

Summary
Increased production of reactive oxygen species (ROS) has long been considered a cause of aging. However, recent studies have implicated ROS as essential secondary messengers. Here we show that the site of ROS production significantly contributes to their apparent dual nature. We report that ROS increase with age as mitochondrial function deteriorates. However, we also demonstrate that increasing ROS production specifically through respiratory complex I reverse electron transport extends Drosophila lifespan. Reverse electron transport rescued pathogenesis induced by severe oxidative stress, highlighting the importance of the site of ROS production in signaling. Furthermore, preventing ubiquinone reduction, through knockdown of PINK1, shortens lifespan and accelerates aging; phenotypes that are rescued by increasing reverse electron transport. These results illustrate that the source of a ROS signal is vital in determining its effects on cellular physiology and establish that manipulation of ubiquinone redox state is a valid strategy to delay aging.
Keywords: aging, coenzyme Q, electron transport chain, mitochondria, reactive oxygen species
Graphical Abstract
Highlights
-
•
Mitochondrial ROS accumulate in the fly brain during aging
-
•
Inducing reverse electron transport (RET) in vivo increases mitochondrial ROS
-
•
RET extends fly lifespan through a ROS-mediated mechanism
-
•
RET improves mitochondrial function in a model of Parkinson’s disease
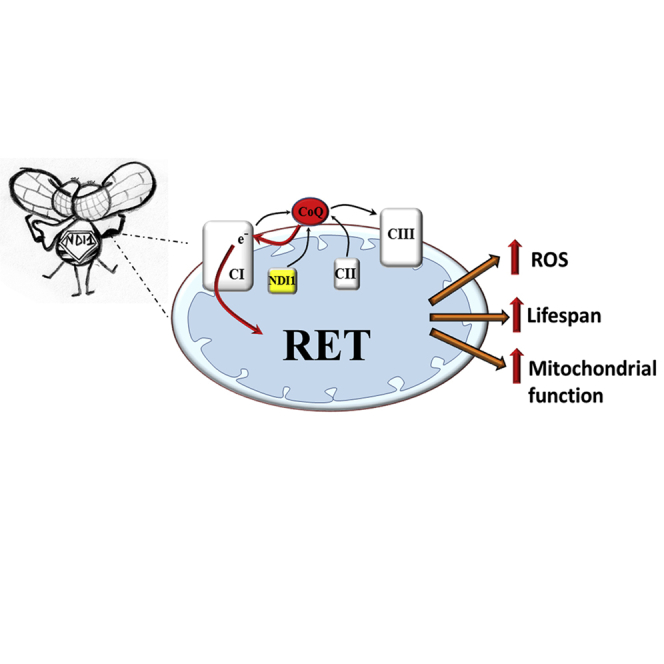
Increasing mitochondrial ROS (mtROS) specifically through complex I improves health and extends lifespan. Scialò et al. show that the site at which mtROS are produced determines their effects and increasing mtROS specifically via reverse electron transport through mitochondrial complex I delays aging and the onset of age-related diseases in flies.
Introduction
Historically, mitochondrial ROS (mtROS) production and oxidative damage have been associated with aging and age-related diseases such as Parkinson’s disease (Morais et al., 2014). In fact, the age-related increase in ROS has been viewed as a cause of the aging process (Forster et al., 1996) while mitochondrial dysfunction is considered a hallmark of aging (López-Otín et al., 2013), as a consequence of ROS accumulation. However, pioneering work in Caenorhabidits elegans has shown that mutations in genes encoding subunits of the electron transport chain (ETC) (Dillin et al., 2002) or genes required for biosynthesis of ubiquinone (Asencio et al., 2003, Wong et al., 1995) extend lifespan despite reducing mitochondrial function. The lifespan extension conferred by many of these alterations is ROS dependent, as reduction of ROS abolishes this effect (Lee et al., 2010, Yang and Hekimi, 2010b). Moreover, chemical inhibition of glycolysis or exposure to metabolic poisons that block respiratory complex I (CI) (rotenone, paraquat, or piericidin A) or complex III (CIII) (e.g., antimycin A) also prolong lifespan in C. elegans in a ROS-dependent manner (Dillin et al., 2002, Schmeisser et al., 2013, Schulz et al., 2007, Yang and Hekimi, 2010a). Various studies have shown that ROS act as secondary messengers in many cellular pathways, including those which protect against or repair damage (Ristow and Schmeisser, 2011, Yee et al., 2014). ROS-dependent activation of these protective pathways may explain their positive effect on lifespan. The confusion over the apparent dual nature of ROS may, in part, be due to a lack of resolution as without focused genetic or biochemical models it is impossible to determine the site from which ROS originate.
A promising path to resolving ROS production in vivo is the use of alternative respiratory enzymes, absent from mammals and flies, to modulate ROS generation at specific sites of the ETC (Rustin and Jacobs, 2009). The alternative oxidase (AOX) of Ciona intestinalis is a cyanide-resistant terminal oxidase able to reduce oxygen to water with electrons from reduced ubiquinone (CoQ), thus bypassing CIII and complex IV (CIV) (Fernandez-Ayala et al., 2009). NDI1 is a rotenone-insensitive alternative NADH dehydrogenase found in plants and fungi, which is present on the matrix-face of the mitochondrial inner membrane where it is able to oxidize NADH and reduce ubiquinone, effectively bypassing CI. Our group and others (Bahadorani et al., 2010, Sanz et al., 2010) have demonstrated that allotopic expression of NDI1 in Drosophila melanogaster can extend lifespan under a variety of conditions and rescue developmental lethality in flies with an RNAi-mediated decrease in CI levels.
To determine the role of increased ROS production in regulating longevity, we utilized allotopic expression of NDI1 and AOX, along with Drosophila genetic tools to regulate ROS production from specific sites in the ETC. We show that NDI1 over-reduces the CoQ pool and increases ROS via reverse electron transport (RET) through CI. Importantly, restoration of CoQ redox state via NDI1 expression rescued mitochondrial function and longevity in two distinct models of mitochondrial dysfunction.
Results and Discussion
ROS Production Increases with Age and Correlates with a Decrease in CI-Linked Respiration
Initially, we asked whether increased mtROS production is a general feature of aging in flies by measuring ROS production in fly brains using two fluorescent probes, MitoSOX (for mitochondrial matrix ROS) and H2DCF (for total cellular ROS levels), and a redox-sensitive GFP based reporter for in vivo mitochondrial H2O2 (mtH2O2) (mtORP1-roGFP) (Albrecht et al., 2011). We observed a consistent increase in ROS in old flies in two wild-type strains (Dahomey and Oregon R) (Figures 1A, 1B, S1A, and S1B). In Dahomey flies, we observed that with age, dorsal flight muscle mitochondrial ultrastructure became increasingly rounded and swollen with the appearance of perturbed cristae structure at 75 days (d) (Figures 1C, S1C, and S1D). Further, in both strains, high-resolution respirometry and enzymatic assays showed a decrease in CI-linked respiration (CI-respiration from here on) and in the enzymatic activity of CI and CIII (Figures 1D and 1E). Aconitase activity initially decreased from 5 to 25 d but remained constant as the flies continued to age (Figure S1E). At this age (25 d), no decrease in locomotive activity (Figure S1F) or increase in ROS (Figures 1A, 1B, S1A, and S1B) was observed. Western blot analysis showed that only the levels of CI and aconitase were significantly affected with age (Figures 1F, 1G, and S1G–S1J). However, CI concentration was decreased at very late (75 and 85 d) ages, suggesting a shift in mitochondrial metabolism supported by an increase in ImpL3 (lactate dehydrogenase A homologue) expression (Figure 1H).
Figure 1.

Increased ROS Production in Aging Flies Correlates with Mitochondrial Dysfunction
(A) Representative images of dissected fly brains stained with MitoSOX from wild-type flies of the indicated ages.
(B) Quantification of (A) (n = 5).
(C) Representative EM images of Dahomey flight muscle sections at 1,000x magnification dissected at the indicated ages (n = 10, 1 muscle per fly; red arrows indicated exemplar swollen, rounded mitochondria, see Figure S1C for quantification).
(D) Mitochondrial respiration in Dahomey and Oregon R flies at the indicated ages (n = 6).
(E) CI and CIII enzymatic activity in wild-type flies of the indicated ages (n = 5).
(F) CI (NDUFS3), CII (SDHB), CIII (CYTOB), CIV (COX4), and CV (ATP5A) levels in wild-type flies. GAPDH is used as a loading control.
(G) Quantification of (F).
(H) ImpL3 expression in wild-type flies of the indicated ages.
Values shown represent means ± SEM of at least three biological replicates, unless otherwise stated. See also Figure S1.
Over-Reduction of the CoQ Pool Increases ROS Production and Extends Lifespan
Based on our previous results, we hypothesized that decreasing ROS and compensating for a loss in CI respiration would extend lifespan. We and others have previously reported that allotopic expression of NDI1 decreases ROS in mitochondria isolated from old individuals (Sanz et al., 2010). Surprisingly, measurement of ROS levels using MitoSOX, H2DCF, and mtORP1-roGPF revealed in fly brains a NDI1-mediated increase in ROS that was partially abolished by AOX expression (Figures 2A, 2B, and S2B). Neither NDI1 nor AOX had any detectable effect on respiration alone (Figure S2C). Strikingly, the increase in ROS elicited by NDI1 was similar to that caused by feeding flies with a dose of rotenone able to significantly inhibit CI (Figures S2D–S2F). AOX expression completely abolished lifespan extension conferred by NDI1 (Figure 2C), suggesting that NDI1 lifespan extension is dependent on over-reduction of the ETC and consequently increased ROS levels. Expression of AOX alone had a mild effect on wild-type ROS production (Figures 2A, 2B, and S2B) and did not shorten lifespan (Figure 2C).
Figure 2.

NDI1 Increases ROS Production via Over-Reduction of CoQ
(A) Representative images of dissected brains from indicated genotypes stained with MitoSOX.
(B) Quantification of (A) (n = 5).
(C) Survival curves for the indicated genotypes (n = 200).
(D) Schematic diagram illustrating effects of expressing two different alternative respiratory enzymes on electron transport: (i) NDI1 generates ROS by over-reducing the CoQ pool; (ii) AOX reverts the effects of NDI1 by re-oxidizing the CoQ pool; (iii) decrease in the levels of CI can prevent reduction of CoQ and subsequent ROS production; (iv) ectopic expression of mtCAT reduces ROS levels without altering mitochondrial respiration or the redox state of CoQ.
(E) Representative images of brains from the indicated genotypes stained with MitoSOX.
(F) Quantification of (E) (n = 5).
(G) Survival curves for the indicated genotypes (n = 160).
(H) In vivo ROS measurements from indicated genotypes in brains dissected from flies expressing a mitochondrially localized redox-sensitive GFP-based reporter.
(I) Quantification of (H) (n = 5–7).
(J) Quantification of brains dissected from flies of the indicated genotypes stained with MitoSOX (n = 5).
(K) Survival curves for the indicated genotypes (n = 200).
(L) Diagram illustrating using metabolic poisons to dissect ROS production: rotenone (ROT), carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), or myxothiazol (MYX). Green dashed arrows indicate the possible flow of electrons following CoQ reduction.
(M) Quantification of brains dissected from NDI1 flies fed with metabolic poisons, stained with MitoSOX (n = 4).
Values shown represent means ± SEM of at least 3 biological replicates, unless otherwise stated.
See also Figure S2 and Table S1 for statistical analysis of survival curves.
Based on previous studies (Owusu-Ansah et al., 2013, Yang and Hekimi, 2010a), we hypothesized that the observed NDI1-mediated increase in ROS production was perhaps a signal that led to an extension in lifespan. To further test this possibility, we generated another model in which we expressed NDI1 but alleviated the over-reduction of the ETC and prevented additional ROS formation by decreasing CI levels (via knockdown of ND-39 (CG6020), the fly homolog of NDUFA9, Figures 2D and S2G). These flies, hereafter referred to as CI > 2;NDI1 > daGAL4 flies, had reduced CI activity (Figure S2H) and, like AOX > 2;NDI1 > daGAL4 flies, had decreased ROS levels compared to NDI1 expressing flies (Figures 2E, 2F, and S2I). As with AOX expression, CI knockdown suppressed lifespan extension conferred by NDI1 (Figure 2G), further supporting the hypothesis that over-reduction of the ETC downstream of CI can increase ROS production and extend lifespan. To verify a change in the redox state of CoQ, we measured the percentage of reduced and oxidized CoQ in NDI1-expressing and controls flies, observing a remarkable increase in the proportion of reduced CoQ in NDI1 flies (Figure S2J). As expected, co-expression of NDI1 with AOX or decreasing CI levels restored the redox state of CoQ to normal (Figure S2J).
To exclude the possibility that the ROS signal originated downstream of CoQ, we knocked down the CIV subunit, levy (CG17280, fly homolog for human COX6A), to over-reduce cytochrome c and CIII in AOX-expressing flies (hereafter referred to as CIV > 2;AOX > daGAL4 flies). AOX was required in this model to maintain the CoQ pool oxidized (Figure S2K) and prevent RET through CI. CIV > 2;AOX > daGAL4 flies displayed significantly decreased CIV-linked respiration (Figure S2L) but normal food intake and body weight (Figures S2M and S2N). However, stabilization of reduced cytochrome c did not produce a ROS signal (Figure S2O) or extend lifespan (Figure S2P), supporting a site specificity for this longevity-extending ROS signal. Finally, to confirm that ROS and not changes in the redox state of CoQ were responsible for NDI1 lifespan extension, we co-expressed NDI1 with a mitochondrially targeted catalase (mtCAT) (Mockett et al., 2003) (Figure 2D; hereafter mtCAT > 2;NDI1 > daGAL4 flies). Expression of mtCAT restored H2O2 levels to normal (Figures 2H and 2I) without, as expected, affecting superoxide (Figure 2J) or the redox state of CoQ (Figure S2R). Since lifespan of mtCAT > 2;NDI1 > daGAL4 flies was similar to control flies, we can conclude that mtROS are responsible for NDI1 lifespan extension (Figure 2K). Significantly, expression of mtCAT alone reduced both mtH2O2 levels (e.g., more than AOX) and lifespan (Figure 2K), indicating that excessive reduction of ROS can be detrimental for lifespan.
To investigate how NDI1 increases ROS, we dissected mtROS production in vivo by adding specific ETC inhibitors to the food of NDI1 flies (Figure 2L). Rotenone is a specific CI Q-site inhibitor that prevents CI binding to CoQ. Rotenone feeding in wild-type flies caused a significant decrease in CI NADH:decylubiquinone (NADH:DQ) activity, while non-physiological NADH:hexaammineruthenium (NADH:HAR) activity, which is not dependent on CoQ binding, was unaffected (Figure S2F), indicating that levels of CI were not reduced. Rotenone feeding significantly decreased ROS production in NDI1 expressing flies (Figures 2M and S2T), while increasing it in wild-type flies (Figures S2D and S2E), suggesting that RET is the source of NDI1-mediated ROS production. RET in vitro is strongly membrane potential-dependent requiring a high enough membrane potential so as to make forward electron transport through CI unfavorable. Collapsing membrane potential by feeding FCCP (a H+ ionophore) like rotenone depleted NDI1-mediated ROS production, whereas myxothiazol (a CIII Qo site inhibitor that blocks entry of electrons into CIII) did not (Figure 2M), strongly supporting the existence of RET in vivo in NDI1-expressing flies. Interestingly, we have recently shown that during ischemia reperfusion RET is the main source of superoxide in vivo (Chouchani et al., 2014).
CoQ-Mediated ROS Signaling Can Rescue Pathology Induced by Oxidative Stress
If ROS signaling is site specific and works to maintain mitochondrial function, we hypothesized that it should be possible for beneficial ROS signaling to rescue deleterious mitochondrial phenotypes induced by oxidative stress. To test this, we knocked down SOD2 (the only mitochondrial matrix superoxide dismutase in Drosophila; Figure S3A) and observed a significant increase in levels of superoxide in fly brains (Figures 3A and 3B) while levels of mtH2O2 were significantly decreased (Figure S3B, right), which is in agreement with the reported positive correlation between SOD2 activity and mtH2O2 levels (Rodríguez et al., 2000). SOD2 knockdown dramatically shortened longevity (Figure 3C) and, in addition, severely affected CI-respiration (Figure 3D), with a significant decrease in the enzymatic activity of CI, CII, and aconitase (Figure 3E). Western blot analysis showed that levels of CI and aconitase were unchanged (Figure S3D), suggesting that, as has been shown for aconitase, high levels of superoxide inhibit the activity of these enzymes (Gardner and Fridovich, 1992). Mitochondrial ultrastructure from dissected dorsal flight muscle was unaffected by SOD2 knockdown (Figures S3E and S3F), showing that high levels of superoxide recapitulate some (e.g., increased mortality and, decreased CI activity), but not all features of aging (e.g., alteration in mitochondrial morphology). Although high levels of superoxide in the absence of SOD2 are detrimental, this situation is unlikely to occur in vivo in aged flies since SOD2 levels (Figures S1G and S1H) and superoxide dismutase activity (Sohal et al., 1990) are reported to increase with age.
Figure 3.

NDI1-Mediated ROS Production Rescues Superoxide-Mediated Mitochondrial Dysfunction
(A) Representative images of fly brains from indicated genotypes stained with MitoSOX.
(B) Quantification of (A) (n = 5).
(C) Survival curves of the indicated genotypes (n = 180–380).
(D) Mitochondrial respiration in flies of the indicated genotypes (n = 6).
(E) CI, CII, and aconitase enzymatic activities in flies of the indicated genotypes (n = 7).
Values shown represent means ± SEM of at least three biological replicates, unless otherwise stated. See also Figure S3 and Table S1 for statistical analysis of survival curves.
To assess the role of CoQ-mediated ROS production, we expressed either AOX or NDI1, which have opposite effects on mtROS and the redox state of the CoQ pool, in SOD2 knockdown flies. AOX expression rescued the increase in superoxide elicited by SOD2 knockdown, but further decreased the concentration of mtH2O2 (Figures 3A, 3B, and S3B), and did not extend lifespan or rescue decreased ETC function (Figures 3C–3E). NDI1 expression partially rescued lifespan (Figure 3C), along with CI respiration and activity (Figures 3D and 3E), despite further increasing ROS levels (Figures 3A, 3B, and S3B). To confirm that NDI1 rescue of lifespan was mediated via RET, we reduced CI levels in SOD2 knockdown flies expressing NDI1 (hereafter SOD2 > CI; NDI1 > ActGS) using the inducible GeneSwitch system (Nicholson et al., 2008). Strikingly, NDI1 rescue of lifespan disappeared when RET was prevented by depletion of CI levels (Figure S3G). These results indicate that SOD2 knockdown perturbs CoQ reduction due to decreased activity of CI and CII, which results in increased superoxide production. In this model, re-establishing RET-derived ROS with NDI1 rescued CI activity, suggesting that RET may be necessary for the maintenance of mitochondrial function and lifespan extension.
Loss of CoQ-Mediated ROS Signaling Accelerates Aging
In Figure 1, we demonstrated that mitochondrial quality was decreased in aged flies. Interestingly, we also found that levels of PINK1 and Parkin decreased in parallel over time (Figures 4A and 4B). Experimental manipulation of PINK1 and Parkin levels has previously been shown to alter Drosophila lifespan (Rana et al., 2013, Todd and Staveley, 2012). Decreases in PINK1 activity have been related to a loss of CI activity (Morais et al., 2014), while Parkin loss of function mutations cause the accumulation of aberrant mitochondria (Greene et al., 2003). Strikingly, decreases in PINK1 and Parkin levels (Figures 4A and 4B) preceded the decline in respiration observed in old flies (Figure 1D). We hypothesized that this may contribute to the mitochondrial dysfunction that occurs with age. It has been recently shown that PINK1 is required for reduction of CoQ by CI via phosphorylation of NDUFA10 (Morais et al., 2014), although this was not confirmed by a later report (Pogson et al., 2014). Since PINK1 protein levels decrease with age, we hypothesized that this decrease could be responsible for the decline in CI-respiration. To test the relevance of CoQ-mediated ROS signaling in a more physiological context, we used the inducible GeneSwitch system to drive expression of an RNAi construct against PINK1 specifically in adult flies (PINK1 > daGS flies, Figure S4A). PINK1 knockdown did not increase ROS levels (Figures 4C and S4B); however, we did observe a significant decrease in CI-linked respiration (Figure 4D) and NADH:DQ CI activity, but not NADH:HAR activity (Figure 4E) or CI protein levels (Figure S4C), showing that reduction of CoQ by CI was impaired, as was observed in SOD2 knockdown flies (Figures 3D and 3E) and 50-d-old wild-type flies (Figures 1D and 1E). Furthermore, PINK1 knockdown flies displayed decreased mobility, time spent flying, and lifespan compared to controls (Figures 4F and 4G).
Figure 4.

Re-Establishing the Redox State of CoQ Prevents Age-Related Pathology
(A) PINK1 and Parkin levels in wild-type flies during aging.
(B) Quantification of (A).
(C) Representative images and quantification of dissected fly brains stained with MitoSOX (quantification n = 7).
(D) Mitochondrial respiration in flies of the indicated genotypes.
(E) CI, CII, and aconitase activities in flies of the indicated genotypes (n = 4–8).
(F) Locomotive activity and flying time in flies of the indicated genotypes (n = 13).
(G) Survival curves for the indicated genotypes (n = 160).
(H) Mitochondrial respiration in flies of the indicated genotypes (n = 5).
(I) Locomotive activity and flying time in flies of the indicated genotypes (n = 6).
(J) Survival curves for the indicated genotypes (n = 160).
Values shown represent means ± SEM of at least three biological replicates, unless otherwise stated. −/+ indicates absence/presence of 500 μM RU-486 during adulthood. See also Figure S4 and Table S1 for statistical analysis of survival curves.
As in SOD2 knockdown flies, NDI1-mediated ROS production (Figure 4C) was able to rescue CI-linked respiration and CI activity in PINK1 knockdown flies (Figures 4D and 4E). However, unlike in SOD2 knockdown flies, CII and aconitase activities were also rescued (Figure 4E), suggesting that SOD2 may be necessary for NDI1-mediated rescue of CII and aconitase. Locomotive activity, time spent flying, and lifespan were also partially or totally rescued by NDI1 expression in PINK1 knockdown flies (Figures 4F and 4G). Interestingly, reduction of CI levels from adulthood decreased CI respiration to levels comparable with 50-d-old wild-type flies and PINK1 knockdown flies (Figure 4H), whereas locomotion and lifespan were similar to that observed in flies with induction of the driver alone (Compare Figures 4I and 4J with Figures S4D–S4G). Moreover, NDI1 did not restore respiration in this background (data not shown), just as it failed to when CI knockdown and NDI1 expression were driven with a stronger driver (Figure S2C). This indicates that the rescue of mitochondrial function conferred by NDI1 in SOD2 and PINK1 knockdown flies is not due to direct compensation by NDI1 of decreased CI activity, rather NDI1 rescues CI function through over-reduction of the CoQ pool. Therefore, we propose that over-reduction of CoQ pool generates, via RET, a ROS signal necessary for homeostasis. Interruption in electron flow through CI would prevent CoQ reduction and thus cause the deleterious phenotypes associated with aging and age-related pathologies.
In summary, we show that mtROS production increases with age and that un-detoxified ROS can be detrimental to Drosophila lifespan, while increasing ROS production specifically from reduced CoQ, possibly via RET, acts as a signal to maintain mitochondrial function (notably CI) and extend lifespan (Figure S4J). These results are similar to those reported in worms where small doses of rotenone (Schmeisser et al., 2013), paraquat, and mutations in CI that increase ROS extend lifespan (Yang and Hekimi, 2010a). However, we did not observe lifespan extension in flies fed with varying doses of rotenone or paraquat (Figures S4H and S4I). It is possible that an intact CI is required for lifespan extension in fruit flies, as metformin, which increases lifespan by blocking CI and increasing ROS in worms (De Haes et al., 2014), fails to do so in fruit flies (Slack et al., 2012). If the mechanism we describe here is conserved in mammals, manipulation of the redox state of CoQ may be a strategy for the extension of both mean and maximum lifespan and the road to new therapeutic interventions for aging and age-related diseases.
Experimental Procedures
Fly Husbandry
Female flies were used in all experiments. See Table S2 for details of the different fly strains used in this study. Flies were maintained at 25°C on standard media in a controlled 12:12 hr light:dark cycle at a density of 20 flies/vial and transferred to new food every 2 to 3 d. For all lifespan experiments, at least 80 flies were used and each experiment was repeated at least two to four times. Statistical analysis for all lifespan experiments can be found in Table S1.
ROS Detection
MitoSOX and dichlorofluorescein (H2DCF) were used to measure either mitochondrial matrix superoxide or total cellular ROS levels, respectively. Following dissection, fly brains were incubated in MitoSOX or H2DCF for 10 min and imaged immediately. For in vivo ROS imaging brains dissected from lines carrying the mtORP1-roGFP reporter construct were imaged under Ex. 488 (reduced) or 405 (oxidized) nm/Em. 510 nm. The total (average) fluorescence intensity of each individual brain imaged was quantified using ImageJ.
Electron Microscopy
Dorsal flight muscles were dissected and suspended in 0.16 M sym-collidine buffer pH 7.4 (Electron Microscopy Sciences) supplemented with 5% glutaraldehyde (fixing solution). Muscle sections were imaged using a JEOL JEM-1400 Plus transmission electron microscope fitted with an 11 Mpx Olympus Quemesa digital camera. The images were manually segmented using GIMP (http://www.gimp.org/), and the resulting segmented image was further analyzed using the ImageJ particle analysis tool.
High-Resolution Respirometry
Respirometry measurements of whole-fly homogenates were performed using an OROBOROS O2k oxygraph. State 4 respiration was initiated by addition of 5 mM pyruvate and 5 mM proline. State 3 was initiated with the addition of 1 mM ADP. CI-linked respiration (CI+CIII+CIV in figures) was inhibited by adding 0.5 μM rotenone, and CIII-linked respiration was stimulated by addition of 20 mM glycerol 3-phosphate (CIII+CIV in figures). CIII-linked respiration was inhibited with the addition of 2.5 μM antimycin A. CIV respiration (CIV in figures) was initiated by the addition of 4 mM ascorbate and 2 mM TMPD. CIV respiration was inhibited by adding 0.5 mM KCN. Data presented correspond to state 3 respiration (i.e., after ADP addition).
Enzymatic Assays
All assays were adapted to a 96-well plate with a final volume of 300 μl. CI (NADH dehydrogenase or NADH: hexaammineruthenium (HAR) oxidoreductase activity), CII (malonate-sensitive succinate:dichlorophenolindophenol (DCPIP) oxidoreductase activity), CIII (antimycin-sensitive decylubiquinol:cytochrome c oxidoreductase activity), CIV (potassium cyanide-sensitive cytochrome c oxidase activity), CV (oligomycin-sensitive NADH-coupled ATPase activity), and mitochondrial aconitase activities were measured according to standard procedures. All ETC activities were normalized to mitochondrial density calculated by citrate synthase (CS).
Behavioral and Locomotive Activity Assays
Live tracking of the flies was performed using a high-sensitivity The Imaging Source USB camera (equipped with a Computar 1/3” Varifocal Lens [2.8–12 mm]), for 60 min per experiment. The recorded video files were used to calculate distance moved per hour for each fly using the EthoVision XT video tracking software (Noldus Information Technology).
Statistical Analysis
Values shown represent means ± SEM. Data were analyzed with Prism 6 (GraphPad) using either one-way ANOVA with Newman-Keul’s post hoc test or using the unpaired Student’s t test. Lifespan data were analyzed using the log-rank Mantel Cox Test. p < 0.05 was taken as statistically significant. In all figures ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, and ∗∗∗∗p < 0.001 denotes significant difference from all other groups unless otherwise indicated by line art. NS, not significantly different. Individual and pooled lifespan experiments with statistical analysis are summarized in Table S1.
Author Contributions
F.S., A. Sriram, D.F.-A., N.G., M.L., G.N., A.L., H.M.C., and P.N. performed experiments and analyzed data. M.P.M. and J.A.E. designed research and co-wrote the manuscript. A. Sanz designed and performed experiments, supervised the project, and co-wrote the manuscript.
Acknowledgments
This work was supported by the European Research Council (ERC Starting Grant to A. Sanz), the Academy of Finland (Academy Research Fellowship to A. Sanz and Postdoctoral Research grant to H.M.C), the BBSRC (Responsive mode grant to A. Sanz), the Centre for International Mobility (Postdoctoral fellowship to N.G.), the Medical Research Council (M.P.M), and the Spanish Ministry of Health and the Instituto de Salud Carlos III (FIS PI14-01962 to P.N.). Electron microscopy image processing was performed at the Laboratory of Electron Microscopy, University of Turku, Finland. We thank Dr Rhoda Stefanatos for help with editing the manuscript. The authors declare no competing financial interests.
Published: April 12, 2016
Footnotes
Supplemental Information includes four figures, two tables, and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.cmet.2016.03.009.
Supplemental Information
References
- Albrecht S.C., Barata A.G., Grosshans J., Teleman A.A., Dick T.P. In vivo mapping of hydrogen peroxide and oxidized glutathione reveals chemical and regional specificity of redox homeostasis. Cell Metab. 2011;14:819–829. doi: 10.1016/j.cmet.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Asencio C., Rodríguez-Aguilera J.C., Ruiz-Ferrer M., Vela J., Navas P. Silencing of ubiquinone biosynthesis genes extends life span in Caenorhabditis elegans. FASEB J. 2003;17:1135–1137. doi: 10.1096/fj.02-1022fje. [DOI] [PubMed] [Google Scholar]
- Bahadorani S., Cho J., Lo T., Contreras H., Lawal H.O., Krantz D.E., Bradley T.J., Walker D.W. Neuronal expression of a single-subunit yeast NADH-ubiquinone oxidoreductase (Ndi1) extends Drosophila lifespan. Aging Cell. 2010;9:191–202. doi: 10.1111/j.1474-9726.2010.00546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani E.T., Pell V.R., Gaude E., Aksentijević D., Sundier S.Y., Robb E.L., Logan A., Nadtochiy S.M., Ord E.N., Smith A.C. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Haes W., Frooninckx L., Van Assche R., Smolders A., Depuydt G., Billen J., Braeckman B.P., Schoofs L., Temmerman L. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc. Natl. Acad. Sci. USA. 2014;111:E2501–E2509. doi: 10.1073/pnas.1321776111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillin A., Hsu A.L., Arantes-Oliveira N., Lehrer-Graiwer J., Hsin H., Fraser A.G., Kamath R.S., Ahringer J., Kenyon C. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- Fernandez-Ayala D.J., Sanz A., Vartiainen S., Kemppainen K.K., Babusiak M., Mustalahti E., Costa R., Tuomela T., Zeviani M., Chung J. Expression of the Ciona intestinalis alternative oxidase (AOX) in Drosophila complements defects in mitochondrial oxidative phosphorylation. Cell Metab. 2009;9:449–460. doi: 10.1016/j.cmet.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Forster M.J., Dubey A., Dawson K.M., Stutts W.A., Lal H., Sohal R.S. Age-related losses of cognitive function and motor skills in mice are associated with oxidative protein damage in the brain. Proc. Natl. Acad. Sci. USA. 1996;93:4765–4769. doi: 10.1073/pnas.93.10.4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner P.R., Fridovich I. Inactivation-reactivation of aconitase in Escherichia coli. A sensitive measure of superoxide radical. J. Biol. Chem. 1992;267:8757–8763. [PubMed] [Google Scholar]
- Greene J.C., Whitworth A.J., Kuo I., Andrews L.A., Feany M.B., Pallanck L.J. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.J., Hwang A.B., Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr. Biol. 2010;20:2131–2136. doi: 10.1016/j.cub.2010.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C., Blasco M.A., Partridge L., Serrano M., Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockett R.J., Bayne A.C., Kwong L.K., Orr W.C., Sohal R.S. Ectopic expression of catalase in Drosophila mitochondria increases stress resistance but not longevity. Free Radic. Biol. Med. 2003;34:207–217. doi: 10.1016/s0891-5849(02)01190-5. [DOI] [PubMed] [Google Scholar]
- Morais V.A., Haddad D., Craessaerts K., De Bock P.J., Swerts J., Vilain S., Aerts L., Overbergh L., Grünewald A., Seibler P. PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science. 2014;344:203–207. doi: 10.1126/science.1249161. [DOI] [PubMed] [Google Scholar]
- Nicholson L., Singh G.K., Osterwalder T., Roman G.W., Davis R.L., Keshishian H. Spatial and temporal control of gene expression in Drosophila using the inducible GeneSwitch GAL4 system. I. Screen for larval nervous system drivers. Genetics. 2008;178:215–234. doi: 10.1534/genetics.107.081968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owusu-Ansah E., Song W., Perrimon N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell. 2013;155:699–712. doi: 10.1016/j.cell.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogson J.H., Ivatt R.M., Sanchez-Martinez A., Tufi R., Wilson E., Mortiboys H., Whitworth A.J. The complex I subunit NDUFA10 selectively rescues Drosophila pink1 mutants through a mechanism independent of mitophagy. PLoS Genet. 2014;10:e1004815. doi: 10.1371/journal.pgen.1004815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana A., Rera M., Walker D.W. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. USA. 2013;110:8638–8643. doi: 10.1073/pnas.1216197110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M., Schmeisser S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 2011;51:327–336. doi: 10.1016/j.freeradbiomed.2011.05.010. [DOI] [PubMed] [Google Scholar]
- Rodríguez A.M., Carrico P.M., Mazurkiewicz J.E., Meléndez J.A. Mitochondrial or cytosolic catalase reverses the MnSOD-dependent inhibition of proliferation by enhancing respiratory chain activity, net ATP production, and decreasing the steady state levels of H(2)O(2) Free Radic. Biol. Med. 2000;29:801–813. doi: 10.1016/s0891-5849(00)00362-2. [DOI] [PubMed] [Google Scholar]
- Rustin P., Jacobs H.T. Respiratory chain alternative enzymes as tools to better understand and counteract respiratory chain deficiencies in human cells and animals. Physiol. Plant. 2009;137:362–370. doi: 10.1111/j.1399-3054.2009.01249.x. [DOI] [PubMed] [Google Scholar]
- Sanz A., Soikkeli M., Portero-Otín M., Wilson A., Kemppainen E., McIlroy G., Ellilä S., Kemppainen K.K., Tuomela T., Lakanmaa M. Expression of the yeast NADH dehydrogenase Ndi1 in Drosophila confers increased lifespan independently of dietary restriction. Proc. Natl. Acad. Sci. USA. 2010;107:9105–9110. doi: 10.1073/pnas.0911539107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser S., Priebe S., Groth M., Monajembashi S., Hemmerich P., Guthke R., Platzer M., Ristow M. Neuronal ROS signaling rather than AMPK/sirtuin-mediated energy sensing links dietary restriction to lifespan extension. Mol. Metab. 2013;2:92–102. doi: 10.1016/j.molmet.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz T.J., Zarse K., Voigt A., Urban N., Birringer M., Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Slack C., Foley A., Partridge L. Activation of AMPK by the putative dietary restriction mimetic metformin is insufficient to extend lifespan in Drosophila. PLoS ONE. 2012;7:e47699. doi: 10.1371/journal.pone.0047699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal R.S., Arnold L., Orr W.C. Effect of age on superoxide dismutase, catalase, glutathione reductase, inorganic peroxides, TBA-reactive material, GSH/GSSG, NADPH/NADP+ and NADH/NAD+ in Drosophila melanogaster. Mech. Ageing Dev. 1990;56:223–235. doi: 10.1016/0047-6374(90)90084-s. [DOI] [PubMed] [Google Scholar]
- Todd A.M., Staveley B.E. Expression of Pink1 with α-synuclein in the dopaminergic neurons of Drosophila leads to increases in both lifespan and healthspan. Genet. Mol. Res. 2012;11:1497–1502. doi: 10.4238/2012.May.21.6. [DOI] [PubMed] [Google Scholar]
- Wong A., Boutis P., Hekimi S. Mutations in the clk-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics. 1995;139:1247–1259. doi: 10.1093/genetics/139.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W., Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010;8:e1000556. doi: 10.1371/journal.pbio.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W., Hekimi S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell. 2010;9:433–447. doi: 10.1111/j.1474-9726.2010.00571.x. [DOI] [PubMed] [Google Scholar]
- Yee C., Yang W., Hekimi S. The intrinsic apoptosis pathway mediates the pro-longevity response to mitochondrial ROS in C. elegans. Cell. 2014;157:897–909. doi: 10.1016/j.cell.2014.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.