

Abstract
The Epstein-Barr virus (EBV) is associated with various lymphoproliferative disorders and lymphomas. We have previously demonstrated that treating wild-type TP53-expressing B cell lines with the TP53 pathway activator nutlin-3 induced apoptosis in EBV-negative and EBV-positive latency I cells whereas EBV-positive latency III cells remained much more apoptosis-resistant. Here, we report a constitutively high level of autophagy in these resistant cells which express high levels of the proautophagic protein BECN1/Beclin 1 based, at least in part, on the activation of the NFKB signaling pathway by the viral protein LMP1. Following treatment with nutlin-3, several autophagy-stimulating genes were upregulated both in EBV-negative and EBV-positive latency III cells. However the process of autophagy was only triggered in the latter and was associated with an upregulation of SESN1/sestrin 1 and inhibition of MTOR more rapid than in EBV-negative cells. A treatment with chloroquine, an inhibitor of autophagy, potentiated the apoptotic effect of nutlin-3, particularly in those EBV-positive cells which were resistant to apoptosis induced by nutlin-3 alone, thereby showing that autophagy participates in this resistant phenotype. Finally, using immunohistochemical staining, clinical samples from various B cell lymphoproliferations with the EBV-positive latency II or III phenotype were found to harbor a constitutively active autophagy.
Keywords: autophagy, BECN1, Burkitt lymphoma, EBV, nulin-3, TP53
Abbreviations
- ACTB
actin beta
- AMPK
AMP-activated protein kinase
- BAF
bafilomycin A1
- BAX
BCL2–associated X protein
- BCL2
B-cell CLL/lymphoma 2
- BECN1
Beclin 1 autophagy related
- BL
Burkitt lymphoma
- DLBCL
diffuse large B cell lymphoma
- EBNA
Epstein-Barr nuclear antigen
- EBV
Epstein-Barr virus
- HRP
horseradish peroxidase
- LCL
lymphoblastoid cell lines
- LMP
latent membrane protein
- mAb
monoclonal antibody
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MDC
monodansylcadaverine
- MDM2
MDM2 proto-oncogene
- E3
ubiquitin protein ligase
- MFI
mean fluorescence intensity
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- NFKB
nuclear factor of kappa light polypeptide gene enhancer in B-cells
- pAb
polyclonal antibody
- PI
propidium iodide
- PTLD
post-transplant lymphoproliferative disorder
- RELA/p65
v-rel avian reticuloendotheliosis viral oncogene homolog A
- RPS6KB/p70S6K
ribosomal protein S6 kinase 70kDa
- SESN1
sestrin 1
- ShRNA
short hairpin RNA
- TP53
tumor protein p53
Introduction
The Epstein-Barr virus (EBV), a γ-Herpes virus, is associated with various B cell malignancies including Burkitt lymphoma (BL) and post-transplant lymphoproliferative disorders (PTLD). Its ability to efficiently immortalize B lymphocytes thus generating lymphoblastoid cell lines (LCL) underlies its oncogenic role. In EBV-infected tumor cells, the virus remains latent, with only a small subset of viral proteins being expressed. These include 6 nuclear antigens (EBNA) and 3 membrane proteins (LMP), which are differentially expressed between the various EBV-associated tumors. For instance, virtually all EBV-positive BL cells only express EBNA1 (latency I phenotype) while most of PTLD cases express the full spectrum of latent proteins (latency III phenotype). However, rare cases of BL with a latency III phenotype have been reported as well as some cases of PTLD with a latency II phenotype (expression of EBNA1, LMP1 and LMP2). Among the viral products, it is mostly the membrane protein LMP1, which has been implicated in tumor formation. This versatile protein initiates the activation of the mitogen-activated protein kinases MAPK8/JNK1 and MAPK14/p38, the Janus kinase-signal transducers and activators of transcription (JAK-STAT) and the NFKB (nuclear factor of kappa light polypeptide gene enhancer in B-cells) signaling cascades, with an overall effect on cell survival.
TP53 (tumor protein p53) is a stress-activated transcription factor which plays a key role in protecting cells against tumor development, either promoting the inhibition of cell proliferation or inducing cell apoptosis.1 Mutation in its coding sequence, sequestration in the cytoplasm or increased interactions with its main cellular regulator MDM2 are but a few of the possible ways whereby the physiological functions of the TP53 protein can be disrupted in tumor cells.2 As recently demonstrated, TP53 is also involved in the regulation of autophagy, a lysosomal degradation pathway that contributes to cell homeostasis. The autophagy process starts with the formation of large double-membrane vesicles called autophagosomes. These in turn fuse with lysosomes to form autolysosomes whose internal content is then degraded by hydrolases, thus clearing the cell from damaged organelles and protein aggregates.3 Numerous proteins have been reported to regulate autophagy. On the negative side, MTOR (mechanistic target of rapamycin [serine/threonine kinase]) is a key factor. On the positive side, BECN1, a critical component in the class III phosphatidylinositol 3-kinase (PtdIns3K) complexes regulates autophagy by inducing the formation of autophagosomes.4 Other positive regulators of autophagy include various transcriptional targets of TP53 such as sestrins whose effects result in MTOR inhibition,5 and DRAM1 (DNA-damage regulated autophagy modulator 1), a lysosomal membrane protein whose mechanism of action remains uncharacterized.6 In contrast, cytoplasmic TP53 inhibits autophagy, thus implying that it plays a dual role in the modulation of this catabolic pathway.7 Given its key function in determining cell fate, several strategies have been developed to restore TP53 activity in tumor cells. Several low molecular mass compounds can disrupt its interaction with MDM2 (MDM2 proto-oncogene, E3 ubiquitin protein ligase) (for a review see refs.8, 9). For example, nutlin-3, a cis-imidazoline, binds the TP53 pocket of MDM2 thereby inducing both the release and the activation of TP53.10 In vitro, nutlin-3 has been found to enhance apoptosis in various experimental systems.11-13 In preclinical studies, a treatment with this drug inhibited tumor growth and exerted a synergistic effect with genotoxic drugs or irradiation. This opened the way to phase I clinical trials (with the second generation nutlin, RG7112) which are currently carried out for various tumor types (reviewed in refs. 14, 15).
In previous studies, we have shown that wild-type (WT) TP53-containing EBV-negative and -positive cells treated with nutlin-3 exhibit TP53 activation as assessed by the induction of CDKN1A/p21 (cyclin-dependent kinase inhibitor 1A [p21, Cip1]), MDM2 and BAX (BCL2–associated X protein). However, whereas nutlin-3-treated EBV-negative BL cells die massively through an apoptotic process involving BAX, EBV-positive latency III BL and LCL cells prove much more resistant,16 in relation with their strong level of expression of BCL2 (BCL2, B-cell CLL/lymphoma 2).17 We also observed that treating latency III cells with an inhibitor of BCL2 only partially restored their sensitivity to nutlin-3, which prompted us to study these cells further. Here, autophagy was found to be constitutively activated in EBV-positive latency III cells and to participate in their mechanisms of resistance to nutlin-3-induced apoptosis. A treatment with chloroquine, an inhibitor of autophagy, provided an apoptosis-inducing effect complementary to the action of nutlin-3. Autophagy was also constitutively activated in EBV-positive latency II and latency III primary B cell lymphoproliferations (diffuse large B cell lymphoma [DLBCL] and PTLD) but neither in EBV-negative nor in EBV-positive latency I BL.
Results
Constitutive levels of autophagy differ between EBV-negative and EBV-positive latency III cells
The MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) protein exists under 2 forms. The cytosolic LC3-I moiety can be converted by lipidation into LC3-II, which can then be recruited into the membrane of phagophores, the precursors to autophagosomes. A western blot analysis revealing the relative importance of these 2 forms in cell populations provides a convenient way to assess their level of basal autophagy. Since EBV is a known activator of the autophagic machinery,18 we tested the presence of the 2 LC3 forms in a panel of EBV-negative BL cell lines (BL2, BL28 and BL40), in their in vitro EBV-infected counterparts (BL2/B95, BL28/B95 and BL40/B95), in LY47, an EBV-positive latency III BL cell line, and in 2 LCL (IARC211, derived from the same patient as BL40, and RPMI8866). As shown in Figure 1A, LC3-II was detected in the EBV-positive cells only, whereas the presence of LC3-I appeared independent of the EBV status of the cell lines. We also used staining of the cells with monodansylcadaverine (MDC) for monitoring autophagy in our cell lines. This autofluorescent compound associates with acidic and lipid-rich compartments and thus gives an indication of the presence of late autophagic vacuoles in the cells.19 As shown in Figure 1B, higher levels of punctate MDC staining were observed in BL2/B95 (23±10%) and RPMI8866 (46±5%), 2 EBV-positive latency III cell lines, than in EBV-negative BL2 cells (11±2%). Together, these data suggest that the level of basal autophagy is higher in EBV-positive latency III cells than in EBV-negative cells.
Figure 1.
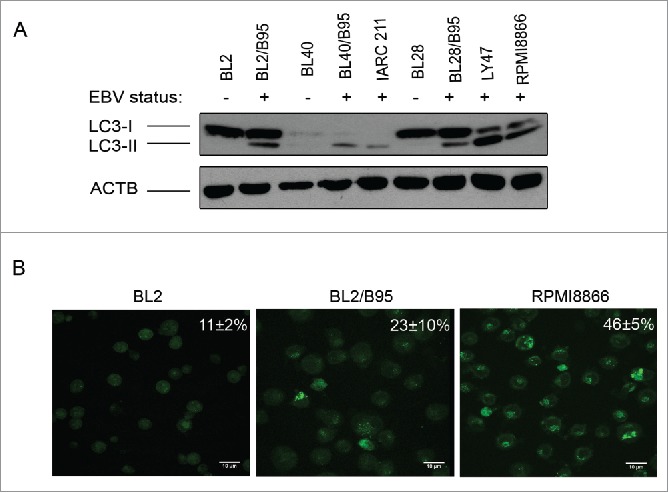
Analysis of constitutive autophagy in EBV-negative and EBV-positive latency III lymphoid cell lines. (A) Whole cell lysates from the indicated cell lines were submitted to western blot analysis for detection of LC3-I, LC3-II and ACTB. (B) Cells stained with MDC were examined by confocal fluorescence microscopy. Mean percentages of MDC-stained cells indicative of autophagy were calculated from 3 independent experiments.
BECN1 and autophagy are upregulated in EBV-positive latency III cells through NFKB activation
The viral LMP1 protein is a known inducer of the NFΚB signaling cascade 20,21 which, under certain circumstances, positively modulates canonical autophagy.22,23 We thus reasoned that the NFKB complex could play a part in the activation of autophagy in these cells. As expected, its RELA/p65 (v-rel avian reticuloendotheliosis viral oncogene homolog A) subunit was found to be more abundant in the EBV-positive latency III than in the EBV-negative cell lines (Fig. 2A). As RELA/p65 binds to and activates the BECN1 gene promoter,24 we then tested the same cell lines for expression of the BECN1 protein which was found to follow that of RELA (Fig. 2A and Fig. S1). To examine RELA expression levels more precisely, cytosolic and nuclear extracts were prepared from both EBV-positive latency III and EBV-negative cell lines. Levels of RELA were found to be higher in the nuclear fraction of EBV-positive cell lines than in their EBV-negative counterparts, contrasting with the cytosolic fractions where no such relation was observed (Fig. S2). This is consistent with RELA playing a role in the process leading to BECN1 expression based on its transcriptional regulatory function. To confirm that LMP1 regulates BECN1 expression through the NFKB pathway we used stable transfectants of DG75 cells, which express LMP1 only in the absence of tetracycline. In these conditions of LMP1 expression, levels of both RELA and BECN1 increased as compared to control cells cultivated in the presence of tetracycline (Fig. 2B). We also used an shRNA approach to test for a direct correlation between the status of the NFKB-BECN1 pathway and the level of autophagy in EBV-positive latency III cells. To this end, RPMI8866 cells were transduced with an shRNA directed against RELA and the levels of expression of RELA, BECN1, LC3-I and LC3-II were tested. As seen in Figure 2C, levels of BECN1 and LC3-II were found strongly decreased in transduced cells where RELA expression was virtually abolished as compared to control cells transduced with an shRNA that does not target any known human gene. LC3-I expression was not affected by inhibition of RELA. Altogether, these data indicate that an LMP1-dependent activation of the NFKB signaling pathway upregulates the expression of BECN1 and the level of autophagy in EBV-positive latency III cells.
Figure 2.
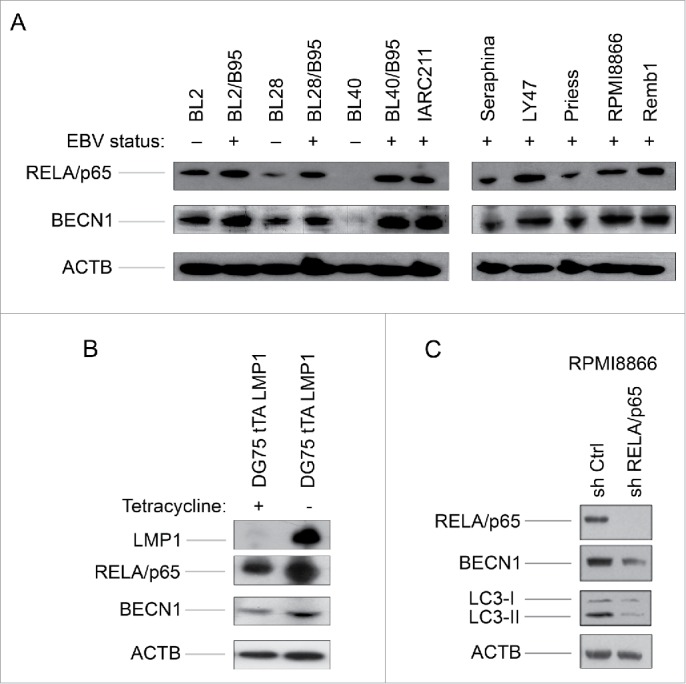
RELA activation and BECN1 expression in EBV-negative and EBV-positive latency III lymphoid cell lines. (A) Whole cell lysates were analyzed by western blotting for RELA and BECN1 expression. (B) Whole cell lysates prepared from DG75 cells, expressing LMP1 in a tetracycline-regulated system, were tested for expression of LMP1, RELA and BECN1. (C) Whole cell lysates prepared from RPMI8866 stably transduced with a RELA-specific shRNA or a control shRNA were submitted to western blot analysis for detection of RELA and BECN1 expression. The western blots shown are representative of 3 independent experiments.
Treatment with nutlin-3 induces the expression of a subset of genes involved in autophagy in EBV-negative and EBV-positive latency III cells
We have previously shown that nutlin-3 similarly induced TP53 activation in EBV-negative and EBV-positive B cells whereas the induction of apoptosis by this compound depended upon their EBV status: EBV-negative and -positive latency I cells are highly sensitive to this antagonist of MDM2 whereas EBV-positive latency III cells are much more resistant.16 Having detected higher levels of basal autophagy in the latter, we decided to examine the transcriptional effect of nutlin-3 treatment on EBV-negative BL2 and EBV-positive latency III BL2/B95 cells. A genome-wide transcriptome analysis was performed at various times of incubation in the presence of 10 µM of nutlin-3. Over time, an increasing number of genes were found to be upregulated in both cell lines (Fig. S3). As expected, these genes encode proteins involved in cellular functions that are regulated by TP53. Among them, 5 genes have been previously implicated in the autophagy process. They are reported in Figure 3A where it can be seen that their mRNA expression levels increased during treatment in both cell types with an apparently more powerful induction in the EBV-converted BL2/B95 cell line than in its EBV-negative parent. These changes in the mRNA expression levels induced by 16 h of treatment with nutlin-3 were then quantified as compared to untreated cells (Fig. 3B). DRAM1, the TP53 target gene most strongly induced in both BL2 and BL2/B95 (fold change 26.8 and 66.8, respectively), encodes a lysosomal protein that induces macroautophagy.25 Treatment of BL2 and BL2/B95 with nutlin-3 also induced higher transcription levels of SESN1 (fold change 6.9 and 13.3), SESN2 (Sestrin 2) (fold change 5.2 and 9.2), PRKAB1, the β-1 regulatory subunit of AMPK (AMP-activated protein kinase) (fold change 2.7 and 3) and TSC2 (tuberous sclerosis 2) (fold change 2, only in BL2/B95). The proteins encoded by these genes are involved in a cascade of events: AMPK is activated by direct interactions with SESN1 and SESN2 and phosphorylates TSC2 which, in turn, inhibits MTOR thus leading to autophagy activation.5 Treatment of BL2 and BL2/B95 with nutlin-3 did not modify the mRNA level of BECN1 (data not shown).
Figure 3.
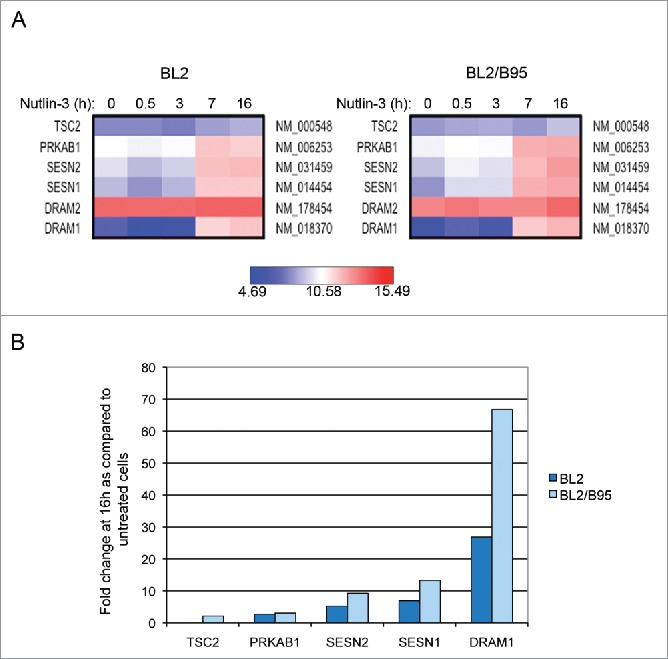
Changes in global gene expression analysis of EBV-negative BL2 and EBV-positive latency III BL2/B95 cells according to nutlin-3 treatments. (A) Probes corresponding to genes relative to autophagy are represented using heatmaps. (B) Fold change in mRNA levels for autophagy-related genes in both cell lines treated for 16 h as compared to untreated cells.
Treatment with nutlin-3 promotes autophagy in EBV-positive latency III cells but not in EBV-negative cells
Since nutlin-3 treatment increases the level of expression of several genes involved in autophagy, we decided to assess if this compound is able to increase the autophagic flux in EBV-negative and -positive cell lines. To this end, BL2, BL2/B95 and RPMI8866 cells were treated with nutlin-3 for 3, 5, 7 or 24 h followed by assessment of autophagy measured by western blot analysis of LC3-I and LC3-II levels. Since LC3-II accumulation may result from either increased autophagosome formation or impaired autophagosome-lysosome fusion, cells were treated either with or without bafilomycin A1 (BAF) which inhibits intralysosomal acidification thereby blocking the degradation of LC3-II.26 It can be seen in Figure 4A that in the absence of BAF, the levels of the autophagosome-specific LC3-II form detected after nutlin-3 treatment differed between cell lines: in BL2 cells, LC3-II remained hardly detectable; in BL2/B95 cells, the amount of LC3-II increased as early as 3 h after treatment and became the major form of LC3 at 24 h; in RPMI 8866 cells, the level of LC3-II, already high in untreated cells, remained stable until 7 h and slightly increased at 24 h. In the latter, the level of the cytosolic LC3-I form decreased progressively all along the 24 h nutlin-3 treatment. When these cell lines were pretreated with BAF (100 nM), the increase of LC3-II levels observed after 3, 5 and 7 h of nutlin-3 treatment was further enhanced particularly in BL2/B95 and RPMI8866 cells (Fig. 4A and Fig. S4). Altogether, these results indicate that in EBV-positive cells, the LC3-II increase induced by nutlin-3 is due to an increase in autophagic activity and not to an impaired autophagosome turnover.
Figure 4.
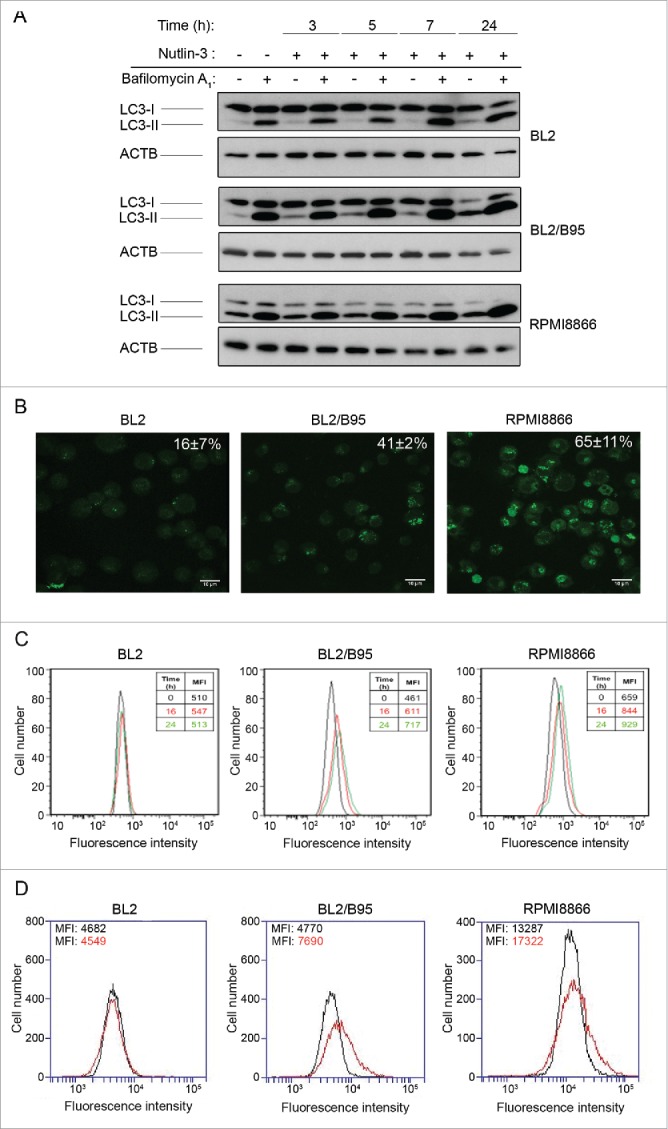
Effect of nutlin-3 treatment on autophagy induction in EBV-negative and EBV-positive latency III lymphoid cell lines. (A) Cells preincubated (30 min) with or without BAF (100 nM) were treated with nutlin-3 for various times. Expression of LC3-I and LC3-II was tested by western blotting of whole cell lysates. (B and C) Cells treated with nutlin-3 for 16 h were stained with MDC. The percentages of cells (means from 3 independent experiments) with MDC stained dots were assessed by fluorescence microscopy (B). The MFI of MDC stained dots was assessed by flow cytometry (C). (D) Cells incubated with or without nutlin-3 for 16 h were stained with the Cyto-ID Autophagy detection kit. The MFI of cyto-ID staining was assessed by flow cytometry.
To further explore this matter, EBV-negative and -positive latency III cells treated with nutlin-3 for 16 h were stained with MDC and observed by fluorescence confocal microscopy (Fig. 4B). From a comparison with results shown in Figure 1B, it can be concluded that the percentage of MDC-stained vacuoles increased after treatment with nutlin-3, and that this increase was much more pronounced in EBV-positive latency III cells (41 ± 2% vs 23 ±10 % and 65 ± 11% vs 46 ± 5% cells stained in BL2/B95 and RPMI8866, respectively) than in EBV-negative BL2 cells (16 ± 7% cells stained vs 11 ± 2%). MDC staining was also measured by flow cytometry after 16 and 24 h of treatment with nutlin-3. Results shown in Figure 4C confirmed that nutlin-3 had a greater effect in EBV-positive than in EBV-negative cells. In addition, induction of autophagy by nutlin-3 treatment was assessed with a new dye (cyto-ID) that makes it possible to selectively stain autophagic compartments. Flow cytometry analysis of cells (Fig. 4D) indicated that treating cells with nutlin-3 for 16 h did not result in increased autophagy in BL2 (mean fluorescence intensity [MFI]: 4549 vs 4682 for untreated cells) whereas such a treatment substantially increased autophagy in BL2/B95 (MFI: 7690 vs 4770) and RPMI8866 (MFI: 17322 vs 13287).
Finally, the EBV-positive BL2/B95 and RPMI8866 cells were submitted to electron microscopy observation. Typical autophagosomes were detected between 7 and 10 h of nutlin-3 treatment (Fig. 5). In contrast, at these time points, such autophagosomes were not detectable in BL2 cells (data not shown). Altogether, these results are thus consistent with a differential effect of nutlin-3 treatment on the level of autophagy in EBV-negative and EBV-positive latency III cells.
Figure 5.

Effect of nutlin-3 treatment on autophagy induction observed by electron microscopy. (A, B and C) BL2/B95 cells, (D, E and F) RPMI8866 cells. B and C, and E and F, are successive magnifications of A and D, respectively. Nucleus (N), multivesicular body (MVB) and autophagosomes (A) as indicated.
Treatment with nutlin-3 inhibits MTOR activity more rapidly in EBV-positive latency III cells than in EBV-negative cells
We next decided to test whether the induction of an autophagic flux after nutlin-3 treatment correlated with MTOR inhibition, which could potentially be induced by activation of the SESN-AMPK-TSC pathway suggested by the transcriptomic analysis. First, BL2 and BL2/B95 as well as LY47 and RPMI8866 cells were treated with 10 µM nutlin-3 and tested at various time points for expression of SESN1 (Fig. 6A). SESN1-specific bands observed on western blots were quantified by densitometry and normalized to ACTB/β-actin levels. In the EBV-positive cells, increased amounts of SESN1 were readily detectable after 1 h (for LY47) or 3 h (for BL2/B95 and RPMI8866) of treatment with nutlin-3. In the EBV-negative BL2 cell line, the accumulation of SESN1 was only detected after 5 h of treatment. To determine whether the induction of SESN1 expression correlated with an inhibition of MTOR kinase activity, we then analyzed the phosphorylation status of its target protein RPS6KB (ribosomal protein S6 kinase, 70kDa)27 using an antibody specific for RPS6KB phosphorylated at Thr389 (the main target of MTOR). In EBV-positive BL2/B95 cells, treatment with nutlin-3 induced a strong inhibition of RPS6KB phosphorylation with kinetics similar to that of SESN1 induction. In contrast, there was virtually no effect in EBV-negative BL2 cells (Fig. 6B). Reblotting with an anti-RPS6KB verified that the treatment with nutlin-3 had not resulted in a decrease in the levels of the protein (Fig. 6B). Since the effects of nutlin-3 appeared to be delayed in EBV-negative as compared to EBV-positive cells, we then tested the expression of phospho-RPS6KB in BL2, BL2/B95, LY47 and RPMI8866 cells after 7 and 24 h of treatment. At these later time points, phospho-RPS6KB was decreased in all 4 cell lines regardless of their EBV status (Fig. 6C). Reblotting with an anti-RPS6KB again verified that the treatment had virtually no effect on the endogenous form of the protein (except for its virtually complete disappearance in BL2 cells treated for 24 h which might be due to the high level of apoptosis observed at that time in these cells). Together, these results are consistent with the interpretation that treatment with nutlin-3 inhibits MTOR activity in both EBV-negative and EBV-positive latency III cells but with more rapid kinetics in the latter.
Figure 6.
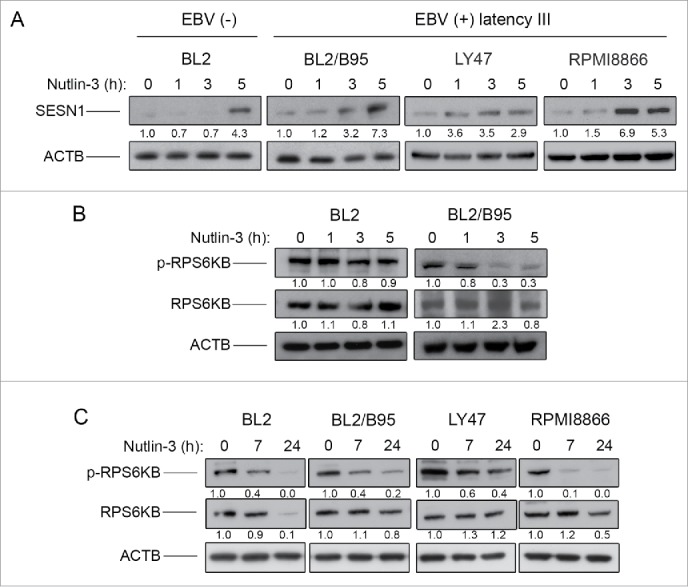
Effect of nutlin-3 treatment on SESN1 expression and MTOR kinase activity in EBV-negative and EBV-positive latency III lymphoid cell lines. SESN1 (A), p70S6K and phospho-P70S6K (B and C) expression was tested by western blot analysis of whole cell lysates at indicated times of treatment. Fold-change values reported under each blot were calculated in reference to untreated (0 h) controls and normalized to ACTB levels. The blots shown are representative of 3 independent experiments.
An inhibitor of autophagy induces apoptosis in EBV-positive latency III cells and enhances the effect of nutlin-3
Having observed that nutlin-3 induced higher levels of autophagy in EBV-positive latency III cells than in EBV-negative cells and since the former are much more resistant to nutlin-3-induced apoptosis than the latter, we wished to test the hypothesis that a high level of autophagy had a causative effect on the resistance to apoptosis induction. To this end, we used chloroquine to inhibit autophagy in EBV-positive latency III BL and LCL cells which were then submitted to nutlin-3 treatment for 24 h. Chloroquine is a lysosomotropic agent that raises intralysosomal pH and impairs autophagic protein degradation, resulting in the accumulation of ineffective autophagosomes and blockade of autophagosomes-lysosomes fusion, with ensuing death in cells relying on autophagy for survival.28 In these experiments the level of apoptosis was assessed by flow cytometry analysis (Fig. 7 and Figure S5 for representative flow cytometry plots). Treatment with chloroquine alone (100 µM) had a variable effect on the 4 cell lines tested. Apoptosis was induced to a very limited extent in BL2 and BL2/B95 cells (20 ± 5% and 27 ± 5%, respectively), RPMI8866 cells were moderately sensitive (38 ± 7%) and LY47 cells proved much more prone to apoptosis (58 ± 5%). Consistent with our previous report,17 a 24-h treatment with nutlin-3 (10 µM) alone also had a variable effect on the various cell lines, with the EBV-negative BL2 cells being very sensitive (64 ± 5% of apoptotic cells) and the EBV-positive latency III cells being more resistant (37 ±4 %, 34 ± 7% and 34 ± 6% in BL2/B95, LY47 and RPMI8866, respectively). When the 2 drugs were used in combination, an additive effect on the induction of apoptosis was observed in all 4 cell lines. Treatment of cells performed with chloroquine alone or nutlin-3 alone were compared to a combination thereof using the Mann-Whitney test and the Bonferroni correction to account for multiple testing. The differences observed between treatments were found statistically significant in all cell lines (P < 0.005 for BL2 and BL2/B95 and P < 0.0005 for LY47 and RPMI8866).
Figure 7.
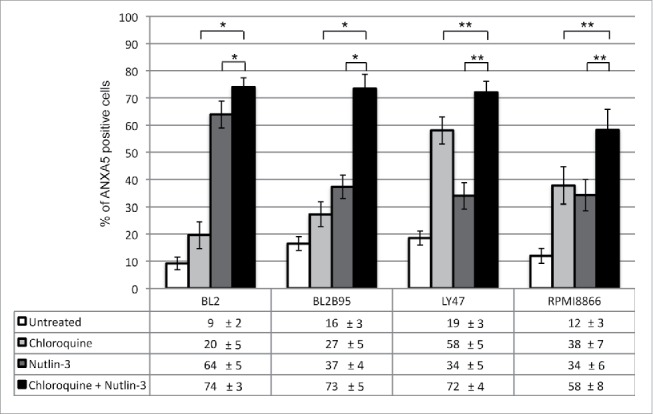
Combined effect of nutlin-3 and chloroquine treatments on apoptosis in EBV-negative and -positive latency III lymphoid cell lines. Cells were incubated with or without chloroquine (100 μM) for 2 h, followed by an incubation in the absence or presence of nutlin-3 for 24 h. Apoptosis was then measured by flow cytometry detection of ANXA5-FITC-positive cells (whether PI-positive or not). Values are means from at least 5 independent experiments, error bars represent standard deviation (SD). Statistical analyses were performed using the nonparametric Mann-Whitney test with Bonferroni correction to compare results obtained after treatment with chloroquine and nutlin-3 to those obtained after treatment with only one of these compounds. Statistical significance is as follows: *, P < 0.005; **P < 0.0005.
Additional experiments were performed with BAF used in place of chloroquine to inhibit autophagy. BL2, BL2/B95, LY47 and RPMI8866 were incubated with or without BAF (100 nM; 2 h) and then in the absence or presence of nutlin-3 (10 μM). The level of apoptosis was assessed 24 h later by flow cytometry (Fig. S6). Unexpectedly, treatment with BAF alone induced a very high level of apoptosis in the EBV-negative cell line BL2 (53 ±11 %), an effect which was more moderate in the 3 EBV-positive cell lines (36 ± 10%, 31 ± 3% and 21 ± 1% for BL2/B95, LY47 and RPMI8866 respectively). A treatment with both drugs induced either no increase of apoptosis (BL2/B95) or a moderate one (BL2, LY47 and RPMI8866) as compared to treatments with either drug alone.
Dose-response curves of the cytotoxic effect of nutlin-3 (5, 10, 20 and 30 µM) and chloroquine (50, 100, 150, and 200 µM) were also measured by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium) assays and the IC50 of each compound were determined (Table S1). The results confirmed that LY47 cells were more sensitive to chloroquine treatment than the other cell lines (IC50: 108 µM for LY47 as compared to 154, 200 and 174 µM for BL2, BL2/B95 and RPMI8866 respectively). They also showed that a lower dose of nutlin-3 was needed to kill the EBV-negative cells than the EBV-positive latency III cells (IC50: 5 µM for BL2 as compared to 14, 26 and 25 µM for BL2/B95 LY47 and RPMI8866, respectively). Cells were then treated with a sublethal dose of choloroquine (50 µM) in combination with the same concentrations of nutlin-3 to determine if chloroquine can sensitize the cells to nutlin-3. Results in Table S1 showed that chloroquine efficiently lowered the IC50 of nutlin-3 for LY47 (from 26 µM to 5 µM) and for RPMI8866 (from 25 µM to 18 µM). Altogether, these results show that in EBV-positive latency III cells which have a constitutive activation of autophagy and are poorly sensitive to nutlin-3 alone, treatment with the autophagy inhibitor chloroquine and nutlin-3 offers an efficient way to trigger cell death. Surprisingly, this additive effect was much less pronounced or even absent with BAF, another inhibitor of autophagy.
EBV-positive latency II and III B cell lymphoproliferations exhibit a constitutively active autophagy
It has been shown by immunohistochemical (IHC) staining of tissue sections that LC3-I displays a diffuse cytoplasmic staining whereas LC3-II appears as distinct puncta.29 To compare the constitutive level of autophagy between EBV-negative or EBV-positive latency I and EBV-positive latency II or III lymphoproliferations, we used IHC to evaluate the expression pattern of LC3 in 3 cases of BL (EBV-negative or EBV-positive latency I), 2 of DLBCL (EBV-positive latency II), 5 of PTLD (EBV-positive latency II or III) and one case of infectious mononucleosis (EBV-positive latency III). All EBV-positive DLBCL and PTLD cases as well as the one infectious mononucleosis case (all LMP1-positive) exhibited low (+) or moderate (++) levels of LC3-positive puncta, indicative of the presence of autophagosomes in these samples. This contrasted with the absence of any puncta in the 3 BL cases which displayed an homogeneous LC3 staining, reflecting its diffuse distribution in the cytoplasm (Table 1 and Fig. S7). A representative staining of EBNA2, LMP1 and LC3 puncta is shown in Figure 8. These in vivo results suggest that LMP1 expression is associated with constitutive autophagy in a subset of EBV-positive latency II and III lymphoproliferations.
Table 1.
Pattern of expression of LC3 by immunohistochemistry in B cell lymphoproliferations.
| Patient | Diagnosis | EBV latency type | EBER expression | EBNA2 expression | LMP1 expression | LC3 puncta |
|---|---|---|---|---|---|---|
| 1 | BL | Not EBV infected | − | − | − | − |
| 2 | BL | I | + | − | − | − |
| 3 | BL | I | + | − | − | − |
| 4 | EBV+ DLBCL of the elderly | II | + | − | + | + |
| 5 | EBV+ DLBCL of the elderly | II | + | − | + | + |
| 6 | Polymorphic PTLD | II | + | − | + | ++ |
| 7 | Polymorphic PTLD | III | + | + | + | + |
| 8 | Monomorphic PTLD | III | + | + | + | ++ |
| 9 | PTLD, early lesion: infectious mononucleosis-like | II`I | + | + | + | + |
| 10 | Infectious mononucleosis | III | + | + | + | + |
Figure 8.

Pattern of expression of EBNA2, LMP1 and LC3 in a case of EBV-positive latency III lymphoproliferation. Immunohistochemical staining obtained from tonsil biopsy. Panels are representative of images taken at 25x (A, B and C) or 40x magnification (D). (A) hematoxylineosin-Safran staining. (B) EBNA2 nuclear staining. (C) LMP1 cell membrane staining. (D) cytosolic LC3 staining with visible puncta.
Discussion
In this study, we have further analyzed molecular mechanisms involved in the resistance to nutlin-3-induced apoptosis in EBV-positive latency III lymphoblastoid cells. We have shown that these cells possess a high level of constitutive autophagy which was further enhanced by nutlin-3 treatment and that autophagy contributes to their resistance to apoptosis. Two distinct pathways are involved in this process. Constitutive autophagy correlates with an increased expression of BECN1 (as compared to EBV-negative cells), which is due, at least partially, to an LMP1-induced activation of NFKB. On the other hand, nutlin-3 treatment, which, as previously shown, induces activation of TP53 in these cells,16 results in: a) an accumulation of SESN1, a protein that can modulate the AMPK-MTOR pathway; b) the inhibition of MTOR kinase activity; c) the delivery of an additional autophagic stimulation. Finally, we have also shown that LMP1-positive B cell lymphoproliferations (latency II or III types) have a high autophagy level, as assessed by the expression of LC3-II in tissue sections.
Lee and Sugden have previously reported that LMP1 enhanced autophagy of EBV-infected cells in a process that regulated its own level of expression.30 Contrasting with our own observations, this upregulation of autophagy by LMP1 was not NFKB-dependent. Indeed, these authors showed that only the transmembrane domains 3 to 6 of the LMP1 protein, which are not involved in the activation of the NFKB signaling pathway, were required for induction of autophagy. This difference between our 2 reports may be due to the distinct cell systems we have been using. In our study, the latency III EBV-positive cell lines which have been used contained the whole EBV genome; in Lee and Sugden's experiments, the effects of the different domains of LMP1 on autophagy are analyzed in cell lines expressing either a mutant or a wild-type version of the LMP1 protein. Furthermore, it is also conceivable that both the C-terminal and the transmembrane domains of LMP1 be involved in the induction of autophagy in EBV-positive cells, the former through activation of NFKB, the latter via an unknown mechanism. Recent data from the Sugden's group have further emphasized the complexity of the signals activated by LMP1.31 Thus, the 6 transmembrane domains of LMP1 were shown to trigger autophagy eventually leading to apoptosis in a process that was blocked by the signaling activated by its C-terminal domain. The study of Sommermann et al., also underlines the importance of the role of LMP1-induced NFKB activation for the fate of the EBV-positive latency III B cells. Indeed in such cells, LMP1 expression increases glucose uptake through NFKB signaling whereas inhibition of NFKB decreased glucose import and induced autophagy. These authors also reported that a treatment of these autophagic cells with chloroquine resulted in cell death thereby suggesting that autophagy is a protective mechanism set up by the cells when NFKB is turned off.32
Other recent studies have examined the role of autophagy during viral infections. For instance, autophagy protects infected cells by degrading intracellular viruses through xenophagy, a form of autophagy whereby foreign molecules are degraded in autophagosomes, or by contributing to both the innate and adaptative immune responses (reviewed in ref. 33). With regard to EBV, it has been shown that autophagy contributes to MHC class II presentation of the viral EBNA1 antigen34 but it remains debated whether CD4-positive T cell clones specific for EBNA1 epitopes efficiently recognize LCL cells or not.35 On the other hand, some viruses utilize mechanisms that counteract autophagy to escape these antiviral processes or may even be able to use autophagy to promote their own replication (reviewed in refs.18, 36). In the present study, we have shown that B cells infected by EBV in vitro have a high constitutive level of autophagy in contrast to EBV-negative cells and, most importantly, we have extended these data on primary EBV-positive and -negative lymphoproliferations. We have also found that treating EBV-positive latency III cell lines with chloroquine, an autophagy inhibitor, induced apoptosis thereby suggesting that autophagy corresponds to a strategy developed by the virus to promote survival of its host cell. This process comes in addition to various other ways EBV utilizes to subvert the cellular machinery and induce transformation in B cells.37
Surprisingly, treatment with another inhibitor of autophagy, BAF, induced only a moderate level of apoptosis in EBV-positive latency III cells but had a strong effect in EBV-negative cells. Moreover, BAF treatment hardly sensitized both types of cells to nutlin-3-induced apoptosis. Since we have shown by various methods that EBV-negative BL2 cells have a much lower level of basal autophagy than EBV-positive latency III cells, this would suggest that the cytotoxicity induced by BAF in our B cell lines is not related to its ability to inhibit autophagy. Further studies are certainly needed to better understand the discrepancies observed between chloroquine and BAF treatments in EBV-positive latency III cells but also to address why inhibition of vacuolar ATP-ases but not deacidification of vacuoles by a weak base induces apoptosis in EBV-negative cells.
We have previously reported that in EBV-positive latency III cells, BCL2 forms complexes with the proapoptotic BAX protein, inhibits its activation and thereby participates in the resistance of these cells to apoptosis.17 Since BCL2 also negatively regulates autophagy by interacting with BECN1, the intriguing question remains: how is a high level of expression of BCL2 in these cells compatible with a high constitutive level of autophagy? The interactions of BCL2 with BAX and BECN1 are both regulated by multisite phosphorylation of BCL2 but it has been proposed by Wei et al. that a low level of BCL2 phosphorylation is sufficient to disrupt BCL2-BECN1 complexes whereas higher levels of phosphorylation are required to disrupt BCL2-BAX complexes.38 Our preliminary results (data not shown) indicate that BCL2 is phosphorylated in EBV-positive latency III cells suggesting that this occurs at a level sufficiently low to preclude interaction with BECN1 but not with BAX.
In a previous study, Chaumorcel et al. have reported that a high level of BCL2 induced by virus infection does not necessarily lead to inhibition of autophagy. In this study, it was shown that human cytomegalovirus (HCMV) infection induced a robust increase of BCL2 protein levels but that this upregulation of BCL2 was not involved in the HCMV-induced inhibition of autophagy. Indeed, in infected cells, BCL2 was phosphorylated by the JUN kinase and thereby unable to associate with BECN1.39 Further experiments including coimmunoprecipitations of BCL2 and BECN1 are clearly needed to determine if the situation is similar in our model. On the other hand, since phosphorylation of BECN1 also regulates its interaction with BCL2,4 it will be necessary to check its level of phosphorylation. Finally, Pattingre et al. have shown that only those BCL2/BECN1 complexes, which are localized to the endoplasmic reticulum effectively inhibit autophagy, suggesting that it would be worth investigating the subcellular localization of BCL2 in EBV-positive latency III cells.40
We have also reported here that nutlin-3 treatment enhanced expression of several genes involved in autophagy in both EBV-negative and -positive latency III cells. However, the nutlin-3 treatment resulted in the induction of very moderate levels of autophagy, if at all, in EBV-negative cells whereas in EBV-positive latency III cells, nutlin-3 strongly enhanced the basal level of autophagy. Part of this differential induction of autophagy could be related to the different kinetics of SESN1 upregulation and MTOR inhibition that we observed between EBV-negative and -positive cells. Recent studies have highlighted the dual role played by TP53 with regard to autophagy: when nuclear, TP53 transactivates target genes thus stimulating autophagy;25,41 when cytoplasmic, TP53 negatively regulates autophagy by interacting with RB1CC1 (RB1-inducible coiled-coil 1), the human ortholog of yeast Atg17 which is involved in autophagosome formation.42 In EBV-negative cells where autophagy is hardly induced, it would be interesting to test the level of RB1CC1 and of cytoplasmic TP53 after nutlin-3 treatment to determine whether their interaction could regulate this catabolic process. Similarly, it would be worth determining the role of DRAM1, the gene most strongly induced by nutlin-3 in both EBV-negative and -positive cells, since this protein is an inducer of autophagy but also contributes to apoptosis.25
The role of autophagy in cancer is complex and controversial. Autophagy can result in tumor suppression through various mechanisms but it can also be involved in tumor survival and in resistance to cancer treatment (reviewed in refs.43, 44). In this study, we have found that using chloroquine, an inhibitor of lysosome and autophagosome fusion, was sufficient to promote apoptosis in EBV-positive latency III cells. Also, pretreating with chloroquine sensitized these cells to nutlin-3-induced apoptosis.
In vitro experiments and preclinical studies in various mouse xenograft tumor models have demonstrated that chloroquine potentiates antitumoral treatments through its capacity to inhibit autophagy.45-47 So far, chloroquine (and its derivative hydroxychloroquine) is the only autophagy inhibitor under clinical testing—in combination with conventional drugs or targeted tools—for the treatment of refractory tumors. Although its mechanisms of action in vivo remain unclear, the first results are encouraging with tumor growth stabilized or median survival times increased observed in patients treated with chloroquine.48
Finally, it is worth noting that in the work reported by Amaravadi et al., the induction of TP53 in mice carrying a Myc-induced lymphoma promotes tumor cell apoptosis but with surviving tumor cells which display a high level of autophagy. In this murine model, inhibiting autophagy using chloroquine or ATG5 shRNA enhances apoptosis and stimulates tumor regression.49 Our in vitro results, fully consistent with those obtained in vivo, confirm that the autophagy induced by TP53 activation can result in tumor cell survival and resistance to treatment. Since EBV-positive latency III cells have a high level of expression of BCL2, our results are also in accordance with those of MacLean et al. showing that chloroquine-induced cell death of Tp53+/+ Eμ-Myc murine B cells is not impaired by overexpression of the antiapoptotic protein BCL2.50 Using chloroquine thus appears interesting to prevent induction of autophagy and enhance cytotoxic efficacy of TP53 activation in cells treated with nutlin-3 or other drugs, which induce a TP53-dependent apoptosis. However, since such an induction of autophagy by TP53 occurs only in cells with a high level of basal autophagy associated with a high expression of BECN1, it could be of interest to test for BECN1 levels prior to any chloroquine treatment.
Materials and Methods
Cell lines
All BL cell lines were originally established from endemic or sporadic cases of BL. BL2, BL28, BL40, BL2/B95, BL28/B95, BL40/B95, and LY47 were kindly provided by the International Agency for Research on Cancer (IARC, Lyon, France); Seraphina was provided by Prof. G. Klein (Stockholm, Sweden); BL2/B95, BL28/B95 and BL40/B95 were generated by stable infection of the original EBV-negative BL2, BL28 and BL40 cells with the B95–8 EBV strain. LCLs were obtained by the in vitro immortalization of normal B lymphocytes. IARC 211 cells were established from the normal B lymphocytes of patient BL40. Priess and Remb1 cells were kindly provided by Dr J.G. Bodmer (London, UK) and RMPI8866 by Dr J. Banchereau (Dardilly, France). DG75 tTA LMP1, a stable transfectant of the DG75 cell line with a tetracycline-regulated LMP1 expression,51 was kindly provided by Dr O. Dellis (Orsay, France). All cell lines, except DG75, express wt TP53. They were cultured in complete RPMI 1640 medium (containing 2 mM L-glutamine, 1 mM sodium pyruvate, 20 mM glucose, 100 U/ml penicillin and 100 μg/ml streptomycin and supplemented with 10% heat-inactivated fetal calf serum). Tetracycline hydrochloride (1 µg/ml; MP Biochemicals, 02194542) was used to silence tetracycline-regulated LMP1 expression in the DG75 tTA LMP1 cell line.
Antibodies and reagents
Nutlin-3 was kindly provided by Roche Laboratories. Chloroquine (C6628) and bafilomycin A1 (B1793) were obtained from Sigma-Aldrich. Anti-RELA/p65 monoclonal antibody (mAb) was purchased from Santa Cruz Biotechnology (sc-8008). Anti-ACTB/β-actin (A5316) mAb was obtained from Sigma-Aldrich. Anti-SESN1 polyclonal antibody (pAb; ab67156) was obtained from Abcam. Anti-BECN1 pAb (3738), anti-RPS6KB/p70S6K pAb (9202), anti-phospho-RPS6KB/p70S6K mAb (Thr389, 9206) and anti-LC3B pAb (2775) were purchased from Cell Signaling technology. Anti-LMP1 mAb (M 0897) was obtained from Dako France SAS. Anti-LC3B mAb (0231–100/LC3–5F10) used for immunohistochemistry was obtained from Nanotools and anti-EBNA2 (NVL-EBV-PE2) from Novocastra. Horseradish peroxidase (HRP)-conjugated rabbit anti-mouse IgG (NA 931) and HRP-conjugated donkey anti-rabbit IgG (NA 934) were purchased from GE Healthcare.
Western blot analysis
A pellet containing 1 × 106 cells was solubilized by incubation in ice-cold lysis buffer (150 mM NaCl, 50 mM Tris. pH 7.4, 5 mM EDTA, 0.1% NP40 (IGEPAL-CA630, Sigma Aldrich, I3021), 0.5% sodium deoxycholate (Sigma Aldrich, D6750), 0.1% SDS (Fluka, 05030), complete protease inhibitor cocktail (Roche, 11 873 580 001) for 15 min or directly in loading buffer (60 mM Tris, pH 6.8, 2% SDS, 10% glycerol, 1% β-mercaptoethanol) for further detection of phosphoproteins. Sample loading buffer was added and the mixture was boiled for 5 min. Proteins were separated by electrophoresis and western blotting was realized as previously described.17
Lentivirus and stable transduction of shRNAs
RPMI8866 cells were cultured in a 96-well plate with lentiviral particules containing a control shRNA (MISSION TRC2 pLKO.5-puro Control Transduction Particle, SHC202V, Sigma Aldrich) or a shRNA targetting RELA/p65 (MISSION Lentiviral Transduction Particles, SHCLNV-NM_021975, clone ID: TRCN0000014686, Sigma Aldrich) at a multiplicity of infection of 5 in complete RPMI medium supplemented with hexadimethrine bromide (4 µg/ml; Sigma Aldrich, D6750). After 24 h, 100 µl of complete RPMI medium was added. After 48 h, transduced cells were washed with phosphate-buffered saline (Life technologies, 18912–014) and then cultured in complete RPMI medium containing puromycin (0.6 µg/ml). Stable transduced cells were obtained after 3 to 4 wk of puromycin selection.
Gene expression analysis
Cells were treated with 10 µM nutlin-3 for various periods of time. Three replicates per condition were done. RNA extracted from each replicate (RNeasy Micro kit, Qiagen, 74004) was used for gene expression with Agilent® 4×44 Human Gene Expression arrays (Agilent Technologies). After single color hybridization and array scanning, performed as recommended by the manufacturer, data were carefully checked using several quality control. Raw data were extracted from image with Feature Extraction (10.5.1.1, Agilent). These data were log2-transformed and then normalized using the quantile method. An unsupervised cluster was computed using Euclidian distance and Ward method for clusterisation, and validated by bootstrapping. PCA analysis was also performed to analyze sources of variance between samples. Supervised analysis of genes differentially expressed between groups of interest was performed using the moderated t test from the LIMMA package. The expression profile of the set of significant differentially expressed genes was used to compute a supervised cluster using the same parameters as for unsupervised cluster.
Detection of autophagy by immunofluorescence staining
EBV-positive latency III and EBV-negative cells were incubated with or without nutlin-3 (10 µM) for 16 h at 37°C. Cells were stained with MDC (100 µM; Sigma-Aldrich, 30432) for 10 min at 37°C. After 2 washes in phosphate-buffered saline, cells were plated on glass slides pretreated with poly-D-lysine. Cells were then fixed in 3% paraformaldehyde for 20 min, washed and examined by fluorescence microscopy (Leica confocal microscope SPE, Leica Microsystèmes SA, Nanterre, France). For each condition, the percentage of cells with characteristic MDC staining dots indicative of autophagy was assessed. MDC staining was also assessed by flow cytometry using an LSR II Becton Dickinson flow cytometer equipped with a 355 nm UV laser (BD Biosciences, Le Pont de Claix, France). Data were analyzed, and MFI values calculated, using the BD FACSDiva software (BD Biosciences). Graphs were done with the FlowJo software (Flow Jo, Tree Star Inc.).
Autophagic vesicles were detected using the CYTO-ID autophagy detection kit according to the manufacturer's instructions (Enzo Life Science, ENZ-51031). The cationic amphilic tracer dye used in this assay selectively labels pre-autophagosomes, autophagosomes and autolysosomes but not lysosomes. Cells incubated with or without nutlin-3 were washed, resuspended in 0.5 ml of freshly diluted Cyto-ID Green stain reagent and incubated at 37°C for 30 min in the dark. Cells were then washed and analyzed by flow cytometry using an Accuri C6 Flow cytometer (BD Biosciences, Le Pont de Claix, France).
Electron microscopy
Cells were incubated with or without 10 µM nutline-3 for 7 h. For Epon embedding, cell pellet fractions were fixed for 1 h at room temperature in 2% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4. Cells pellets were postfixed for 1 h with 1% osmium tetroxide in 0.1M cacodylate buffer and 1.5% potassium ferrocyanide. Cells were dehydrated in increasing concentrations of ethanol and embedded in Epon812 (EMS, 14120). Polymerization was carried out for 72 h at 56°C. Ultrathin sections (70 nm) were collected on collodion-carbon-coated copper grids (EMS, G200-Cu). Sections were contrasted by uranyl acetate and lead citrate. Cells were observed with a Zeiss 902 electron microscope (Carl Zeiss microscopy GmbH, Jena, Germany) in filtered zero-loss mode using a CCD Megaview III camera and a SIS system (Olympus Soft Imaging Solutions GmbH, Münster, Germany).
Induction and quantification of apoptosis
5 × 105 cells were treated for 24 h, at 37°C, with nutlin-3 (10 μM), chloroquine (100 µM) or both of these compounds. Cells were stained with FITC-labeled ANXA5/annexin-V (Roche Applied Science, 11–828–681–001) and propidium iodide (PI) and analyzed by flow cytometry as previously described.17 Apoptosis is assessed by the number of ANXA5-positive cells whether PI-positive or not.
Statistical analysis
Data are expressed as mean ± standard deviation (SD). Statistical differences between each treatment alone (chloroquine or nutlin-3) and the combination of these drugs were analyzed by the nonparametric Mann-Whitney 2-tailed test. A Bonferroni correction was applied to account for multiple testing (8 pair-wise comparisons performed) and tests with a P value < 0.006 were considered statistically different (α = 0.05/8).
Patients
Lymphoma cases were retrieved from the pathology departments in 2 French university hospitals (Créteil and Kremlin-Bicêtre). All cases were independently reviewed by at least 2 pathologists specialized in hematology and were classified as EBV (+) DLBCL of the elderly (n = 2), BL (n = 3), infectious mononucleosis (n = 1) or PTLD (n = 4). In accordance to French laws, all patients have given informed consent.
Immunohistochemistry
Staining was performed as described in ref.29 Briefly, paraffin-embedded tissue sections were deparaffinized with xylene, dehydrated through graded alcohols and subjected to antigen retrieval using 10 mM Tris-EDTA buffer (Diapath, T0013) for 25 min at 98°C. After cooling, sections were incubated in BLOXALL blocking solution (Vector Laboratories, SP6000) for 5 min to block for endogenous peroxidase. Sections were then washed with TBS and incubated with primary antibodies (anti-LC3, anti-EBNA2, anti-LMP1 mAbs) for 45 min at room temperature. After 2 washes in TBS-Tween for 5 min, sections were incubated with secondary anti-mouse antibody (Dako EnVision + System-HRP labeled polymer, K4000) for 30 min at room temperature. After washing, sections were incubated with DAB (3,3′-diaminobenzidine, Vector Laboratories, SK-4100) for 10 min at room temperature and then washed in TBS. Sections were then counter-stained with Mayer hematoxylin, mounted (Shandon, Immu-Mount, 9990402) and observed under a microscope Axioskop 2 (Zeiss, Marly le Roi, France).
Disclosure of Potential Conflict of interest
The authors declare no conflict of interest.
Acknowledgments
We thank Sophie Salome-Desnoulez and Yann Lécluse (Imaging and Cytometry Platform, Institut Gustave Roussy) for expert technical assistance in performing confocal microscopy and flow cytometry analyses. We are very grateful to Agnès Laplanche (Department of Biostatistics and Epidemiology, Institut Gustave-Roussy) for her great help in statistical analyses and to Evelyne May (UMR 8126 CNRS) for helpful discussions and suggestions.
Supplemental material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was supported by grants from the Fondation de France 00012093 (JW), the Cancéropole and Région Ile-de-France (ERABL, IF09–2092/R), the Fondation pour la Recherche Médicale (doctoral fellowship to AP) and the Université Paris-Sud (BQR 2009).
References
- 1.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997; 88:323-31; PMID:9039259; http://dx.doi.org/ 10.1016/S0092-8674(00)81871-1 [DOI] [PubMed] [Google Scholar]
- 2.Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer 2002; 2:594-604; PMID:12154352; http://dx.doi.org/ 10.1038/nrc864 [DOI] [PubMed] [Google Scholar]
- 3.Mizushima N. Autophagy: process and function. Genes Dev 2007; 21:2861-73; PMID:18006683; http://dx.doi.org/ 10.1101/gad.1599207 [DOI] [PubMed] [Google Scholar]
- 4.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011; 18:571-80; PMID:21311563; http://dx.doi.org/ 10.1038/cdd.2010.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008; 134:451-60; PMID:18692468; http://dx.doi.org/ 10.1016/j.cell.2008.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu EY, Ryan KM. Autophagy and cancer–issues we need to digest. J Cell Sci 2012; 125:2349-58; PMID:22641689; http://dx.doi.org/ 10.1242/jcs.093708 [DOI] [PubMed] [Google Scholar]
- 7.Tasdemir E, Chiara Maiuri M, Morselli E, Criollo A, D'Amelio M, Djavaheri-Mergny M, Cecconi F, Tavernarakis N, Kroemer G. A dual role of p53 in the control of autophagy. Autophagy 2008; 4:810-4; PMID:18604159; http://dx.doi.org/ 10.4161/auto.6486 [DOI] [PubMed] [Google Scholar]
- 8.Dickens MP, Fitzgerald R, Fischer PM. Small-molecule inhibitors of MDM2 as new anticancer therapeutics. Semin Cancer Biol 2010; 20:10-8; PMID:19897042; http://dx.doi.org/ 10.1016/j.semcancer.2009.10.003 [DOI] [PubMed] [Google Scholar]
- 9.Vu BT, Vassilev L. Small-molecule inhibitors of the p53-MDM2 interaction. Curr Top Microbiol Immunol 2011; 348:151-72; PMID:21046355 [DOI] [PubMed] [Google Scholar]
- 10.Vassilev LT, Vu BT,Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N,Kammlott U, Lukacs C,Klein C,et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303:844-8; PMID:14704432; http://dx.doi.org/ 10.1126/science.1092472 [DOI] [PubMed] [Google Scholar]
- 11.Stuhmer T, Chatterjee M, Hildebrandt M, Herrmann P, Gollasch H, Gerecke C, Theurich S, Cigliano L, Manz RA, Daniel PT, et al. Nongenotoxic activation of the p53 pathway as a therapeutic strategy for multiple myeloma. Blood 2005; 106:3609-17; PMID:16081689; http://dx.doi.org/ 10.1182/blood-2005-04-1489 [DOI] [PubMed] [Google Scholar]
- 12.Sarek G, Kurki S, Enback J, Iotzova G, Haas J, Laakkonen P, Laiho M, Ojala PM. Reactivation of the p53 pathway as a treatment modality for KSHV-induced lymphomas. J Clin Invest 2007; 117:1019-28; PMID:17364023; http://dx.doi.org/ 10.1172/JCI30945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Secchiero P, Barbarotto E, Tiribelli M, Zerbinati C, di Iasio MG, Gonelli A, Cavazzini F, Campioni D, Fanin R, Cuneo A, et al. Functional integrity of the p53-mediated apoptotic pathway induced by the nongenotoxic agent nutlin-3 in B-cell chronic lymphocytic leukemia (B-CLL). Blood 2006; 107:4122-9; PMID:16439677; http://dx.doi.org/ 10.1182/blood-2005-11-4465 [DOI] [PubMed] [Google Scholar]
- 14.Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol Med 2007; 13:23-31; PMID:17126603; http://dx.doi.org/ 10.1016/j.molmed.2006.11.002 [DOI] [PubMed] [Google Scholar]
- 15.Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov 2014; 13:217-36; PMID:24577402; http://dx.doi.org/ 10.1038/nrd4288 [DOI] [PubMed] [Google Scholar]
- 16.Renouf B, Hollville E, Pujals A, Tetaud C, Garibal J, Wiels J. Activation of p53 by MDM2 antagonists has differential apoptotic effects on Epstein-Barr virus (EBV)-positive and EBV-negative Burkitt's lymphoma cells. Leukemia 2009; 23:1557-63; PMID:19421231; http://dx.doi.org/ 10.1038/leu.2009.92 [DOI] [PubMed] [Google Scholar]
- 17.Pujals A, Renouf B, Robert A, Chelouah S, Hollville E, Wiels J. Treatment with a BH3 mimetic overcomes the resistance of latency III EBV (+) cells to p53-mediated apoptosis. Cell Death Dis 2011; 2:e184; PMID:21796156; http://dx.doi.org/ 10.1038/cddis.2011.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cavignac Y, Esclatine A. Herpesviruses and autophagy: catch me if you can! Viruses 2010; 2:314-33; PMID:21994613; http://dx.doi.org/ 10.3390/v2010314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/ 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huen DS, Henderson SA, Croom-Carter D, Rowe M. The Epstein-Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-kappa B and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene 1995; 10:549-60; PMID:7845680 [PubMed] [Google Scholar]
- 21.Laherty CD, Hu HM, Opipari AW, Wang F, Dixit VM. The Epstein-Barr virus LMP1 gene product induces A20 zinc finger protein expression by activating nuclear factor kappa B. J Biol Chem 1992; 267:24157-60; PMID:1332946 [PubMed] [Google Scholar]
- 22.Nivon M, Richet E, Codogno P, Arrigo AP, Kretz-Remy C. Autophagy activation by NFkappaB is essential for cell survival after heat shock. Autophagy 2009; 5:766-83; PMID:19502777; http://dx.doi.org/ 10.4161/auto.8788 [DOI] [PubMed] [Google Scholar]
- 23.Ryu HJ, Kim JE, Yeo SI, Kang TC. p65/RelA-Ser529 NF-kappaB subunit phosphorylation induces autophagic astroglial death (Clasmatodendrosis) following status epilepticus. Cell Mol Neurobiol 2011; 31:1071-8; PMID:21598036; http://dx.doi.org/ 10.1007/s10571-011-9706-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Copetti T, Demarchi F, Schneider C. p65/RelA binds and activates the beclin 1 promoter. Autophagy 2009; 5:858-9; PMID:19458474; http://dx.doi.org/ 10.4161/auto.8822 [DOI] [PubMed] [Google Scholar]
- 25.Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006; 126:121-34; PMID:16839881; http://dx.doi.org/ 10.1016/j.cell.2006.05.034 [DOI] [PubMed] [Google Scholar]
- 26.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/ 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Isotani S, Hara K, Tokunaga C, Inoue H, Avruch J, Yonezawa K. Immunopurified mammalian target of rapamycin phosphorylates and activates p70 S6 kinase alpha in vitro. J Biol Chem 1999; 274:34493-8; PMID:10567431; http://dx.doi.org/ 10.1074/jbc.274.48.34493 [DOI] [PubMed] [Google Scholar]
- 28.Glaumann H, Ahlberg J. Comparison of different autophagic vacuoles with regard to ultrastructure, enzymatic composition, and degradation capacity–formation of crinosomes. Exp Mol Pathol 1987; 47:346-62; PMID:3678466; http://dx.doi.org/ 10.1016/0014-4800(87)90018-9 [DOI] [PubMed] [Google Scholar]
- 29.Rosenfeldt MT, Nixon C, Liu E, Mah LY, Ryan KM. Analysis of macroautophagy by immunohistochemistry. Autophagy 2012; 8:963-9; PMID:22562096; http://dx.doi.org/ 10.4161/auto.20186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee DY, Sugden B. The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 2008; 27:2833-42; PMID:18037963; http://dx.doi.org/ 10.1038/sj.onc.1210946 [DOI] [PubMed] [Google Scholar]
- 31.Pratt ZL, Zhang J, Sugden B. The latent membrane protein 1 (LMP1) oncogene of Epstein-Barr virus can simultaneously induce and inhibit apoptosis in B-cells. J Virol 2012; 86:4380-93; PMID:22318153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sommermann TG, O'Neill K, Plas DR, Cahir-McFarland E. IKKbeta and NF-kappaB transcription govern lymphoma cell survival through AKT-induced plasma membrane trafficking of GLUT1. Cancer Res 2011; 71:7291-300; PMID:21987722; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jackson WT. Viruses and the autophagy pathway. Virology 2015; 479-480:450-6; PMID:25858140; http://dx.doi.org/ 10.1016/j.virol.2015.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Munz C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005; 307:593-6; PMID:15591165; http://dx.doi.org/ 10.1126/science.1104904 [DOI] [PubMed] [Google Scholar]
- 35.Leung CS, Haigh TA, Mackay LK, Rickinson AB, Taylor GS. Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4 epitope display. Proc Natl Acad Sci U S A 2010; 107:2165-70; PMID:20133861; http://dx.doi.org/ 10.1073/pnas.0909448107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kirkegaard K. Subversion of the cellular autophagy pathway by viruses. Curr Top Microbiol Immunol 2009; 335:323-33; PMID:19802573 [DOI] [PubMed] [Google Scholar]
- 37.Klein G, Klein E, Kashuba E. Interaction of Epstein-Barr virus (EBV) with human B-lymphocytes. Biochem Biophys Res Commun 2010; 396:67-73; PMID:20494113; http://dx.doi.org/ 10.1016/j.bbrc.2010.02.146 [DOI] [PubMed] [Google Scholar]
- 38.Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008; 4:949-51; PMID:18769111; http://dx.doi.org/ 10.4161/auto.6788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaumorcel M, Lussignol M, Mouna L, Cavignac Y, Fahie K, Cotte-Laffitte J, Geballe A, Brune W, Beau I, Codogno P, et al. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1. J Virol 2012; 86:2571-84; PMID:22205736; http://dx.doi.org/ 10.1128/JVI.05746-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927-39; PMID:16179260; http://dx.doi.org/ 10.1016/j.cell.2005.07.002 [DOI] [PubMed] [Google Scholar]
- 41.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A 2005; 102:8204-9; PMID:15928081; http://dx.doi.org/ 10.1073/pnas.0502857102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morselli E, Shen S, Ruckenstuhl C, Bauer MA, Marino G, Galluzzi L, Criollo A, Michaud M, Maiuri MC, Chano T, et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle 2011; 10:2763-9; PMID:21775823; http://dx.doi.org/ 10.4161/cc.10.16.16868 [DOI] [PubMed] [Google Scholar]
- 43.Eskelinen EL. The dual role of autophagy in cancer. Curr Opin Pharmacol 2011; 11:294-300; PMID:21498118; http://dx.doi.org/ 10.1016/j.coph.2011.03.009 [DOI] [PubMed] [Google Scholar]
- 44.Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis 2011; 32:955-63; PMID:21317301; http://dx.doi.org/ 10.1093/carcin/bgr031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo XL, Li D, Hu F, Song JR, Zhang SS, Deng WJ, Sun K, Zhao QD, Xie XQ, Song YJ, et al. Targeting autophagy potentiates chemotherapy-induced apoptosis and proliferation inhibition in hepatocarcinoma cells. Cancer Lett 2012; 320:171-9. [DOI] [PubMed] [Google Scholar]
- 46.Sasaki K, Tsuno NH, Sunami E, Tsurita G, Kawai K, Okaji Y, Nishikawa T, Shuno Y, Hongo K, Hiyoshi M, et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010; 10:370; PMID:20630104; http://dx.doi.org/ 10.1186/1471-2407-10-370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 2011; 10:1533-41; PMID:21878654; http://dx.doi.org/ 10.1158/1535-7163.MCT-11-0047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maes H, Rubio N, Garg AD, Agostinis P. Autophagy: shaping the tumor microenvironment and therapeutic response. Trends Mol Med 2013; 19:428-46; PMID:23714574; http://dx.doi.org/ 10.1016/j.molmed.2013.04.005 [DOI] [PubMed] [Google Scholar]
- 49.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117:326-36; PMID:17235397; http://dx.doi.org/ 10.1172/JCI28833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maclean KH, Dorsey FC, Cleveland JL, Kastan MB. Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. J Clin Invest 2008; 118:79-88; PMID:18097482; http://dx.doi.org/ 10.1172/JCI33700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Floettmann JE, Ward K, Rickinson AB, Rowe M. Cytostatic effect of Epstein-Barr virus latent membrane protein-1 analyzed using tetracycline-regulated expression in B cell lines. Virology 1996; 223:29-40; PMID:8806537; http://dx.doi.org/ 10.1006/viro.1996.0452 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.