

Abstract
Therapeutic interventions that lower LDL-cholesterol effectively reduce the risk of coronary artery disease (CAD). However, statins, the most widely prescribed LDL-cholesterol lowering drugs, increase diabetes risk. We used genome-wide association study (GWAS) data in the public domain to investigate the relationship of LDL-C and diabetes and identify loci encoding potential drug targets for LDL-cholesterol modification without causing dysglycemia. We obtained summary-level GWAS data for LDL-C from GLGC, glycemic traits from MAGIC, diabetes from DIAGRAM and CAD from CARDIoGRAMplusC4D consortia. Mendelian randomization analyses identified a one standard deviation (SD) increase in LDL-C caused an increased risk of CAD (odds ratio [OR] 1.63 (95 % confidence interval [CI] 1.55, 1.71), which was not influenced by removing SNPs associated with diabetes. LDL-C/CAD-associated SNPs showed consistent effect directions (binomial P = 6.85 × 10−5). Conversely, a 1-SD increase in LDL-C was causally protective of diabetes (OR 0.86; 95 % CI 0.81, 0.91), however LDL-cholesterol/diabetes-associated SNPs did not show consistent effect directions (binomial P = 0.15). HMGCR, our positive control, associated with LDL-C, CAD and a glycemic composite (derived from GWAS meta-analysis of four glycemic traits and diabetes). In contrast, PCSK9, APOB, LPA, CETP, PLG, NPC1L1 and ALDH2 were identified as “druggable” loci that alter LDL-C and risk of CAD without displaying associations with dysglycemia. In conclusion, LDL-C increases the risk of CAD and the relationship is independent of any association of LDL-C with diabetes. Loci that encode targets of emerging LDL-C lowering drugs do not associate with dysglycemia, and this provides provisional evidence that new LDL-C lowering drugs (such as PCSK9 inhibitors) may not influence risk of diabetes.
Electronic supplementary material
The online version of this article (doi:10.1007/s00439-016-1647-9) contains supplementary material, which is available to authorized users.
Keywords: Coronary Artery Disease, Ezetimibe, Coronary Artery Disease Risk, Mendelian Randomization, Glycemic Status
Introduction
LDL-cholesterol (LDL-C) is a recognized causal risk factor for coronary artery disease (CAD) (Cholesterol Treatment Trialists et al. 2012; Holmes et al. 2015). Meta-analysis of randomized clinical trials (RCTs) shows a 1 mmol/l reduction in LDL-C results in 25 % reduction in risk of CAD (Cholesterol Treatment Trialists et al. 2010). Indeed, statins remain the drug of choice to achieve LDL-C reduction, as they have proven long-term efficacy for reducing risk of cardiovascular disease and overall mortality. However, statins have been linked to increased risk of type 2 diabetes (T2D), (Preiss et al. 2011; Sattar et al. 2010) with recent evidence indicating this is mediated by an on-target effect (specifically through inhibition of 3-hydroxy-3-methylglutaryl-CoA reductase, HMGCR, the intended target of statins) (Swerdlow et al. 2015).
Whether the T2D effects of statins are specific to HMGCR inhibition or a general characteristic of LDL-C modification is of considerable importance given the ongoing development of drugs designed to reduce LDL-C. These include: (1) monoclonal antibody inhibitors of proprotein convertase subtilisin/kexin type 9 (PCSK9, encoded by the PCSK9 gene) such as evolocumab and alirocumab (Stein et al. 2012); (2) antisense inhibitors of apolipoprotein B (apoB-100, encoded by APOB), such as mipomersen (Akdim et al. 2010), and; (3) the antisense inhibitor ISIS APO(a)Rx, which reduces lipoprotein(a) (Lp(a), encoded by LPA). These compounds are now in phase II (APO(a)Rx: NCT02160899) and phase III (mipomersen: NCT01475825; evolocumab: NCT01764633l) randomized clinical trials (RCTs) for CAD events. It is, therefore, important to characterize any glycemia-modifying properties of drugs that target protein products of PCSK9, APOB and LPA and to identify and prioritize additional potential therapeutic targets that alter LDL-C and risk of CAD but without causing dysglycemia.
Genetic studies provide unique opportunities to inform our understanding of disease etiology, causal mechanisms and potential therapeutic targets. Recently, data from a variety of GWAS studies have become available in the public domain, and by integrating multiple such data sets, it should become possible to obtain novel information on the potential intended and unintended consequences of drug therapy. Furthermore, these GWAS data can be exploited for Mendelian randomization analyses to generate unbiased, causal effect estimates that are free from reverse causality and confounding (Lawlor et al. 2008).
In this study, we clarify the relationship of LDL-C, CAD and dysglycemia through integrative analyses of GWAS datasets. This involves investigating: (1) whether, risk of T2D is altered as a consequence of LDL-C modification; (2) whether CAD prevention by LDL-C modification is dependent on the effect of LDL-C on diabetes; (3) whether pharmacological targets of emerging LDL-C lowering drugs associate with dysglycemia, and; (4) discovery of potential therapeutic targets for LDL-C lowering and CAD prevention that do not result in dysglycemia.
Methods
We obtained summary-level data for: (1) LDL-C from the Global Lipids Genetics Consortium (GLGC); (2) glycemic traits from the Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC), (3) T2D from the DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) consortium, and (4) CAD from the Coronary ARtery DIsease Genome-wide Replication And Meta Analysis (CARDIoGRAM) plus The Coronary Artery Disease (C4D) Genetics, collectively known as CARDIoGRAMplusC4D consortium. The consortia provide these data openly on their respective websites: GLGC: http://www.sph.umich.edu/csg/abecasis/public/lipids2013; MAGIC: http://www.magicinvestigators.org; DIAGRAM: http://diagram-consortium.org; and, CARDIoGRAMplusC4D: http://www.cardiogramplusc4d.org. All datasets were limited to individuals of European ancestry.
We used data from GLGC as a means to harmonize estimates across the consortia. We limited our focus to SNPs that were genome-wide significant for their association with LDL-C in GLGC (at P < 5 × 10−8). We made SNPs directionally consistent across the datasets so that the effect alleles increased LDL-C. This was done by inverting alleles and corresponding beta coefficients where necessary. SNPs were mapped to the nearest loci using RefSeq (http://www.ncbi.nlm.nih.gov/refseq).
We used these data to investigate the shared association of LDL-C-related SNPs with risk of CAD, T2D and concentrations of fasting glucose. We used a nominal significance threshold of P < 0.05, on the basis that LDL-C is recognized as causal for CHD, and that some LDL-C loci also modify glycemic traits; thus, a Bonferroni-adjusted P value threshold would be too stringent in this scenario.
Mendelian randomization analysis was conducted by identifying SNPs in independent loci and R 2 < 0.8 that associated with LDL-C at P < 5 × 10−8. As a sensitivity analysis, we used a stricter R 2 threshold of <0.2. Corresponding beta coefficients or log odds (together with their standard errors) were obtained for CAD, T2D and fasting glucose and we arranged SNPs so that the estimates corresponded to the same reference allele. Using the summary estimates for each of the traits, we synthesized instrumental variable estimates for each SNP by dividing the SNP-outcome association by the SNP-LDL-C association and using the delta method to approximate the standard error (Thomas et al. 2007). This generated an instrumental variable estimate for each SNP, which we pooled using fixed-effects meta-analysis to yield a summary causal effect of the association of LDL-C with risk of CAD, T2D and concentrations of fasting glucose. To investigate the independence of effect of LDL-C on risk of CAD, we removed SNPs that associated with T2D at P < 0.05 and subsequently at P < 0.01 and repeated the analysis focusing on only the remaining SNPs. Effect estimates were compared using the three different analyses (i.e. using all SNPs, excluding SNPs at P < 0.01 for T2D and excluding SNPs at P < 0.05 for T2D).
To increase power from the published datasets, we performed a ‘glycemic burden composite’ GWAS, by meta-analyzing the SNP beta coefficients and corresponding standard errors for glycemic traits and T2D risk in MAGIC and DIAGRAM. This was conducted in METAL. The GWAS meta-analysis approach is an established technique to increase power to detect associations of SNPs with clinically related phenotypes (Ellinghaus et al. 2012; McGeachie et al. 2014; Zhernakova et al. 2011). We meta-analyzed four out of the six glycemic traits in MAGIC (fasting glucose, fasting insulin, fasting proinsulin and HbA1c) and T2D risk from DIAGRAM. We excluded HOMA-B and HOMA-IR from MAGIC based on the high correlation between the Z-scores of those traits with that of fasting insulin (Kendall’s tau rank correlation 0.95 and 0.75 for fasting insulin with HOMA-IR and HOMA-B, respectively; Supplementary Table 4), which provided little additional information for the meta-analysis results and resulted in excessive genomic inflation due to the same individuals being analyzed multiple times (including HOMA-B/HOMA-IR λ = 1.91; excluding HOMA-B/HOMA-IR λ = 1.24).
We followed two approaches to identify loci associated with differences in LDL-C and a corresponding difference in risk of CAD that were (1) related and (2) unrelated to the glycemic burden composite. First, SNP pruning was conducted by LD pruning under an r 2 < 0.5 threshold and using the “clumping” function in Plink to prioritize lead SNPs based on P value with the glycemic burden composite. This reduced the number of SNPs from ~2.6 M to ~440 k SNPs. SNPs that associated with LDL-C (P < 5 × 10−8), CAD (P < 0.05) and did not associate with the glycemic burden composite (P > 0.05) were taken forward. Moreover, we selected only genes that did not have an over-representation of SNPs associated with the glycemic burden (Fisher’s exact P > 0.05). Second, we then repeated this step looking for loci associating with LDL-C, CAD and the glycemic burden—these loci were taken forward and we selected only those that had an over-representation of SNPs that associated with the glycemic burden composite (Fisher’s exact P < 0.05). We calculated Fisher's exact tests with EVA (available at http://www.exploratoryvisualanalysis.org/).
To investigate whether drugs exist that target the proteins encoded by these genes, we used publicly available drug-gene interaction databases. For small molecules, we used chEMBL (Gaulton et al. 2012), a repository of experimental molecules (most of which have not been fully developed and are of unknown efficacy), developed mainly by the pharmaceutical industry. For already marketed drugs, we used an integrated database, DGIdb (http://dgidb.genome.wustl.edu), which incorporates several drug-gene interaction databases, such as DrugBank (Law et al. 2014) and PharmGKB (Thorn et al. 2010). Loci that were pharmacodynamic targets of drugs were identified through online searches (including DrugBank http://www.drugbank.ca, GeneCards http://www.genecards.org, PubMed and Google Scholar).
Finally, we investigated PCSK9, APOB and LPA for their association with LDL-C, CAD and glycemic burden composite. This was to investigate the likely impact on glycemic status of emerging LDL-C lowering agents at intermediate or advanced stages of clinical development. For a positive control, we examined SNPs in HMGCR, given their known causal effects on LDL-C, CAD and T2D (Swerdlow et al. 2015). We identified SNPs in PCSK9, APOB, LPA and HMGCR associating with LDL-C at GWAS significance in the GWAS catalog (http://www.genome.gov/gwastudies accessed October 1st, 2014) and took these forward to investigate their associations with LDL-C, CAD and glycemic burden composite in our datasets. To investigate the preponderance for SNPs in these loci to associate with the glycemic burden composite, we synthesized a Circos plot.
Analyses were conducted in R version 2.15.2, Stata version 13.1 (College Station, Texas) and METAL (http://www.sph.umich.edu/csg/abecasis/metal).
Results
We identified SNPs from the Global Lipids Genetics Consortium [GLGC, including data from up to 95,454 individuals of European ancestry (Global Lipids Genetics et al. 2013)] that surpassed the significance threshold (P < 5 × 10−8) with LDL-C and took these forward to interrogate their relationship with CAD, T2D and fasting glucose. This resulted in 2966 SNPs associated with LDL-C, corresponding to 197 independent SNPs at 172 distinct loci.
SNPs associated with LDL-C and CAD
84 of the 2966 LDL-C associated SNPs (25 of 172 loci) had nominally significant associations (at P < 0.05) with CAD risk in CARDIoGRAMplusC4D (including 63,746 cases and 130,681 controls of European ancestry) (Supplementary Fig. 1) (Consortium et al. 2013). Of these 25 loci, 22 (88 %) showed the same direction of effect for LDL-C and CAD (i.e., were associated both with higher LDL-C concentration and with higher risk of CAD; binomial P = 6.85 × 10−5) (Fig. 1).
Fig. 1.

Relationship of LDL-C-associated loci with risk of CAD. The majority (22 of 25) of loci showed a consistent direction of effect with risk of CAD. LDL-C effect estimates are per SD; whiskers represent 95 % CI
Mendelian randomization analysis of 197 independent SNPs associated with LDL-C yielded a causal OR for CAD of 1.63 (95 % confidence interval [CI] 1.55, 1.71; P = 8.0 × 10−83) per one standard deviation (SD) increase in LDL-C (Fig. 2). Using a stricter R 2 threshold (<0.2) for SNP inclusion identified 145 independent SNPs and did not materially alter the findings (Supplementary Figure 4).
Fig. 2.

Mendelian randomization to investigate the causal relationship of a one standard deviation genetically-instrumented increase in LDL-C with risk of coronary artery disease (CAD), type 2 diabetes (T2D) and levels of fasting glucose. Single nucleotide polymorphisms (SNPs) were initially selected based on their independent association with LDL-C at R 2 < 0.8 (n = 197; “All SNPs” stratum). Thereafter, we removed SNPs that associated with T2D risk at P < 0.01 (15 SNPs removed) and P < 0.05 (34 SNPs removed). Findings for the analysis using a stricter R 2 threshold (<0.2) are presented in Supplementary Figure 4
SNPs associated with LDL-C and T2D
61 of the 2966 LDL-C SNPs (15 of 172 loci) were nominally significant (at P < 0.05) for T2D in DIAGRAM (34,840 cases, 114,981 controls of European ancestry) (Supplementary Fig. 1). However, there was no clear relationship between LDL-C and T2D: of the 15 loci, 6 (40 %) showed the same direction of effect (binomial P = 0.15; Fig. 3).
Fig. 3.

Relationship of LDL-C-associated loci with risk of T2D. Six of the 15 loci showed a positive association with T2D risk. LDL-C effect estimates are per SD; whiskers represent 95 % CI
Mendelian randomization analysis incorporating 197 independent LDL-C associated SNPs yielded a causal OR for T2D of 0.86 (95 % CI 0.81, 0.91 P = 2.1 × 10−7) per 1-SD increase in LDL-C. Removal of 15 SNPs that associated with T2D at P < 0.01 and 34 SNPs that associated with T2D at P < 0.05 resulted in a diminution of the causal OR for T2D to 0.89 (95 % CI: 0.83, 0.94; P = 0.002) and 0.94 (95 % CI: 0.88, 1.01; P = 0.10), respectively. The corresponding causal OR for CAD when the 34 SNPs associated with T2D (at P < 0.05) were removed remained unaltered at 1.61 (95 % CI 1.52, 1.70; P = 3.3 × 10−61) (Fig. 2). As before, using a stricter R 2 threshold (of <0.2) did not materially alter the findings (Supplementary Figure 4).
SNPs associated with LDL-C and fasting glucose
In the MAGIC consortium dataset (133,010 individuals of European ancestry), 58 SNPs (19 of 172 loci) were nominally associated (at P < 0.05) with fasting glucose (Supplementary Fig. 1). As with T2D, there was no clear consistency in direction of effect, with 9 of 19 loci (47 %) showing concordant directions of effect for LDL-C and fasting glucose (binomial P = 0.17; Fig. 4).
Fig. 4.

Relationship of LDL-C-associated loci with fasting glucose. Nine of 19 loci showed a positive association with fasting glucose. LDL-C effect estimates are per SD; Fasting glucose effect estimates are in mmol/l; whiskers represent 95 % CI
Mendelian randomization using 197 LDL-C-associated SNPs showed that a 1-SD increase in LDL-C had no clear effect on fasting glucose (0.009 mmol/l; 95 % CI −0.001, 0.020; P = 0.08).
SNPs associated with LDL-C, CAD, fasting glucose and T2D
We next integrated data from the four traits (LDL-C, CAD, fasting glucose and T2D) using a subset of the 2966 SNPs associated with LDL-C (at P < 5 × 10−8) that also showed a nominally significant association (P < 0.05) with CAD risk (n = 84). Of these 84 SNPs, 17 were associated with fasting glucose and 13 with T2D risk. Six SNPs were associated with both T2D risk and fasting glucose. Of note, these six independent SNPs that associated with all four traits were consistent in their associations with higher LDL-C, higher CAD risk, lower fasting glucose and lower T2D risk (binomial P = 0.016) (Supplementary Fig. 2).
Eight of 17 SNPs associating with LDL-C, CAD and fasting glucose were located at the HMGCR locus. Other loci that were associated with LDL-C and CAD, and with fasting glucose and T2D included CELSR2, PSRC1, APOC1 and SUGP1.
Synthesis of a glycemic burden composite trait
To increase power to detect associations of loci with glycemic status, we developed a “glycemic burden composite”, which involved meta-analysis of associations of four glycemic traits (fasting glucose, fasting insulin, fasting proinsulin, HbA1c) together with T2D risk and included over 2.5 million SNPs in the MAGIC and DIAGRAM consortia datasets. We excluded HOMA-B and HOMA-IR from MAGIC based on the high correlation (see “Methods” and Supplementary Table 4 for more details), This identified 306 SNPs with significant associations with the glycemic burden composite (at Bonferroni-corrected P < 5 × 10−8) (Supplementary Fig. 3 and Data file S1).
We evaluated suitable loci that altered LDL-C levels and CAD risk that were free from dysglycemic effects, following three routes:
Loci that encode established or emerging LDL-C drug targets for CAD prevention
We focused our attention on four loci encoding targets for existing or emerging lipid-lowering agents: HMGCR (the intended target of the statin drugs), PCSK9, APOB and LPA (targets of drugs currently in phase II and phase III RCTs). We identified SNPs in the GWAS catalog (http://www.genome.gov/gwastudies, accessed October 1st 2014) in these loci that associated with LDL-C at GWAS significance.
For HMGCR (which served as a positive control), 5 SNPs were identified (rs12916, rs3846662, rs3846663, rs7703051, rs12654264, Table 1): all 5 HMGCR SNPs associated with the glycemic burden composite. The direction of effect was as expected: SNPs associated with lower LDL-C levels, lower risk of CAD and higher values for the glycemic burden composite.
Table 1.
Association of variants in HMGCR, PCSK9, APOB and LPA with LDL-C, CAD risk and glycemic burden composite for GWAS Catalog SNPs (color figure online)
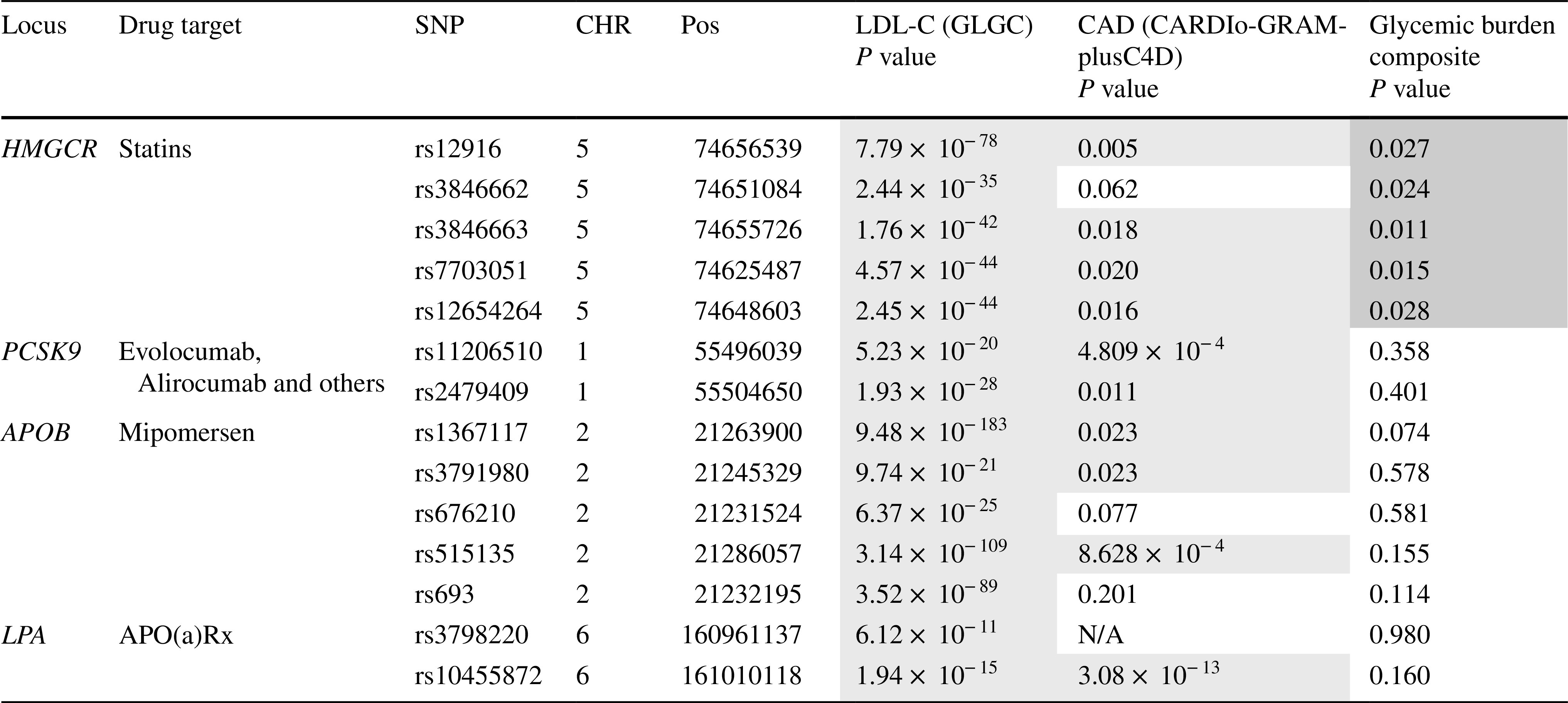
Blue = lower level of trait or risk of outcome; red = higher level of trait or risk of outcome
For PCSK9, two SNPs were identified (rs11206510, rs2479409). Both PCSK9 SNPs associated with CAD, yet neither of them associated with the glycemic burden composite.
For APOB, five SNPs were identified (rs1367117, rs3791980, rs676210, rs515135, rs693), three of which associated with CAD. Again, no association of these five APOB SNPs was identified with the glycemic burden composite.
For LPA, two SNPs were identified (rs3798220, rs10455872), one of which (rs3798220) was not present in CARDIoGRAMplusC4D and no suitable proxy was available. The other SNP (rs10455872) associated with CAD. Neither of the two SNPs associated with the glycemic burden composite.
To exploit all available data, we focused on SNPs in the same four loci (PCSK9, APOB, LPA and HMGCR) and evaluated the physical distribution and associations of these SNPs with the glycemic burden composite (Supplementary Fig. 3). The majority of SNPs in HMGCR associated with the glycemic burden composite, in contrast to the SNPs in PCSK9, APOB or LPA (Fig. 5).
Fig. 5.

Circos diagram to show association of SNPs in PCSK9, APOB, LPA, LDLR and HMGCR with glycemic burden composite. The outer ring represents the genomic/chromosomal location. Each SNP is a green, orange or red point in the graph. Green dots in green shaded ring represent SNPs with 1 > P ≥ 0.05; orange circles in orange shaded ring correspond to SNPs within 0.05 > P ≥ 0.001 and; red triangles in red shaded ring represent SNPs with P < 0.001. 61 % of HMGCR SNPs associated with the glycemic burden composite (at P < 0.05) vs. less than 5 % for SNPs in PCSK9, APOB and LPA (color figure online)
Loci that associate with LDL-C and CAD, but do not associate with the glycemic burden composite
To identify potential drug targets that alter LDL-C and CAD risk with no consequence on glycemic traits, we examined SNPs in loci that associated with LDL-C (at P < 5 × 10−8) and CAD (at P < 0.05) but did not associate with the glycemic burden composite (P > 0.05). This yielded 74 loci.
Because these loci may still harbor SNPs that associate with the glycemic composite, we investigated the proportion of independent SNPs in these loci that associated with the glycemic burden composite. Loci that did not show an excess of independent SNPs associating with the glycemic burden composite (at Fisher’s exact P < 0.05) were investigated for druggability. In this context, “druggable” relates to a locus that encodes a protein targeted by an existing therapeutic (see “Methods” for more details).
Of the 74 loci, 62 were identified that did not harbor an excessive proportion of SNPs associating with the glycemic burden composite (Supplementary Table 1).
The protein products of 23 of the 62 loci were identified as targets of existing medications (Supplementary Table 2), see “Methods” for more details. Seven of the 23 loci were identified as pharmacodynamic targets for drugs (Table 2): PCSK9, APOB, CETP, PLG, NPC1L1, LPA and ALDH2. PCSK9, APOB and LPA have been discussed above.
Table 2.
Loci that are pharmacodynamic targets of existing drugs identified from integrative analysis of the datasets
| Gene | Drug that acts pharmacodynamically on gene protein product | Gene effect | ||
|---|---|---|---|---|
| LDL-C level | CAD risk | Glycemic burden composite | ||
| Drug-loci combinations with no effect on glycemic status | ||||
| PCSK9 | ALN-PCS, BMS-PCSK9, ALN-PCS01, SPC5001 | ↓ | ↓ | No effect predicted |
| APOB | SPC4955 | ↓ | ↓ | No effect predicted |
| CETP | Dalcetrapib, Anacetrapib, Torcetrapib, R7232, PF-3185043, CP-800569 | ↓ | ↓ | No effect predicted |
| PLG | Tenecteplase, Alteplase, Reteplase, Urokinase, Aminocaproic acid, Anistreplase, Streptokinase | ↓ | ↓ | No effect predicted |
| NPC1L1 | Ezetimibe | ↓ | ↓ | No effect predicted |
| ALDH2 | Disulfiram | ↑ | ↓ | No effect predicted |
| Daidzin | ||||
| LPA | ISIS APO(a)Rx | ↓ | ↓ | No effect predicted |
| Drug-loci combinations that impact glycemic status | ||||
| HMGCR | Statins | ↓ | ↓ | ↑ |
| SLC22A3 | Metformin | ↓ | ↓ | ↓ |
We identified CETP as a druggable locus (targeted by CETP inhibitors such as anacetrapib) that has associations with LDL-C and CAD risk and an absence of association with the glycemic burden composite.
PLG encodes plasminogen, an enzyme that degrades plasma proteins including thrombin clots. Plasminogen is also associated with circulating lipid levels (Crutchley et al. 1989). The drugs that target the protein product of PLG, namely tissue plasminogen activators (e.g., streptokinase), are used clinically to degrade coronary artery thrombi in the setting of acute coronary syndrome with treatment efficacy demonstrated in RCTs (Baigent et al. 1998). Their adverse effect profile, however, includes higher risk of serious bleeding (a direct consequence of their mechanism of action), rendering them unsuitable for use in primary prevention of CAD. Furthermore, their primary mode of action is not lipid reduction, but thrombolysis.
ALDH2 was identified to associate with LDL-C and CAD in the absence of modifying glycemic status. Interestingly, and in contrast to the other loci, ALDH2 associated with directionally opposite effects on LDL-C and risk of CAD (Table 2). ALDH2 encodes aldehyde dehydrogenase, responsible for metabolizing acetaldehyde, a breakdown product of alcohol. Disulfiram, a drug currently used to treat alcohol dependence, directly inhibits aldehyde dehydrogenase.
NPC1L1 was identified to alter LDL-C and CAD risk but did not associate with the glycemic burden composite. NPC1L1 encodes Niemann-Pick C1-Like 1 protein, a transmembrane protein that is inhibited by ezetimibe (Garcia-Calvo et al. 2005).
Loci that associate with LDL-C, CAD and glycemic burden composite
These loci affect glycemic status in addition to their effects on LDL-C and CAD risk, and therefore drugs modulating their encoded proteins may cause adverse dysglycemic effects. Forty independent loci were found to associate with LDL-C (P < 5 × 10−8), CAD and the glycemic burden composite (both P < 0.05). Of these, 11 loci were shown to have a higher than expected proportion of SNPs associated with the glycemic burden composite (Fisher’s exact P < 0.05), of which five loci (HMGCR, SLC22A3, FADS2, ABO and PTPN11) were druggable (Supplementary Table 3). Two of these loci (HMGCR and SLC22A3) have existing drugs that target them pharmacodynamically. SLC22A3 is gaining recognition as the target of metformin, (Chen et al. 2010) a drug used to treat diabetes (by reducing blood glucose concentration), which also reduces levels of LDL-C (Keidan et al. 2002; Pentikainen et al. 1990; Robinson et al. 1998; Salpeter et al. 2008; Wulffele et al. 2004) and risk of CAD (Lamanna et al. 2011). Unlike HMGCR, where SNPs in the locus reduce LDL-C and CAD risk yet increase glycemic burden, SLC22A3 SNPs reduce all three traits (LDL-C, CAD risk and glycemic burden).
Discussion
We sought to clarify the relationship between LDL-C, dysglycemia and risk of CAD and shed light on potential therapeutic targets for CAD prevention that are free from dysglycemic effects. To this end, we exploited the public availability of data from several large-scale genetic consortia. Using genetic data can reliably guide which therapeutic targets should be prioritized (Holmes et al. 2013; Nelson et al. 2015).
In this study, we found that the vast majority of SNPs that influence both LDL-C and risk of CAD have the same direction of effect, that is, alleles associated with higher LDL-C levels increase CAD risk. This is further underscored by the causal effect estimate derived from Mendelian randomization, consistent with the known causal relationship between LDL-C and CAD. Both contribute further evidence in support of the so-called “LDL hypothesis”: regardless of the means, a reduction in LDL-C results in a corresponding reduction in risk of CAD (Jarcho and Keaney 2015). In contrast to the relationship of LDL-C with risk of CAD, we observed no clear patterns of association for SNPs that influence LDL-C, risk of T2D or concentrations of fasting glucose. This is despite our Mendelian randomization analysis that revealed a protective causal effect of LDL-C on the risk of T2D [directionally consistent with the relationship seen with statins and T2D risk in randomized clinical trials (Preiss et al. 2011)]. Even so, there are many loci (including druggable loci) that alter LDL-C and CAD risk that are expected to have no substantive effect on glycemic status: these include targets of novel therapies that are protein products of PCSK9, APOB and LPA. These findings are reinforced by the persistence of the causal relationship between LDL-C and risk of CAD even after excluding SNPs associated with T2D. Importantly, this demonstrates that the underlying causal association of LDL-C SNPs with risk of CAD remains intact, irrespective of whether SNPs also associate with T2D. Real potential therefore exists in identifying LDL-C targets that alter risk of CAD and do not impact upon glycemic status.
Of particular importance was our analysis of four candidate loci: HMGCR, PCSK9, APOB and LPA. HMGCR encodes 3-hydroxy-3-methyl-glutaryl-CoA reductase, the intended pharmacological target of statins, and is recognized to increase risk of T2D, both from randomized clinical trials (Preiss et al. 2011; Sattar et al. 2010) and from a recent large-scale Mendelian randomization study (Swerdlow et al. 2015). HMGCR SNPs that associated with LDL-C and CAD had a strong association with our glycemic burden composite. In contrast, SNPs in APOB and PCSK9 that associated with LDL-C and CAD did not associate with the glycemic burden composite. These findings were reciprocated when we analyzed all available SNPs in these loci—there was a clustering of HMGCR SNPs associated with the glycemic burden trait that was not found for PCSK9, APOB or LPA. Thus, the overwhelming evidence, from several independent sources, is that drugs that target protein products of PCSK9, APOB or LPA should not impact upon glycemia. This is important as on-going phase III clinical trials of PCSK9 inhibitory monoclonal antibodies (e.g. evolocumab in NCT01764633; RN316 in NCT01975389 and NCT01975376; and, alirocumab in NCT01617655) and APOB mRNA antisense oligonucleotide inhibitor (mipomersen in NCT01475825) will most likely show beneficial effects on major clinical outcomes [as evidenced by strong genetic associations with CAD and extremely encouraging findings from large, individual (Koren et al. 2014) and pooled analysis of phase II RCTs of PCSK9 inhibition (Stein et al. 2014)]. Our findings indicate these emerging drugs are unlikely to be hampered by mechanism-based effects on glycemic status. It is therefore possible that these emerging drugs may, in future, replace statins as the drug of choice for LDL-C lowering and CAD prevention, although this is likely to follow several years of safety monitoring and patent expiration to reduce costs.
Our multi-trait meta-GWAS to quantify a glycemic burden composite enabled us to investigate potential druggable genes that alter LDL-C and CAD risk but have no appreciable effect on glycemic status. In addition to PCSK9, LPA and APOB loci, we identified the druggable loci CETP, NPC1L1, ALDH2 and PLG. CETP is particularly interesting and controversial (Hewing and Fisher 2012; Miller 2014; Mohammadpour and Akhlaghi 2013). CETP inhibitors were developed principally to raise HDL-C with the aim of reducing CAD risk, but potent examples of these drugs also reduce LDL-C (Bloomfield et al. 2009). Phase III clinical trials of CETP inhibitors for clinical events have been conducted, the largest being dal-OUTCOMES (Schwartz et al. 2012), which randomized 15,871 patients to dalcetrapib or placebo for 31 months and was terminated early because of futility. Furthermore, meta-analyses of several phase III RCTs have failed to show cardiovascular benefit (Kaur et al. 2014; Keene et al. 2014). Of note, meta-analyses may be flawed by including torcetrapib, a CETP inhibitor that had ‘off-target’, deleterious hypertensive effects (Gutstein et al. 2012; Sofat et al. 2010) and has since been abandoned (Diener et al. 2012). Furthermore, therapeutic effects of dalcetrapib (used in dal-OUTCOMES) on LDL-C were small (Schwartz et al. 2012). Our data suggest that more potent CETP inhibitors, such as anacetrapib, that lower LDL-C (in addition to raising HDL-C) (Bloomfield et al. 2009) are likely to reduce CAD risk without any consequence on glycemic status. While ACCELERATE (NCT01687998), a phase III placebo-controlled RCT of 12,000 individuals with existing vascular disease randomized to evacetrapib has been halted for futility, (Lilly 2015) REVEAL (NCT01252953), with 30,000 individuals randomized to anacetrapib or placebo remains on-going and is anticipated to provide definitive evidence.
NPC1L1 encodes the pharmacodynamic target of ezetimibe, an LDL-C lowering therapeutic that has, similar to CETP inhibitors, had a controversial history. Despite the effective lowering of LDL-C by ezetemibe, initial RCTs had not demonstrated its efficacy for surrogate markers of CHD or CHD events (Kastelein et al. 2008; Taylor et al. 2009). However, recent genetic studies (Myocardial Infarction Genetics Consortium I et al. 2014; Ference et al. 2015) and findings from a phase III RCT [IMPROVE-IT (Cannon et al. 2015)] provide evidence that ezetimibe is efficacious at reducing risk of CVD (McPherson and Hegele 2015). Our findings extend current knowledge to suggest that pharmacological lowering of LDL-C by ezetimibe with corresponding CAD prevention is unlikely to be accompanied by dysglycemia.
ALDH2 is also of considerable interest. Given the recent large-scale Mendelian randomization analysis of a SNP in ADH1B that indicates alcohol consumption alters LDL-C and CAD risk, (Holmes et al. 2014) we have the corollary of ALDH2, encoding aldehyde dehydrogenase, another key enzyme in the primary metabolic pathway of alcohol. SNPs in ALDH2 associate with an increase in LDL-C concentration and yet a reduction in CAD risk. Importantly, drugs that specifically target the protein product of ALDH2, used to treat alcohol dependence, such as disulfiram, should be further investigated for their effect on LDL-C and CAD risk. Preliminary studies suggest that disulfiram increases total cholesterol (Major and Goyer 1978), thus the drug may associate with a reduction in risk of CAD (in keeping with the expected pattern of association as reported in Table 2). The association of PLG with LDL-C and CAD is interesting: prospective studies and clinical trials show consistent associations of plasminogen with lipid levels (Crutchley et al. 1989) and CAD risk (Baigent et al. 1998; Lowe et al. 2004; Sakkinen et al. 1999). However, the mechanism-based risk of bleeding that exists with plasminogen activators renders their widespread use for CAD prevention unlikely.
Our study has several advantages. First, it demonstrates the value of exploiting data available in the public domain to conduct original analyses and answer important questions on the causal relationships between traits and diseases. In this respect, the Mendelian randomization analysis for CAD limited to SNPs not associating with T2D risk provides novel insights into disentangling the relationships between LDL-C, glycemic status and risk of CAD. Second, the now well-characterized associations of the HMGCR locus, selected as a positive control, with the glycemic burden composite, LDL-C and CAD were confirmed, further validating the techniques we used. The SLC22A3 locus, of which the protein product is reported as the pharmacological target of metformin, is also noteworthy. Metformin retains a special place in the management of T2D as the only oral hypoglycemic agent that benefits both glycemic status and risk of CAD. We show that variants in SLC22A3 alter glycemia, LDL-C and risk of CAD in a fashion that reflects the profile of actions seen with metformin in randomized trials, (Lamanna et al. 2011; Salpeter et al. 2008; Wulffele et al. 2004) providing further evidence that SLC22A3 may well be the pharmacological target of metformin. It is intriguing that SLC22A3 shares the same LDL-C and CAD modifying properties as HMGCR whereas the opposite effect on glycemia. One could speculate that metformin co-prescribed with statins could offset the diabetogenic effects of statins, whilst providing an additional means to reduce LDL-C (and CAD risk) that is independent of HMGCR.
Our study also has several limitations. First, use of summary-level data prevented more intricate analysis including use of covariates and conditioning, and was limited to the models used in the original analyses. However, use of summary estimates from published GWAS consortia maximizes use of all available data, thereby mitigating against publication bias, increasing power and enhancing generalizability of findings (Lin and Zeng 2010). Second, our threshold for the glycemic burden composite (P < 0.05) in SNPs associating with LDL-C (P < 5 × 10−8) and CAD (P < 0.05) may be interpreted as insufficiently stringent, given the multiple tests conducted. However, we followed up investigations of all loci for the glycemic burden composite with a Fisher’s test to identify independent loci that harbored SNPs associated with the glycemic burden composite—and in doing so, we minimized any false positives (or negatives) using this approach. The choice of a P value threshold of P < 0.05 for CAD is justifiable given the known causal association of LDL-C SNPs with CAD together with the directions of effect of SNPs on both traits, and given that all SNPs associated at GWAS significance for LDL-C. Third, gene–gene interactions may make an important contribution to the genetic architecture of disease, and such interactions may not display so-called “marginal effects”, meaning that associations arising from interactions would not be detected in conventional associated analyses (Cordell 2009; De et al. 2015). Follow-up studies could investigate the role of gene–gene interactions in this setting. Fourth, the association of HMGCR loci with glycemic burden composite means that, if this information was known a few decades ago, statins may not have been developed for CAD prevention. However, this is often the case as drug discovery progresses, and early drugs are superseded by drugs with a more favorable adverse effect profile, or a broader therapeutic index (Diener et al. 2012). Finally, our Mendelian randomization analyses used a “conventional” ratio approach that does not take into account potential pleiotropy of the genetic instruments. Further studies are needed to investigate whether these findings are influenced by unbalanced pleiotropy using emerging approaches such as multivariate and/or Egger-Mendelian randomization (Bowden et al. 2015; Burgess et al. 2015).
The integrative use of multiple GWAS datasets, as we report, represents a novel approach to answering critical questions on disease etiology and to inform on intended and unintended consequences of pharmacological modification of biomarkers. Real opportunities exist for academia to work together with pharmaceutical industry to translate GWAS data and maximize understanding of which therapeutic targets to prioritize based on robust, large-scale, integrative genomic analyses (Kathiresan 2015). This would facilitate discovery of safe, efficacious new therapeutics and potentially offset the exuberant costs of drug development. Indeed, drug mechanisms that have genetic support are more than twice as likely to succeed in clinical trials, (Nelson et al. 2015) and GWAS plus Mendelian randomization have been identified as key solutions to revitalizing drug development in cardiovascular disease (Fordyce et al. 2015).
In conclusion, we used publicly available data to interrogate the relationship of LDL-C-associated SNPs for their associations with CAD risk, glycemic traits and T2D risk. We identify several potential therapeutic targets that influence LDL-C and risk of CAD that do not alter glycemic status. We provide evidence that emerging drugs that target protein products of PCSK9, APOB and LPA are unlikely to impact upon glycemic status, and in that regard, may have advantages over statins for LDL-C lowering and prevention of CAD.
Data and materials availability
Data retrieved from sources, as listed in Materials and Methods. Summary estimates from glycemic burden composite GWAS meta-analysis are provided in the Data file S1.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 2 (DOCX 202 kb) Data File S1: Summary estimates from GWAS meta-analysis of glycemic traits (fasting glucose, fasting insulin, fasting proinsulin and HbA1c) and type 2 diabetes to yield a glycemic burden composite
Compliance with ethical standards
Funding
FWA is supported by a Dekker scholarship-Junior Staff Member 2014T001—Netherlands Heart Foundation. The research leading to these results has received funding from the European Union Seventh Framework Programme FP7/2007-2013 under grant agreement n° HEALTH-F2-2013-601456 (CVgenes-at-target).
Conflict of interest
DIS is a consultant to Pfizer.
Contributor Information
Folkert W. Asselbergs, Email: f.w.asselbergs@umcutrecht.nl
Michael V. Holmes, Email: michael.holmes@ndph.ox.ac.uk
References
- Akdim F, Stroes ES, Sijbrands EJ, Tribble DL, Trip MD, Jukema JW, Flaim JD, Su J, Yu R, Baker BF, Wedel MK, Kastelein JJ. Efficacy and safety of mipomersen, an antisense inhibitor of apolipoprotein B, in hypercholesterolemic subjects receiving stable statin therapy. J Am Coll Cardiol. 2010;55:1611–1618. doi: 10.1016/j.jacc.2009.11.069. [DOI] [PubMed] [Google Scholar]
- Baigent C, Collins R, Appleby P, Parish S, Sleight P, Peto R. ISIS-2: 10 year survival among patients with suspected acute myocardial infarction in randomised comparison of intravenous streptokinase, oral aspirin, both, or neither. The ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. BMJ. 1998;316:1337–1343. doi: 10.1136/bmj.316.7141.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomfield D, Carlson GL, Sapre A, Tribble D, McKenney JM, Littlejohn TW, 3rd, Sisk CM, Mitchel Y, Pasternak RC. Efficacy and safety of the cholesteryl ester transfer protein inhibitor anacetrapib as monotherapy and coadministered with atorvastatin in dyslipidemic patients. Am Heart J. 2009;157(352–360):e2. doi: 10.1016/j.ahj.2008.09.022. [DOI] [PubMed] [Google Scholar]
- Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512–525. doi: 10.1093/ije/dyv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess S, Thompson SG. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol. 2015;15(181):251–260. doi: 10.1093/aje/kwu283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon Christopher P., Blazing Michael A., Giugliano Robert P., McCagg Amy, White Jennifer A., Theroux Pierre, Darius Harald, Lewis Basil S., Ophuis Ton Oude, Jukema J. Wouter, De Ferrari Gaetano M., Ruzyllo Witold, De Lucca Paul, Im KyungAh, Bohula Erin A., Reist Craig, Wiviott Stephen D., Tershakovec Andrew M., Musliner Thomas A., Braunwald Eugene, Califf Robert M. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. New England Journal of Medicine. 2015;372(25):2387–2397. doi: 10.1056/NEJMoa1410489. [DOI] [PubMed] [Google Scholar]
- Chen L, Pawlikowski B, Schlessinger A, More SS, Stryke D, Johns SJ, Portman MA, Chen E, Ferrin TE, Sali A, Giacomini KM. Role of organic cation transporter 3 (SLC22A3) and its missense variants in the pharmacologic action of metformin. Pharmacogenet Genomics. 2010;20:687–699. doi: 10.1097/FPC.0b013e32833fe789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholesterol Treatment Trialists C, Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, Simes J, Collins R. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholesterol Treatment Trialists C, Mihaylova B, Emberson J, Blackwell L, Keech A, Simes J, Barnes EH, Voysey M, Gray A, Collins R, Baigent C. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet. 2012;380:581–590. doi: 10.1016/S0140-6736(12)60367-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deloukas Panos, Kanoni Stavroula, Willenborg Christina, Farrall Martin, Assimes Themistocles L, Thompson John R, Ingelsson Erik, Saleheen Danish, Erdmann Jeanette, Goldstein Benjamin A, Stirrups Kathleen, König Inke R, Cazier Jean-Baptiste, Johansson Åsa, Hall Alistair S, Lee Jong-Young, Willer Cristen J, Chambers John C, Esko Tõnu, Folkersen Lasse, Goel Anuj, Grundberg Elin, Havulinna Aki S, Ho Weang K, Hopewell Jemma C, Eriksson Niclas, Kleber Marcus E, Kristiansson Kati, Lundmark Per, Lyytikäinen Leo-Pekka, Rafelt Suzanne, Shungin Dmitry, Strawbridge Rona J, Thorleifsson Gudmar, Tikkanen Emmi, Van Zuydam Natalie, Voight Benjamin F, Waite Lindsay L, Zhang Weihua, Ziegler Andreas, Absher Devin, Altshuler David, Balmforth Anthony J, Barroso Inês, Braund Peter S, Burgdorf Christof, Claudi-Boehm Simone, Cox David, Dimitriou Maria, Do Ron, Doney Alex S F, Mokhtari NourEddine El, Eriksson Per, Fischer Krista, Fontanillas Pierre, Franco-Cereceda Anders, Gigante Bruna, Groop Leif, Gustafsson Stefan, Hager Jörg, Hallmans Göran, Han Bok-Ghee, Hunt Sarah E, Kang Hyun M, Illig Thomas, Kessler Thorsten, Knowles Joshua W, Kolovou Genovefa, Kuusisto Johanna, Langenberg Claudia, Langford Cordelia, Leander Karin, Lokki Marja-Liisa, Lundmark Anders, McCarthy Mark I, Meisinger Christa, Melander Olle, Mihailov Evelin, Maouche Seraya, Morris Andrew D, Müller-Nurasyid Martina, Nikus Kjell, Peden John F, Rayner N William, Rasheed Asif, Rosinger Silke, Rubin Diana, Rumpf Moritz P, Schäfer Arne, Sivananthan Mohan, Song Ci, Stewart Alexandre F R, Tan Sian-Tsung, Thorgeirsson Gudmundur, Schoot C Ellen van der, Wagner Peter J, Wells George A, Wild Philipp S, Yang Tsun-Po, Amouyel Philippe, Arveiler Dominique, Basart Hanneke, Boehnke Michael, Boerwinkle Eric, Brambilla Paolo, Cambien Francois, Cupples Adrienne L, de Faire Ulf, Dehghan Abbas, Diemert Patrick, Epstein Stephen E, Evans Alun, Ferrario Marco M, Ferrières Jean, Gauguier Dominique, Go Alan S, Goodall Alison H, Gudnason Villi, Hazen Stanley L, Holm Hilma, Iribarren Carlos, Jang Yangsoo, Kähönen Mika, Kee Frank, Kim Hyo-Soo, Klopp Norman, Koenig Wolfgang, Kratzer Wolfgang, Kuulasmaa Kari, Laakso Markku, Laaksonen Reijo, Lee Ji-Young, Lind Lars, Ouwehand Willem H, Parish Sarah, Park Jeong E, Pedersen Nancy L, Peters Annette, Quertermous Thomas, Rader Daniel J, Salomaa Veikko, Schadt Eric, Shah Svati H, Sinisalo Juha, Stark Klaus, Stefansson Kari, Trégouët David-Alexandre, Virtamo Jarmo, Wallentin Lars, Wareham Nicholas, Zimmermann Martina E, Nieminen Markku S, Hengstenberg Christian, Sandhu Manjinder S, Pastinen Tomi, Syvänen Ann-Christine, Hovingh G Kees, Dedoussis George, Franks Paul W, Lehtimäki Terho, Metspalu Andres, Zalloua Pierre A, Siegbahn Agneta, Schreiber Stefan, Ripatti Samuli, Blankenberg Stefan S, Perola Markus, Clarke Robert, Boehm Bernhard O, O'Donnell Christopher, Reilly Muredach P, März Winfried, Collins Rory, Kathiresan Sekar, Hamsten Anders, Kooner Jaspal S, Thorsteinsdottir Unnur, Danesh John, Palmer Colin N A, Roberts Robert, Watkins Hugh, Schunkert Heribert, Samani Nilesh J. Large-scale association analysis identifies new risk loci for coronary artery disease. Nature Genetics. 2012;45(1):25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordell HJ. Detecting gene-gene interactions that underlie human diseases. Nat Rev Genet. 2009;10:392–404. doi: 10.1038/nrg2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crutchley DJ, McPhee GV, Terris MF, Canossa-Terris MA. Levels of three hemostatic factors in relation to serum lipids. Monocyte procoagulant activity, tissue plasminogen activator, and type-1 plasminogen activator inhibitor. Arteriosclerosis. 1989;9:934–939. doi: 10.1161/atv81.9.6.2511826. [DOI] [PubMed] [Google Scholar]
- De R, Verma SS, Drenos F, Holzinger ER, Holmes MV, Hall MA, Crosslin DR, Carrell DS, Hakonarson H, Jarvik G, Larson E, Pacheco JA, Rasmussen-Torvik LJ, Moore CB, Asselbergs FW, Moore JH, Ritchie MD, Keating BJ, Gilbert-Diamond D. Identifying gene-gene interactions that are highly associated with Body Mass Index using Quantitative Multifactor Dimensionality Reduction (QMDR) BioData Min. 2015;8:41. doi: 10.1186/s13040-015-0074-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diener HC, Eikelboom J, Granger CB, Hacke W. The king is dead (warfarin): direct thrombin and factor Xa inhibitors: the next Diadochian War? Int J Stroke. 2012;7:139–141. doi: 10.1111/j.1747-4949.2011.00739.x. [DOI] [PubMed] [Google Scholar]
- Ellinghaus D, Ellinghaus E, Nair RP, Stuart PE, Esko T, Metspalu A, Debrus S, Raelson JV, Tejasvi T, Belouchi M, West SL, Barker JN, Koks S, Kingo K, Balschun T, Palmieri O, Annese V, Gieger C, Wichmann HE, Kabesch M, Trembath RC, Mathew CG, Abecasis GR, Weidinger S, Nikolaus S, Schreiber S, Elder JT, Weichenthal M, Nothnagel M, Franke A. Combined analysis of genome-wide association studies for Crohn disease and psoriasis identifies seven shared susceptibility loci. Am J Hum Genet. 2012;90:636–647. doi: 10.1016/j.ajhg.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ference BA, Majeed F, Penumetcha R, Flack JM, Brook RD. Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both: a 2 × 2 Factorial Mendelian Randomization Study. J Am Coll Cardiol. 2015;65:1552–1561. doi: 10.1016/j.jacc.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordyce CB, Roe MT, Ahmad T, Libby P, Borer JS, Hiatt WR, Bristow MR, Packer M, Wasserman SM, Braunstein N, Pitt B, DeMets DL, Cooper-Arnold K, Armstrong PW, Berkowitz SD, Scott R, Prats J, Galis ZS, Stockbridge N, Peterson ED, Califf RM. Cardiovascular drug development: is it dead or just hibernating? J Am Coll Cardiol. 2015;65:1567–1582. doi: 10.1016/j.jacc.2015.03.016. [DOI] [PubMed] [Google Scholar]
- Garcia-Calvo M, Lisnock J, Bull HG, Hawes BE, Burnett DA, Braun MP, Crona JH, Davis HR, Jr, Dean DC, Detmers PA, Graziano MP, Hughes M, Macintyre DE, Ogawa A, O’Neill KA, Iyer SP, Shevell DE, Smith MM, Tang YS, Makarewicz AM, Ujjainwalla F, Altmann SW, Chapman KT, Thornberry NA. The target of ezetimibe is Niemann-Pick C1-Like 1 (NPC1L1) Proc Natl Acad Sci USA. 2005;102:8132–8137. doi: 10.1073/pnas.0500269102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer Cristen J, Schmidt Ellen M, Sengupta Sebanti, Peloso Gina M, Gustafsson Stefan, Kanoni Stavroula, Ganna Andrea, Chen Jin, Buchkovich Martin L, Mora Samia, Beckmann Jacques S, Bragg-Gresham Jennifer L, Chang Hsing-Yi, Demirkan Ayşe, Den Hertog Heleen M, Do Ron, Donnelly Louise A, Ehret Georg B, Esko Tõnu, Feitosa Mary F, Ferreira Teresa, Fischer Krista, Fontanillas Pierre, Fraser Ross M, Freitag Daniel F, Gurdasani Deepti, Heikkilä Kauko, Hyppönen Elina, Isaacs Aaron, Jackson Anne U, Johansson Åsa, Johnson Toby, Kaakinen Marika, Kettunen Johannes, Kleber Marcus E, Li Xiaohui, Luan Jian'an, Lyytikäinen Leo-Pekka, Magnusson Patrik K E, Mangino Massimo, Mihailov Evelin, Montasser May E, Müller-Nurasyid Martina, Nolte Ilja M, O'Connell Jeffrey R, Palmer Cameron D, Perola Markus, Petersen Ann-Kristin, Sanna Serena, Saxena Richa, Service Susan K, Shah Sonia, Shungin Dmitry, Sidore Carlo, Song Ci, Strawbridge Rona J, Surakka Ida, Tanaka Toshiko, Teslovich Tanya M, Thorleifsson Gudmar, Van den Herik Evita G, Voight Benjamin F, Volcik Kelly A, Waite Lindsay L, Wong Andrew, Wu Ying, Zhang Weihua, Absher Devin, Asiki Gershim, Barroso Inês, Been Latonya F, Bolton Jennifer L, Bonnycastle Lori L, Brambilla Paolo, Burnett Mary S, Cesana Giancarlo, Dimitriou Maria, Doney Alex S F, Döring Angela, Elliott Paul, Epstein Stephen E, Eyjolfsson Gudmundur Ingi, Gigante Bruna, Goodarzi Mark O, Grallert Harald, Gravito Martha L, Groves Christopher J, Hallmans Göran, Hartikainen Anna-Liisa, Hayward Caroline, Hernandez Dena, Hicks Andrew A, Holm Hilma, Hung Yi-Jen, Illig Thomas, Jones Michelle R, Kaleebu Pontiano, Kastelein John J P, Khaw Kay-Tee, Kim Eric, Klopp Norman, Komulainen Pirjo, Kumari Meena, Langenberg Claudia, Lehtimäki Terho, Lin Shih-Yi, Lindström Jaana, Loos Ruth J F, Mach François, McArdle Wendy L, Meisinger Christa, Mitchell Braxton D, Müller Gabrielle, Nagaraja Ramaiah, Narisu Narisu, Nieminen Tuomo V M, Nsubuga Rebecca N, Olafsson Isleifur, Ong Ken K, Palotie Aarno, Papamarkou Theodore, Pomilla Cristina, Pouta Anneli, Rader Daniel J, Reilly Muredach P, Ridker Paul M, Rivadeneira Fernando, Rudan Igor, Ruokonen Aimo, Samani Nilesh, Scharnagl Hubert, Seeley Janet, Silander Kaisa, Stancáková Alena, Stirrups Kathleen, Swift Amy J, Tiret Laurence, Uitterlinden Andre G, van Pelt L Joost, Vedantam Sailaja, Wainwright Nicholas, Wijmenga Cisca, Wild Sarah H, Willemsen Gonneke, Wilsgaard Tom, Wilson James F, Young Elizabeth H, Zhao Jing Hua, Adair Linda S, Arveiler Dominique, Assimes Themistocles L, Bandinelli Stefania, Bennett Franklyn, Bochud Murielle, Boehm Bernhard O, Boomsma Dorret I, Borecki Ingrid B, Bornstein Stefan R, Bovet Pascal, Burnier Michel, Campbell Harry, Chakravarti Aravinda, Chambers John C, Chen Yii-Der Ida, Collins Francis S, Cooper Richard S, Danesh John, Dedoussis George, de Faire Ulf, Feranil Alan B, Ferrières Jean, Ferrucci Luigi, Freimer Nelson B, Gieger Christian, Groop Leif C, Gudnason Vilmundur, Gyllensten Ulf, Hamsten Anders, Harris Tamara B, Hingorani Aroon, Hirschhorn Joel N, Hofman Albert, Hovingh G Kees, Hsiung Chao Agnes, Humphries Steve E, Hunt Steven C, Hveem Kristian, Iribarren Carlos, Järvelin Marjo-Riitta, Jula Antti, Kähönen Mika, Kaprio Jaakko, Kesäniemi Antero, Kivimaki Mika, Kooner Jaspal S, Koudstaal Peter J, Krauss Ronald M, Kuh Diana, Kuusisto Johanna, Kyvik Kirsten O, Laakso Markku, Lakka Timo A, Lind Lars, Lindgren Cecilia M, Martin Nicholas G, März Winfried, McCarthy Mark I, McKenzie Colin A, Meneton Pierre, Metspalu Andres, Moilanen Leena, Morris Andrew D, Munroe Patricia B, Njølstad Inger, Pedersen Nancy L, Power Chris, Pramstaller Peter P, Price Jackie F, Psaty Bruce M, Quertermous Thomas, Rauramaa Rainer, Saleheen Danish, Salomaa Veikko, Sanghera Dharambir K, Saramies Jouko, Schwarz Peter E H, Sheu Wayne H-H, Shuldiner Alan R, Siegbahn Agneta, Spector Tim D, Stefansson Kari, Strachan David P, Tayo Bamidele O, Tremoli Elena, Tuomilehto Jaakko, Uusitupa Matti, van Duijn Cornelia M, Vollenweider Peter, Wallentin Lars, Wareham Nicholas J, Whitfield John B, Wolffenbuttel Bruce H R, Ordovas Jose M, Boerwinkle Eric, Palmer Colin N A, Thorsteinsdottir Unnur, Chasman Daniel I, Rotter Jerome I, Franks Paul W, Ripatti Samuli, Cupples L Adrienne, Sandhu Manjinder S, Rich Stephen S, Boehnke Michael, Deloukas Panos, Kathiresan Sekar, Mohlke Karen L, Ingelsson Erik, Abecasis Gonçalo R. Discovery and refinement of loci associated with lipid levels. Nature Genetics. 2013;45(11):1274–1283. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutstein DE, Krishna R, Johns D, Surks HK, Dansky HM, Shah S, Mitchel YB, Arena J, Wagner JA. Anacetrapib, a novel CETP inhibitor: pursuing a new approach to cardiovascular risk reduction. Clin Pharmacol Ther. 2012;91:109–122. doi: 10.1038/clpt.2011.271. [DOI] [PubMed] [Google Scholar]
- Hewing B, Fisher EA. Rationale for cholesteryl ester transfer protein inhibition. Curr Opin Lipidol. 2012;23:372–376. doi: 10.1097/MOL.0b013e328353ef1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MV, Simon T, Exeter HJ, Folkersen L, Asselbergs FW, Guardiola M, Cooper JA, Palmen J, Hubacek JA, Carruthers KF, Horne BD, Brunisholz KD, Mega JL, van Iperen EP, Li M, Leusink M, Trompet S, Verschuren JJ, Hovingh GK, Dehghan A, Nelson CP, Kotti S, Danchin N, Scholz M, Haase CL, Rothenbacher D, Swerdlow DI, Kuchenbaecker KB, Staines-Urias E, Goel A, van’t Hooft F, Gertow K, de Faire U, Panayiotou AG, Tremoli E, Baldassarre D, Veglia F, Holdt LM, Beutner F, Gansevoort RT, Navis GJ, Mateo Leach I, Breitling LP, Brenner H, Thiery J, Dallmeier D, Franco-Cereceda A, Boer JM, Stephens JW, Hofker MH, Tedgui A, Hofman A, Uitterlinden AG, Adamkova V, Pitha J, Onland-Moret NC, Cramer MJ, Nathoe HM, Spiering W, Klungel OH, Kumari M, Whincup PH, Morrow DA, Braund PS, Hall AS, Olsson AG, Doevendans PA, Trip MD, Tobin MD, Hamsten A, Watkins H, Koenig W, Nicolaides AN, Teupser D, Day IN, Carlquist JF, Gaunt TR, Ford I, Sattar N, Tsimikas S, Schwartz GG, Lawlor DA, Morris RW, Sandhu MS, Poledne R, Maitland-van der Zee AH, Khaw KT, Keating BJ, van der Harst P, Price JF, Mehta SR, Yusuf S, Witteman JC, Franco OH, Jukema JW, de Knijff P, Tybjaerg-Hansen A, Rader DJ, Farrall M, Samani NJ, et al. Secretory phospholipase A(2)-IIA and cardiovascular disease: a mendelian randomization study. J Am Coll Cardiol. 2013;62:1966–1976. doi: 10.1016/j.jacc.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MV, Dale CE, Zuccolo L, Silverwood RJ, Guo Y, Ye Z, Prieto-Merino D, Dehghan A, Trompet S, Wong A, Cavadino A, Drogan D, Padmanabhan S, Li S, Yesupriya A, Leusink M, Sundstrom J, Hubacek JA, Pikhart H, Swerdlow DI, Panayiotou AG, Borinskaya SA, Finan C, Shah S, Kuchenbaecker KB, Shah T, Engmann J, Folkersen L, Eriksson P, Ricceri F, Melander O, Sacerdote C, Gamble DM, Rayaprolu S, Ross OA, McLachlan S, Vikhireva O, Sluijs I, Scott RA, Adamkova V, Flicker L, Bockxmeer FM, Power C, Marques-Vidal P, Meade T, Marmot MG, Ferro JM, Paulos-Pinheiro S, Humphries SE, Talmud PJ, Mateo Leach I, Verweij N, Linneberg A, Skaaby T, Doevendans PA, Cramer MJ, van der Harst P, Klungel OH, Dowling NF, Dominiczak AF, Kumari M, Nicolaides AN, Weikert C, Boeing H, Ebrahim S, Gaunt TR, Price JF, Lannfelt L, Peasey A, Kubinova R, Pajak A, Malyutina S, Voevoda MI, Tamosiunas A, Maitland-van der Zee AH, Norman PE, Hankey GJ, Bergmann MM, Hofman A, Franco OH, Cooper J, Palmen J, Spiering W, de Jong PA, Kuh D, Hardy R, Uitterlinden AG, Ikram MA, Ford I, Hypponen E, Almeida OP, Wareham NJ, Khaw KT, Hamsten A, Husemoen LL, Tjonneland A, Tolstrup JS, Rimm E, Beulens JW, Verschuren WM, et al. Association between alcohol and cardiovascular disease: Mendelian randomisation analysis based on individual participant data. BMJ. 2014;349:g4164. doi: 10.1136/bmj.g4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MV, Asselbergs FW, Palmer TM, Drenos F, Lanktree MB, Nelson CP, Dale CE, Padmanabhan S, Finan C, Swerdlow DI, Tragante V, van Iperen EP, Sivapalaratnam S, Shah S, Elbers CC, Shah T, Engmann J, Giambartolomei C, White J, Zabaneh D, Sofat R, McLachlan S, on behalf of the Uc. Doevendans PA, Balmforth AJ, Hall AS, North KE, Almoguera B, Hoogeveen RC, Cushman M, Fornage M, Patel SR, Redline S, Siscovick DS, Tsai MY, Karczewski KJ, Hofker MH, Verschuren WM, Bots ML, van der Schouw YT, Melander O, Dominiczak AF, Morris R, Ben-Shlomo Y, Price J, Kumari M, Baumert J, Peters A, Thorand B, Koenig W, Gaunt TR, Humphries SE, Clarke R, Watkins H, Farrall M, Wilson JG, Rich SS, de Bakker PI, Lange LA, Davey Smith G, Reiner AP, Talmud PJ, Kivimaki M, Lawlor DA, Dudbridge F, Samani NJ, Keating BJ, Hingorani AD, Casas JP. Mendelian randomization of blood lipids for coronary heart disease. Eur Heart J. 2015;36:539–550. doi: 10.1093/eurheartj/eht571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarcho John A., Keaney John F. Proof That Lower Is Better — LDL Cholesterol and IMPROVE-IT. New England Journal of Medicine. 2015;372(25):2448–2450. doi: 10.1056/NEJMe1507041. [DOI] [PubMed] [Google Scholar]
- Kastelein JJ, Akdim F, Stroes ES, Zwinderman AH, Bots ML, Stalenhoef AF, Visseren FL, Sijbrands EJ, Trip MD, Stein EA, Gaudet D, Duivenvoorden R, Veltri EP, Marais AD, de Groot E, Investigators E. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008;358:1431–1443. doi: 10.1056/NEJMoa0800742. [DOI] [PubMed] [Google Scholar]
- Kathiresan S. Developing medicines that mimic the natural successes of the human genome: lessons from NPC1L1, HMGCR, PCSK9, APOC3, and CETP. J Am Coll Cardiol. 2015;65:1562–1566. doi: 10.1016/j.jacc.2015.02.049. [DOI] [PubMed] [Google Scholar]
- Kaur N, Pandey A, Negi H, Shafiq N, Reddy S, Kaur H, Chadha N, Malhotra S. Effect of HDL-raising drugs on cardiovascular outcomes: a systematic review and meta-regression. PLoS One. 2014;9:e94585. doi: 10.1371/journal.pone.0094585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene D, Price C, Shun-Shin MJ, Francis DP. Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: meta-analysis of randomised controlled trials including 117,411 patients. BMJ. 2014;349:g4379. doi: 10.1136/bmj.g4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keidan B, Hsia J, Katz R. Plasma lipids and antidiabetic agents: a brief overview. Br J Diabetes Vasc Dis. 2002;2:40–43. doi: 10.1177/14746514020020011801. [DOI] [Google Scholar]
- Koren MJ, Giugliano RP, Raal FJ, Sullivan D, Bolognese M, Langslet G, Civeira F, Somaratne R, Nelson P, Liu T, Scott R, Wasserman SM, Sabatine MS, Investigators O. Efficacy and safety of longer-term administration of evolocumab (AMG 145) in patients with hypercholesterolemia: 52-week results from the Open-Label Study of Long-Term Evaluation Against LDL-C (OSLER) randomized trial. Circulation. 2014;129:234–243. doi: 10.1161/CIRCULATIONAHA.113.007012. [DOI] [PubMed] [Google Scholar]
- Lamanna C, Monami M, Marchionni N, Mannucci E. Effect of metformin on cardiovascular events and mortality: a meta-analysis of randomized clinical trials. Diabetes Obes Metab. 2011;13:221–228. doi: 10.1111/j.1463-1326.2010.01349.x. [DOI] [PubMed] [Google Scholar]
- Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, Maciejewski A, Arndt D, Wilson M, Neveu V, Tang A, Gabriel G, Ly C, Adamjee S, Dame ZT, Han B, Zhou Y, Wishart DS. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27:1133–1163. doi: 10.1002/sim.3034. [DOI] [PubMed] [Google Scholar]
- Lilly (2015) Lilly to discontinue development of evacetrapib for high-risk atherosclerotic cardiovascular disease. https://investor.lilly.com/releasedetail.cfm?ReleaseID=936130. Accessed 5 Nov 2015
- Lin DY, Zeng D. Meta-analysis of genome-wide association studies: no efficiency gain in using individual participant data. Genet Epidemiol. 2010;34:60–66. doi: 10.1002/gepi.20435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe GD, Danesh J, Lewington S, Walker M, Lennon L, Thomson A, Rumley A, Whincup PH. Tissue plasminogen activator antigen and coronary heart disease. Prospective study and meta-analysis. Eur Heart J. 2004;25:252–259. doi: 10.1016/j.ehj.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Major LF, Goyer PF. Effects of disulfiram and pyridoxine on serum cholesterol. Ann Intern Med. 1978;88:53–56. doi: 10.7326/0003-4819-88-1-53. [DOI] [PubMed] [Google Scholar]
- McGeachie MJ, Clemmer GL, Lasky-Su J, Dahlin A, Raby BA, Weiss ST. Joint GWAS Analysis: comparing similar GWAS at different genomic resolutions identifies novel pathway associations with six complex diseases. Genomics Data. 2014;2:202–211. doi: 10.1016/j.gdata.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson R, Hegele RA. Ezetimibe: rescued by randomization (clinical and mendelian) Arterioscler Thromb Vasc Biol. 2015;35:e13–e15. doi: 10.1161/ATVBAHA.114.305012. [DOI] [PubMed] [Google Scholar]
- Miller NE. CETP inhibitors and cardiovascular disease: time to think again. F1000Res. 2014;3:124. doi: 10.12688/f1000research.4396.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadpour AH, Akhlaghi F. Future of cholesteryl ester transfer protein (CETP) inhibitors: a pharmacological perspective. Clin Pharmacokinet. 2013;52:615–626. doi: 10.1007/s40262-013-0071-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myocardial Infarction Genetics Consortium I. Stitziel NO, Won HH, Morrison AC, Peloso GM, Do R, Lange LA, Fontanillas P, Gupta N, Duga S, Goel A, Farrall M, Saleheen D, Ferrario P, Konig I, Asselta R, Merlini PA, Marziliano N, Notarangelo MF, Schick U, Auer P, Assimes TL, Reilly M, Wilensky R, Rader DJ, Hovingh GK, Meitinger T, Kessler T, Kastrati A, Laugwitz KL, Siscovick D, Rotter JI, Hazen SL, Tracy R, Cresci S, Spertus J, Jackson R, Schwartz SM, Natarajan P, Crosby J, Muzny D, Ballantyne C, Rich SS, O’Donnell CJ, Abecasis G, Sunyaev S, Nickerson DA, Buring JE, Ridker PM, Chasman DI, Austin E, Ye Z, Kullo IJ, Weeke PE, Shaffer CM, Bastarache LA, Denny JC, Roden DM, Palmer C, Deloukas P, Lin DY, Tang ZZ, Erdmann J, Schunkert H, Danesh J, Marrugat J, Elosua R, Ardissino D, McPherson R, Watkins H, Reiner AP, Wilson JG, Altshuler D, Gibbs RA, Lander ES, Boerwinkle E, Gabriel S, Kathiresan S. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N Engl J Med. 2014;371:2072–2082. doi: 10.1056/NEJMoa1405386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, Floratos A, Sham PC, Li MJ, Wang J, Cardon LR, Whittaker JC, Sanseau P. The support of human genetic evidence for approved drug indications. Nat Genet. 2015;47:856–860. doi: 10.1038/ng.3314. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, Davenport AP, McGrath JC, Peters JA, Southan C, Spedding M, Yu W, Harmar AJ, Nc I. The IUPHAR/BPS guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res. 2014;42:D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentikainen PJ, Voutilainen E, Aro A, Uusitupa M, Penttila I, Vapaatalo H. Cholesterol lowering effect of metformin in combined hyperlipidemia: placebo controlled double blind trial. Ann Med. 1990;22:307–312. doi: 10.3109/07853899009147912. [DOI] [PubMed] [Google Scholar]
- Preiss D, Seshasai SR, Welsh P, Murphy SA, Ho JE, Waters DD, DeMicco DA, Barter P, Cannon CP, Sabatine MS, Braunwald E, Kastelein JJ, de Lemos JA, Blazing MA, Pedersen TR, Tikkanen MJ, Sattar N, Ray KK. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. JAMA. 2011;305:2556–2564. doi: 10.1001/jama.2011.860. [DOI] [PubMed] [Google Scholar]
- Rask-Andersen M, Masuram S, Schioth HB. The druggable genome: evaluation of drug targets in clinical trials suggests major shifts in molecular class and indication. Annu Rev Pharmacol Toxicol. 2014;54:9–26. doi: 10.1146/annurev-pharmtox-011613-135943. [DOI] [PubMed] [Google Scholar]
- Robinson AC, Burke J, Robinson S, Johnston DG, Elkeles RS. The effects of metformin on glycemic control and serum lipids in insulin-treated NIDDM patients with suboptimal metabolic control. Diabetes Care. 1998;21:701–705. doi: 10.2337/diacare.21.5.701. [DOI] [PubMed] [Google Scholar]
- Sakkinen PA, Cushman M, Psaty BM, Rodriguez B, Boineau R, Kuller LH, Tracy RP. Relationship of plasmin generation to cardiovascular disease risk factors in elderly men and women. Arterioscler Thromb Vasc Biol. 1999;19:499–504. doi: 10.1161/01.ATV.19.3.499. [DOI] [PubMed] [Google Scholar]
- Salpeter SR, Buckley NS, Kahn JA, Salpeter EE. Meta-analysis: metformin treatment in persons at risk for diabetes mellitus. Am J Med. 2008;121(149–157):e2. doi: 10.1016/j.amjmed.2007.09.016. [DOI] [PubMed] [Google Scholar]
- Sattar N, Preiss D, Murray HM, Welsh P, Buckley BM, de Craen AJ, Seshasai SR, McMurray JJ, Freeman DJ, Jukema JW, Macfarlane PW, Packard CJ, Stott DJ, Westendorp RG, Shepherd J, Davis BR, Pressel SL, Marchioli R, Marfisi RM, Maggioni AP, Tavazzi L, Tognoni G, Kjekshus J, Pedersen TR, Cook TJ, Gotto AM, Clearfield MB, Downs JR, Nakamura H, Ohashi Y, Mizuno K, Ray KK, Ford I. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375:735–742. doi: 10.1016/S0140-6736(09)61965-6. [DOI] [PubMed] [Google Scholar]
- Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, Leitersdorf E, McMurray JJ, Mundl H, Nicholls SJ, Shah PK, Tardif JC, Wright RS, Dal OI. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–2099. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- Sofat R, Hingorani AD, Smeeth L, Humphries SE, Talmud PJ, Cooper J, Shah T, Sandhu MS, Ricketts SL, Boekholdt SM, Wareham N, Khaw KT, Kumari M, Kivimaki M, Marmot M, Asselbergs FW, van der Harst P, Dullaart RP, Navis G, van Veldhuisen DJ, Van Gilst WH, Thompson JF, McCaskie P, Palmer LJ, Arca M, Quagliarini F, Gaudio C, Cambien F, Nicaud V, Poirer O, Gudnason V, Isaacs A, Witteman JC, van Duijn CM, Pencina M, Vasan RS, D’Agostino RB, Sr, Ordovas J, Li TY, Kakko S, Kauma H, Savolainen MJ, Kesaniemi YA, Sandhofer A, Paulweber B, Sorli JV, Goto A, Yokoyama S, Okumura K, Horne BD, Packard C, Freeman D, Ford I, Sattar N, McCormack V, Lawlor DA, Ebrahim S, Smith GD, Kastelein JJ, Deanfield J, Casas JP. Separating the mechanism-based and off-target actions of cholesteryl ester transfer protein inhibitors with CETP gene polymorphisms. Circulation. 2010;121:52–62. doi: 10.1161/CIRCULATIONAHA.109.865444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein EA, Mellis S, Yancopoulos GD, Stahl N, Logan D, Smith WB, Lisbon E, Gutierrez M, Webb C, Wu R, Du Y, Kranz T, Gasparino E, Swergold GD. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 2012;366:1108–1118. doi: 10.1056/NEJMoa1105803. [DOI] [PubMed] [Google Scholar]
- Stein EA, Giugliano RP, Koren MJ, Raal FJ, Roth EM, Weiss R, Sullivan D, Wasserman SM, Somaratne R, Kim JB, Yang J, Liu T, Albizem M, Scott R, Sabatine MS, Investigators P. Efficacy and safety of evolocumab (AMG 145), a fully human monoclonal antibody to PCSK9, in hyperlipidaemic patients on various background lipid therapies: pooled analysis of 1359 patients in four phase 2 trials. Eur Heart J. 2014;35:2249–2259. doi: 10.1093/eurheartj/ehu085. [DOI] [PubMed] [Google Scholar]
- Swerdlow Daniel I, Preiss David, Kuchenbaecker Karoline B, Holmes Michael V, Engmann Jorgen E L, Shah Tina, Sofat Reecha, Stender Stefan, Johnson Paul C D, Scott Robert A, Leusink Maarten, Verweij Niek, Sharp Stephen J, Guo Yiran, Giambartolomei Claudia, Chung Christina, Peasey Anne, Amuzu Antoinette, Li KaWah, Palmen Jutta, Howard Philip, Cooper Jackie A, Drenos Fotios, Li Yun R, Lowe Gordon, Gallacher John, Stewart Marlene C W, Tzoulaki Ioanna, Buxbaum Sarah G, van der A Daphne L, Forouhi Nita G, Onland-Moret N Charlotte, van der Schouw Yvonne T, Schnabel Renate B, Hubacek Jaroslav A, Kubinova Ruzena, Baceviciene Migle, Tamosiunas Abdonas, Pajak Andrzej, Topor-Madry Romanvan, Stepaniak Urszula, Malyutina Sofia, Baldassarre Damiano, Sennblad Bengt, Tremoli Elena, de Faire Ulf, Veglia Fabrizio, Ford Ian, Jukema J Wouter, Westendorp Rudi G J, de Borst Gert Jan, de Jong Pim A, Algra Ale, Spiering Wilko, der Zee Anke H Maitland-van, Klungel Olaf H, de Boer Anthonius, Doevendans Pieter A, Eaton Charles B, Robinson Jennifer G, Duggan David, Kjekshus John, Downs John R, Gotto Antonio M, Keech Anthony C, Marchioli Roberto, Tognoni Gianni, Sever Peter S, Poulter Neil R, Waters David D, Pedersen Terje R, Amarenco Pierre, Nakamura Haruo, McMurray John J V, Lewsey James D, Chasman Daniel I, Ridker Paul M, Maggioni Aldo P, Tavazzi Luigi, Ray Kausik K, Seshasai Sreenivasa Rao Kondapally, Manson JoAnn E, Price Jackie F, Whincup Peter H, Morris Richard W, Lawlor Debbie A, Smith George Davey, Ben-Shlomo Yoav, Schreiner Pamela J, Fornage Myriam, Siscovick David S, Cushman Mary, Kumari Meena, Wareham Nick J, Verschuren W M Monique, Redline Susan, Patel Sanjay R, Whittaker John C, Hamsten Anders, Delaney Joseph A, Dale Caroline, Gaunt Tom R, Wong Andrew, Kuh Diana, Hardy Rebecca, Kathiresan Sekar, Castillo Berta A, van der Harst Pim, Brunner Eric J, Tybjaerg-Hansen Anne, Marmot Michael G, Krauss Ronald M, Tsai Michael, Coresh Josef, Hoogeveen Ronald C, Psaty Bruce M, Lange Leslie A, Hakonarson Hakon, Dudbridge Frank, Humphries Steve E, Talmud Philippa J, Kivimäki Mika, Timpson Nicholas J, Langenberg Claudia, Asselbergs Folkert W, Voevoda Mikhail, Bobak Martin, Pikhart Hynek, Wilson James G, Reiner Alex P, Keating Brendan J, Hingorani Aroon D, Sattar Naveed. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. The Lancet. 2015;385(9965):351–361. doi: 10.1016/S0140-6736(14)61183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AJ, Villines TC, Stanek EJ, Devine PJ, Griffen L, Miller M, Weissman NJ, Turco M. Extended-release niacin or ezetimibe and carotid intima-media thickness. N Engl J Med. 2009;361:2113–2122. doi: 10.1056/NEJMoa0907569. [DOI] [PubMed] [Google Scholar]
- Thomas DC, Lawlor DA, Thompson JR, et al. Re: estimation of bias in nongenetic observational studies using “Mendelian triangulation” by Bautista et al. Ann Epidemiol. 2007;17:511–513. doi: 10.1016/j.annepidem.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Thorn CF, Klein TE, Altman RB. Pharmacogenomics and bioinformatics: PharmGKB. Pharmacogenomics. 2010;11:501–505. doi: 10.2217/pgs.10.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulffele MG, Kooy A, de Zeeuw D, Stehouwer CD, Gansevoort RT. The effect of metformin on blood pressure, plasma cholesterol and triglycerides in type 2 diabetes mellitus: a systematic review. J Intern Med. 2004;256:1–14. doi: 10.1111/j.1365-2796.2004.01328.x. [DOI] [PubMed] [Google Scholar]
- Zhernakova A, Stahl EA, Trynka G, Raychaudhuri S, Festen EA, Franke L, Westra HJ, Fehrmann RS, Kurreeman FA, Thomson B, Gupta N, Romanos J, McManus R, Ryan AW, Turner G, Brouwer E, Posthumus MD, Remmers EF, Tucci F, Toes R, Grandone E, Mazzilli MC, Rybak A, Cukrowska B, Coenen MJ, Radstake TR, van Riel PL, Li Y, de Bakker PI, Gregersen PK, Worthington J, Siminovitch KA, Klareskog L, Huizinga TW, Wijmenga C, Plenge RM. Meta-analysis of genome-wide association studies in celiac disease and rheumatoid arthritis identifies fourteen non-HLA shared loci. PLoS Genet. 2011;7:e1002004. doi: 10.1371/journal.pgen.1002004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 2 (DOCX 202 kb) Data File S1: Summary estimates from GWAS meta-analysis of glycemic traits (fasting glucose, fasting insulin, fasting proinsulin and HbA1c) and type 2 diabetes to yield a glycemic burden composite