

Abstract
A key point in starvation-induced autophagy occurs at the end of the process, where lysosomes are regenerated from autolysosomes through a pathway termed autophagic lysosome reformation (ALR). ALR occurs when autolysosomal MTOR becomes reactivated by amino acids derived from the autophagic delivery of protein cargo. This activation not only turns off autophagosome formation but also leads to reformation of lysosomes, ready for the next round of autophagy, through a series of events involving autolysosomal tubulation. We have now found that MTOR regulates multiple steps of ALR including direct activation of the PIK3C3-UVRAG lipid kinase complex to enable autolysosomal tubules to break away and regenerate lysosomes.
Keywords: autophagic lysosome reformation, lysosome, MTOR, phosphatidylinositol 3-phosphate, tubulation, UVRAG, VPS34
The ability of a healthy cell to induce autophagy offers a beneficial stress response, enabling survival under nutrient starvation or in clearing toxic components such as aggregates or intracellular pathogens. Due to this, autophagy has become an attractive therapeutic target. To date, much of our endeavors to understand autophagy have focused on the signals and components required for its activation. However, it is now clear that the terminal stage of autophagy, that is, turnover of autolysosomal contents and regeneration of lysosomes, is also highly regulated and, as we have found, its normal progression is essential for cell survival. This ouroboric-like lysosomal cycle therefore offers another therapeutic target area. Recent findings from our lab identified the lipid phosphatidylinositol-3-phosphate (PtdIns3P), the product of the lipid kinase PIK3C3/VPS34 as a novel regulator of lysosomal regeneration. Additionally, this highlights the conservation of factors, namely MTOR and PIK3C3, that are involved at multiple stages of autophagy regulation.
Inhibition of the amino acid-responsive MTOR kinase complex is a key signal for autophagosome biogenesis, primarily through activation of the ULK kinase complex that occurs in conjunction with that of the PIK3C3-BECN1-ATG14 complex. Autophagosomes then fuse with endosomes and/or lysosomes to form autolysosomes, allowing the degradation of cytoplasmic cargo. Macromolecular building blocks relinquished by this degradative activity are subsequently transported out of the autolysosome and reused by the cell. During a sustained starvation response, this release of lysosomal nutrients is sufficient to reactivate MTOR, providing negative feedback to inhibit further autophagosome formation and prevent the breakdown of more components than is necessary for survival. The cell, however, is faced with an additional problem as lysosomes must be reformed from the large amount of autolysosomes produced during starvation. This MTOR reactivation also plays a critical role here.
MTOR reactivation leads to the formation of tubules that extend from the autolysosome, which then undergo scission by the small GTPase DNM2 (dynamin 2). These small tubular ‘proto-lysosome’ structures contain several lysosomal resident membrane proteins, such as LAMP1, and go on to acquire the necessary hydrolases during their progression to mature lysosomes. This process has been termed autophagic lysosomal reformation (ALR). While previously known to require MTOR activity, our work has now dissected further the molecular details involved in ALR regulation. We uncovered a small but significant pool of PtdIns3P on lysosomal membranes and found that loss of this, by specific pharmacological inhibition of PIK3C3, results in numerous tubular lysosome extensions that persist without undergoing scission. The tubular phenotype was only observed with active MTOR, suggesting MTOR is required and is upstream of PIK3C3 activity. In support of this, we found that the UVRAG-containing PIK3C3 complex is indeed an MTOR substrate. UVRAG can be directly phosphorylated by MTOR in vitro and in cells on 2 serine residues, serine 550 and 571, and a major consequence of this is an increase in associated PIK3C3 activity and concomitant PtdIns3P production. Abolishing UVRAG phosphorylation by mutation of the serine residues to alanine, leads to loss of lysosomal PtdIns3P and generates long, persistent lysosomal tubules. This effect is specific to lysosomal tubulation and almost identical to that observed with pharmacological PIK3C3 inhibition. Together this suggests that lysosomal MTOR activation of PIK3C3 drives local production of PtdIns3P to recruit or activate the DNM/dynamin machinery required for tubule scission and proto-lysosome formation. It is important to note that inhibition of MTOR blocks lysosomal tubule formation in general, implying MTOR initiates tubule formation as well regulates its PIK3C3-mediated scission.
Disruption of MTOR-UVRAG phosphorylation afforded us the ability to specifically stall ALR midway, which revealed insights to the importance of this process. Short-term autophagy induction is not significantly affected by loss of UVRAG phosphorylation; however, sustained serum starvation induces cell death much more rapidly in UVRAG mutant cells, which undergo apoptotic cell death shortly after reactivation of MTOR. Interestingly, pharmacologically inhibiting MTOR reactivation prevents cell death, implying the inability to complete ALR once tubules have formed has severe consequences for cell survival. Our data suggest that persistent tubulation renders the cell more sensitive to lysosomal-mediated cell death.
Figure 1 summarizes the induction of autophagy and lysosomal recycling, highlighting the MTOR-PIK3C3-UVRAG role in lysosomal tubulation. It is abundantly clear that autophagy is regulated at both its start and end points by several of the same mediators and that the autolysosome, often considered the terminal organelle of autophagy, is a key signaling platform for this initiation and termination control. Inhibition of MTOR drives autophagy initiation, but further blocking its reactivation prevents resolution of autophagy through ALR. Depending on the stage of ALR disruption, we have found this can lead to cell death: while it may be beneficial to take advantage of this phenomenon in cancer cells, in healthy cells this could have dire consequences. The genetic condition hereditary spastic paraplegia, for example, is defined by the loss of motor neurons and is most commonly associated with mutations in the PtdIns3P binding and lysosome-localized protein ZFYVE26. It is tempting to speculate that this increased neuronal death may be attributable to defects in the ALR process. Importantly, further understanding of the terminal stages of autophagy and lysosomal signaling may provide novel targets for regulating autophagy and treating disease.
Figure 1.
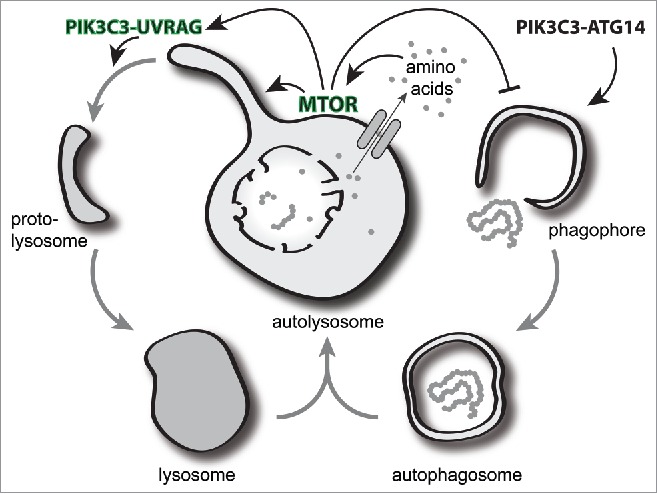
The autolysosome plays a central role as a signaling platform for the resolution of autophagic responses. Following fusion of an autophagosome with the lysosome, the autolysosome is generated and degrades cellular material. The release of newly acquired nutrients from the autolysosome locally activates MTOR at the lysosome and is critical for a 2-part response. First, the activation of MTOR can inhibit the autophagy initiation machinery to prevent further autophagosome formation. Second, MTOR activity drives autolysosome tubulation and phosphorylates the PIK3C3-UVRAG complex to generate localized PtdIns3P required for the subsequent scission of tubules. The cleaved tubules or proto-lysosomes are then able to mature by acquisition of hydrolases into new lysosomes primed for further fusion.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.