

Summary
In this work, different approaches were investigated to enhance the expression rabies virus glycoprotein (RABV‐G) in the yeast Pichia pastoris; this membrane protein is responsible for the synthesis of rabies neutralizing antibodies. First, the impact of synonymous codon usage bias was examined and an optimized RABV‐G gene was synthesized. Nevertheless, data showed that the secretion of the optimized RABV‐G gene was not tremendously increased as compared with the non‐optimized one. In addition, similar levels of RABV‐G were obtained when α‐factor mating factor from Saccharomyces cerevisiae or the acid phosphatase PHO1 was used as a secretion signal. Therefore, sequence optimization and secretion signal were not the major bottlenecks for high‐level expression of RABV‐G in P. pastoris. Unfolded protein response (UPR) was induced in clones containing high copy number of RABV‐G expression cassette indicating that folding was the limiting step for RABV‐G secretion. To circumvent this limitation, co‐overexpression of five factors involved in oxidative protein folding was investigated. Among these factors only PDI1,ERO1 and GPX1 proved their benefit to enhance the expression. The highest expression level of RABV‐G reached 1230 ng ml−1. Competitive neutralizing assay confirmed that the recombinant protein was produced in the correct conformational form in this host.
Introduction
The methylotrophic yeast Pichia pastoris (Komagataella sp.) has become a substantial workhorse for biotechnology, especially for heterologous protein production (Kurtzman, 2009; Ahmad et al., 2014). Using this system, a variety of proteins of different origins (human, animal, plant, fungal, bacterial and viral) has been produced with varying degrees of success (Sreekrishna et al., 1997; Leonardo et al., 2012).
However, not all recombinant proteins are efficiently secreted. High‐level expression in P. pastoris could face some potential bottlenecks, such as limitations in gene dosage, mRNA transcription, protein processing and folding in the endoplasmic reticulum (ER) (Agaphonov et al., 2002) and translocation which is depending on the secretion signal peptide (Koganesawa et al., 2001).
Genetic modification and molecular biotechnology proved to be most useful and effective tools for high‐level expression (Bai et al., 2011). Several genetic factors can be modified to enhance protein expression such as sequence optimization (Bai et al., 2011), gene copy number (Norden et al., 2011; Shen et al., 2012), promoter selection (Shen et al., 1998; Hohenblum et al., 2004), secretion signal (Koganesawa et al., 2001) and coexpression of folding‐assistant proteins (Li et al., 2010; Shen et al., 2012).
Previous investigations have demonstrated that the limiting steps are dependent on several factors such the protein to be expressed, the promoter and the host strain employed (Cereghino and Cregg, 2000; Li et al., 2007; Ashe and Bill, 2011). Generally, sequence optimization to adapt the codon usage of the gene of interest to the preferred host codon usage has been identified as one of the important factors influencing heterologous expression in P. pastoris (Cereghino and Cregg, 2000). For instance production of Aspergillus niger lipase (Yang and Liu, 2010), yeast multidrug resistance protein MDR1 (Bai et al., 2011) and Streptomyces rimosus GDS(L)‐lipase (Vujaklija et al., 2002) in P. pastoris were increased through codon optimization by 5.3‐fold, threefold and 22‐fold respectively. In addition, numerous studies have shown that gene dosage of the foreign protein has a high impact on recombinant protein production (Yu et al., 2009; Zhu et al., 2009). Yu et al. (2010) have demonstrated that increasing the gene copy number of lip2 from Yarrowia lipolytica enhanced the protein expression level by twofold. In many cases, increasing the target gene copy number dramatically enhances the production of foreign protein in P. pastoris. Although in some cases, such as human trypsinogen (Hohenblum et al., 2004) and Na‐ASPI (Inan et al., 2006) opposite results were obtained; increased gene dosage led to a reduction of the expression level. This was attributed to limitations in folding and secretion or a saturation of the secretory pathway. Secretory protein production usually requires the presence of a secretion signal sequence at the N‐terminus of the foreign protein to target it to the secretory pathway; different secretion signals are available to target protein secretion in P. pastoris, such as PHO1 secretion signal (Chang et al., 1986) and Saccharomyces cerevisiae α‐mating factor secretion signal (Payne et al., 1995). Each signal has its particular advantage, and there is no common rule which allows the identification of the most effective sequence (Hashimoto et al., 1998; Damasceno et al., 2012; Gasser et al., 2013).
During the journey of a protein through different cellular compartments, namely the ER, the Golgi apparatus, and finally, vesicular transport to the extracellular environment several post‐translational modifications occur (Vanz et al., 2014). However, not all recombinant proteins are efficiently secreted and ER retention during high‐level production can be a problem. In particular, aberrant folding properties of the target protein and/or high‐level production can lead to the accumulation of unfolded or even aggregated proteins in the ER (Inan et al., 2006; Hesketh et al., 2013) which can initiate the unfolded protein response (UPR) (Hohenblum et al., 2004; Whyteside et al., 2011; Zhu et al., 2011) and ER‐associated degradation (ERAD) (Whyteside et al., 2011; Vanz et al., 2012).
The ER contains several chaperones and foldases such as protein disulfide isomerase (PDI), the hsp70 member Kar2/BiP, calreticulin and calnexin, which are mainly involved in protein folding processes. Their co‐overexpression with the product gene has significantly improved the productivity of several secreted proteins (Inan et al., 2006; Damasceno et al., 2007; Li et al., 2010). Protein folding and ER stress have a severe impact on the redox state of the ER as well as on the cytosolic redox balance which can affect the process of folding its self as described in Delic et al. (2012). By altering the levels of redox active enzymes such as glutathione peroxidase (GPX) or the antioxidant transcription factor YAP1, the amount of a secreted heterologous protein was improved (Delic et al., 2012, 2014).
In this work, we report our efforts to enhance the heterologous production of rabies virus glycoprotein (RABV‐G) in P. pastoris. This protein which is exposed on the surface of rabies virus has been identified as the major antigen that induces protective immunity and thereby affords complete protection against rabies virus challenge (Cox et al., 1977). To identify the limiting steps in RABV‐G expression in this yeast we investigated the effect of different factors that can have a significant impact on heterologous gene expression; these factors are: gene optimization, secretion signal sequence, gene copy number and the coexpression of different proteins. In particular, we studied the effect of the coexpression of proteins which are involved in the oxidative protein folding process (PDI1 and ERO1), those related to glutathione (GPX1 and GLR1) and the transcriptional regulator factor YAP1, which responds to oxidative stress and was shown to be induced during methanol growth (Yano et al., 2009). YAP1 activates the expression of the glutathione redox system including glutathione reductase (GLR1).
To explore the importance of codon bias and sequence optimization on the expression of RABV‐G in P. pastoris, the sequence of wild‐type (wt)‐RABV‐G gene was optimized for P. pastoris and the expression of optimized (opt)‐RABV‐G in the selected transformants was monitored by Western blot and enzyme linked immunosorbent assay (ELISA). To understand the effect of RABV‐G gene dosage, the copy number of the expression cassette inserted into host genome was determined by RT‐qPCR in the transformed clones and correlated with the expression level. Two secretion signals (α factor and PHO1) were also studied. Finally the effect of coexpression of five intracellular proteins (PDI1, ERO1, GPX1, GLR1 and YAP1) on RABV‐G production level was investigated. The results of these explorations are presented in this study.
Results
Effect of gene optimization, signal sequence and gene dosage on RABV‐G production
Rabies virus glycoprotein is a type I membrane glycoprotein containing 505 amino acids in the native form, and it is the mediator of binding to cellular receptors and entry to host cells (Anilionis et al., 1981). It is composed of a cytoplasmatic domain, a transmembrane domain, and an ectodomain; the protein forms homotrimers and is anchored on the membrane envelope of the virion (Gaudin et al., 1992). RABV‐G has seven disulfide bonds and three potential N‐glycosylation sites (Gaudin et al., 1992; Walker and Kongsuwan, 1999). The important immunogenic property of RABV‐G makes it as an attractive alternative that can be used as a vaccine or as diagnostic antigen in ELISA for detecting anti‐glycoprotein antibodies in immunized host.
The expression of native RABV‐G gene in P. pastoris under the control of AOX1 promoter and using α‐factor as a secretion signal, has led a low expression level around 60 ng ml−1 (Ben Azoun et al., in press). It is worth to note that the expression of this membrane protein in this host was not reported so far in the literature.
In an effort to improve the expression level of RABV‐G by increasing translational efficiency, an optimized gene of RABV‐G (opt‐RABV‐G) supplied by GeneCust was used to transform P. pastoris cells. The sequence of wt‐RABV‐G was optimized with an appropriate algorithm that replaces rare codons by preferred ones in P. pastoris and optimizes other aspects of mRNA structure for optimal expression in this host. Some codons used in RABV‐G were converted to the high frequency codon preferences in P. pastoris summarized in Table S1. A total of 188 codons in the wt gene were changed to codon preferred by P. pastoris, 317 codons remained unchanged. To design the codon‐optimized gene, two restriction endonuclease sites were introduced at 5′ and 3′ ends of the coding sequence for an easy cloning.
To determine the effect of sequence optimization on the yield of RABV‐G two expression plasmids were constructed, the optimized sequence of RABV‐G gene was cloned in pPICZαA containing the α‐MF‐prepro secretion signal of S. cerevisiae and in pHIL‐S1 containing the acid phosphatase PHO1 signal sequence of P. pastoris. The expression plasmids containing the opt‐RABV‐G gene were linearized and transformed into the P. pastoris strains KM71H (pPICZαA) or GS115His− (pHIL‐S1). The expression cassette was integrated into the P.pastoris genome at the 5′AOX1 locus via homologous recombination, giving rise to (His+; MutS) and (His+; Mut+) phenotype in the transformants of KM71H and GS115 His− respectively.
For each construction, we selected six recombinant strains and determined the copy number of the integrated expression cassette by real‐time q‐PCR (Fig. 1A). For clones transformed with pPICZαA‐opt‐RABV‐G, two clones named α‐8, harbouring eight copies of RABV‐G gene, and α‐7 containing seven copies were isolated. Clones bearing intermediate number of the expression cassette were also identified, and named α‐3 and α‐4. Clones with low copy were isolated, and designated as α‐1 and α‐2.
Figure 1.
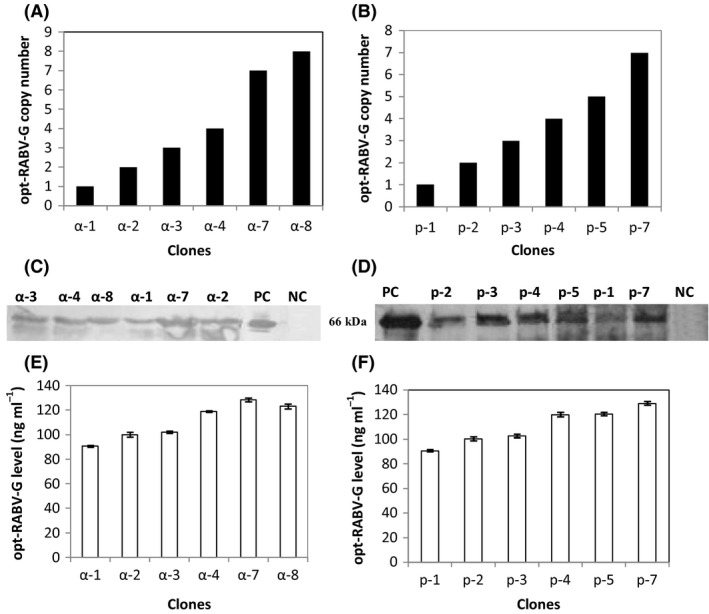
RABV‐G protein expression in the selected recombinant Pichia pastoris strains. Copy number of selected recombinant P. pastoris strains of (A) pPICZαA‐RABV‐G and (B) pHIL‐S1‐RABV‐G transformants. Western blot analysis of 15 μl of culture supernatants (concentrated 10‐fold by centricon) of (C) pPICZαA‐RABV‐G, and (D) pHIL‐S1‐RABV‐G transformants. PC (positive control): inactivated and purified rabies virus. NC (negative control): KM71H and GS115His− strains transformed with empty pPICZαA and pHIL‐S1 vectors respectively. The amount of RABV‐G produced by the different clones with α‐factor (E) or PHO1 (F) signal sequence at 72 h of induction as determined by ELISA.
A similar approach was applied for clones transformed with pHIL‐S1‐opt‐RABV‐G and resulted in the isolation of clones containing different copies of the expression cassette. p‐7 clone contains seven copies, whereas p‐4 and p‐5 harbour intermediate copy number of the expression cassettes: four and five copies respectively. Clones with low copy number were isolated, and named p‐1, p‐2 and p‐3. These clones contain one copy, two copies and three copies of the expression cassette respectively (Fig. 1B).
The selected clones were cultivated in deep well plates, after induction of RABV‐G by methanol during 72 h; RABV‐G levels in culture medium were determined by Western blot using anti‐rabies polyclonal antibodies as shown in Fig. 1C and D. Only a protein band of 66 kDa corresponding to RABV‐G was clearly found in all culture supernatants of the recombinant strains. No band was detected in the transformed clones with empty vectors (pPICZαA and pHIL‐S1), suggesting that the RABV‐G protein was successfully secreted into the culture medium after methanol induction. The expression level of RABV‐G by the different clones was measured by ELISA. Figure 1E and F shows that RABV‐G level depends on the number of integrated expression cassette, but not on the type of signal secretion. Clones α‐7 and α‐8 bearing seven and eight copies, respectively, showed an expression level of 128.5 ng ml−1. For clones containing lower RABV‐G gene copies, we observed a correlation between the inserted copies of the expression cassette and the expression level; although no linear correlation was observed between these factors. Therefore, these data show that there is an optimal copy number of the inserted expression cassette beyond which the efficiency of folding is limited (Fig. 1E). p‐7 clone showed an expression level of 129 ng ml−1 which was similar to that obtained with α‐7 clone, harbouring the same copy number of the expression cassette but using PHO1 as a signal secretion.
These data indicate that sequence optimization had increased the expression level only by 2.1‐fold; nevertheless the use of different secretion signals did not show a marked impact on product yield. Therefore, these factors do not seem to be major bottlenecks for high‐level expression of RABV‐G. On the contrary, the improvement of the RABV‐G level seems to be due to the enhancement of RABV‐G transcription; the correlation between mRNA of RABV‐G transcript level, gene dosage and protein level is shown in Fig. S1.
The transcription level of the RABV‐G gene increased with the increase of gene dosage in both constructs (Fig. S1). The transcription levels of RABV‐G in clones containing equal copy numbers of α‐factor‐opt‐RABV‐G and PHO1‐opt‐RABV‐G were compared with respective clones harbouring one copy of each expression cassette. The transcription level of RABV‐G increased in clones containing two gene copies (α‐2 and p‐2) by 1.12‐fold when compared with the single copy clones (α‐1 and p‐1). For clones harbouring three and four copies of RABV‐G gene, namely α‐3, α‐4, p‐3 and p‐4, the enhancement of RABV‐G transcript level was higher and was in the range of 1.5‐fold for clones with three copies to 2.3 for clones with four gene copies.
Clones containing high copy number of each expression cassette showed a remarkable increase of mRNA level of RABV‐G gene. α‐7 and p‐7 clones which integrated seven copies of RABV‐G gene exhibited an enhancement factor of 4.7‐fold and 4.3‐fold respectively. Clone α‐8 which contains eight copies of the expression cassette reached the highest increase of RABV‐G transcript (5.5‐fold). These data show that there is a linear correlation between the mRNA level of the gene of interest and the expression level. The increase of gene dosage resulted in higher mRNA levels but did not significantly enhance the secreted RABV‐G protein level. This can be due to depletion of precursors and energy (Baumann et al., 2011) or to an accumulation of the protein in the cell caused by limitations in folding and/or secretion. These data also indicate that mRNA levels of RABV‐G gene were not the limiting factor for high‐level expression of RABV‐G, thus we thought that RABV‐G could be retained in the cell.
Determination of intracellular RABV‐G protein in different recombinant clones
To check if RABV‐G protein was retained within the cell, cell extracts were divided into soluble and membrane‐associated fractions (including the secretory organelles) according to Hohenblum et al. (2004) and analysed by Western blot (Fig. 2). The intensity of bands obtained by different clones was more pronounced in the soluble fraction than in the membrane‐associated fraction for clones utilizing either α‐factor or PHO1 as a secretion signal. Figure 2 shows that the gene copy number impacts the number and the intensity of the intracellular product bands. High copy strains such as α‐8 and α‐7 showed more intense bands than intermediate copy strains (α‐4 and α‐3). By contrast, for low copy strains (α‐1 and α‐2), minor level of RABV‐G was seen in the soluble fraction (Fig. 2). In addition, for high copy strains (as α‐8 and α‐7), a band with a molecular weight lower than 66 kDa was observed. This band could be due to degradation of the protein of interest. For clones where the secretion of RABV‐G protein was directed by PHO1 signal, the accumulation of full length RABV‐G protein in the soluble fraction was lower than that seen for α‐factor clones (Fig. 2). Nevertheless, the degradation of RABV‐G protein was more prominent even for the low copy strains. However for α‐factor clones, the proportion of RABV‐G retained in the membrane‐associated fraction was more prominent than that obtained for PHO1‐strains. This effect was prominent for the high copy strains (Fig. 2C and D).
Figure 2.
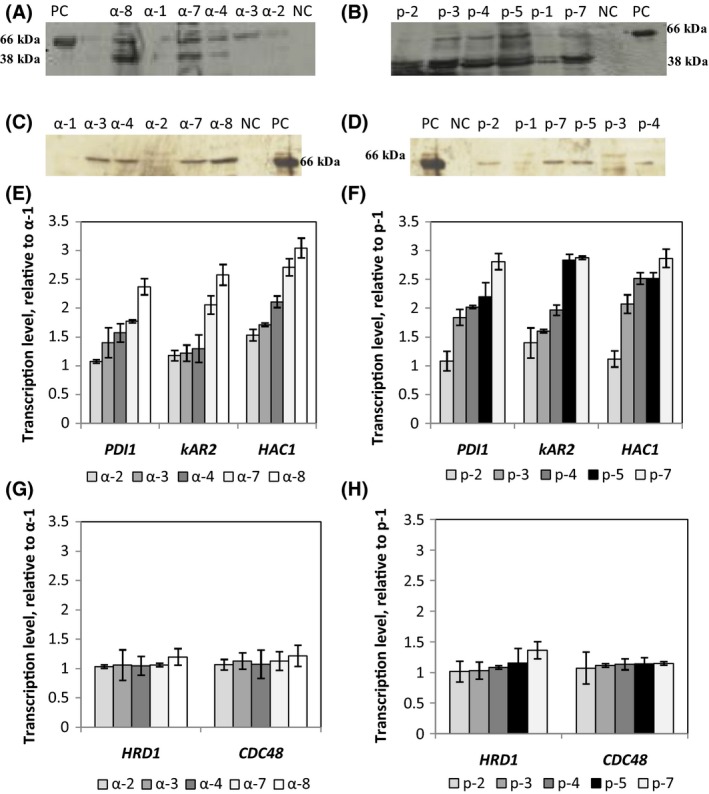
Soluble intracellular RABV‐G protein accumulation in clones with (A) α‐factor and (B) clones with PHO1 signal sequence. Membrane‐associated RABV‐G protein level in clones with (C) α‐factor and (D) in clones with PHO1 signal secretion. Transcription levels of selected UPR‐related genes (PDI1,KAR2,HAC1) in clones with (E) α‐factor and (F) PHO1 as a signal secretion. Transcription levels of ERAD‐related genes (HRD1 and CDC48) in the different Pichia pastoris recombinant strains with α‐factor (G) and (H) PHO1 signal secretion. Gene transcript levels were normalized relative to α‐1 or p‐1 strains containing single copy of RABV‐G gene. α‐2, α‐3, α‐4, α‐7 and α‐8 correspond to selected clones of recombinant P. pastoris strains harbouring different copies of RABV‐G gene and where RABV‐G secretion was driven by the α‐factor. p‐2, p‐3, p‐4, p‐5 and p‐7: selected clones where RABV‐G was directed by PHO1 signal secretion and containing different copies of RABV‐G gene.
Transcription study of key genes involved in UPR and ERAD pathway
To determine if the retention of RABV‐G inside the cell was due to folding limitations in the ER, the transcription levels of three typical UPR signal proteins (HAC1, PDI1 and KAR2) were determined. In addition, the transcript levels of two key proteins involved in ERAD pathway (HRD1 and CDC48) were measured, to determine if the UPR response had triggered an ERAD response.
The transcription levels of HAC1 and the two chaperones located in the ER, KAR2 and PDI1 determined in all clones after methanol induction for 72 h are displayed in Fig. 2E and F. As compared with the strain containing one copy of RABV‐G gene, the transcription level of HAC1 was 2.7‐fold and 2.8‐fold higher in α‐7 and p‐7, respectively, but lower in the low and medium copy number strains. On the other side HAC1 transcript level elevated to threefold in the α‐8 strain which contains eight copies of the RABV‐G gene.
Therefore, increasing the RABV‐G copy number resulted in an expected increase in the HAC1 mRNA level. These results suggest that RABV‐G over expression had induced the UPR in all strains. The transcription level of the other two UPR related genes confirmed this hypothesis. PDI1 and KAR2 transcripts showed a similar trend, with an increase of 1.7‐fold in α‐7 and 2.8‐fold in p‐7 for PDI1 mRNA level, and 2.05‐fold in α‐7 and 2.8‐fold in p‐7 for KAR2 transcript (Fig. 2E and F).
To determine the destiny of unfolded RABV‐G, the transcription level of two key genes involved in ERAD pathway were determined in clones with either the α‐factor or PHO1 secretion signal. Figure 2G and H shows the relative transcription levels of HRD1 and CDC48 in the different strains. No significant difference in transcription of these two genes in all clones was seen as compared with α‐1 or p‐1; this demonstrates that the ERAD pathway was not activated.
These data clearly suggest a folding limitation of the expressed protein. Therefore, we attempted to coexpress five proteins to improve the folding of RABV‐G protein. These proteins are (i) PDI1 and ERO1 which are the main players of the oxidative protein folding machinery in the ER; (ii) Glutathione‐related genes such as GLR1 which is responsible for converting oxidized glutathione to reduced glutathione and GPX1 involved in the detoxification of reactive oxygen species (ROS) at the expense of reduced glutathione, and (iii) Yap1which is the transcription factor of the oxidative stress response.
Coexpression of the oxidative protein folding (PDI1 and ERO1)
Protein disulfide isomerase (PDI1) is an ER‐resident foldase which plays a crucial role in the formation, isomerization and reduction of disulfide bonds. During disulfide bond formation, PDI1 accepts electrons from cysteine residues in nascent proteins, leading to the reduction of PDI1 (Delic et al., 2012). ER oxido‐reduction (ERO1), on the other hand, is an enzyme which interplays with PDI1 during this process and transfers the electrons to molecular oxygen or other electron acceptors to restore the oxidized form of PDI1 (Tu et al., 2000).
Plasmids overexpressing PDI1or ERO1 under control of glyceraldehyde‐3‐phosphate dehydrogenase (GAP) promoter were transformed by electroporation into α‐7/KM71H and p‐7/GS115− strains that contained seven copies of RABV‐G gene.
Six clones from each construction were selected and their copy number was estimated by real‐time q‐PCR (Table S2). α‐7 clones containing low, medium and high copy numbers of PDI1 gene, namely α‐7/P1, α‐7/P3 and α‐7/P6 were selected. p‐7/P1, p‐7/P6 and p‐7/P3 bearing different copy numbers of PDI1 were also isolated. In addition, α‐7 and p‐7 clones harbouring different copy numbers of ERO1 were selected. The production of RABV‐G by the selected clones was studied and compared with α‐7 and p‐7 strains which contain only seven copies of RABV‐G gene. The expression level was monitored by Western blot and ELISA of culture supernatants after 72 h of methanol induction.
Western blot analysis of culture supernatants of the selected recombinant strains expressing PDI1or ERO1 shows the presence of a single band at the expected size of RABV‐G (Fig. 3A‐1). The intensity of the band depends on the chaperone coexpressed. Coexpression of PDI1 dramatically improved the level of secreted RABV‐G, independent of the secretion signal used; this effect increased with higher gene copy number of PDI1. Insertion of six copies of PDI1 resulted in 9.6‐fold enhancement of secreted RABV‐G when compared with α‐7 and p‐7 strains. Insertion of three or one copy of PDI1 increased the production level 8.6‐fold and 7.9‐fold respectively (Fig. 3A‐2).
Figure 3.
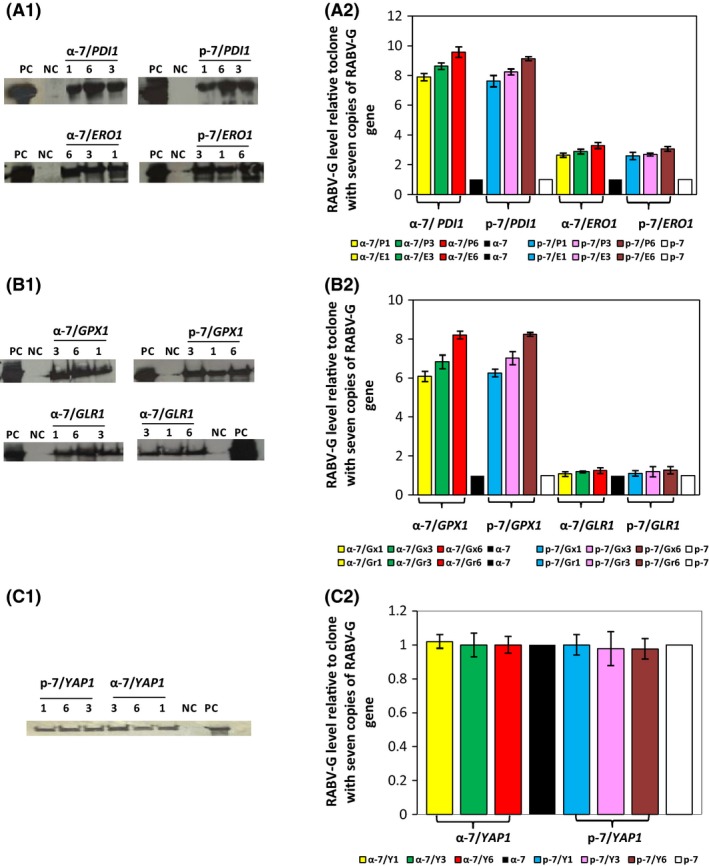
Effect of coexpression of five factors involved in oxidative protein folding on the expression level of RABV‐G. Western blot analysis of 15 μl of culture supernatants (concentrated 10‐fold by centricon) of yeast clones with α‐factor or PHO1 sequence leader to direct RABV‐G protein secretion coexpressing (A‐1), PDI1 or ERO1 genes, (B‐1) GPX1 or GLR1 and (C‐1) YAP1 genes. PC (positive control): inactivated and purified rabies virus. NC (negative control): KM71H and GS115His− transformed with an empty pPICZαA and pHIL‐S1 respectively. The numbers 1, 3 and 6 correspond to copy number of different genes contained in each clone. The amount of RABV‐G coexpressed with (A‐2) PDI1 or ERO1, (B‐2) GPX1 or GLR1 and (C‐2) YAP1 produced by the different clones with α‐factor or PHO1 as a signal sequence at 72 h of methanol induction as determined by ELISA. Clone abbreviations are explained in Tables S2, S3 and S4.
ERO1 coexpression improved RABV‐G expression in all clones, as seen in Western blot analysis of culture supernatants. However, when compared with PDI1 expression, the effect was lower. Here, we observe an enhancement factor around three independent of the ERO1 gene copy number (Fig. 3A‐1 and A2).
These results clearly demonstrate that PDI1 overexpression led to a significantly higher level of RABV‐G when compared with ERO1 strains.
Coexpression of glutathione‐related genes (GPX1 and GLR1)
Glutathione reductase is a key enzyme in the conversion of oxidized GSSG to its reduced form GSH and is crucial for the maintenance of the cellular glutathione redox potential (Toledano et al., 2013). GPX1, on the other hand, is an enzyme involved in the detoxification of ROS in particular H2O2, at the expense of reduced glutathione, thereby generating GSSG (Toledano et al., 2013). The effect of these two enzymes on the RABV‐G production was evaluated, and the selection of α‐7 and p‐7 clones harbouring low, medium and high copy number of GPX1 and GLR1 genes was performed (Table S3).
The expression of the RABV‐G protein by the selected clones was analysed by Western blot and ELISA (Fig. 3B‐1 and B2). We showed that GPX1coexpression had a positive effect on the expression level of RABV‐G protein. A single protein band with the molecular weight of 66 kDa was revealed in all culture supernatants; its intensity increased with GPX1 gene copy number (Fig. 3B‐1). This result was also confirmed by ELISA (Fig. 3B‐2); the insertion of six copies of GPX1 gene in α‐7 and p‐7 strains enhanced the expression of RABV‐G protein by 8.2‐fold. The insertion of one or three copies of GPX1 gene improved the expression level by six‐ and 6.8‐fold respectively. Coexpression of GLR1 with RABV‐G had only a minor effect on the expression level of RABV‐G protein (Fig. 3B‐1 and B‐2). Gene dosage of GLR1 does not seem to have a significant impact on the expression level for α‐7 and p‐7 strains, as GLR1 coexpression enhanced RABV‐G production by 1.1‐ to 1.28‐fold for all clones. These data indicate that the coexpression of GPX1 has a large impact on RABV‐G when compared with GLR1.
Coexpression of the transcription factor in stress response (YAP1)
The transcription factor YAP1 is a major oxidative stress regulator. During growth on methanol, YAP1 activates the expression of the glutathione redox system and upregulates GLR1 (Yano et al., 2009). The impact of the coexpression of this protein on RABV‐G production was evaluated in strains with low, medium and high copy number of YAP1, named as α‐7/Y1, α‐7/Y3, α‐7/Y6 and p‐7/Y1, p‐7/Y3, p‐7/Y6. The expression of RABV‐G was compared with α‐7 and p‐7 containing seven copies of RABV‐G gene (Table S4).
Overexpression of YAP1 had no effect on the level of RABV‐G when compared with their respective controls (α‐7 and p‐7), as determined by Western bolt and ELISA (Fig. 3C‐1 and C‐2). RABV‐G expression level reached after 72 h of methanol induction remained unchanged for all clones.
Competition of neutralization activity of rabies‐immune serum by RFFIT
To prove that the RABV‐G expressed by clone α‐7/P6 was secreted in its native form and can compete with rabies virus to react with anti‐rabies neutralizing serum and to block cell infection, the RFFIT test was used with the modifications introduced by Li et al. (2010). The neutralizing activity of rabies‐immune serum was evaluated in the presence of different levels of recombinant RABV‐G varying from 0.8 to 49 μg ml−1. As shown in Fig. 4, the neutralizing titre of the rabies‐immune serum was significantly reduced in the presence of RABV‐G compared with that seen in the absence of recombinant RABV‐G. These results suggest that the RABV‐G produced by P. pastoris was recognized by rabies virus neutralizing antibodies.
Figure 4.
Competition of the neutralizing activity of the rabies‐immune serum by recombinant RABV‐G. The neutralizing titres (IU ml−1) were determined in the presence of varying amounts of RABV‐G expressed by α‐7/P6 recombinant clone, NC (negative control): culture supernatant of KM71H strain transformed with empty pPICZαA vector.
Discussion
High‐level expression of a given protein in a heterologous system can be affected by several factors; a practical solution is to identify major bottlenecks which are in general, host‐ and product‐dependent. Codon optimization, signal sequence and gene dosage are the main factors studied to improve the expression of a target protein. Several examples have been mentioned in literature that demonstrate that the use of an optimized sequence, the increase of gene copy number and the choice of leader sequence can significantly enhance protein productivity (Vujaklija et al., 2002; Zhao et al., 2009; Shen et al., 2012).
In this work, we first studied the effect of gene optimization on the expression of RABV‐G in P. pastoris. We showed that the expression of opt‐RABV‐G led to approximately 2.1‐fold increase in RABV‐G production compared with wt‐RABV‐G (S. Ben Azoun, unpublished). These data indicate that sequence optimization was not a critical parameter for RABV‐G expression in P. pastoris, as gene optimization sequence did not result in a tremendous increase of the expression level.
Numerous studies have demonstrated that the choice of secretion signal had a profound impact on the expression level of heterologous proteins (Ide et al., 2007; Ghosalkar et al., 2008). It controls the entry of proteins into the oxidative protein folding compartments such the ER in eukaryotes and the periplasm in prokaryotes (Killian et al., 1990; Von Heijne, 1990; Rapoport, 1992). For P. pastoris, few secretion leader sequences are known and applied for the secretion of heterologous proteins (Damasceno et al., 2012; Gasser et al., 2013). The most widely used secretion signal is the S. cerevisiae α‐mating factor leader sequence, whereas the P. pastoris acid phosphatase PHO1 signal sequence has been used in few cases (Heimo et al., 1997; Romero et al., 1997). The S. cerevisiae SUC2 gene signal sequence was used occasionally (Paifer et al., 1994). It was reported that the expression level of hIFN‐α2b in P. pastoris was higher with α‐factor signal sequence than with its native signal sequence (Ghosalkar et al., 2008). Expression of ocanase in P. pastoris under the direction of the PHO1 secretion signal was less effective than α‐factor (Zhao et al., 2009). While Koganesawa et al. (2001) demonstrated that the silkworm lysozyme expression using α‐factor leader was so unstable that it could be easily attacked by proteases, they suggested that the expression level and the stability of secreted heterologous protein were greatly affected by the selection of the appropriate secretion signal sequence.
PHO1 and α‐factor secretion leader were evaluated in this work for the secretory expression of RABV‐G in P. pastoris. We found that the expression level of secreted RABV‐G was similar in clones containing equal copies of the product gene from both constructs. However, intracellular RABV‐G retention profile was different; a band with a molecular weight lower than 66 kDa was observed. This band could be due to degradation of the protein of interest; this band was more important in clones where the secretion of RABV‐G protein was directed by PHO1 signal. These clones are derived from the strain P. pastoris GS115, they have the phenotype Mut+. Hence, they have faster cellular machinery on methanol compared with KM71H recombinant clones which are Muts (clones with α‐factor sequence leader).
The optimization of the gene copy number is another factor that can be modulated to improve the expression level in P. pastoris. Several studies have shown that this parameter is critical for heterologous protein expression in P. pastoris (Shen et al., 2012; Yang et al., 2013). We demonstrated that increasing RABV‐G copy number resulted in an increased RABV‐G transcript level as well as secreted RABV‐G level. This positive correlation between the copy number and the expression was only observed in clones bearing less than eight copies of RABV‐G gene, despite the high transcriptional rate observed in this clone. This can be due to the saturation of the folding capacity in this high copy number clone, and to the activation of UPR mechanism as described for secreted heterologous proteins in S. cerevisiae (Kauffman et al., 2002) or in P. pastoris (Inan et al., 2006).
The retention of RABV‐G in the P. pastoris strains was investigated, we have found that RABV‐G protein was partially accumulated in intracellular compartments; this accumulation increased with the gene copy number, implying that the secretory protein pathway might be saturated in high copy strains.
One can assume that the product is retained in the ER due to limitations in folding and/or disulfide bond formation and the set up of ER stress as a consequence. This triggers the activation UPR pathway which aims at reducing ER stress conditions by induction of genes involved in protein folding (Hoseki et al., 2010; Kohno, 2010). To test whether the UPR was induced, the transcript levels of three key genes (HAC1, PDI1, KAR2) involved in UPR pathway were analysed. Seventy‐two hours after induction of RABV‐G expression lead to a significant increase of HAC1, PDI1 and KAR2 transcripts and an accumulation of intracellular RABV‐G (Fig. 2). These data clearly indicate that UPR was induced upon induction of RABV‐G expression. Often the UPR is followed by ERAD (Lünsdorf et al., 2011; Vanz et al., 2012; Lin et al., 2013). The ERAD pathway has been investigated through the monitoring of the transcripts of two key genes (HRD1and CDC48). There was no significant difference between the recombinant strains and the negative control indicating that ERAD was not triggered in RABV‐G expressing P. pastoris (Fig. 2).
Protein folding in the ER is a critical step, both, in mammals and yeasts and is a prerequisite for secretion (Helenius et al., 1992; Inan et al., 2006). The newly synthesized heterologous protein enters the lumen of the ER; it encounters a change in the redox environment that ultimately promotes the formation of intra‐chain and/or inter‐chain disulfide bonds (Inan et al., 2006). Chaperone proteins play a critical role in protein folding in the ER and efficient processing is necessary to obtain high levels of proteins (Ngiam et al., 2000).
Coexpression of the oxidative protein folding factors PDI1 or ERO1 has been successful in increasing the amount of some heterologous proteins in P. pastoris (Lodi et al., 2005; Baumann et al., 2011; Vad et al., 2005; Inan et al., 2012) although in some cases this approach was not effective to improve heterologous protein expression (Damasceno et al., 2007).
In the current study, coexpression of the two folding factors PDI1 or ERO1 remarkably increased the expression level of RABV‐G by 9.5‐fold and 3.3‐fold, respectively, in the high copy of RABV‐G gene strains (Fig. 3). This indicates that restriction in folding or misfolding of RABV‐G are the major bottlenecks for optimal expression of this protein. Recently, Delic et al. (2012) proved that protein folding and ER stress have a severe impact on the cytosolic redox balance which may be a major factor during folding.
In P. pastoris, several genes of gluthatione redox system, such as the gluthatione reductase gene (GLR1), the GPX1, are induced by the transcriptional regulator YAP1 which responds to oxidative stress (Yano et al., 2009).
In this study, we coexpressed the glutathione‐related genes GPX1 and GLR1 and the transcription factor involved in stress response YAP1. It has been reported by Delic et al. (2012) that coexpression of GPX1 has improved the level of trypsinogen 1.6‐fold in glucose‐driven protein production conditions compared with GLR1 which failed to increase the production of this protein in P. pastoris. GPX1 overexpression correlated with more oxidizing redox conditions in the ER, as did PDI1 overexpression but not ERO1 or GLR1 overexpression (Delic et al., 2012). On the other hand, YAP1 overexpression increased trypsinogen secretion by twofold and re‐established cytosolic redox state to the state of glucose‐grown wt strains (Delic et al., 2014).
The coexpression of GPX1, GLR1 or YAP1 genes in high RABV‐G copy strains yielded different results. The highest relative improvement was observed with clones that overexpressed GPX1, which showed 8.2‐fold increase of RABV‐G expression level, whereas the coexpression of GLR1 slightly increased the RABV‐G expression (1.2‐fold). These results clearly show that cell engineering towards conditions previously shown to be beneficial for protein secretion by enhancing ER oxidation in glucose‐grown P. pastoris is also improving production of secreted product in methanol grown cells.
Surprisingly, the coexpression of YAP1 has failed to enhance RABV‐G expression; conversely the yield of RABV‐G protein was slightly decreased in the clone α‐7/Y6 (Fig. 3). Hence, overexpression of YAP1 gene is not required to enhance RABV‐G expression, the available YAP1 level allows the cells to manage with oxidative stress. This result is in line with those reported by Yano et al. (2009) who identified the P. pastoris YAP1 homologue and showed the involvement of this transcription factor in the detoxification of formaldehyde and ROS in P. pastoris cells grown on methanol. Delic et al. (2014) also showed that YAP1 is involved in physiological detoxification of ROS formed upon oxidative folding in the ER. YAP1 is required to activate the antioxidant enzymes, which are quenching ROS. Cells with significantly lowered YAP1 level react to increased secretory folding load with accumulation of ROS and strong flocculation.
Therefore, YAP1 level is already high in methanol grown cells (Yano et al., 2009); this might explain why the increase of YAP1 gene copies does not elicit any further antioxidant response.
However, it still remains to be analysed in future if the reduction of the cytosolic redox state upon production of secretory proteins and ER stress observed in glucose conditions (Delic et al., 2012, 2014) is also occurring in methanol grown P. pastoris, or if the constant high YAP1 level on methanol prevent or diminish this response.
The RABV‐G protein produced by P. pastoris was able to significantly reduce the neutralizing activity of the human immune rabies serum, thus demonstrating that the recombinant protein was produced in the correct conformational form.
In conclusion, in the current study we investigated different approaches to enhance RABV‐G expression in P. pastoris. The data obtained showed that using an optimized gene sequence, different signal sequence or increasing gene copy number were not the major bottlenecks to improve the amount of recombinant RABV‐G. Coexpression of oxidative folding proteins such as PDI1 or ERO1 and the glutathione‐related genes (GPX1 or GLR1) improved the secretion of RABV‐G by 9.5‐fold, 3.3‐fold, 8.2‐fold and 1.2‐fold respectively. The secreted RABV‐G protein was also able to react with neutralizing antirabies serum, demonstrating therefore a correct folding of the recombinant protein. Overall, these results demonstrate through combined engineering of the expression construct and the cellular oxidative protein folding machinery, P. pastoris can produce sufficient amounts of functional recombinant RABV‐G which can be used as diagnostic antigen for detecting rabies virus neutralizing antibodies in immunized hosts or as a vaccine.
Experimental procedures
Strains, plasmids and media
Pichia pastoris KM71H and GS115His− (Invitrogen, Carlsbad, CA, USA) were used as host strains for protein expression. Escherichia coli DHB10 (Invitrogen) was used for the propagation of recombinant vectors.
The plasmids pPICZαA with alpha factor secretion signal (Invitrogen) and pHIL‐S1 with PHO1 signal sequence were used for cloning and expression of opt‐RABV‐G protein (GenBank accession number KT878717). The plasmid Ppuzzle (Delic et al., 2012) was used for cloning and expression of five folding‐assisting factors: PDI1 (GenBank accession number EU_805807.1), ERO1 (GenBank accession number XM_002489600.1), GPX1 (GenBank accession number AB_472088.1), GLR1 (GenBank accession number AB_472087.1) and YAP1 (GenBank accession number AB_472084.1). Gene expression was under control of the glycolytic glyceraldehyde‐3‐phosphate dehydrogenase promoter PGAP.
Yeast peptone dextrose (YPD) agar (2% peptone, 1% yeast extract, 2% glucose and 20 g l−1 agar) with zeocin (100 mg ml−1) was used to select the positive clones which contain the recombinant plasmid pPICZαA‐opt‐RABV‐G.
RDB (Regeneration Dextrose Medium + His) agar lacking histidine (1 M sorbitol, 1% glucose, 1.34% yeast nitrogen base, 4 × 10−5% biotin, 0.005% of L‐glutamic acid, L‐methionine, L‐lysine, L‐leucine and L‐isoleucine, and 20 g l−1 agar) was employed to select the His‐prototrophic clones transformed with the recombinant plasmid pHIL‐S1‐opt‐RABV‐G.
BMGY (Buffered Glycerol‐complex Medium) (1 mM potassium phosphate pH 6, 2% peptone, 1% yeast extract, 1.34% yeast nitrogen base, 1% glycerol and 4 × 10−5% biotin) was used for cell growth before the induction of recombinant clones of P. pastoris (KM71H, GS115His−) strains.
BMMY (Buffered Methanol‐complex medium) (1 mM potassium phosphate pH 6, 2% peptone, 1% yeast extract, 1.34% yeast nitrogen base, 1% methanol and 4 × 10−5% biotin) containing methanol as a carbon source was used to induce the expression of RABV‐G protein.
Construction of expression vectors containing the optimized rabies virus glycoprotein sequence
Codon‐opt‐RABV‐G was obtained from GeneCust (Dudelange, Luxembourg). The opt‐RABV‐G lacking its native signal peptide sequence was cloned in two plasmids containing two different secretion signal sequences: pPICZαA with S. cerevisiae α‐mating factor as secretion signal and pHIL‐S1 with P. pastoris PHO1 as signal sequence.
The opt‐RABV‐G was amplified by PCR from the recombinant plasmid PUC 19 (GeneCust) using two different primer pairs listed in Table S5. αG‐F/αG‐R primers were used to clone RABV‐G gene into pPICZαA, the second pair pG‐F/pG‐R was employed for the cloning of RABV‐G gene into pHIL‐S1.
After amplification, PCR products were purified and ligated into pPICZαA or pHIL‐S1 and transformed into E. coli; positive clones from the ligation reactions were analysed by restriction digestion and sequencing of the insert fragment.
Construction of expression vectors containing PDI1, ERO1, GPX1, GLR1 and YAP1sequences
P. pastoris genes PDI1, ERO1, GPX1, GLR1 and YAP1 were isolated from the recombinant pPuzzle vectors (Delic et al., 2012) which contain a zeocin selection marker. The genes were cloned into a pPuzzle vector under control of the GAP promoter and with KanMX as selection marker. The vectors were integrated into the 5′AOX1 region of P. pastoris recombinant clones harbouring seven copies of RABV‐G gene, after linearization of the respective sequences. After transformation by electroporation, positive transformants of P. pastoris were selected on YPD plates with zeocin and G418 or YPD plates with G418.
Expression of recombinant Opt‐RABV‐G protein in deep 24‐well cell culture plates
Recombinant P. pastoris clones were grown in YPD agar plates or RDB agar plates at 30°C. For expression studies, 2 ml of BMGY in culture plate (Dominique Dutscher, Brumath, France) were grown overnight at 30°C and 250 r.p.m. After 14–16 h, optical density was measured at 600 nm, and cells were resuspended in 2 ml of fresh BMMY medium to an initial OD600 of 1. Cultures were performed at 250 r.p.m., 30°C up to 72 h with methanol added to 1% every 24 h. Aliquots were taken every 24 h, cells were pelleted; supernatants and cells were stored at −20°C for further analysis.
Lysis of cells and detection of proteins
Cell pellets (the equivalent of 1 ml at an OD600 of ~50) were washed twice in phosphate buffered saline, then resuspended in 500 μl of yeast breaking buffer (50 mM sodium phosphate, 1 mM PMSF (Phenylmethylsulfonyl floride), 1 mM EDTA (Ethylenediaminetetraacetic acid) and 5% (v/v) glycerol), as described by Shen et al. (2012) and mixed with an equal volume of acid‐washed glass beads (Sigma Aldrich, St Louis, MO, USA). The yeast cell wall was broken by vortexing 10 times for 1 min with 1 min chilling on ice. The lysate was centrifuged at 16 000 g for 20 min at 4°C. The supernatants containing the cytosolic proteins were collected. The pellet containing the membrane proteins was further treated with 400 μl yeast breaking buffer plus 2% (w/v) SDS. After centrifugation at 4000 g for 5 min at 4°C, the supernatants containing the membrane proteins were collected and analysed by Western blot.
Western blot analysis
Secreted proteins, soluble proteins and membrane proteins were analysed on a 12% SDS‐polyacrylamide gel using Bio‐Rad cell system. Proteins were transferred to a nitrocellulose membrane (GE Healthcare, Uppsala, Sweden). The membrane was incubated overnight at 4°C and then incubated with horse anti‐rabies virus polyclonal antibody for 1 h. After washing, the membrane was incubated with HRP‐conjugated polyclonal anti‐horse IgG (antibodies online, Germany). The membrane was finally incubated for two minutes with ECL solution (GE Healthcare).
Enzyme‐linked immunosorbent assay test
An indirect ELISA was performed to quantify of RABV‐G expressed by the different clones. About 100 μl per well of either the sample or the standard (inactivated and purified rabies virus) were incubated for 2 h at 37°C. Thereafter, monoclonal antibody anti‐glycoprotein TW1 (NIBSC, Hertfordshire, UK) was added to the wells and incubated for 1 h at 37°C. Finally, anti‐human antibody coupled to peroxidase (Sigma Aldrich) were added and incubated 30 min at 37°C. After tetramethylbenzidine addition, the reaction intensity was measured at 450 nm. OD values higher than 0.150 were considered as positive.
Competitive neutralization assay
The neutralizing titre of rabies‐immune serum was determined by RFFIT according to the standard methodology (Smith et al., 1973; Li et al., 2010). To determine whether RABV‐G expressed in P. pastoris reacted with neutralizing antibodies present in the human immune rabies serum, serial dilutions of recombinant RABV‐G were mixed with 50 μl rabies human immune serum (European Pharmacopeia, Strasbourg, France) and incubated at 37°C for 1 h. The serum and RABV‐G mixtures were then incubated at 37°C for 1 h with a constant dose of challenge rabies virus that causes infection in 80% of BHK‐21 cells. BHK‐21 cells were then added to the samples and incubated for 20 h at 37°C, 5% CO2. BHK‐21cells were fixed in 80% acetone stained with fluorescein‐labelled anti‐rabies nucleocapsid immunoglobulins (Sanofi Diagnostic Pasteur, Marnes la Coquette, France) to detect the presence of non‐neutralized virus (fluorescent foci).
RNA extraction
For transcript quantification, frozen cells were resuspended in Trizol reagent (Invitrogen) and disrupted with glass beads in FastPrep™ cell homogenizer (Thermo Scientific, Waltham, MA, USA). Total RNA was then extracted using the RNeasy Kit from Qiagen (Hilden, Germany), following the manufacturer's instructions. RNA was tested in 1% agarose gel, and was quantified by measuring OD 230/260/280 using a NanoDrop (Thermo Scientific).
Quantitative real‐time PCR to determine transgene copy number and gene transcriptional level
Individual reactions were carried out in 10 μl containing 5 μl Maxima™ SYBR® Green qPCR Master Mix (Roche, Mannheim, Germany), 0.25 μl fw‐ and rev‐primers (listed in Table S5) and 4 ng template genomic DNA or 2.5 μg cDNA. RT‐qPCR was performed using a thermal cycler (Bio‐Rad, Hercules, CA, USA). PCR conditions were as followed: pre‐incubation at 95°C for 10 min, the thermal cycler was programmed to perform 45 cycles of: 15 s at 95°C; 20 s at 60°C; 15 s at 72°C. A melting curve was carried out to ensure that only a specific amplification product was obtained. RT‐qPCR was run in duplicate with biological replicates (independent experiments) to allow for the statistical confidence in differential gene expression.
Data analysis
The relative gene expression was calculated for each sample; biological replicates (independent experiments) were conducted for all studies. As amplification efficiencies of the target and reference genes were not the same, Pfaffl method (Pfaffl, 2005) was chosen for the relative quantification of RT‐qPCR results.
Conflict of Interest
The authors declare that they have no conflict of interest.
Supporting information
Fig. S1. (A‐1) Correlation between mRNA level of RABV‐G gene, gene copy number and RABV‐G protein level (α‐RABV‐G and p‐RABV‐G correspond to the expression level of RABV‐G in P. pastoris clones expressing RABV‐G protein with α‐factor or PHO1 as signal sequence; α‐RNA, p‐RNA correspond to mRNA levels of RABV‐G gene in P. pastoris clones, where RABV‐G secretion was directed by α‐factor or PHO1 signal sequence).
Table S1. Codon preference in P. pastoris and comparison of codon usage frequency (%) in the wild type and the optimized synthetic RABV‐G genes.
Table S2. Copy number of helper factor genes (PDI1, ERO1) of P. pastoris clones coexpressed with RABV‐G.
Table S3. Copy number of helper factor genes (GPX1, GLR1) of P. pastoris clones coexpressing RABV‐G gene and containing seven copies of the expression cassette.
Table S4. Copy number of the helper factor YAP1gene of P. pastoris clones coexpressing RABV‐G sequence.
Table S5. List of oligonucleotides used in this study. Underlined sequences indicate restriction sites used for gene construction.
Acknowledgements
This work is supported by the Tunisian Ministry of Scientific Research and Higher Education (grants LR11IPT01 and “Valorisation des Résultats de la Recherche”).
Microbial Biotechnology (2016) 9(3), 355–368
References
- Agaphonov, M.O. , Romanova, N.V. , Trushkina, P.M. , Smirnov, V.N. , and Ter‐Avanesyan, M.D. (2002) Aggregation and retention of human urokinase type plasminogen activator in the yeast endoplasmic reticulum. BMC Mol Biol, 3: 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad, M. , Hirz, M. , Pichler, H. , and Schhwab, H. (2014) Protein expression in Pichia pastoris: recent achievements and perspectives for heterologous protein production. Appl Microbiol Biotechnol, 12: 5301–5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anilionis, A. , Wunner, W.H. , and Curtis, P.J. (1981) Structure of the glycoprotein gene in rabies virus. Nature, 294: 275–278. [DOI] [PubMed] [Google Scholar]
- Ashe, M.P. , and Bill, R.M. (2011) Mapping the yeast host cell response to recombinant membrane protein production relieving the biological bottlenecks. Biotechnol J, 6: 707–714. [DOI] [PubMed] [Google Scholar]
- Bai, J. , Swartz, D.J. , Protasevich, I. , Brouillette, C. , Harrell, P. , Gasser, B. , et al (2011) A gene optimization strategy that enhances production of fully functional P‐glycoprotein in Pichia pastoris . PlosOne, 6: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann, K. , Adelantado, N. , Lang, C. , Mattanovich, D. , and Ferrer, P. (2011) Protein trafficking, ergosterol biosynthesis and membrane physics impact recombinant protein secretion in Pichia pastoris . Microb Cell Fact, 10: 93–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Azoun, S. , Ben Zakour, M. , Sghaeir, S. and Kallel, H. (2016) Expression of rabies virus glycoprotein in the methylotrophic yeast Pichia pastoris . Biotechnol Appl Biochem. DOI: 10.1002/bab.1471 (in press) [DOI] [PubMed] [Google Scholar]
- Cereghino, J.L. , and Cregg, J.M. (2000) Heterologous protein expression in the methylotrophic yeast Pichia pastoris . FEMS Microbiol Rev, 24: 45–66. [DOI] [PubMed] [Google Scholar]
- Chang, C.N. , Matteucci, M. , Perry, J. , Wulf, J.J. , Chen, C.Y. , and Hitzeman, R.A. (1986) Saccharomyces cerevisiae secretes and correctly processes human interferon hybrid proteins containing yeast invertase signal peptides. Mol Cell Biol, 6: 1812–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, J.H. , Dietzschold, B. , and Schneider, L.G. (1977) Rabies virus glycoprotein. II. Biological and serological characterization. Infect Immun, 16: 754–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damasceno, L.M. , Anderson, K.A. , Ritter, G. , Cregg, J.M. , Old, L.J. , and Batt, C.A. (2007) Cooverexpression of chaperones for enhanced secretion of a single‐chain antibody fragment in Pichia pastoris . Appl Microbiol Biotechnol, 74: 381–389. [DOI] [PubMed] [Google Scholar]
- Damasceno, L.M. , Huang, C.J. , and Batt, C.A. (2012) Protein secretion in Pichia pastoris and advances in protein production. Appl Microbiol Biotechnol, 93: 31–39. [DOI] [PubMed] [Google Scholar]
- Delic, M. , Rebnegger, C. , Wanka, F. , Puxbaum, V. , Troyer, C. , Hann, S. , et al (2012) Oxidative protein folding and unfolded protein response elicit differing redox regulation in endoplasmic reticulum and cytosol of yeast. Free Radic Biol Med, 52: 2000–2012. [DOI] [PubMed] [Google Scholar]
- Delic, M. , Graf, A.B. , Koellensperger, G. , Haberhauer‐Troyer, C. , Hann, S. , Mattanovich, D. , and Gasser, B. (2014) Overexpression of the transcription factor Yap1 modifies intracellular redox conditions and enhances recombinant protein secretion. Microb Cell., 1: 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser, B. , Prielhofer, R. , Marx, H. , Maurer, M. , Nocon, J. , Steiger, M. , et al (2013) Pichia pastoris ‐ protein production host and model organism for biomedical research. Future Microbiol, 8: 191–208. [DOI] [PubMed] [Google Scholar]
- Gaudin, Y. , Ruigrok, R. , Tuffereau, C. , Knossow, M. , and Flamand, A. (1992) Rabies glycoprotein is a trimer. Virology, 187: 627–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosalkar, A. , Sahai, V. , and Srivastava, A. (2008) Secretory expression of interferon‐alpha 2b in recombinant Pichia pastoris using three different secretion signals. Protein Exp Purif, 60: 103–109. [DOI] [PubMed] [Google Scholar]
- Hashimoto, Y. , Koyabu, N. , and Imoto, T. (1998) Effects of signal sequences on the secretion of hen lysozyme by yeast: construction of four secretion cassette vectors. Protein Eng, 11: 75–77. [DOI] [PubMed] [Google Scholar]
- Heimo, H. , Palmu, K. , and Suominen, I. (1997) Expression in Pichia pastoris and purification of Aspergillus awamori glucoamylasecatalytic domain. Protein Exp Purif, 10: 70–79. [DOI] [PubMed] [Google Scholar]
- Helenius, A. , Marquardt, T. , and Braakman, I. (1992) The endoplasmic reticulum as a protein‐folding compartment. Trends Cell Biol, 2: 227–231. [DOI] [PubMed] [Google Scholar]
- Hesketh, A.R. , Castrillo, J.I. , Sawyer, T. , Archer, D.B. , and Oliver, S.G. (2013) Investigating the physiological response of Pichia (Komagataella) pastoris GS115 to the heterologous expression of misfolded proteins using chemostat cultures. Appl Microbiol Biotechnol, 97: 9747–9762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenblum, H. , Gasser, B. , Maurer, M. , Borth, N. , and Mattanovich, D. (2004) Effects of gene dosage, promoters, and substrates on unfolded proteinstress of recombinant Pichia pastoris . Biotechnol Bioeng, 85: 367–375. [DOI] [PubMed] [Google Scholar]
- Hoseki, J. , Ushioda, R. , and Nagata, K. (2010) Mechanism and components of endoplasmic reticulum‐associated degradation. J Biochem, 147: 19–25. [DOI] [PubMed] [Google Scholar]
- Ide, N. , Masuda, T. , and Kitabatake, N. (2007) Effects of pre‐ and pro‐sequence of thaumatin on the secretion by Pichia pastoris . Biochem Biophys Res Commun, 363: 708–714. [DOI] [PubMed] [Google Scholar]
- Inan, M. , Aryasomayajula, M. , Sinha, J.D. , and Meagher, M.M. (2006) Enhancement of protein secretion in Pichia pastoris by overexpression of protein disulfide isomerase. Biotechnol Bioeng, 93: 771–778. [DOI] [PubMed] [Google Scholar]
- Kauffman, K.J. , Pridgen, E.M. , Doyle, F.J. , Dhurjati, P.S. , and Robinson, A.S. (2002) Decreased protein expression and intermittent recoveries in BiP levels result from cellular stress during heterologous protein expression in Saccharomyces cerevisiae . Biotechnol Prog, 18: 942–950. [DOI] [PubMed] [Google Scholar]
- Killian, J.A. , de Jong, A.M. , Bijvelt, J. , Verklei, A.J. , and de Kruijfff, B. (1990) Induction of non‐bilayer lipid structures by functional signal peptides. EMBO J, 9: 815–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koganesawa, N. , Aizawa, T. , Masaki, K. , Matsuura, A. , Nimori, T. , Bando, H. , et al (2001) Construction of an expression system of insect lysozyme lacking thermal stability: the effect of selection of signal sequence on level of expression in the Pichia pastoris expression system. Prot Eng, 14: 705–710. [DOI] [PubMed] [Google Scholar]
- Kohno, K. (2010) Stress‐sensing mechanisms in the unfolded protein response: similarities and differences between yeast and mammals. J Biochem, 147: 27–33. [DOI] [PubMed] [Google Scholar]
- Kurtzman, C.P. (2009) Biotechnological strains of Komagataella (Pichia) pastoris are Komagataella phaffii as determined from multigene sequence analysis. J Ind Microbio lBiotechnol, 36: 1435–1438. [DOI] [PubMed] [Google Scholar]
- Leonardo, M. , Damasceno, C. , and Batt, C. (2012) Protein secretion in Pichia pastoris and advances in protein production. Appl Microbiol Biotechnol, 93: 31–39. [DOI] [PubMed] [Google Scholar]
- Li, P. , Anumanthan, A. , Gao, X. , Ilangovan, K. , Suzara, V. , Duzgunes, N. , and Renugopalakrishnan, V. (2007) Expression of recombinant proteins in Pichia pastoris . Appl Biochem Biotechnol, 142: 105–124. [DOI] [PubMed] [Google Scholar]
- Li, X. , Luo, J. , Wang, S. , Shen, Y. , Qiu, Y. , Wang, X. , et al (2010) Engineering, expression, and immune‐characterization of recombinant protein comprising multi‐neutralization sites of rabies virus glycoprotein. Prot Exp Purif, 70: 183. [DOI] [PubMed] [Google Scholar]
- Lin, X.Q. , Liang, S.L. , Han, S.Y. , Zheng, S.P. , Ye, Y.R. , and Lin, Y. (2013) Quantitative iTRAQ LC‐MS/MS proteomic reveals the cellular response to heterologous protein overexpression and the regulation of HAC1 in Pichia pastoris . J Proteomics, 91: 58–72. [DOI] [PubMed] [Google Scholar]
- Lodi, T. , Neglia, B. , and Donnini, C. (2005) Secretion of human serumalbumin by Kluyveromyces lactis overexpressing KlPDI1 and KlERO1. Appl Environ Microbiol, 71: 4359–4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lünsdorf, H. , Gurramkonda, C. , Adnan, A. , Khanna, N. , and Rinas, U. (2011) Virus‐like particle production with yeast: ultrastructural and immunocytochemical insights into Pichia pastoris producing high levels of the Hepatitis B surfaceantigen. Microb Cell Fact, 10: 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngiam, C. , Jeenes, D.J. , Punt, P.J. , Hondel, C.A. , and Archer, D.B. (2000) Characterisation of a foldase, protein disulphide isomerase A, in the proteinsecretory pathway of Aspergillus Niger . Appl Environ Microbiol, 66: 775–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norden, K. , Agemark, M. , Danielson, J. , Alexandresson, E. , Kjelbom, P. , and Johanson, U. (2011) Increasing gene dosage greatly enhances recombinant expression of aquaporins in Pichia pastoris . BMC Biotechnol, 11: 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paifer, E. , Margolles, E. , Cremata, J. , Montesino, R. , Herrera, L. , and Delgado, J.M. (1994) Efficient expression and secretion of recombinant alpha amylase in Pichia pastoris using two different signal sequences. Yeast, 10: 1415–1419. [DOI] [PubMed] [Google Scholar]
- Payne, W.E. , Gannon, P.M. , and Kaiser, C.A. (1995) An inducible acidphosphatase from the yeast Pichia pastoris: characterization of the gene and its product. Gene, 163: 19–26. [DOI] [PubMed] [Google Scholar]
- Pfaffl, M. (2005) A new mathematical model for relative quantification in real‐time RT‐PCR. Nucleic Acids Res, 29: 2003–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport, T.A. (1992) Transport of proteins across the endoplasmic reticulum membrane. Science, 258: 931–936. [DOI] [PubMed] [Google Scholar]
- Romero, P.A. , Lussier, M. , Sdicu, A.M. , Bussey, H. , and Herscovics, A. (1997) Ktr1p is an a‐1, 2‐mannosyltransferase of Saccharomyces cerevisiae: comparison of the enzymatic properties of soluble recombinant Ktr1p and Kre2p/Mnt1p produced in Pichia pastoris . Biochem J, 321: 289–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, S. , Sulter, G. , and Cregg, J. (1998) A strong nitrogen source‐regulated promoter for controlled expression of foreign genes in the yeast Pichia pastoris . Gene, 216: 93–102. [DOI] [PubMed] [Google Scholar]
- Shen, Q. , Ming, W. , Hai‐Bin, W. , Hua, N. , and Shu‐Qing, C. (2012) The effect of gene copy number and co‐expression of chaperone on production of albumin fusion proteins in Pichia pastoris . Appl Gen Mol Biotechnol, 96: 763–772. [DOI] [PubMed] [Google Scholar]
- Smith, J.S. , Yager, P.A. and Baer, G.M. (1973) A rapid tissue culture test for determining rabies neutralizing antibody In: Laboratory Techniques in Rabies. Kaplan M.M. and Koprowski H. (eds). Geneva: WHO, pp. 354–357. [PubMed] [Google Scholar]
- Sreekrishna, K. , Brankamp, R.G. , Kropp, K.E. , Blankenship, D.T. , Tsay, J.T. , Smith, P.L. , et al (1997) Strategies for optimal synthesis and secretion of heterologous proteins in the methylotrophic yeast Pichia pastoris . Gene, 190: 55–62. [DOI] [PubMed] [Google Scholar]
- Toledano, M.B. , Delaunay‐Moisan, A. , Outten, C.E. , and Igbaria, A. (2013) Functions and cellular compartmentation of the thioredoxin and glutathione pathways in yeast. Antioxid Redox Signal, 18: 1699–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu, B.P. , Ho‐Schleyer, S.C. , Traves, K.J. and Weissman, J.S. (2000) Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science, 290: 1571–1574. [DOI] [PubMed] [Google Scholar]
- Vad, R. , Nafstad, E. , Dahl, L. , and Gabrielsen, O. (2005) Engineering of a Pichia pastoris expression system for secretion of high amounts of intact human parathyroid hormone. J Biotechnol, 116: 251–260. [DOI] [PubMed] [Google Scholar]
- Vanz, A.L. , Lunsdorf, H. , Adnan, A. , Nimtz, M. , Gurramkonda, C. , Khanna, N. , and Rinas, U. (2012) Physiological response of Pichia pastoris GS115 to methanol‐induced high level production of the Hepatitis B surface antigen: catabolic adaptation, stress responses, and autophagic processes. Microb Cell Factories, 11: 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanz, A.L. , Nimtz, M. , and Rinas, U. (2014) Decrease of UPR‐ and ERAD‐related proteins in Pichia pastoris during methanol‐induced secretory insulin precursor production in controlled fed‐batch cultures. Microb Cell Factories, 13: 23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Heijne, G. (1990) The signal peptide. J Memb Biol, 115: 195–201. [DOI] [PubMed] [Google Scholar]
- Vujaklija, D. , Schro, W. , and Abramic, M. (2002) A novel streptomycete lipase: cloning, sequencing and high‐level expression of the Streptomyces rimosus GDS(L)‐lipase gene. Arch Microbiol, 178: 124–130. [DOI] [PubMed] [Google Scholar]
- Walker, P.J. , and Kongsuwan, K. (1999) Deduced structural model for animal rhabdovirus glycoproteins. J Gen Virol, 80: 1211–1220. [DOI] [PubMed] [Google Scholar]
- Whyteside, G. , Nor, R.M. , Alcocer, M.J. , and Archer, D.B. (2011) Activation of the unfolded protein response in Pichia pastoris requires splicing of a HAC1 mRNA intron and retention of the C‐terminal tail of Hac1p. FEBS Lett, 585: 1037–1041. [DOI] [PubMed] [Google Scholar]
- Yang, J.K. , and Liu, L.Y. (2010) Codon optimization through a two‐step gene synthesis leads to a high‐level expression of Aspergillus niger lip 2 gene in Pichia pastoris . J Mol Cat B Enzymatic, 63: 164–169. [Google Scholar]
- Yang, S. , Kuang, Y. , Li, H. , Liu, Y. , Hui, X. , Li, P. , et al (2013) Enhanced production of recombinant secretory proteins in Pichia pastoris by optimizing Kex2 P1site. PLoS ONE, 8: 75347–75358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano, T. , Takigami, E. , Yurimoto, H. , and Sakai, Y. (2009) Yap1‐regulated glutathione redox system curtails accumulation of formaldehyde and reactive oxygen species in methanol metabolism of Pichia pastoris . Eukaryot Cell, 8: 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, X. , Wang, L. , and Xu, Y. (2009) Rhizopuschinensis lipase: gene cloning, expression in Pichia pastoris andproperties. J Mol Cat B Enzymatic, 57: 304–311. [Google Scholar]
- Yu, M. , Wen, S. , and Tan, T. (2010) Enhancing production of Yarrowia lipolytica lipase Lip2 in Pichia pastoris . Eng Life Sci, 10: 458–464. [Google Scholar]
- Zhao, H. , He, Q. , Xue, C. , Sun, B. , Yao, X. , and Liu, Z. (2009) Secretory expression of glycosylated and aglycosylated mutein of onconase from Pichia pastoris using different secretion signals and their purification and characterization. FEMS Yeast Res, 9: 591–599. [DOI] [PubMed] [Google Scholar]
- Zhu, T. , Guo, M. , Tang, Z. , Zhang, M. , Zhuang, Y. , Chu, J. , and Zhang, S. (2009) Efficient generation of multi‐copy strains for optimizing secretory expression of porcine insulin precursor in yeast Pichia pastoris . J Appl Microbiol, 107: 954–963. [DOI] [PubMed] [Google Scholar]
- Zhu, T. , Guo, M. , Zhuang, Y. , Chu, J. , and Zhang, S. (2011) Understanding the effect of foreign gene dosage on the physiology of Pichia pastoris by transcriptional analysis of key genes. Appl Microbiol Biotechnol, 89: 1127–1135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. (A‐1) Correlation between mRNA level of RABV‐G gene, gene copy number and RABV‐G protein level (α‐RABV‐G and p‐RABV‐G correspond to the expression level of RABV‐G in P. pastoris clones expressing RABV‐G protein with α‐factor or PHO1 as signal sequence; α‐RNA, p‐RNA correspond to mRNA levels of RABV‐G gene in P. pastoris clones, where RABV‐G secretion was directed by α‐factor or PHO1 signal sequence).
Table S1. Codon preference in P. pastoris and comparison of codon usage frequency (%) in the wild type and the optimized synthetic RABV‐G genes.
Table S2. Copy number of helper factor genes (PDI1, ERO1) of P. pastoris clones coexpressed with RABV‐G.
Table S3. Copy number of helper factor genes (GPX1, GLR1) of P. pastoris clones coexpressing RABV‐G gene and containing seven copies of the expression cassette.
Table S4. Copy number of the helper factor YAP1gene of P. pastoris clones coexpressing RABV‐G sequence.
Table S5. List of oligonucleotides used in this study. Underlined sequences indicate restriction sites used for gene construction.