

Abstract
Study Objectives:
Sleep rebound—the increase in sleep that follows sleep deprivation—is a hallmark of homeostatic sleep regulation that is conserved across the animal kingdom. However, both the mechanisms that underlie sleep rebound and its relationship to habitual daily sleep remain unclear. To address this, we developed an efficient thermogenetic method of inducing sleep deprivation in Drosophila that produces a substantial rebound, and applied the newly developed method to assess sleep rebound in a screen of 1,741 mutated lines. We used data generated by this screen to identify lines with reduced sleep rebound following thermogenetic sleep deprivation, and to probe the relationship between habitual sleep amount and sleep following thermogenetic sleep deprivation in Drosophila.
Methods:
To develop a thermogenetic method of sleep deprivation suitable for screening, we thermogenetically stimulated different populations of wake-promoting neurons labeled by Gal4 drivers. Sleep rebound following thermogenetically-induced wakefulness varies across the different sets of wake-promoting neurons that were stimulated, from very little to quite substantial. Thermogenetic activation of neurons marked by the c584-Gal4 driver produces both strong sleep loss and a substantial rebound that is more consistent within genotypes than rebound following mechanical or caffeine-induced sleep deprivation. We therefore used this driver to induce sleep deprivation in a screen of 1,741 mutagenized lines generated by the Drosophila Gene Disruption Project. Flies were subjected to 9 h of sleep deprivation during the dark period and released from sleep deprivation 3 h before lights-on. Recovery was measured over the 15 h following sleep deprivation. Following identification of lines with reduced sleep rebound, we characterized baseline sleep and sleep depth before and after sleep deprivation for these hits.
Results:
We identified two lines that consistently exhibit a blunted increase in the duration and depth of sleep after thermogenetic sleep deprivation. Neither of the two genotypes has reduced total baseline sleep. Statistical analysis across all screened lines shows that genotype is a strong predictor of recovery sleep, independent from effects of genotype on baseline sleep.
Conclusions:
Our data show that rebound sleep following thermogenetic sleep deprivation can be genetically separated from sleep at baseline. This suggests that genetically controlled mechanisms of sleep regulation not manifest under undisturbed conditions contribute to sleep rebound following thermogenetic sleep deprivation.
Citation:
Dubowy C, Moravcevic K, Yue Z, Wan JY, Van Dongen HP, Sehgal A. Genetic dissociation of daily sleep and sleep following thermogenetic sleep deprivation in Drosophila. SLEEP 2016;39(5):1083–1095.
Keywords: sleep deprivation, sleep rebound, thermogenetics, Drosophila
Significance.
Sleep drive builds up during both spontaneous waking and enforced sleep deprivation. The mechanisms that underlie the build up of sleep drive in these two conditions are often presumed to be the same. However, this premise has not been tested in a large-scale genetic screen. Here, we enforce wakefulness in the fruit fly Drosophila melanogaster by activating wake-promoting neurons in the brain and test 1,741 mutated lines to identify lines with aberrant responses to sleep deprivation. We find that genetic differences can have a large impact on sleep after sleep deprivation in ways that cannot be predicted by differences in habitual sleep amount, suggesting that accumulation of sleep drive under these two conditions is differentially sensitive to genetic perturbations.
INTRODUCTION
Sleep is a fundamental biological phenomenon important for both survival and proper brain function; however, we are just beginning to identify its molecular underpinnings.1 A physiological model of sleep regulation proposes that sleep is regulated by two independent processes: a circadian process, which regulates sleep based on time of day, and a homeostatic process, which regulates sleep based on accumulated sleep need.2,3 The molecules that drive the circadian process were first identified in Drosophila with forward genetic screens,4,5 and conserved mechanisms were subsequently found in mammals.6,7 The genes identified in these screens exhibit cycles in expression and activity over the course of the day and their cycling drives a diverse set of circadian behaviors and physiological processes.8 However, identifying equivalent molecules that can fully explain homeostatic sleep regulation has been challenging.
Homeostatic sleep regulation is reflected both in the normal build-up of sleep pressure during spontaneous wakefulness, and in the further increase or “rebound” sleep after sleep deprivation. The widely acknowledged two-process model proposed by Borbély, Daan and colleagues predicts that the same mechanisms should drive sleep pressure under both conditions.2,3 Indeed, electroencephalogram (EEG) slow wave activity (SWA), a widely used marker of sleep need, builds up with similar dynamics during undisturbed wake and acute sleep deprivation conditions, supporting this idea.9 However, the relevance of SWA remains unclear,10–13 and there are conflicting accounts regarding the increase in SWA and sleep amount under conditions of chronic sleep restriction or deprivation.10,14–19 Different types of sleep deprivation producing equivalent sleep loss have also been shown to result in differential homeostatic responses in mice, as evidenced by different responses in multiple sleep latency tests despite equivalent SWA responses during recovery sleep.20 Neurobehavioral performance after sleep deprivation can also be described by the two-process model,21 but as with sleep regulation, unexpected results have also emerged from chronic sleep restriction studies.21–23
Attempts to identify molecular substrates of sleep homeostasis in mammals have not yet provided a mechanistic account of sleep drive.24 Adenosine, as well as its upstream activators prostaglandin D and nitrous oxide, growth hormone-releasing factor, tumor necrosis factor-α, and interleukin-1β, meet the minimal criteria of a sleep homeostasis substrate: these molecules increase during sleep deprivation, and are sufficient to drive sleep when infused into the brains of mammals.25,26 However, the effects of knocking down the receptors for these molecules or pharmacologically inhibiting these pathways tend to be either subtle or restricted to specific aspects of sleep homeostasis, i.e., EEG parameters or sleep following sleep deprivation, suggesting that none of these alone can account for the entire homeostatic component of the two-process model.27–35 This raises the possibility that there exist multiple mechanisms of homeostatic sleep regulation,36 which account for different aspects of the proposed homeostatic process.
Unbiased genetic studies in Drosophila have identified mutants with extremely low habitual sleep amounts.37–44 Many of these mutants have reduced rebound, although these results can be difficult to interpret because extreme short sleepers have less sleep to lose during sleep deprivation.38–43 Moreover, for at least some short sleepers there is evidence that sleep drive remains high: many of these mutants have an increased number of sleep bouts and upregulated biomarkers of sleep need.37,39,41,42,45 Thus, the deficit seems to be in the ability to maintain sleep rather than the ability to sense prior wakefulness. Studying sleep rebound in Drosophila may be a more direct way to probe the genetics that underlie the build-up of sleep need.
To date, there is little information on mutants from unbiased screens based on sleep deprivation, and so the mutations with the most extreme sleep rebound phenotypes following sleep deprivation have likely not yet been found. Moreover, the relationship between sleep at baseline and sleep during recovery has not been well characterized for either wild-type or mutant Drosophila. Thus, it is unclear whether baseline sleep and rebound sleep are closely related across different genotypes or if these two phenomena are largely independent.
In this study we develop a thermogenetic tool for sleep deprivation in Drosophila that enables high-throughput screening to identify lines with reduced sleep rebound. This method produces a strong and consistent sleep rebound compared with other thermogenetic methods, and results in less within-genotype variance compared to sleep rebound following mechanical and caffeine-induced sleep deprivation. In the course of developing this tool, we find that activation of some populations of neurons produces strong sleep loss with no apparent homeostatic compensation the following day. We used thermogenetic stimulation of a population of neurons that does produce a homeostatic response to perform a screen on a collection of mutant insertion lines generated by the Genome Disruption Project46,47 and identify two lines with low rebound, reflected by a blunted increase in both sleep amount and depth after sleep deprivation compared to a control line. Neither line shows evidence of a decrease in the duration, consolidation, or depth of sleep at baseline. Furthermore, statistical analysis shows that across our screen data set, genotype can explain much of the variance in recovery sleep that is not explained by linear relationships with baseline sleep parameters. Taken together, these findings suggest that regulation of sleep amount under baseline and recovery conditions can be controlled by independent genetic mechanisms.
METHODS
Fly Stocks and Crosses
Fly stocks and crosses were maintained at room temperature or 18°C on standard cornmeal-molasses medium. Mutant lines carrying MI{MiC} (“MI”) and P{SUPor-P} (“KG”) insertions generated by the Gene Disruption Project were obtained from Bloomington Stock Center at Indiana University. Lines with transposon insertion sites within genes expressed in the central nervous system were selected for screening (Flybase. org). UAS-TrpA1 and MJ63-Gal4 were a gift from L. Griffith. 53b-Gal4 line was a gift from R. Greenspan. c305-Gal4 was a gift from S. Waddell. 36y-Gal4 and NPF-Gal4 were gifts from P. Taghert. c584-Gal4, c739-Gal4, and Ddc-Gal4 were ordered from the Bloomington Stock Center. 103808-Gal4 and 104906-Gal4 lines were ordered from the Drosophila Genetic Resource Center. The c584-Ga14 and UAS-TrpA1 stocks were each outcrossed into an isogenic background, and a c584-Gal4, UAS-TrpA1 stock was made from these outcrossed lines by allowing meiotic recombination in c584-Gal4/UAS-TrpA1 parents. Progeny carrying a recombined chromosome with both transgenes were identified by polymerase chain reaction and then crossed to a balancer stock to generate a stable line.
Sleep Assays
Sleep was monitored using the Drosophila Activity Monitoring (DAM) System (TriKinetics, Waltham, MA) in glass locomotor tubes containing 5% sucrose / 2% agarose food. Activity data were collected in 1-min bins. All behavioral experiments were conducted in a 12 h:12 h light-dark (LD) cycle. To test potential thermogenetic methods of sleep deprivation, flies were raised at 18°C until they were 1 to 9 days of age. To test the effects of thermogenetic neuronal stimulation, flies were loaded into the DAM system and placed at 21°C, entrained for 2 to 4 days, then subjected to a full day at 28°C starting at Zeitgeber time (ZT)0. For caffeine-induced sleep deprivation, flies were raised to 3 to 6 days old at 25°C, then loaded into the DAM system and flipped to food containing 0.5 mg/mL of caffeine for 24 h starting at ZT0 on day 5. For the pilot mechanical sleep deprivation screen and subsequent mechanical sleep deprivation experiments, flies were raised to 4 to 7 days old at 25°C, then loaded into the DAM system and sleep deprived from ZT18–24 on day 4 or day 5 by shaking on an adapted vortex (TriKinetics, Waltham, MA) for 2 sec every 20 sec. In the primary thermogenetic screen and in subsequent experiments with the c584-Gal4, UAS-TrpA1 thermogenetic method of sleep deprivation, transposon insertion lines were crossed into the c584-Gal4, UAS-TrpA1 background. For heterozygous insertions, progeny of the cross between the insertion stock and the c584-Gal4, UAS-TrpA1 stock were tested. For homozygous insertions, balancers were used to track the insertion in two- to three-generation crossing schemes. For testing responses to thermogenetic sleep deprivation, flies were raised at 18°C to 7 to 13 days old, loaded into DAMS tubes, and entrained for 4 days at 21°C. Sleep deprivation was induced by raising the temperature to 29°C from ZT12–ZT21 on day five. Five to eight female flies per genotype were tested in the primary screen. Total sleep times were obtained from DAMS data using PySolo,48 and sleep consolidation data was obtained using either PySolo or Excel Macros generated by the Allada laboratory.49
Arousal Threshold Assays
For arousal threshold assays, female flies were raised as described previously for the thermogenetic screen and loaded into DAMS monitors. Arousability was assessed at ZT23 for both undisturbed flies, kept at constant 21°C, and sleep deprivation flies, subjected to 9 h of thermogenetic sleep deprivation from ZT12-ZT21. The stimulus was generated by dropping a 12 oz. rubber weight from a 4.5-inch height onto the rack supporting DAMS monitors. Sleeping flies, with no activity in the 5 min prior to the stimulus, were counted as aroused if they exhibited beam crossings in the 2 min following the stimulus.
Immunohistochemistry
Fly heads were opened and fixed in 4% paraformaldehyde (in phosphate buffered saline, PBS) for 15–20 min before brains were dissected. All dissection, washing, and immunostaining was done in PBS with 0.1% Triton-X100. Following dissection, brains were washed three times, incubated 30 min in blocking buffer (5% normal goat serum) and incubated overnight at 4°C in primary antibody solution of 1:300 rabbit anti-tyrosine hydroxylase (TH) AB152 (Millipore, Darmstadt, Germany) and 1:500 chicken anti-green fluorescent protein (GFP) GFP-1020 (Aves Labs, Tigard, OR) in blocking buffer. The following day brains were washed three times, incubated 90 min in secondary antibody solution of 1:400 Alexa Fluor 488 goat anti-chicken and 1:400 Alexa Fluor 680 goat anti-rabbit (Life Technologies, Carlsbad, CA) or 1:400 Cy5 goat anti-rabbit (Rockland Immunochemicals, Pottstown, PA) in blocking buffer, washed three times, then mounted in Vectashield. Brains were imaged on a Leica TCS SP5 confocal microscope (Leica Microsystems, Wetzlar, Germany).
Statistics
Statistics were performed using the base package in R version 3.1.2 (R Foundation for Statistical Computing, Vienna, Austria). For multiple linear regression, variables were added to the model hierarchically in a predetermined order based on expected biological relationships. The analysis of variance (anova) function was used to perform a variance ratio test comparing each new model to the previous model to assess the significance of the new variables.
RESULTS
Development of a Novel Thermogenetic Method to Induce Sleep Deprivation in Drosophila
We tested thermogenetic methods of sleep deprivation to identify an approach that could be used as an efficient screening tool (Figure 1A). For the thermogenetic methods, we selected candidate Gal4 drivers thought to express in wake-promoting neurons and used these to drive expression of the heat-sensitive cation channel TrpA1. Candidate Gal4 drivers were selected based on data generated in a recent Gal4 screen for circadian output neurons,50 in which TrpA1 was used to drive depolarization of Gal4-labeled neurons for 5 days in constant darkness. To assess induced wakefulness and subsequent recovery in these same lines, we employed conditions typically used to study sleep and sleep rebound—12:12 light:dark cycles (LD) with a single day of deprivation. We crossed candidate lines with Gal4 drivers on chromosomes II or III to lines with a UAS-TrpA1 transgene on the same chromosome. Progeny from these crosses were subjected to a baseline day at 21°C, at which there is no TrpA1 activation,51 followed by a day at the TrpA1 activation temperature of 28°C, and a subsequent recovery day at 21°C. Sleep loss and sleep rebound were assessed by comparing the 24-h TrpA1 activation and recovery periods with the baseline day.
Figure 1.

Development of a novel thermogenetic tool to induce sleep deprivation and rebound in Drosophila. (A) Gal4 lines were screened to identify drivers that produce strong sleep loss and subsequent rebound when coupled with the heat-activated cation channel TrpA1. Each candidate Gal4 driver was paired with a UAS-TrpA1 transgene on the same chromosome as the Gal4 driver. A full day of baseline data were collected at 21°C, followed by 24 h of TrpA1 activation at 28°C (ZT0-ZT24) and a subsequent recovery day where flies were returned to 21°C. Error bars represent standard deviation. Significance was assessed with a one-sample Student t-test with a Bonferroni correction for multiple testing. P = 0.05. n = 11–52 per genotype. (B) GFP expression in c584-Gal4/UAS-nGFP flies shows relatively sparse expression in the brain driven by c584-Gal4. Immunohistochemistry with anti-TH and anti-GFP antibodies reveals clustering and costaining of c584-expressing neurons with dopaminergic neurons. GFP expression in c584-Gal4/UAS-nGFP flies includes non-dopaminergic neurons around the dopaminergic PPL1 cluster and co-staining with TH in neurons of the PPM3 cluster. Scale bar = 100 μm. ZT, Zeitgieber Time.
There is a wide range of effectiveness and consistency in thermogenetically induced wakefulness across Gal4 drivers (Figure 1A). Moreover, drivers that produced equivalent amounts of sleep loss can produce highly divergent amounts of rebound the following day. In particular, c584-Gal4, 104906-Gal4, MJ63-Gal4, and c453-Gal4 all produce substantial sleep loss, but whereas c584-Gal4 and 104906-Gal4 produce signifi-cant rebound, MJ63-Gal4 and c453-Gal4 display little to no evidence of a rebound, suggesting that these drivers produce wakefulness via a mechanism that circumvents or counteracts sleep homeostasis.
The Wake-Promoting c584-Gal4 Driver is Expressed in Brain Regions Implicated in Drosophila Sleep
We used c584-Gal4 in subsequent experiments to thermogenetically induce sleep deprivation because it produces a consistent rebound and has relatively restricted expression in the fly brain (Figure 1B). We were unable to determine a precise genomic insertion site for the c584-Gal4 P-element due to the repetitive nature of DNA sequences surrounding the insertion site (data not shown). However, coupling c584-Gal4 with a UAS-nuclear green fluorescent protein (nGFP) reporter reveals that c584-Gal4 drives expression in the pars intercerebralis (PI) and in neurons with projections to the fan-shaped body (Figure 1B); in addition, previous work has shown that c584-Gal4 labels neurons expressing short neuropeptide F (sNPF).52,53 All of these regions have been previously implicated in sleep control, although the reported roles for the PI and sNPF include both sleep promoting and wake-promoting functions.43,50,54–56 Previous work identified wake-promoting neurons with projections to the fan-shaped body in the dopaminergic PPM3 and PPL1 clusters,57,58 so we performed experiments to determine whether c584-Gal4 co-localizes with tyrosine hydroxylase (TH), a marker of dopaminergic neurons. Co-staining brains of c584-Gal4 > UAS-nGFP animals with the TH antibody reveals overlap between c584 neurons and a subset of dopaminergic neurons in the PPM3 cluster, and close proximity between c584 neurons and dopaminergic neurons of the PPL1 cluster (Figure 1B). 104906-Gal4, although more widespread than c584-Gal4 with staining that appears to includes Kenyon cells, also labels the PPM3 and PPL1 clusters, making those dopaminergic clusters good candidates for the wake-promoting effects of these drivers (Figure S1, supplemental material). To facilitate screening, we generated a c584-Gal4, UAS-TrpA1 stock with both transgenes on the same chromosome, into which we could cross transposon insertion mutations generated by the Gene Disruption Project.46,47
Thermogenetic Sleep Deprivation Produces a More Consistent Sleep Rebound with Less Within-Genotype Variance Compared to Caffeine or Mechanical Sleep Deprivation
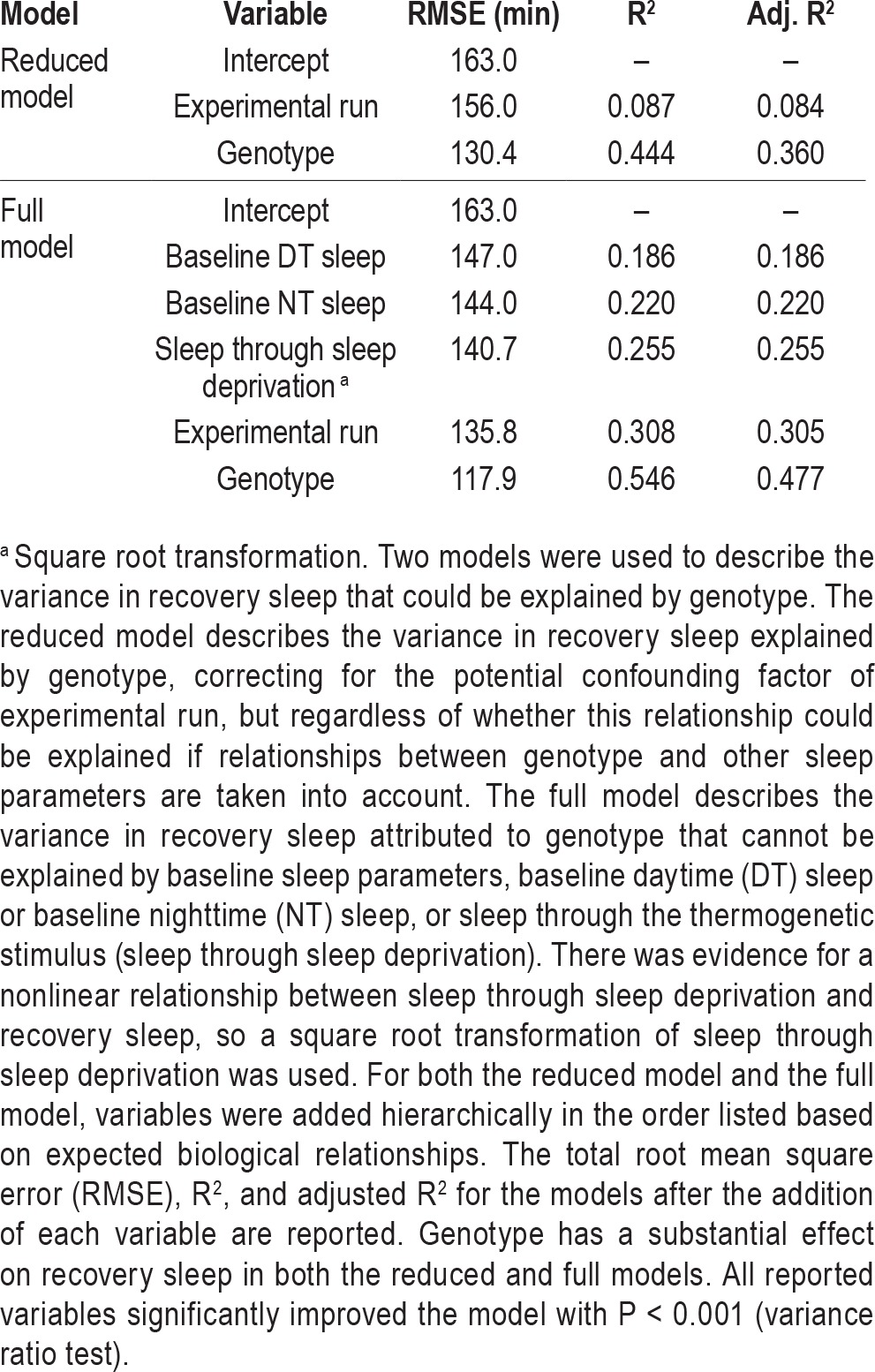
Following development of a thermogenetic method of inducing sleep deprivation, pilot screens were conducted using caffeine, mechanical sleep deprivation, and the c584-Gal4 driven thermogenetic approach to compare suitability for screening. For the caffeine pilot screen, flies were fed caffeine at a concentration previously shown to produce sleep loss59 for 24 h from ZT0-ZT24, then returned to regular food to assess rebound. For the mechanical sleep deprivation screen, flies were sleep deprived by shaking on an adapted vortex for 6 h from ZT18-ZT24. Both sleep deprivation protocols were applied to homozygous MiMIC stocks ordered from the Bloomington stock center. The thermogenetic screening protocol is described in the next paragraph (Figure 2A). For the thermogenetic pilot screen, lethal or second chromosome MiMIC insertions were tested in the heterozygous condition by crossing the MiMIC stock to the c584-Gal4, UAS-TrpA1 stock. Importantly, although our c584-Gal4, UAS-TrpA1 line was backcrossed to an isogenic background, transposon insertion lines generated by the Gene Disruption Project are not generated in isogenic backgrounds, so there may be multiple genetic differences between stocks. Caffeine and mechanical sleep deprivation pilot screens also allowed for any recessive differences between stocks to be revealed, so the genetic diversity of animals tested in these screens should be greater than the genetic diversity of the heterozygous animals tested in the thermogenetic pilot screen. Despite this, genotype is a stronger determinant of recovery sleep in the thermogenetic pilot screen than either the mechanical sleep deprivation screen or the caffeine screen (Table 1). Moreover, the remaining Root Mean Square Error (RMSE) not explained by genotype is smaller in the thermogenetic screen than the pilot screens with caffeine or mechanical sleep deprivation. This suggests that rebound following thermogenetic sleep deprivation presents a more consistent behavior, suitable for genetic screening.
Figure 2.

Thermogenetic screen for mutants with reduced sleep rebound. (A) Screen protocol: Insertion lines were crossed into the c584-Gal4, UASTrpA1 background and the flies were entrained at 21°C. Sleep deprivation was induced for 9 h (ZT12-ZT21) at 29°C, after which the flies were allowed to recover at 21°C. The PySolo sleep profile presented is average sleep of all flies from a representative group of lines run in the screen. Y-axis represents fraction of time asleep in a 30-min bin. (B) Overall screen schematic: Flow chart describing the number of insertion lines selected at each screening stage. (C) Histogram showing rebound sleep (sleep amount on the recovery day subtracted from sleep amount on the baseline day Zeitgeber time (ZT)21-ZT12) for all the lines tested in the screen. Candidates for rescreening (< 2.5 percentile) are boxed. (D) Sleep rebound hits: After rescreening, two lines, MI00323/+ and MI00393/+, show reduced rebound after sleep deprivation. Plotted are sleep loss (SL) and sleep recovered (SR) from four independent experiments for MI00323/+ and MI00393/+ compared to all MiMIC insertions tested as heterozygotes in the screen. Error bars represent standard deviation. (E) Sleep profile for MI00323/+ and MI00393/+ during the thermogenetic sleep deprivation protocol in a representative experiment.
Table 1.
Comparison of mechanical, caffeine-induced, and thermogenetic sleep deprivation in pilot screens.
Screen For Mutants with Reduced Sleep Rebound
To ensure that the sleep rebound we measured in our screen was the result of accumulated sleep loss and not an acute response to the retraction of the wake-promoting stimulus, we chose a protocol for screening where sleep deprivation takes place within the first 9 h of the night (ZT12–ZT21), allowing recovery from the temperature shift to begin 3 h before lights-on (Figure 2A). Rebound is defined as the difference in the duration of sleep between the recovery period and the baseline period during the 15 h following sleep deprivation (ZT21– ZT12), but because most flies sleep through the last 3 h of the night under baseline conditions, a substantial increase in the duration of sleep typically does not occur until the daytime period following sleep deprivation (ZT0–ZT12). Thus, our protocol favors quantification of residual sleep need that can be attributed to the net sleep loss in sleep deprived flies. In addition to changes in sleep amount following sleep deprivation, changes in sleep bout architecture can also be observed; however, these changes are less consistent, with significant heterogeneity across flies (Figure S2, supplemental material).
The overall screen schematic is presented in Figure 2B. We obtained previously mapped in-gene transposon element insertion lines from the MI and KG collections generated by the Genome Disruption Project.46,47 Both types of transposons are predicted to act as loss-of-function mutations by knocking down gene expression at the site of their insertion. In the primary screen, we tested 1,741 transposon insertion lines, homozygous when possible and heterozygous when the insertion was lethal or on the second chromosome. We focused on lines with reduced rebound as these results were easier to interpret; although we do observe outliers with increased rebound in the screen, these lines tend to have low baseline daytime sleep, creating a greater opportunity to rebound compared to a fly with a more prominent siesta. To identify lines with reduced rebound, we excluded the lines that had high baseline daytime sleep, which created a ceiling effect resulting in lower rebound, and lines in which thermogenetic stimulation did not produce significant sleep loss. Of 1,539 lines that remained, we rescreened lines that fell into the lowest 2.5 percentile in terms of their rebound sleep, approximately 50 min or less (Figure 2C). There were two lines, both tested as heterozygotes, for which we were able to recapitulate low rebound below the 10th percentile (∼100 min) in four independent experiments: MI00323/+ and MI00393/+ (Figure 2D–2E).
Lines with Reduced Sleep Rebound Have Normal Baseline Sleep
MiMIC insertions in the two mutant lines, MI00323 and MI00393, were previously mapped to Pka-R1, the regulatory subunit of protein kinase A (PKA), and N-Cadherin,46 respectively. It should be noted that the PKA pathway has previously been implicated in sleep maintenance in Drosophila.60 However, preliminary genetic mapping experiments suggest that the sleep rebound phenotype does not map to the MIMIC insertions suggesting a contribution of other unknown genetic variations in each of these lines.
If there exists a single homeostatic mechanism that governs sleep need in both undisturbed conditions and after a perturbation, animals with a reduced rebound might be expected to have reduced sleep at baseline as well as after sleep deprivation. However, this does not appear to be true for the top hits in our screen. During the primary screen in which we observed reduction in sleep rebound with both MI00323/+ and MI00393/+, overall baseline sleep duration appears to be similar to all other heterozygous MiMIC insertion lines tested (Figure S3, supplemental material).
In order to confirm this observation, we measured the baseline sleep parameters for MI00323/+ and MI00393/+ alongside MI00386/+, a control MiMIC insertion that exhibited average amount of rebound in the primary screen. Although baseline sleep is inconsistent across experiments, we do not observe an overall decrease in baseline sleep for MI00323/+ and MI00393/+ compared to MI00386/+ (Figure 3A). For MI00323/+, in most experiments there is an increase in daytime sleep amount relative to MI00386/+ that is accompanied by an increase in daytime sleep consolidation, with fewer bouts of greater length (Figure 3B). In MI00393/+, there is a shift in the timing of sleep compared to MI00386/+, with shorter daytime sleep and longer nighttime sleep accompanied by greater nighttime sleep consolidation. Overall, these findings do not suggest an overall reduction in sleep amount or consolidation at baseline for lines with reduced rebound sleep; rather, baseline sleep appears to be unchanged or increased for these lines relative to the control.
Figure 3.

Baseline sleep is not reduced in lines with reduced rebound. (A) Sleep rebound following thermogenetic sleep deprivation and baseline sleep amount (± standard error of the mean) and (B) sleep episode data for a representative experiment are shown for MI00323/+ and MI00393/+ (lines with reduced rebound) compared to MI00386/+, which had an average amount of rebound sleep following sleep deprivation in the primary screen. For MI00323/+, rebound was significantly reduced in four of four experiments with n = 28–32 per genotype. Increase in baseline daytime (DT) sleep episode (SE) length was significant in five of seven experiments, and increase in baseline DT sleep amount, and decrease in baseline DT SE number were significant in four of seven experiments. For MI00393/+, rebound was significantly reduced in three of three experiments. Increase in baseline nighttime (NT) SE length and decrease in baseline NT SE number was significant in four of six experiments, and increase in baseline NT sleep amount and decrease in baseline DT sleep amount (not significant in the representative experiment shown) were significant in three of six experiments. Significance for sleep rebound and sleep amount data was assessed with Welch t-test, P < 0.05. Significance for sleep episode data was assessed with Wilcoxon rank-sum test, P < 0.05. n = 28–32 per genotype in each experiment.
Because daytime sleep amount was sometimes higher in MI00323/+, we wondered if the reduced rebound in this line could be explained by a ceiling effect, wherein this line is unable to recover sleep because baseline daytime sleep is already very high. However, even when compared to the distribution of recovery sleep for all screened heterozygous MiMIC insertion lines with daytime sleep above 300 min, MI00323/+ would still be classified as an outlier (Figure S4, supplemental material).
Another possible explanation for reduced rebound in our hits is that instead of recovering lost sleep by sleeping longer, these lines recover lost sleep with deeper sleep immediately after sleep deprivation. To test this hypothesis, we performed an arousal threshold assay at ZT23 for both un-deprived flies and for flies 2 h following thermogenetic sleep deprivation. For undeprived flies, there are no significant differences in arousability between MI00323/+, MI00393/+, and the control MI00386/+ (Figure 4A). After sleep deprivation, arousal threshold is increased in all three lines, but this increase is blunted for MI00323/+ and MI00393/+, such that more animals respond to the arousing stimulus. This suggests that sleep depth, like sleep amount, increases less in our hits compared to our control line following sleep deprivation.
Figure 4.
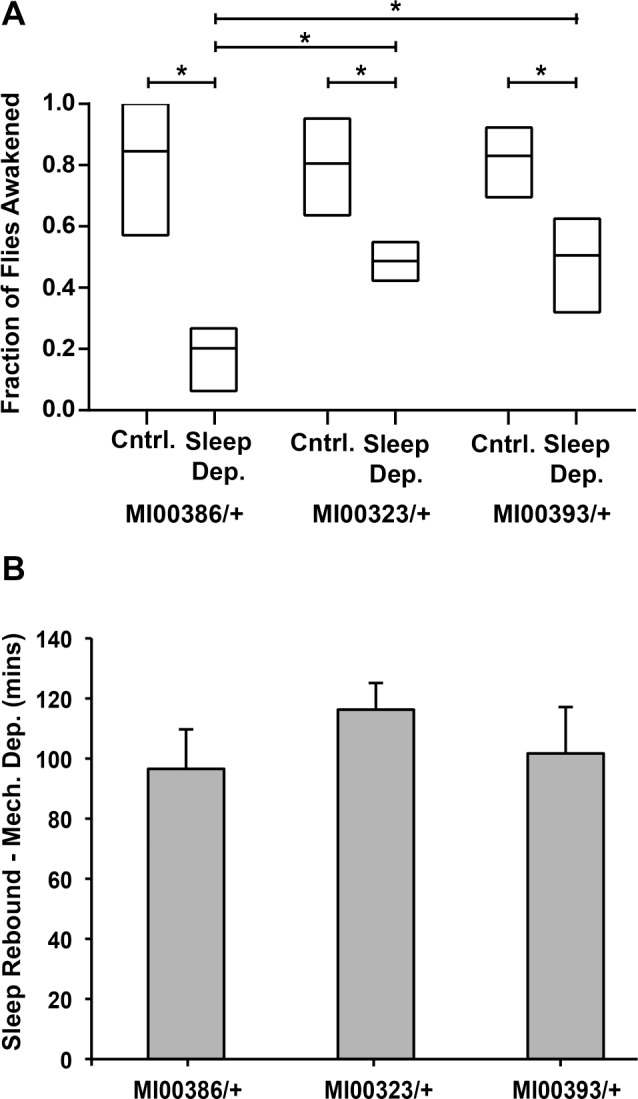
Reduction in sleep rebound following thermogenetic sleep deprivation extends to sleep depth/arousal threshold but is not observed following mechanical sleep deprivation. (A) Arousal threshold for the control MI00386/+ and lines with low sleep rebound, MI00323/+ and MI00393/+, under undeprived conditions (Cntrl) or after thermogenetic sleep deprivation (Sleep Dep). Mechanical stimulus was applied at Zeitgeber time (ZT)23, 2 h after the temperature was returned to 21°C for the sleep deprived groups. Flies that were asleep at the time the stimulus was applied were marked as responding if they showed movement within 2 min following stimulus. Plotted data are the mean and range of fraction of flies awoken in four independent experiments (n = 12–32 sleeping flies in each experiment). A two-way analysis of variance with experimental run as an additional blocking variable indicates main effects of sleep deprivation and genotype on arousal threshold as well as a significant interaction between sleep deprivation and genotype, P < 0.05. Tukey honest significant difference test is used for individual comparisons between groups in post hoc analysis. (B) Sleep rebound following mechanical sleep deprivation for the lines identified as hits from the thermogenetic sleep deprivation screen, with comparison to MI00386/+ as a control. Data are plotted ± standard error of the mean from three combined experiments, n = 24–32 per genotype in each experiment. No significant reduction of sleep rebound is observed for either MI00323/+ or MI00393/+ in three of three experiments assessed by Welch t-test, P < 0.05.
Lines with Reduced Rebound with Thermogenetic Sleep Deprivation Do Not Exhibit Reduced Rebound with Mechanical Sleep Deprivation
To determine whether the lines identified as hits in our screen exhibit reduced rebound with mechanical sleep deprivation as well as thermogenetic sleep deprivation, we subjected heterozygous flies to mechanical sleep deprivation for 6 h from ZT18–ZT24, as done previously in our pilot screen. In contrast with our findings with thermogenetic sleep deprivation, we find that sleep rebound is not reduced in these lines following mechanical sleep deprivation (Figure 4B).
Baseline Sleep and Recovery Sleep are Genetically Separable
The data for baseline and recovery sleep we obtained in the screen of baseline and recovery sleep in ∼1,750 lines allowed us to probe the relationships between genotype, recovery sleep, and baseline sleep not just for our hits with the most extreme phenotypes, but also more broadly across the entire screening dataset. We first used a nested ANOVA model with experimental run as a blocking variable to assess the magnitude of the effect of genotype on recovery sleep in our screen. To avoid confounding our subsequent analyses, we used total sleep through the recovery period (not “rebound” as defined to select hits) as the dependent variable in these models. We find in the nested ANOVA that genotype has a significant effect on recovery sleep, with an increase in R2 of 0.35 when genotype is added (“Reduced Model” in Table 2, Figure 5).
Table 2.
Variance in recovery sleep explained by predictor variables in a hierarchical multiple linear regression model.
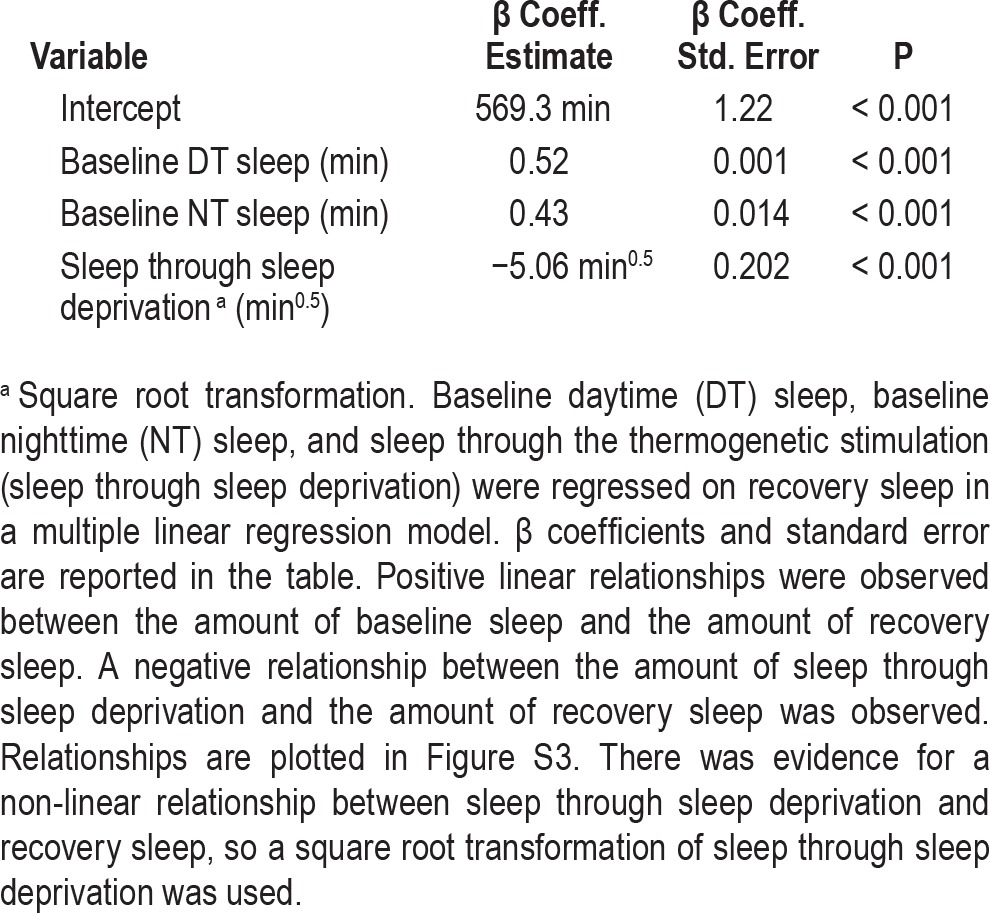
Figure 5.
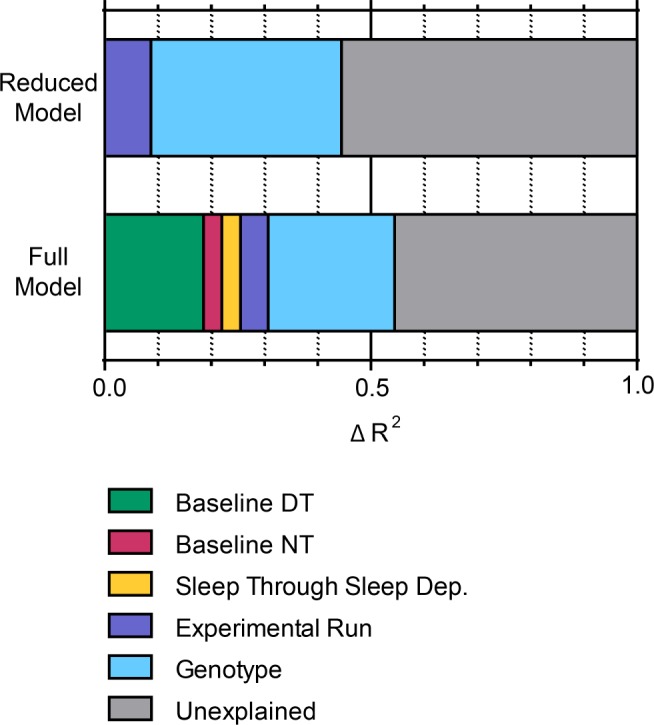
Δ R2 in hierarchical multiple linear regression models shows contribution of genotype cannot be explained by effect of genotype on baseline sleep. The Δ R2 is plotted for each variable from both the reduced model and the full model, described in Table 2. Variables were added hierarchically to the models in the order depicted (left to right). DT, daytime; NT, nighttime.
We next explored the relationship between baseline sleep and recovery sleep in our screen. Most conceptual frameworks for sleep homeostasis predict that baseline sleep and the amount of sleep loss should affect the amount of sleep during recovery, and indeed, there are significant relationships between baseline sleep, sleep loss, and recovery sleep in our data. The relationships between baseline sleep variables and sleep through the recovery period are adequately described by linear relationships, with both daytime and nighttime baseline sleep positively correlated with recovery sleep (Table 3, Figure S5, supplemental material). Sleep through the thermogenetic sleep deprivation period is, as expected, negatively correlated with recovery sleep. A square root transformation of the sleep during thermogenetic deprivation variable produces a better fit than the untransformed variable, so this transformation is used in this and subsequent models.
Table 3.
Regression of recovery sleep against daytime and nighttime baseline sleep and sleep through thermogenetic sleep deprivation.
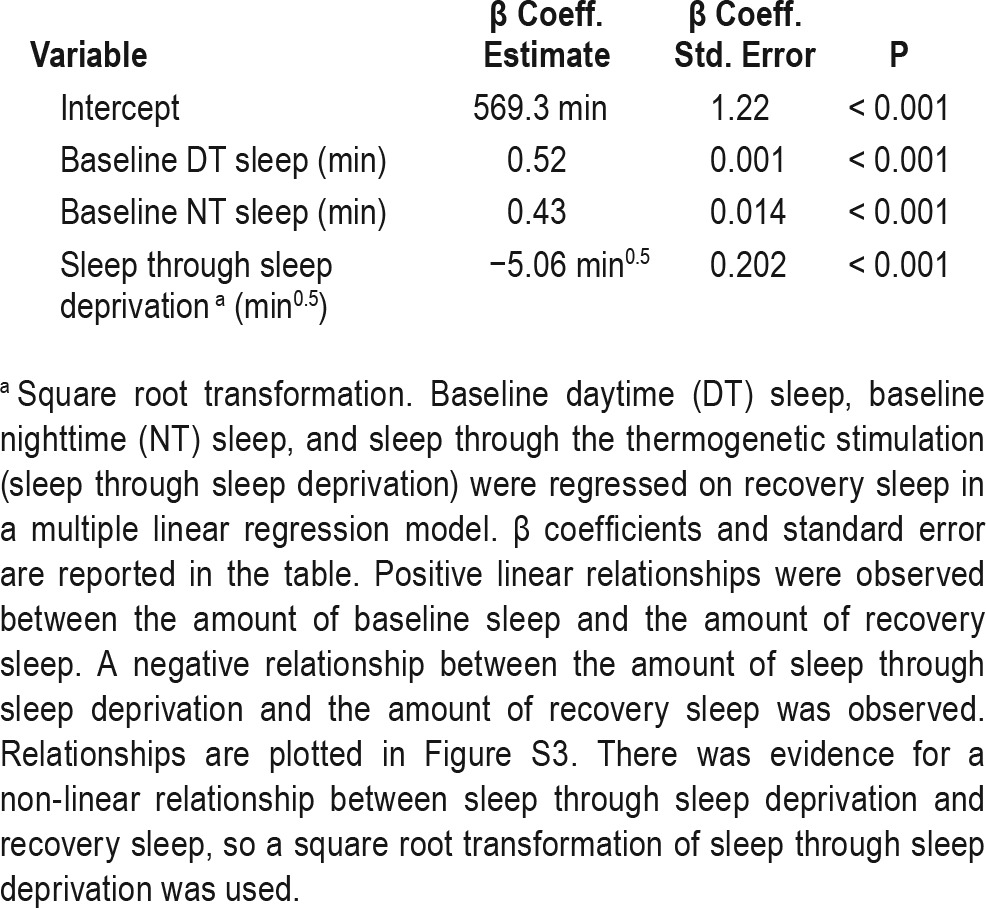
A model including all three of these variables (daytime baseline sleep, nighttime baseline sleep, and sleep through sleep deprivation) has an R2 value of 0.255, indicating modest predictive value. Adding variables reflecting sleep episode length and number at baseline do not significantly improve the fit of the model, suggesting that these are not meaningful determinants of recovery sleep in Drosophila when sleep amount has already been taken into account.
Given these relationships between baseline sleep and recovery sleep, two different ways that genotype might contribute to sleep rebound can be distinguished: (1) by altering the amount of baseline sleep or sleep loss, secondarily affecting rebound sleep, or (2) by specifically affecting recovery sleep in a way that is independent from effects on baseline sleep or sleep loss. As noted previously, baseline sleep for hits MI00323/+ and MI00393/+ is only minimally different from the control line MI00386/+, and these lines have near-complete sleep loss, supporting the second possibility. To address whether this finding in our hits would extend to the entire set of screening data, we constructed a hierarchical multiple linear regression model that includes predictor variables reflecting baseline sleep and the amount of sleep through the thermogenetic stimulation (as well as the potential confounding factor of experimental run, included in the reduced model/nested ANOVA) and asked whether the effect of genotype persists even when these variables have been accounted for.
Predictor variables were added to the multiple linear regression model sequentially in the order listed (Table 2). Without genotype, the model with the predictor variables (baseline sleep, sleep during deprivation and experimental run) has a total R2 value of 0.308, suggesting these variables could account for a substantial amount of variance in the data. Most of this effect is due to the correlations with baseline sleep parameters discussed above, although experimental run also has a significant effect.
When genotype is added to the model that includes baseline sleep and sleep through the thermogenetic stimulus, the R2 value increases by 0.24. Compared to 0.35, the change in R2 when genotype is added to the reduced model, this is a somewhat smaller effect (Table 2, Figure 5). This suggests that some of the effect of genotype on recovery sleep can be thought of as secondary to effects of genotype on baseline sleep or sleep through the thermogenetic stimulus. Nonetheless, the larger part of the effect of genotype on recovery sleep persists in the full model. Thus, to the extent that the linear modeling reflects the true relationship between baseline sleep and recovery sleep in Drosophila, our data support the idea that much of the effect of genotype on recovery sleep is direct and cannot be explained by indirect effects resulting from relationships with baseline sleep or sleep loss.
To ensure that the effect of genotype is not due to broad differences in genetic background resulting from the different collections we screened, but rather to specific differences between individual lines within a collection of insertions, we applied the same modeling approach separately to each type of insertion we screened (Tables S1, S2, and S3, supplemental material). Although the relative contributions of baseline sleep and sleep loss vary among the different collections, in all collections the effect of genotype on recovery sleep persists even when variables reflecting baseline sleep and sleep through thermogenetic sleep deprivation are included in the model.
DISCUSSION
Sleep homeostasis is often described as a single process that regulates sleep both when animals are left undisturbed and when animals are kept awake for extended periods.61 Disparate molecules have been implicated in regulating sleep amount and intensity, but this work has not yet yielded a coherent mechanism to explain all aspects of the proposed homeostatic “Process S”.24 A growing body of evidence suggests that responses to sleep deprivation, sleep restriction, or disruption expose mechanisms regulating sleep homeostasis that are not observed under undisturbed conditions, and conversely there are manipulations that substantially affect daily sleep amount without producing a subsequent homeostatic response. This may explain why attempts to identify a unified molecular mechanism for sleep homeostasis have thus far not been fruitful.
Here, we have developed a thermogenetic method of inducing sleep deprivation that produces a more uniform response and is more subject to influences from genotype than mechanical or caffeine-based approaches. In the course of developing a thermogenetic method to induce sleep deprivation, we find that manipulations of some neuronal populations produce strong reductions in sleep followed by a strong rebound, whereas other populations of neurons produce strong sleep loss without any rebound the next day (Figure 1A). This finding is reminiscent of the observation that certain environmental factors are able to provoke changes in habitual sleep amount in organisms without apparent homeostatic compensation. Although these findings have been somewhat controversial, food availability, mating status, light, and seasonal migration have all been reported to suppress sleep without a subsequent rebound.62–66 Our work suggests that there are neural substrates for wake-promoting mechanisms that are able to bypass or counteract the accumulation of sleep need, which may explain how environmental factors are able to provoke changes in sleep that appear to circumvent a homeostatic response.
We also describe genetic manipulations that specifically affect sleep during recovery from sleep deprivation but have little apparent effect on sleep at baseline. Our unbiased screen yielded two lines that show no evidence of reduced total baseline sleep, despite having little to no sleep rebound and a blunted increase in sleep depth after sleep deprivation (Figures 2, 3, and 4). Multiple linear regression analysis of our data suggests that these observations can be generalized to our entire screening data set: although we do observe positive correlations between baseline and recovery sleep, genotype has a strong effect on sleep after sleep deprivation that is not explained by baseline sleep parameters (Tables 2 and 3, Figure 5).
Despite a robust phenotype with thermogenetic sleep deprivation, our hits do not show reduced rebound with mechanical sleep deprivation (Figure 4B). Given our results showing that the response to mechanical sleep deprivation is less susceptible to genetic perturbation (Table 1), this is not necessarily surprising. It is possible that mechanical sleep deprivation invokes multiple neural circuits to produce sleep rebound, whereas our thermogenetic approach invokes a specific neuronal mechanism. Similar findings have been observed in other organisms; recent work in Caenorhabditis elegans shows that distinct genetic mechanisms regulate sleep after strong disruptions compared to microhomeostatic regulation of quiescent bouts under undisturbed or “low-noise” conditions.67 Taken together, these studies suggest that sleep rebound is a phenomenon that is mechanistically distinct from sleep at baseline, and suggest that there are multiple mechanisms that calibrate sleep to different types of environmental conditions and perturbations.
The findings presented here highlight the potential of the model organism Drosophila to elucidate mechanisms that underlie sleep and other behaviors. The ability to identify mutants with highly extreme phenotypes in large genetic screens allowed us to identify two lines with little to no sleep rebound following thermogenetic sleep deprivation that nonetheless exhibit normal sleep at baseline. It is currently unclear whether the phenotypes will map to single genes. Further work will be important to determine whether these animals are sensitive to other behavioral consequences of sleep deprivation— for example, whether learning and memory are affected in the absence of sleep rebound—or if they are truly resilient. Nonetheless, the lines identified in our unbiased genetic screen demonstrate that extreme phenotypes specific to sleep deprivation can result from genetic differences between animals, and provide the field with valuable tools for identifying mechanisms that underlie the response to sleep deprivation.
NOTE ADDED IN PROOF
While this manuscript was in revision, Seidner et al. reported similar results regarding differing amounts of sleep rebound following thermogenetic activation of different populations of wake-promoting neurons in Drosophila. (Seidner et al. Identification of neurons with a privileged role in sleep homeostasis in Drosophila melanogaster. Curr Biol. 2015;25(22):2928–38.)
DISCLOSURE STATEMENT
This was not an industry supported study. This work was supported by a grant from the Ellison Medical Foundation awarded to Dr. Sehgal and a grant from the Office of Naval Research awarded to Dr. Van Dongen. Dr. Sehgal is an HHMI investigator. Dr. Moravcevic was supported by a fellowship from the Damon Runyon Cancer Research Foundation. Christine Dubowy was supported by institutional T32 NIH training grants. Dr. Van Dongen has consulted for Pulsar Informatics and FedEx Express. The other authors have indicated no financial conflicts of interest.
ACKNOWLEDGMENTS
The authors thank David Raizen and Bill Joiner for critical reading of the manuscript and Michael Gulledge for assistance with immunohistochemistry experiments.
REFERENCES
- 1.Sehgal A, Mignot E. Genetics of sleep and sleep disorders. Cell. 2011;146:194–207. doi: 10.1016/j.cell.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borbély AA. A two process model of sleep regulation. Hum Neurobiol. 1982;1:195–204. [PubMed] [Google Scholar]
- 3.Daan S, Beersma DG, Borbely AA. Timing of human sleep: recovery process gated by a circadian pacemaker. Am J Physiol. 1984;246:R161–83. doi: 10.1152/ajpregu.1984.246.2.R161. [DOI] [PubMed] [Google Scholar]
- 4.Konopka RJ, Benzer S. Clock mutants of Drosophila melanogaster. Proc Natl Acad Sci U S A. 1971;68:2112–6. doi: 10.1073/pnas.68.9.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sehgal A, Price JL, Man B, Young MW. Loss of circadian behavioral rhythms and per RNA oscillations in the Drosophila mutant timeless. Science. 1994;263:1603–6. doi: 10.1126/science.8128246. [DOI] [PubMed] [Google Scholar]
- 6.Antoch MP, Song EJ, Chang AM, et al. Functional identification of the mouse circadian Clock gene by transgenic BAC rescue. Cell. 1997;89:655–67. doi: 10.1016/s0092-8674(00)80246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tei H, Okamura H, Shigeyoshi Y, et al. Circadian oscillation of a mammalian homologue of the Drosophila period gene. Nature. 1997;389:512–6. doi: 10.1038/39086. [DOI] [PubMed] [Google Scholar]
- 8.Zheng X, Sehgal A. Speed control: cogs and gears that drive the circadian clock. Trends Neurosci. 2012;35:574–85. doi: 10.1016/j.tins.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franken P, Chollet D, Tafti M. The homeostatic regulation of sleep need is under genetic control. J Neurosci. 2001;21:2610–21. doi: 10.1523/JNEUROSCI.21-08-02610.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rechtschaffen A, Bergmann BM, Gilliland MA, Bauer K. Effects of method, duration, and sleep stage on rebounds from sleep deprivation in the rat. Sleep. 1999;22:11–31. doi: 10.1093/sleep/22.1.11. [DOI] [PubMed] [Google Scholar]
- 11.Qiu MH, Chen MC, Lu J. Cortical neuronal activity does not regulate sleep homeostasis. Neuroscience. 2015;297:211–8. doi: 10.1016/j.neuroscience.2015.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis CJ, Clinton JM, Jewett KA, Zielinski MR, Krueger JM. Delta wave power: an independent sleep phenotype or epiphenomenon? J Clin Sleep Med. 2011;7:S16–8. doi: 10.5664/JCSM.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tucker AM, Dinges DF, Van Dongen HP. Trait interindividual differences in the sleep physiology of healthy young adults. J Sleep Res. 2007;16:170–80. doi: 10.1111/j.1365-2869.2007.00594.x. [DOI] [PubMed] [Google Scholar]
- 14.Kim Y, Laposky AD, Bergmann BM, Turek FW. Repeated sleep restriction in rats leads to homeostatic and allostatic responses during recovery sleep. Proc Natl Acad Sci U S A. 2007;104:10697–702. doi: 10.1073/pnas.0610351104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deurveilher S, Rusak B, Semba K. Time-of-day modulation of homeostatic and allostatic sleep responses to chronic sleep restriction in rats. Am J Physiol Regul Integr Comp Physiol. 2012;302:R1411–25. doi: 10.1152/ajpregu.00678.2011. [DOI] [PubMed] [Google Scholar]
- 16.Clasadonte J, McIver SR, Schmitt LI, Halassa MM, Haydon PG. Chronic sleep restriction disrupts sleep homeostasis and behavioral sensitivity to alcohol by reducing the extracellular accumulation of adenosine. J Neurosci. 2014;34:1879–91. doi: 10.1523/JNEUROSCI.2870-12.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leemburg S, Vyazovskiy VV, Olcese U, Bassetti CL, Tononi G, Cirelli C. Sleep homeostasis in the rat is preserved during chronic sleep restriction. Proc Natl Acad Sci U S A. 2010;107:15939–44. doi: 10.1073/pnas.1002570107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akerstedt T, Kecklund G, Ingre M, Lekander M, Axelsson J. Sleep homeostasis during repeated sleep restriction and recovery: support from EEG dynamics. Sleep. 2009;32:217–22. doi: 10.1093/sleep/32.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Dongen HP, Maislin G, Mullington JM, Dinges DF. The cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep. 2003;26:117–26. doi: 10.1093/sleep/26.2.117. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki A, Sinton CM, Greene RW, Yanagisawa M. Behavioral and biochemical dissociation of arousal and homeostatic sleep need influenced by prior wakeful experience in mice. Proc Natl Acad Sci U S A. 2013;110:10288–93. doi: 10.1073/pnas.1308295110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCauley P, Kalachev LV, Smith AD, Belenky G, Dinges DF, Van Dongen HP. A new mathematical model for the homeostatic effects of sleep loss on neurobehavioral performance. J Theoret Biol. 2009;256:227–39. doi: 10.1016/j.jtbi.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grant DA, Van Dongen HPA. Individual differences in sleep duration and responses to sleep loss. In: Shaw PJ, Tafti M, Thorpy MJ, editors. The genetic basis of sleep and sleep disorders. Cambridge, England: Cambridge University Press; 2013. pp. 189–96. [Google Scholar]
- 23.Basner M, Rao H, Goel N, Dinges DF. Sleep deprivation and neurobehavioral dynamics. Curr Opin Neurobiol. 2013;23:854–63. doi: 10.1016/j.conb.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mang GM, Franken P. Genetic dissection of sleep homeostasis. In: Meerlo P, Benca RM, Abel T, editors. Sleep, neuronal plasticity and brain function. Vol. 25. Heidelberg, Germany: Springer; 2015. pp. 25–63. [DOI] [PubMed] [Google Scholar]
- 25.Porkka-Heiskanen T, Kalinchuk AV. Adenosine, energy metabolism and sleep homeostasis. Sleep Med Rev. 2011;15:123–35. doi: 10.1016/j.smrv.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 26.Obal F, Jr., Krueger JM. Biochemical regulation of non-rapid-eye-movement sleep. Front Biosci. 2003;8:d520–50. doi: 10.2741/1033. [DOI] [PubMed] [Google Scholar]
- 27.Stenberg D, Litonius E, Halldner L, Johansson B, Fredholm BB, Porkka-Heiskanen T. Sleep and its homeostatic regulation in mice lacking the adenosine A1 receptor. J Sleep Res. 2003;12:283–90. doi: 10.1046/j.0962-1105.2003.00367.x. [DOI] [PubMed] [Google Scholar]
- 28.Bjorness TE, Kelly CL, Gao T, Poffenberger V, Greene RW. Control and function of the homeostatic sleep response by adenosine A1 receptors. J Neurosci. 2009;29:1267–76. doi: 10.1523/JNEUROSCI.2942-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang ZL, Qu WM, Eguchi N, et al. Adenosine A2A, but not A1, receptors mediate the arousal effect of caffeine. Nat Neurosci. 2005;8:858–9. doi: 10.1038/nn1491. [DOI] [PubMed] [Google Scholar]
- 30.Mizoguchi A, Eguchi N, Kimura K, et al. Dominant localization of prostaglandin D receptors on arachnoid trabecular cells in mouse basal forebrain and their involvement in the regulation of non-rapid eye movement sleep. Proc Natl Acad Sci U S A. 2001;98:11674–9. doi: 10.1073/pnas.201398898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalinchuk AV, Lu Y, Stenberg D, Rosenberg PA, Porkka-Heiskanen T. Nitric oxide production in the basal forebrain is required for recovery sleep. J Neurochem. 2006;99:483–98. doi: 10.1111/j.1471-4159.2006.04077.x. [DOI] [PubMed] [Google Scholar]
- 32.Opp MR, Krueger JM. Anti-interleukin-1 beta reduces sleep and sleep rebound after sleep deprivation in rats. Am J Physiol. 1994;266:R688–95. doi: 10.1152/ajpregu.1994.266.3.R688. [DOI] [PubMed] [Google Scholar]
- 33.Obal F, Jr, Alt J, Taishi P, Gardi J, Krueger JM. Sleep in mice with nonfunctional growth hormone-releasing hormone receptors. Am J Physiol Regul Integr Comp Physiol. 2003;284:R131–9. doi: 10.1152/ajpregu.00361.2002. [DOI] [PubMed] [Google Scholar]
- 34.Obal F, Jr., Fang J, Taishi P, Kacsoh B, Gardi J, Krueger JM. Deficiency of growth hormone-releasing hormone signaling is associated with sleep alterations in the dwarf rat. J Neurosci. 2001;21:2912–8. doi: 10.1523/JNEUROSCI.21-08-02912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deboer T, Fontana A, Tobler I. Tumor necrosis factor (TNF) ligand and TNF receptor deficiency affects sleep and the sleep EEG. J Neurophysiol. 2002;88:839–46. doi: 10.1152/jn.2002.88.2.839. [DOI] [PubMed] [Google Scholar]
- 36.Krueger JM, Rector DM, Roy S, Van Dongen HP, Belenky G, Panksepp J. Sleep as a fundamental property of neuronal assemblies. Nat Rev Neurosci. 2008;9:910–9. doi: 10.1038/nrn2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi M, Yue Z, Kuryatov A, Lindstrom JM, Sehgal A. Identification of Redeye, a new sleep-regulating protein whose expression is modulated by sleep amount. eLife. 2014;3:e01473. doi: 10.7554/eLife.01473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koh K, Joiner WJ, Wu MN, Yue Z, Smith CJ, Sehgal A. Identification of SleepLESS, a sleep-promoting factor. Science. 2008;321:372–6. doi: 10.1126/science.1155942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cirelli C, Bushey D, Hill S, et al. Reduced sleep in Drosophila Shaker mutants. Nature. 2005;434:1087–92. doi: 10.1038/nature03486. [DOI] [PubMed] [Google Scholar]
- 40.Kume K, Kume S, Park SK, Hirsh J, Jackson FR. Dopamine is a regulator of arousal in the fruit fly. J Neurosci. 2005;25:7377–84. doi: 10.1523/JNEUROSCI.2048-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stavropoulos N, Young MW. insomniac and Cullin-3 regulate sleep and wakefulness in Drosophila. Neuron. 2011;72:964–76. doi: 10.1016/j.neuron.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rogulja D, Young MW. Control of sleep by Cyclin A and its regulator. Science. 2012;335:1617–21. doi: 10.1126/science.1212476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Donlea JM, Pimentel D, Miesenbock G. Neuronal machinery of sleep homeostasis in Drosophila. Neuron. 2014;81:860–72. doi: 10.1016/j.neuron.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Afonso DJ, Liu D, Machado DR, et al. TARANIS Functions with Cyclin A and Cdk1 in a Novel Arousal Center to Control Sleep in Drosophila. Curr Biol. 2015;25:1717–26. doi: 10.1016/j.cub.2015.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pfeiffenberger C, Allada R. Cul3 and the BTB adaptor insomniac are key regulators of sleep homeostasis and a dopamine arousal pathway in Drosophila. PLoS Genet. 2012;8:e1003003. doi: 10.1371/journal.pgen.1003003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Venken KJ, Schulze KL, Haelterman NA, et al. MiMIC: a highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nat Meth. 2011;8:737–43. doi: 10.1038/nmeth.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bellen HJ, Levis RW, He Y, et al. The Drosophila gene disruption project: progress using transposons with distinctive site specificities. Genetics. 2011;188:731–43. doi: 10.1534/genetics.111.126995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gilestro GF, Cirelli C. pySolo: a complete suite for sleep analysis in Drosophila. Bioinformatics. 2009;25:1466–7. doi: 10.1093/bioinformatics/btp237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pfeiffenberger C, Lear BC, Keegan KP, Allada R. Processing sleep data created with the Drosophila Activity Monitoring (DAM) System. Cold Spring Harb Protoc. 2010;2010:pdb.prot5520. doi: 10.1101/pdb.prot5520. [DOI] [PubMed] [Google Scholar]
- 50.Cavanaugh DJ, Geratowski JD, Wooltorton JR, et al. Identification of a circadian output circuit for rest:activity rhythms in Drosophila. Cell. 2014;157:689–701. doi: 10.1016/j.cell.2014.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pulver SR, Pashkovski SL, Hornstein NJ, Garrity PA, Griffith LC. Temporal dynamics of neuronal activation by Channelrhodopsin-2 and TRPA1 determine behavioral output in Drosophila larvae. J Neurophysiol. 2009;101:3075–88. doi: 10.1152/jn.00071.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kahsai L, Martin JR, Winther AM. Neuropeptides in the Drosophila central complex in modulation of locomotor behavior. J Exp Biol. 2010;213:2256–65. doi: 10.1242/jeb.043190. [DOI] [PubMed] [Google Scholar]
- 53.Kahsai L, Winther AM. Chemical neuroanatomy of the Drosophila central complex: distribution of multiple neuropeptides in relation to neurotransmitters. The Journal of Comparative Neurology. 2011;519:290–315. doi: 10.1002/cne.22520. [DOI] [PubMed] [Google Scholar]
- 54.Crocker A, Shahidullah M, Levitan IB, Sehgal A. Identification of a neural circuit that underlies the effects of octopamine on sleep:wake behavior. Neuron. 2010;65:670–81. doi: 10.1016/j.neuron.2010.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shang Y, Donelson NC, Vecsey CG, Guo F, Rosbash M, Griffith LC. Short neuropeptide F is a sleep-promoting inhibitory modulator. Neuron. 2013;80:171–83. doi: 10.1016/j.neuron.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen W, Shi W, Li L, et al. Regulation of sleep by the short neuropeptide F (sNPF) in Drosophila melanogaster. Insect Biochem Mol Biol. 2013;43:809–19. doi: 10.1016/j.ibmb.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 57.Liu Q, Liu S, Kodama L, Driscoll MR, Wu MN. Two dopaminergic neurons signal to the dorsal fan-shaped body to promote wakefulness in Drosophila. Curr Biol. 2012;22:2114–23. doi: 10.1016/j.cub.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ueno T, Tomita J, Tanimoto H, et al. Identification of a dopamine pathway that regulates sleep and arousal in Drosophila. Nat Neurosci. 2012;15:1516–23. doi: 10.1038/nn.3238. [DOI] [PubMed] [Google Scholar]
- 59.Wu MN, Ho K, Crocker A, Yue Z, Koh K, Sehgal A. The effects of caffeine on sleep in Drosophila require PKA activity, but not the adenosine receptor. J Neurosci. 2009;29:11029–37. doi: 10.1523/JNEUROSCI.1653-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hendricks JC, Williams JA, Panckeri K, et al. A non-circadian role for cAMP signaling and CREB activity in Drosophila rest homeostasis. Nat Neurosci. 2001;4:1108–15. doi: 10.1038/nn743. [DOI] [PubMed] [Google Scholar]
- 61.Borbély AA, Achermann P. Sleep homeostasis and models of sleep regulation. J Biol Rhythms. 1999;14:557–68. doi: 10.1177/074873099129000894. [DOI] [PubMed] [Google Scholar]
- 62.Thimgan MS, Suzuki Y, Seugnet L, Gottschalk L, Shaw PJ. The Perilipin homologue, Lipid Storage Droplet 2, regulates sleep homeostasis and prevents learning impairments following sleep loss. PLoS Biol. 2010;8:e1000466. doi: 10.1371/journal.pbio.1000466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Isaac RE, Li C, Leedale AE, Shirras AD. Drosophila male sex peptide inhibits siesta sleep and promotes locomotor activity in the post-mated female. Proc Biol Soc. 2010;277:65–70. doi: 10.1098/rspb.2009.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rattenborg NC, Mandt BH, Obermeyer WH, et al. Migratory sleeplessness in the white-crowned sparrow (Zonotrichia leucophrys gambelii) PLoS Biol. 2004;2:e212. doi: 10.1371/journal.pbio.0020212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Berger RJ, Phillips NH. Constant light suppresses sleep and circadian rhythms in pigeons without consequent sleep rebound in darkness. Am J Physiol. 1994;267:R945–52. doi: 10.1152/ajpregu.1994.267.4.R945. [DOI] [PubMed] [Google Scholar]
- 66.Yokogawa T, Marin W, Faraco J, et al. Characterization of sleep in zebrafish and insomnia in hypocretin receptor mutants. PLoS Biol. 2007;5:e277. doi: 10.1371/journal.pbio.0050277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nagy S, Tramm N, Sanders J, et al. Homeostasis in C. elegans sleep is characterized by two behaviorally and genetically distinct mechanisms. eLife. 2014;3:e04380. doi: 10.7554/eLife.04380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.