents. J. Med. Chem. 2013, 56, 7651–7668. 10.1021/jm401067s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan P. J. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis 2003, 83, 91–97. 10.1016/S1472-9792(02)00089-6. [DOI] [PubMed] [Google Scholar]
- De Rossi E.; Aínsa J. A.; Riccardi G. Role of mycobacterial efflux transporters in drug resistance: An unresolved question. FEMS Microbiol. Rev. 2006, 30, 36–52. 10.1111/j.1574-6976.2005.00002.x. [DOI] [PubMed] [Google Scholar]
- Martin D. P.; Blachly P. G.; Marts A. R.; Woodruff T. M.; de Oliveira C. A. F.; McCammon J. A.; Tierney D. L.; Cohen S. M. Unconventional” coordination chemistry by metal chelating fragments in a metalloprotein active site. J. Am. Chem. Soc. 2014, 136, 5400–5406. 10.1021/ja500616m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson S. A.; Mashalidis E. H.; Bender A.; McLean K. J.; Munro A. W.; Abell C. Biofragments: An approach towards predicting protein function using biologically related fragments and its application to mycobacterium tuberculosis CYP126. ChemBioChem 2014, 15, 549–555. 10.1002/cbic.201300697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.